

Abstract
Sirtuin1 (Sirt1), a mammalian homolog of yeast Sir2, deacetylates the tumor suppressor protein p53 and attenuates p53-mediated cell death. Necdin, a p53-interacting protein expressed predominantly in postmitotic neurons, is a melanoma antigen family protein that promotes neuronal differentiation and survival. In mammals, the necdin gene (Ndn) is maternally imprinted, and mutant mice carrying mutated paternal Ndn show abnormalities of neuronal development. Here we report that necdin regulates the acetylation status of p53 via Sirt1 to suppress p53-dependent apoptosis in postmitotic neurons. Double-immunostaining analysis demonstrated that necdin colocalizes with Sirt1 in postmitotic neurons of mouse embryonic forebrain in vivo. Coimmunoprecipitation and in vitro binding analyses revealed that necdin interacts with both p53 and Sirt1 to potentiate Sirt1-mediated p53 deacetylation by facilitating their association. Primary cortical neurons prepared from paternal Ndn-deficient mice have high p53 acetylation levels and are sensitive to the DNA-damaging compounds camptothecin and hydrogen peroxide. Moreover, DNA transfection per se increases p53 acetylation and apoptosis in paternal Ndn-deficient neurons, whereas small interfering RNA-mediated p53 knockdown completely blocks these changes. However, Sirt1 knockdown increases both acetylated p53 level and apoptosis in wild-type neurons but fails to affect them in paternal Ndn-deficient neurons. In organotypic forebrain slice cultures treated with hydrogen peroxide, p53 is accumulated and colocalized with necdin and Sirt1 in cortical neurons. These results suggest that necdin downregulates p53 acetylation levels by forming a stable complex with p53 and Sirt1 to protect neurons from DNA damage-induced apoptosis.
Keywords: necdin, Sirt1, p53, acetylation, apoptosis, DNA damage
Introduction
Postmitotic neurons must survive against various intrinsic and extrinsic stresses throughout the lifetime of the organism. The tumor suppressor protein p53 has been implicated as a key regulator of neuronal apoptosis in response to neuropathological and neurodegenerative conditions (Culmsee and Mattson, 2005). p53 invariably undergoes acetylation in cells exposed to a variety of cellular and environmental insults (Ito et al., 2001). Acetylation of p53 activates transcription of downstream targets with proapoptotic properties (Gu and Roeder, 1997). Moreover, p53 acetylation is downregulated through nerve growth factor (NGF) signaling pathway (Vaghefi and Neet, 2004). Therefore, the acetylation status of p53 may be crucial for life and death decisions of neurons in developmental and pathological conditions.
Sirtuin1 (Sirt1) is a mammalian nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase (HDAC) that downregulates the acetylation levels of many regulatory proteins involved in energy homeostasis, DNA repair, cell survival, and lifespan extension (Haigis and Guarente, 2006). A study using Sirt1-defective mice has shown that Sirt1 is indispensable for normal development (McBurney et al., 2003). Sirt1 deacetylates p53 and represses its activity to protect various cells from DNA damage-induced apoptosis (Luo et al., 2001; Vaziri et al., 2001). Sirt1 is expressed predominantly in developing nervous systems (Sakamoto et al., 2004). Moreover, Sirt1 overexpression confers a significant protection against neurodegeneration (Kim et al., 2007a). These findings suggest that Sirt1 plays a key role in the regulation of p53 acetylation in neurons under various developmental and neuropathological conditions.
Necdin, a melanoma antigen (MAGE) family protein, interacts with p53 and represses p53-mediated apoptosis in transformed cell lines (Taniura et al., 1999, 2005). Because necdin is expressed in virtually all postmitotic neurons, but not in cell lines derived from neuroblastoma, pheochromocytoma, and glioma (Aizawa et al., 1992; Uetsuki et al., 1996; Kobayashi et al., 2002), the regulation of p53 activities by endogenous necdin in postmitotic neurons is poorly understood. In mammals, expression of the necdin gene (Ndn) is regulated by genomic imprinting (Jay et al., 1997; MacDonald and Wevrick, 1997). Because Ndn is maternally imprinted and expressed from the paternal allele, necdin is absent from neurons in mice carrying mutated paternal Ndn. These mutant mice show abnormalities and apoptosis of central and peripheral neurons (Muscatelli et al., 2000; Ren et al., 2003; Kuwako et al., 2005; Andrieu et al., 2006; Kurita et al., 2006; Kuwajima et al., 2006; Tennese et al., 2008). These findings suggest that paternally expressed necdin is required for neuronal differentiation and survival. However, it is unclear whether p53-dependent apoptosis is enhanced in necdin-defective postmitotic neurons.
We here demonstrate a close interplay between necdin and Sirt1 to regulate the acetylation status of neuronal p53. We also show that DNA damage-induced p53 acetylation and apoptosis are significantly enhanced in necdin-deficient postmitotic neurons. The present study suggests that necdin is an intrinsic regulator of the acetylation status of neuronal p53 via Sirt1 to promote neuronal survival against DNA-damaging insults.
Materials and Methods
Antibodies.
Primary antibodies used for immunohistochemistry are as follows: rabbit polyclonal antibody against Sirt1 (07-131; 1:200; Upstate Biotechnology), acetyl-p53 (Lys373) (06-916; 1:1000; Upstate Biotechnology), mouse monoclonal antibody against βIII-tubulin (1:1000; Promega), p53 (DO-1; 1:200; Santa Cruz Biotechnology), and guinea pig polyclonal antibody against necdin (GN1; 1:500) (Kuwako et al., 2005). The secondary antibodies cyanine 2-conjugated anti-rabbit IgG (1:500), cyanine 3-conjugated anti-rabbit IgG (1:500), cyanine 3-conjugated anti-mouse IgG (1:500), cyanine 2-conjugated anti-mouse IgG (1:500), and cyanine 3-conjugated anti-guinea pig IgG (1:500) are from Jackson ImmunoResearch. Antibodies used for Western blotting are Myc (9E10; 1:10), necdin (NC243; 1:3000) (Niinobe et al., 2000), FLAG (M2; 1:500; Sigma-Aldrich), Sirt1 (07-131; 1:1000; Upstate Biotechnology), class I HDAC (HDAC1; C19; 1:200; Santa Cruz Biotechnology), proliferating cell nuclear antigen (PCNA) (PC10; 1:1000; Santa Cruz Biotechnology), acetyl-lysine (06-933; 1:500; Upstate Biotechnology), acetylated-p53 (Lys382) (2525; 1:500; Cell Signaling Technology), acetyl-p53 (Lys373) (06-916; 1:500; Upstate Biotechnology), p53 (sc-6243-G; 1:200; Santa Cruz Biotechnology), actin (JLA20; 1:50; Sigma-Aldrich), and peroxidase-conjugated IgGs (Cappel). The antibodies used for coimmunoprecipitation assay are necdin (NC243; 1:100), Myc (9E10; 1:4), FLAG (M2; 1:50), and necdin (GN1; 1:100). Primary antibodies used for immunocytochemistry are guinea pig polyclonal antibody against necdin (GN1; 1:500), rabbit polyclonal antibody against FLAG (F 7425; 1:200; Sigma-Aldrich), microtubule-associated protein 2 (MAP2) (1:1000) (a gift from Dr. M. Niinobe, Osaka University, Osaka, Japan), activated caspase-3 p20/p17 subunits activated caspase 3 (ACP3) (1:1000) (Uetsuki et al., 1999), and green fluorescent protein (GFP) (1:500; MBL), and goat polyclonal antibody against p53 (sc-6243-G; 1:200; Santa Cruz Biotechnology). Secondary antibodies are cyanine 2-conjugated anti-guinea pig IgG (1:500), cyanine 2-conjugated anti-rabbit IgG (1:500), cyanine 2-conjugated anti-goat IgG (1:500), cyanine 3-conjugated anti-mouse IgG (1:500), cyanine 3-conjugated anti-guinea pig IgG (1:500) are from Jackson ImmunoResearch, and Alexa Fluor 350-conjugated anti-rabbit IgG (1:500) is from Invitrogen.
Ndn knock-out mice.
Ndn knock-out mice (Ndntm1Ky) were generated and maintained as described previously (Kuwako et al., 2005). Heterozygous male mice (Ndn+/−) (>15 generations in ICR background) were crossed with wild-type female mice (Ndn+/+) to obtain wild-type (Ndn+m/+p) and paternal Ndn-deficient (Ndn+m/−p) mice as siblings. Genotypes of all mice used were analyzed by PCR for mutated Ndn locus. Experiments using gene-targeted mice were approved by the Recombinant DNA and Animal Experiment Committees of the Institute for Protein Research, Osaka University, and performed in accordance with institutional guidelines and regulations.
Immunohistochemistry.
Frozen 12-μm-thick tissue sections were prepared from mouse embryos at embryonic day 14.5 (E14.5) and immunostained as described previously (Kuwajima et al., 2004). The sections were incubated at 4°C overnight with primary antibodies and fluorescence dye-conjugated secondary antibodies at room temperature for 90 min. The images were observed with a fluorescence microscope (BX-50-34-FLAD1; Olympus Optical) (MZ16 F; Leica Microsystems), taken by charge-coupled device camera system (DP70; Olympus Optical), and processed using Adobe PhotoShop CS2 software.
Coimmunoprecipitation assay.
Human lung carcinoma H1299 cells were transfected with cDNAs as described previously (Kuwako et al., 2004). cDNA encoding mouse full-length Sirt1 (a gift from Dr. Y. Horio, Sapporo Medical University, Sapporo, Japan) was subcloned into p3xFLAG-CMV14 (Sigma-Aldrich) for FLAG tagging. pRc-necdin, Myc-tagged p53, and Myc-tagged p53ΔN were as described previously (Taniura et al., 1998, 1999). The cells were harvested 24 h after transfection and lysed in 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 10% glycerol, and protease inhibitors (Complete; Roche). Proteins in the lysates (200 μg) were incubated at 4°C for 2 h with antibodies, pelleted with protein A-Sepharose (GE Healthcare), separated by 7.5% SDS-PAGE, and detected by Western blotting. For analysis of endogenous interaction between necdin and Sirt1, proteins in forebrain tissue lysates of E14.5 mice (1 mg) were incubated with guinea pig anti-necdin antibody (GN1; 1:100), pelleted with protein A-Sepharose, separated by 10% SDS-PAGE, and detected by Western blotting. The protein concentration was determined by the Bradford method (Bio-Rad).
Immunocytochemistry.
H1299 cells transfected with expression vectors for necdin, Myc-p53, and Sirt1-FLAG were immunostained as described previously (Kuwajima et al., 2004). Dispersed cortical neurons were cultured for 48 h and then treated with 10 μm camptothecin (CPT) (Sigma-Aldrich) or 200 μm hydrogen peroxide (H2O2; Wako Pure Chemicals) for the indicated duration. These cells were fixed with 10% formalin solution at room temperature for 20 min and then permeabilized with methanol at room temperature for 20 min. Fixed cells were incubated at 4°C overnight with primary antibodies and fluorescence dye-conjugated secondary antibodies at room temperature for 90 min. Immunofluorescence images were obtained by confocal laser-scanning microscopy (LSM5 PASCAL; Zeiss) and a fluorescence microscope (BX-50-34-FLAD1; Olympus Optical) and processed using Adobe PhotoShop CS2 software.
In vitro binding assay.
The fragments of Sirt1 and its deletion mutants were subcloned into pMAL-C2 (New England Biolabs) to make maltose-binding protein (MBP) fusion proteins. The MBP fusion proteins were affinity purified with amylose resin (New England Biolabs). Purified MBP–Sirt1 fusion proteins (5 μg) bound to amylose resin (40 μl) were incubated with His-tagged necdin (200 ng) (Matsumoto et al., 2001) at 4°C for 30 min in 0.5 ml of the binding buffer containing 20 mm Tris-HCl, pH 7.5, 200 mm NaCl, and 1 mm EDTA. After the resin was washed with the binding buffer, bound proteins were eluted with 20 mm maltose and detected by Western blotting. The protein levels were quantified with an image analyzer (LAS-1000 Plus; Fuji Film).
In vitro deacetylation assay.
pMAL-C2 plasmid carrying cDNA for full-length p53 (amino acids 1–393), its N terminal deletion mutant p53 (amino acids 75–393) and CREB-binding protein (CBP)-histone acetyltransferase (HAT) (amino acids 1099–1758), were constructed for MBP fusion proteins (Taniura et al., 1999). MBP fusion proteins were expressed in Escherichia coli strain BL21. Purified MBP-p53 fusion proteins (1 μg) were incubated at 30°C for 1 h with purified MBP-CBP (HAT) fusion protein (0.5 μg) in the presence or absence of 0.1 mm acetyl coenzyme A (Ac-CoA; Sigma-Aldrich) in the HAT assay buffer containing 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.1 mm EDTA, 1 mm DTT, 20 mm nicotinamide (Sigma-Aldrich), 3 μm trichostatin A (Wako Pure Chemicals), 10 mm sodium butyrate (Wako Pure Chemicals), and 10% glycerol. Reaction products were analyzed by Coomassie brilliant blue staining and Western blotting. Acetylated MBP-p53 fusion proteins were incubated with purified MBP-Sirt1 fusion protein (0.5 μg), purified His-tagged necdin (3.6 μg), 1 mm NAD (Sigma-Aldrich), and 20 mm nicotinamide at 30°C for 1 h in the HDAC assay buffer containing 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 10% glycerol. Reaction products were analyzed by Coomassie brilliant blue staining and Western blotting.
Primary neuron cultures.
Dispersed cortical neurons were prepared from E14.5 ICR mice (SLC). The cortex was dissected and incubated in 0.5 ml of Ca2+/Mg2+-free, glucose-supplemented HBSS with 0.05% trypsin for 5 min at 37°C. The tissues were dissociated in DMEM supplemented with 10% fetal bovine serum, and then resulting suspension was centrifuged at 1000 rpm for 3 min. The cell pellet was resuspended in Neurobasal medium (Invitrogen), 2 mm l-glutamine, penicillin/kanamycin and 1:50 dilution of B-27 supplement (Invitrogen), and placed at a density of 8 × 104 cells/cm2 in culture dishes precoated with poly-dl-ornithine.
Cell death analysis.
Dispersed cortical neurons were cultured for 48 h and treated with 10 μm CPT (Sigma-Aldrich) or 200 μm H2O2 (Wako Pure Chemicals) for the indicated duration. Pifithrin-α (BIOMOL Research Laboratories) and resveratrol (RSV) (Sigma-Aldrich) were added to the medium 1 h before the addition of CPT or H2O2. Cultures were then treated with Hoechst 33342 (Sigma-Aldrich) for 5 min before fixation, and cortical neurons carrying condensed or fragmented nuclei were judged to be apoptotic cells.
RNA interference.
The sequences of custom small interfering RNA (siRNA) duplex for p53 (GeneDesign) and Sirt1 (B-Bridge International) are sense 5′-ccacuugauggagaguauu-3′ and antisense 5′-aauacucucuccaucaagugg-3′ (p53 siRNA) and sense 5′-gugagaaaaugcuggccua-3′ and antisense 5′-uaggccagcauuuucucac-3′ (Sirt1 siRNA). The sequence of p53 and Sirt1 siRNAs correspond to nucleotides 955–973 of p53 mRNA and 1828–1846 of Sirt1 mRNA, respectively. The RNA duplex sense 5′-guaagcggcuugaggguaa-3′ and antisense 5′-uuacccucaagccgcuuac-3′ (corresponding to Sirt1 mRNA nucleotides 1871–1889) had no interfering effect on the expression of p53 and Sirt1 and was used for both p53 and Sirt1 siRNA analyses as negative siRNA controls. Dispersed cortical neurons were plated in a 12-well plate (4 × 105 cells per well), cultured for 24 h, and transiently transfected with 0.2 μg of pEGFP-C2 (Clontech) and siRNA (40 pmol) using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instruction. After 24 h from siRNA transfection, cultures were treated with Hoechst 33342 (Sigma-Aldrich) for 5 min before fixation, and GFP-positive neurons carrying condensed or fragmented nuclei were judged to be apoptotic cells. Efficiencies of p53 and Sirt1 knockdown were assessed by Western blotting.
Organotypic culture.
Organotypic slice cultures of embryonic mouse forebrain were prepared as described previously (Kuwajima et al., 2006). Briefly, embryonic brain was removed and embedded in 1.3% low-melting agarose (Cambrex) in Hanks buffer, pH 7.4. Coronal sections (150 μm thick) were cut using the McIlwain tissue chopper (Geneq) and transferred to the Hanks buffer. The sections were transferred to collagen-coated polytetrafluoroethylene membranes (diameter, 12 mm; pore size, 3 μm) (Corning Life Sciences) in 12-well cell culture plate containing Neurobasal/B27 medium. After incubation (1 h), slices were treated with H2O2 (200 μm) for 6 h, and fixed with 4% paraformaldehyde in PBS for immunohistochemistry.
Statistical tests.
Statistical significance was tested using an unpaired Student's t test or one-way ANOVA, followed by the Tukey's post hoc test. A significance of p < 0.05 was required for rejection of the null hypothesis.
Results
Necdin is coexpressed with Sirt1 in embryonic forebrain neurons in vivo
We first examined the expression patterns of Sirt1 and βIII-tubulin, a young postmitotic neuron marker, in E14.5 mouse forebrain by immunohistochemistry. Sirt1 was strongly expressed in the striatum, septum, and cortical layer consisting of the marginal zone, cortical plate, and subplate (Fig. 1A). A moderate Sirt1 immunoreactivity was also detected in the ventricular zone of the cortex, in which neural stem cells undergo mitosis, whereas Sirt1 was weakly expressed in the intermediate zone. Double-immunostaining analysis revealed that the cells with strong Sirt1 immunoreactivity overlap with βIII-tubulin-immunopositive cells in the cortex, suggesting that Sirt1 is expressed preferentially in young postmitotic neurons. The Sirt1 immunoreactivity was detected in the nucleus and cytoplasm of cortical neurons. We then compared the distribution pattern of Sirt1 with that of necdin in embryonic neocortex (Fig. 1B,C). Sirt1 and necdin were strongly expressed in the subplate, and most of the Sirt1-immunopositive cells overlapped with necdin-immunopositive cells (Fig. 1B, arrow). Necdin, like Sirt1, was distributed in the nucleus and cytoplasm of cortical neurons. These results suggest that necdin and Sirt1 are coexpressed in cortical neurons at early stages of development.
Figure 1.
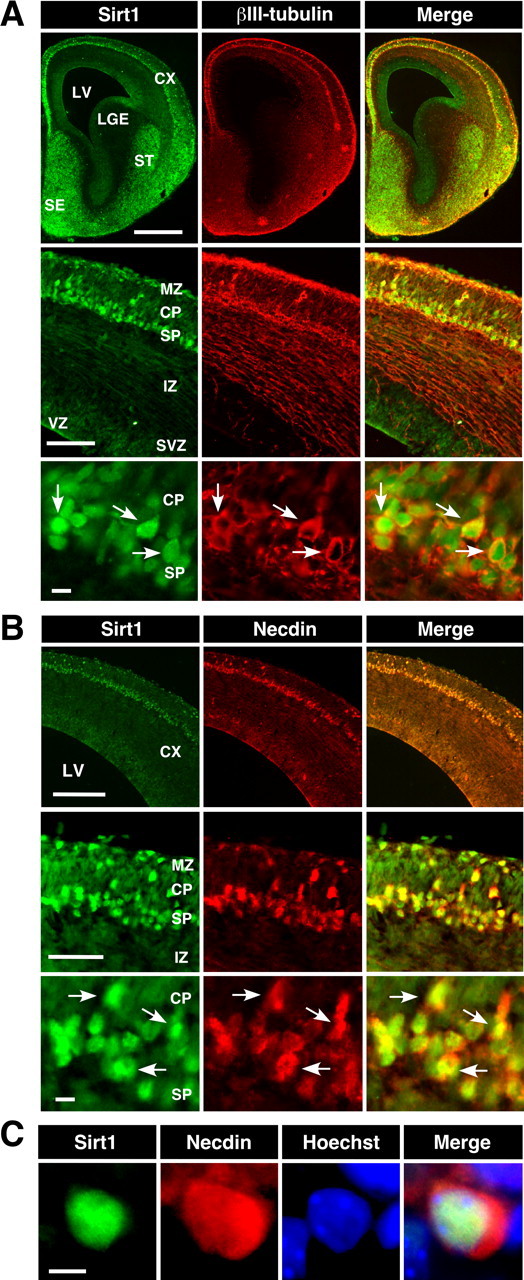
Distribution of Sirt1 and necdin in mouse embryonic forebrain. A, Double immunostaining for Sirt1 and βIII-tubulin. Frozen forebrain sections of E14.5 mouse embryo were double immunostained for Sirt1 (green) and βIII-tubulin (red), and two images were merged (yellow). CX, Cortex; LV, lateral ventricle; LGE, lateral ganglionic eminence; ST, striatum; SE, septum; MZ, marginal zone; CP, cortical plate; SP, subplate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone. Arrows point to representative double-immunopositive cells. B, Double immunostaining for Sirt1 and necdin. Frozen sections were double immunostained for Sirt1 (green), necdin (red), and two images were merged (yellow). Arrows point to representative double-immunopositive cells. C, Triple staining for Sirt1, necdin, and Hoechst. Frozen sections were triple immunostained for Sirt1 (green), necdin (red), Hoechst 33342 (blue), and three images were merged. Scale bars: (in A) top, 500 μm; middle, 100 μm; bottom, 10 μm; (in B) top, 200 μm; middle, 50 μm; bottom, 10 μm; (in C) 5 μm.
Necdin interacts with Sirt1 in vivo and in vitro
We then examined the association between necdin and Sirt1 by coimmunoprecipitation assay using human lung carcinoma H1299 cells, a cell line lacking p53 expression. We first examined the interactions of p53 with necdin and Sirt1 using H1299 cells transfected with cDNAs for necdin, Myc-tagged p53, and FLAG-tagged Sirt1. Consistent with previous reports (Taniura et al., 1999; Luo et al., 2001; Vaziri et al., 2001), p53 was able to interact with necdin (Fig. 2A) and Sirt1 (Fig. 2B) in this assay system. We then examined whether necdin interacts with Sirt1 (Fig. 2C). Sirt1 was coprecipitated with necdin, which was conversely coprecipitated with Sirt1. To determine the direct interaction between necdin and Sirt1, we next performed in vitro binding assay using MBP-fused Sirt1 deletion mutants and His-tagged necdin (Fig. 2D). Necdin bound to full-length Sirt1 (amino acids 1–737), the N-terminal domain of Sirt1 (amino acids 1–235) and the sirtuin homology domain (amino acids 236–490), but not to the C-terminal domain of Sirt1 (amino acids 491–737). We also performed the coimmunoprecipitation assay to detect a complex of necdin and Sirt1 in embryonic forebrain extracts (Fig. 2E). Sirt1 was coprecipitated with necdin from the wild-type extract but not from the necdin-deficient extract. Necdin failed to interact with HDAC1 or PCNA. These results suggest that endogenous necdin forms a complex with Sirt1 in neurons.
Figure 2.

Necdin interacts with both p53 and Sirt1 in vivo and in vitro. A–C, Interactions between necdin, p53, and Sirt1. Lysates (200 μg of protein) of H1299 cells transfected with expression vectors for necdin, Myc-p53, and Sirt1-FLAG were immunoprecipitated (IP) with anti-necdin antibody (αNecdin) (A, C) or anti-FLAG antibody (αFLAG) (B) and immunoblotted (IB) with anti-Myc antibody (αMyc) (A, B) or anti-FLAG antibody (C) (top panels). The antibodies for IP and IB were conversely used to confirm the interactions (third panels). Expressed proteins in cell lysates (10 μg) are shown (second and bottom panels). Molecular sizes are in kilodaltons (kDa). D, In vitro binding assay. Purified MBP-Sirt1 deletion mutants immobilized on amylose were incubated with His-tagged necdin (His-Necdin). Bound His-necdin was detected by immunoblotting with anti-necdin antibody (top). MBP-Sirt1 deletion mutants were separated by 7.5% SDS-PAGE and stained with Coomassie brilliant blue (bottom). FL, full-length; CT, C-terminal domain; NT, N-terminal domain; SH, sirtuin homology domain. E, Immunoprecipitation of the endogenous complex containing necdin and Sirt1. Tissue lysate (1 mg) of E14.5 mouse forebrain was immunoprecipitated with guinea pig anti-necdin IgG GN1 (αNecdin IgG) or control preimmune IgG (preimmune IgG) and immunoblotted with antibodies against Sirt1, HDAC1, PCNA, and necdin. WT, Ndn+m/+p; KO, Ndn+m/−p. Lysate, Tissue lysate (10 μg).
Necdin potentiates p53 deacetylation through interaction with Sirt1
We next examined whether necdin simultaneously interacts with p53 and Sirt1 to form a ternary complex by coimmunoprecipitation assay using transfected H1299 cells. Necdin was coprecipitated with p53 but not with p53ΔN, a deletion mutant lacking the N-terminal necdin-binding region (Taniura et al., 1999) (Fig. 3A, top panel, lanes 2, 6). However, necdin interacted with p53ΔN in the presence of coexpressed Sirt1 (Fig. 3A, top panel, lane 7), indicating that necdin forms a complex with p53ΔN via Sirt1. Sirt1 was coprecipitated with both wild-type p53 and p53ΔN even in the absence of necdin, and coexpression of necdin increased the amount of Sirt1 bound to p53 (Fig. 3A, second panel) (2.1 ± 0.19 times, lane 3 vs lane 4; n = 3; p < 0.02). These data suggest that necdin and Sirt1 interact with p53 via different binding sites to form a ternary complex. Furthermore, we examined whether necdin colocalizes with Sirt1 and p53 in specific subcellular compartments by immunocytochemistry. Sirt1 and p53 were localized mainly to the nucleus of H1299 cells, whereas necdin was distributed in the nucleus and cytoplasm (Fig. 3B, top and middle). Triple staining revealed that the three proteins are colocalized in the nucleus (Fig. 3B, bottom).
Figure 3.
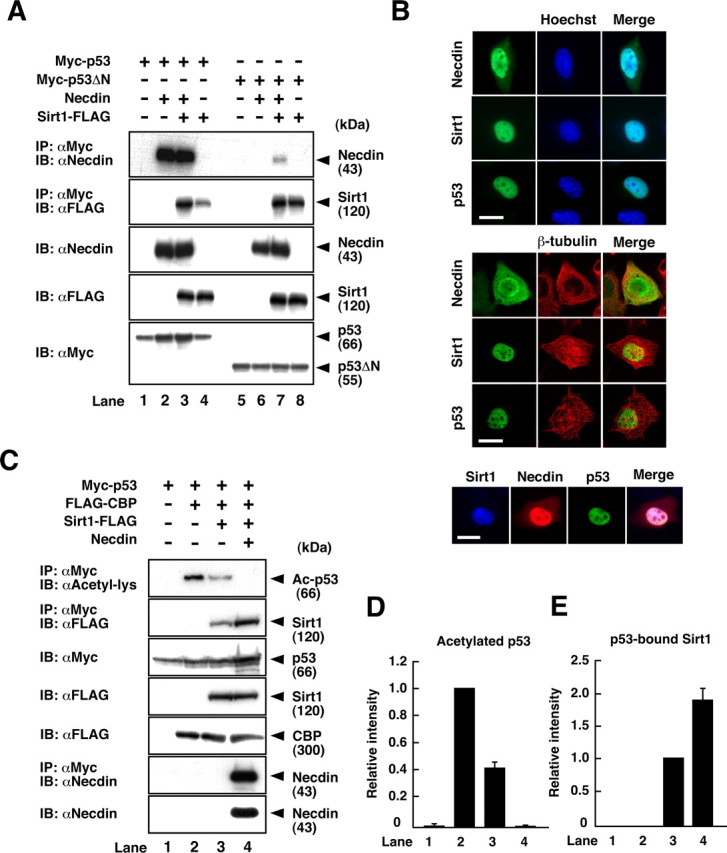
Necdin potentiates p53 deacetylation via Sirt1. A, Immunoprecipitation of the ternary complex. H1299 cells were transfected with expression vectors for necdin, Myc-p53, Myc-p53ΔN, and Sirt1-FLAG, and the lysates (200 μg) were immunoprecipitated (IP) with anti-Myc antibody (αMyc) and immunoblotted (IB) with antibodies to necdin (αNecdin) and FLAG (αFLAG). Expressed proteins (5 μg) are shown in the bottom three panels. B, Immunocytochemistry. H1299 cells were transfected with expression vectors for necdin, Myc-p53, and Sirt1-FLAG, and FLAG (for Sirt1), necdin, and p53 were double stained with the nuclear marker Hoechst 33342 (top) or with the cytoplasmic marker β-tubulin (middle). The cells were triple stained for colocalization of the three proteins (bottom). Scale bars, 20 μm. C, Detection of p53 acetylation. H1299 cells were transfected with expression vectors for Myc-p53, FLAG-CBP, Sirt1-FLAG, and necdin. Cell lysates (200 μg) were immunoprecipitated with anti-Myc antibody (αMyc) and immunoblotted with anti-acetyl lysine antibody (αAcetyl-lys) (top). To detect the interactions between p53, Sirt1, and necdin, the lysates were immunoprecipitated with anti-Myc antibody and immunoblotted with antibodies to FLAG (second panel) and necdin (sixth panel). Expressed proteins (5 μg) are shown in the third, fourth, fifth, and bottom panels. D, E, Quantification of acetylated p53 and p53-bound Sirt1. Acetylated p53 (C, top) and Sirt1 coprecipitated with p53 (C, second panel) were quantified (mean ± SEM of 3 independent experiments).
To elucidate the biological significance of the ternary complex formation, we examined the modulatory effects of necdin on p53 deacetylation. Acetylated p53 levels in cDNA-transfected cells were analyzed by Western blotting using anti-acetyl lysine antibody (Fig. 3C). Sirt1 reduced the amount of acetylated p53 to 42% of the control level, and coexpression of necdin further reduced it to an almost undetectable level (Fig. 3C, top panel; D). Necdin increased the amount of p53-bound Sirt1 1.9 times the control level (Fig. 3C, second panel; E), indicating that necdin facilitates the association between p53 and Sirt1. Necdin also increased the amount of p53 (deacetylated form) when coexpressed with p53 and Sirt1 (Fig. 3C, third panel), suggesting that deacetylated p53 is stabilized in the complex. In this assay system, necdin did not interact with CBP (data not shown).
We next investigated whether necdin enhances Sirt1-mediated p53 deacetylation by in vitro acetylation/deacetylation assay using acetylated recombinant p53 and p53ΔN proteins. Purified MBP-Sirt1 reduced the acetylation level of p53 in the presence of NAD (Fig. 4A,C, lane 3), consistent with the fact that Sirt1 requires NAD for its enzyme activity. His-tagged necdin failed to deacetylate p53 in the absence of MBP-Sirt1 (Fig. 4A,C, lane 4), indicating that necdin itself has no p53 deacetylation activity. His-tagged necdin in combination with MBP-Sirt1 markedly reduced the acetylated level of p53, in-dicating that necdin enhances Sirt1-mediated p53 deacetylation (Fig. 4A,C, lane 5). Nicotinamide, a Sirt1 inhibitor, abrogated the necdin- and Sirt1-dependent p53 deacetylation (Fig. 4A,C, lane 6). We then examined whether necdin directly enhances Sirt1 activity (Fig. 4B). MBP-Sirt1 was able to deacetylate p53ΔN, a mutant bound to Sirt1 but not to necdin (Fig. 4B,D, lane 2), and necdin did not further reduce the acetylation level of p53ΔN (Fig. 4B,D, lane 3). These results suggest that necdin is unable to activate Sirt1 directly but only potentiates p53 deacetylation by facilitating the accessibility of Sirt1 to its substrate p53.
Figure 4.

Necdin promotes p53 deacetylation by bridging Sirt1 and p53. A, B, In vitro deacetylation assay. Acetylated MBP-p53 (A) and acetylated MBP-p53ΔN (B) were incubated with combinations of MBP-Sirt1, 1 mm NAD, 20 mm nicotinamide, and His-tagged necdin (His-Necdin). The levels of acetylated p53 (Ac-p53; A) and acetylated p53ΔN (Ac-p53ΔN; B) were analyzed by Western blotting using anti-acetylated-p53 (Lys382) antibody (top). Recombinant proteins were stained with Coomassie brilliant blue (bottom). C, D, Quantification of acetylated MBP-p53 and acetylated MBP-p53ΔN. Acetylated p53 (Ac-p53; A) and acetylated p53ΔN (Ac-p53ΔN; B) were quantified (mean ± SEM of 3 independent experiments) and presented in C and D, respectively. Note that necdin potentiates deacetylation of p53 (lane 5 vs lane 3; C) but not that of p53ΔN (lane 3 vs lane 2; D), which lacks the necdin-binding domain.
Necdin deficiency enhances p53-induced apoptosis in primary cortical neurons
To examine whether endogenous necdin suppresses p53-dependent apoptosis, primary cortical neurons were prepared from paternal Ndn-deficient (Ndn+m/−p) and wild-type (Ndn+m/+p) mice at E14.5 and treated with the topoisomerase-I inhibitor CPT, a strong DNA-damaging compound that induces p53-mediated apoptosis (Morris and Geller, 1996). Cultured cortical cells were immunostained for the differentiated neuron marker MAP2 (Fig. 5A, inset). In these cultures, >97% of cells were postmitotic neurons. In necdin-deficient cultures, the number of apoptotic cells was significantly increased by CPT treatment as examined up to 12 h. At 24 h of CPT treatment, no significant difference was observed between wild-type and necdin-deficient neurons because almost all neurons underwent apoptosis (data not shown). There was no difference in neuronal apoptosis between wild-type and necdin-deficient neurons under unstimulating conditions (0 h control). Similar findings were observed in necdin-deficient cortical neurons treated with etoposide, a topoisomerase-II inhibitor that also induces p53-dependent apoptosis (data not shown). We also performed immunocytochemistry for ACP3 in CPT-treated cultures (Fig. 5B). The number of ACP3-positive neurons was significantly increased in necdin-deficient cultures. These results suggest that endogenous necdin suppresses CPT-induced apoptosis.
Figure 5.
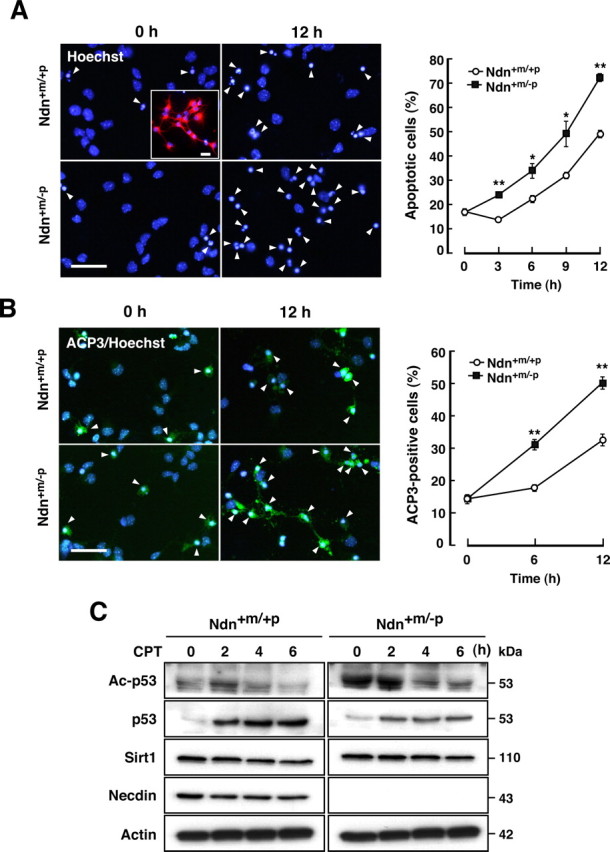
Necdin deficiency enhances CPT-induced apoptosis of cortical neurons. A, Apoptosis of CPT-treated neurons. Primary neurons were prepared from the cerebral cortex of wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/−p) mouse forebrain at E14.5. Dissociated cell cultures were treated with CPT (10 μm) and stained with Hoechst 33342. Arrowheads indicate the apoptotic cells. Neuronal enrichment was evaluated by immunostaining MAP2 (inset). Scale bars, 20 μm. Neurons with condensed or fragmented nuclei were counted at the indicated times (>250 cells; mean ± SEM; n = 3) (graph). *p < 0.05; **p < 0.01. B, Neurons containing ACP3. Primary neurons were treated with CPT as in A and stained with an antibody to ACP3. Scale bar, 20 μm. ACP3-immunopositive cells were counted (>220 cells; n = 3) (graph). **p < 0.01. C, Western blot analysis. Neuronal lysates (7 μg per lane) were prepared at the indicated times of CPT treatment and analyzed by Western blotting with antibodies against acetyl-p53 (Lys373) (Ac-p53), p53, Sirt1, necdin, and actin.
We next examined the expression levels of acetylated p53 and total p53 in CPT-treated cultures by Western blot analysis (Fig. 5C). In the paternal Ndn-deficient (Ndn+m/−p) cultures, expression of endogenous necdin was undetected (Fig. 5C, fourth panel). Because acetylation of human p53 K373 (corresponding to mouse p53 K370) links to apoptosis (Knights et al., 2006), we analyzed the acetylated K370 level by Western blotting with an antibody that specifically recognizes human p53 acetyl-K373. Nec-din-deficient neurons had high levels of acetylated p53 under unstimulating and CPT-treated conditions, suggesting that endogenous necdin suppresses p53 acetylation. Total p53 levels were increased 2–6 h after CPT treatment in wild-type and necdin-deficient neurons, and wild-type neurons contained higher total p53 levels than necdin-deficient neurons (Fig. 5C, second panel). CPT exerted no appreciable effects on the levels of Sirt1 and necdin (in wild-type neurons) (Fig. 5C, third and fourth panels).
Necdin specifically suppresses p53-dependent neuronal apoptosis
To examine whether necdin suppresses p53-mediated apoptosis, we used siRNA to knockdown endogenous p53 expression in primary neurons. p53 siRNA was efficiently suppressed p53 expression (Fig. 6A, top panel, lanes 5, 6). Unexpectedly, acetylated p53 levels in necdin-deficient neurons were elevated even by mock (GFP alone) and control siRNA transfections (Fig. 6A, second panel, lanes 2, 4), whereas wild-type neurons had low acetylated p53 levels (Fig. 6A, lanes 1, 3). This suggests that lipofection per se acts as a stress to increase p53 acetylation in necdin-null neurons. The acetylated p53 levels of wild-type and necdin-deficient neurons transfected with p53 siRNA were markedly decreased (Fig. 6A, second panel, lanes 5, 6). We then analyzed the effects of p53 siRNA on neuronal apoptosis by immunocytochemical analysis (Fig. 6B). In parallel with the acetylated p53 levels, the number of apoptotic neurons treated with control siRNA was significantly increased in necdin-deficient mice (2.3× the wild-type control) (Fig. 6C), whereas p53 siRNA significantly reduced the number of apoptotic necdin-deficient neurons. No significant difference was noted in the number of apoptotic neurons between wild-type and necdin-deficient neurons when treated with p53 siRNA (Fig. 6C), indicating that necdin specifically represses p53-dependent apoptosis. To further confirm whether CPT-induced neuronal apoptosis is dependent on p53, the p53 inhibitor pifithrin-α was used to inhibit p53-mediated apoptosis. Treatment of wild-type and necdin-deficient neurons with pifithrin-α significantly attenuated CPT-induced neuronal apoptosis (Fig. 6D). No significant difference was noted in the number of apoptotic neurons between necdin-deficient and wild-type neurons treated with both CPT and pifithrin-α. These results suggest that endogenous necdin efficiently suppresses p53-dependent apoptosis in cortical neurons.
Figure 6.
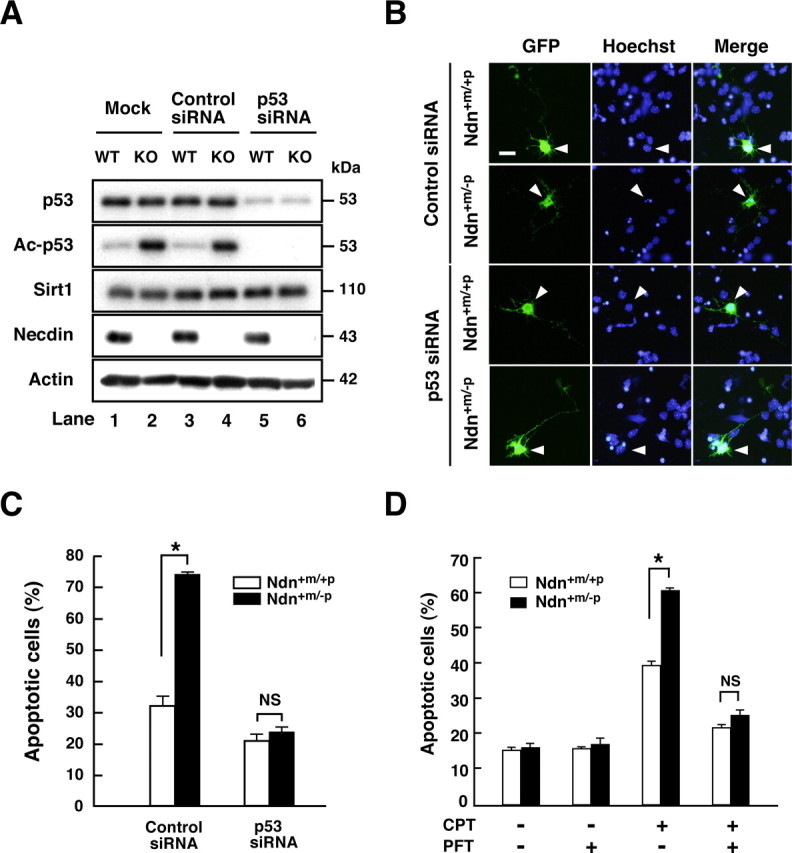
Necdin specially suppresses p53-dependent neuronal apoptosis. A, Western blot analysis. Cortical neurons were prepared from wild-type (Ndn+m/+p, WT) and necdin-deficient (Ndn+m/−p, KO) mouse forebrain and transfected with p53 siRNA and negative control siRNA (Control siRNA). Neurons were harvested 24 h later, and the lysates were immunoblotted with antibodies to p53, acetyl-p53 (Lys373) (Ac-p53), Sirt1, necdin, and actin. B, Immunostaining. Cortical neurons transfected with negative control siRNA (Control siRNA), and p53 siRNA were visualized by cotransfected GFP and immunostained with Hoechst 33342 (arrowheads). Scale bar, 10 μm. C, Quantification of apoptotic neurons. Neurons with condensed or fragmented nuclei among >30 GFP-positive cells were counted (mean ± SEM; n = 3). D, Inhibition of p53-dependent apoptosis by pifithrin-α (PFT). Neurons were treated with PFT (200 nm) and CPT (10 μm) for 9 h and stained with Hoechst 33342. Neurons with condensed or fragmented nuclei were counted (>200 cells; mean ± SEM; n = 3). *p < 0.01. NS, Not significant (p > 0.05).
Necdin deficiency reduces RSV-mediated Sirt1 activation
We next examined the effects of RSV, a small molecule polyphenolic Sirt1 activator, on CPT-induced neuronal apoptosis. We first analyzed the expression levels of acetylated p53 in RSV-treated cortical neurons (Fig. 7A). RSV markedly decreased CPT-induced p53 acetylation in wild-type neurons (Fig. 7A, top panel, lane 2), whereas it failed to reduce it in necdin-deficient neurons (Fig. 7A, lane 4). RSV had no effect on the levels of p53 and Sirt1 (Fig. 7A, second and third panels). We then examined the effects of RSV on neuronal apoptosis in the presence and absence of endogenous necdin (Fig. 7B). In wild-type neurons, RSV markedly reduced CPT-induced apoptosis (Ndn+m/+p/RSV− vs Ndn+m/+p/RSV+), whereas RSV failed to reduce CPT-induced apoptosis in necdin-null neurons (Ndn+m/−p/RSV− vsNdn+m/−p/RSV+). We also observed that neurons underwent apoptosis when treated with H2O2, an oxidative stress-inducing agent that also activates the p53 pathway (Fig. 7C). H2O2-induced apoptosis was also markedly increased in necdin-deficient neurons (Ndn+m/+p/CPT+ vs Ndn+m/−p/CPT+). In these cultures, RSV reduced the apoptotic population of H2O2-treated wild-type neurons (Ndn+m/+p/RSV− vs Ndn+m/+p/RSV+) but had no effect on that of necdin-deficient neurons (Ndn+m/−p/RSV− vs Ndn+m/−p/RSV+). These results suggest that endogenous necdin confers the RSV responsiveness on Sirt1 in neurons under DNA damaging conditions.
Figure 7.
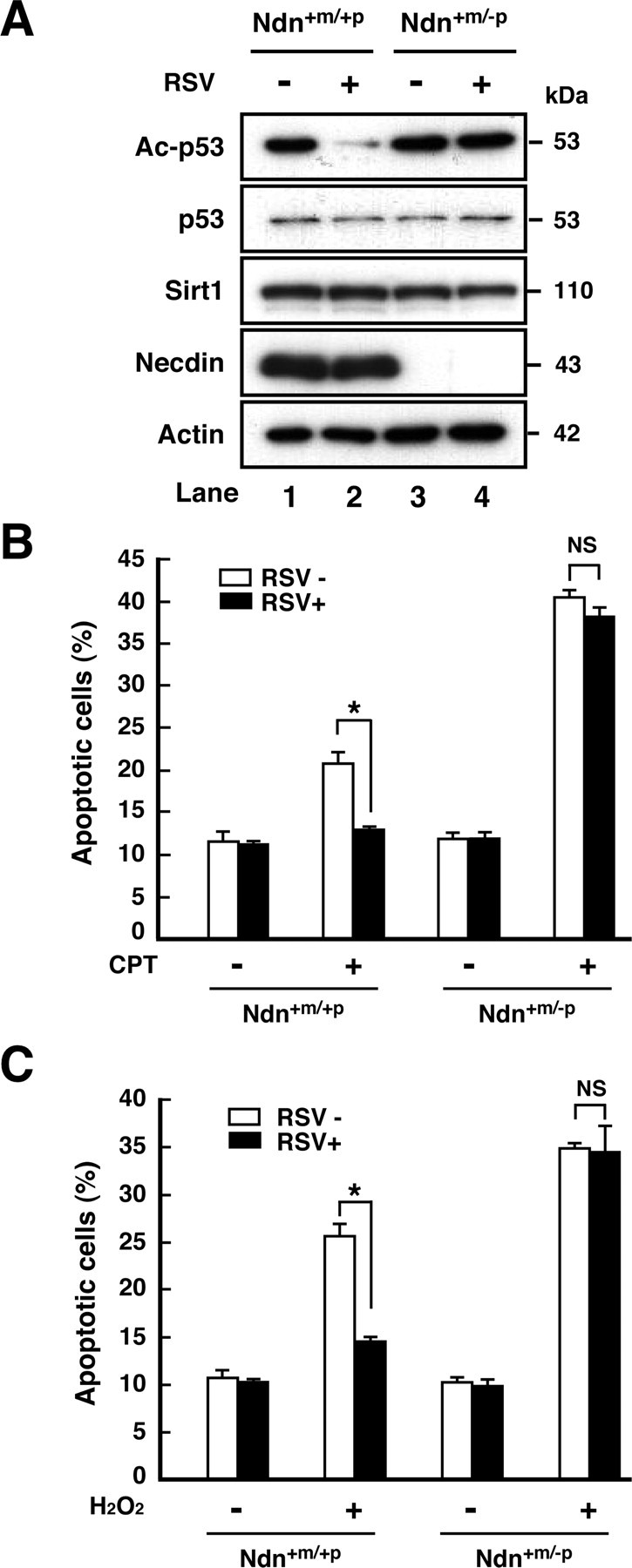
Necdin deficiency diminishes RSV-induced Sirt1 activation. A, Western blot analysis. Primary neurons were prepared from the cerebral cortex of wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/−p) mouse forebrain at E14.5. Neurons were cultured in the presence of RSV (10 μm) and CPT (10 μm) for 6 h, and the lysates (7 μg) were analyzed by Western blotting with antibodies to acetyl-p53 (Lys373) (Ac-p53), p53, Sirt1, necdin, and actin. B, Effects of RSV on CPT-induced apoptosis. Neurons were treated with CPT (10 μm) for 6 h in the presence (RSV+) or absence (RSV−) of RSV (10 μm). Neurons were stained with Hoechst 33342, and those with condensed or fragmented nuclei were counted (>240 cells; mean ± SEM; n = 3). C, Effects of RSV on H2O2-induced apoptosis. Neurons were treated with H2O2 (200 μm) for 6 h in the presence or absence of RSV (10 μm), and those with apoptotic nuclei were counted (>250 cells; mean ± SEM; n = 3). *p < 0.01. NS, Not significant (p > 0.05).
Necdin downregulates p53 acetylation and apoptosis via Sirt1
To examine whether necdin enhances p53 deacetylation and represses apoptosis in Sirt1-dependent manners, we used siRNA to knockdown endogenous Sirt1 in primary neurons (Fig. 8). Sirt1 siRNA, which efficiently suppressed Sirt1 expression (Fig. 8A, top panel, lanes 5, 6), increased acetylated p53 levels (Fig. 8A, second panel, lane 3 vs lane 5), indicating that p53 deacetylation is dependent on endogenous Sirt1 in neurons. Consistent with the findings of p53 siRNA analysis, acetylated p53 levels in necdin-deficient neurons were elevated even by mock and control siRNA transfections (Fig. 8A, second panel, lanes 2, 4), whereas wild-type neurons had low acetylated p53 levels (Fig. 8A, lanes 1, 3). The acetylated p53 level of wild-type neurons transfected with Sirt1 siRNA was similar to that of necdin-null neurons transfected with control siRNA (Fig. 8A, second panel, lane 4 vs lane 5). We then analyzed the effects of Sirt1 siRNA on neuronal apoptosis by immunocytochemical analysis (Fig. 8B). In parallel with the acetylated p53 levels, the population of apoptotic wild-type neurons treated with Sirt1 siRNA was significantly increased (2.0 times the control siRNA) (Fig. 8C) (Ndn+m/+p/control siRNA vs Ndn+m/+p/Sirt1 siRNA). Wild-type neurons treated with Sirt1 siRNA underwent apoptosis in an extent similar to control siRNA-treated necdin-null neurons (Ndn+m/+p/Sirt1siRNA vs Ndn+m/−p/control siRNA), and Sirt1 knockdown exerted no appreciable effect on the apoptotic population of necdin-null neurons (Ndn+m/−p/control siRNA vs Ndn+m/−p/Sirt1siRNA). These results suggest that endogenous necdin in cortical neurons suppresses both p53 acetylation and p53-induced apoptosis in Sirt1-dependent manners.
Figure 8.
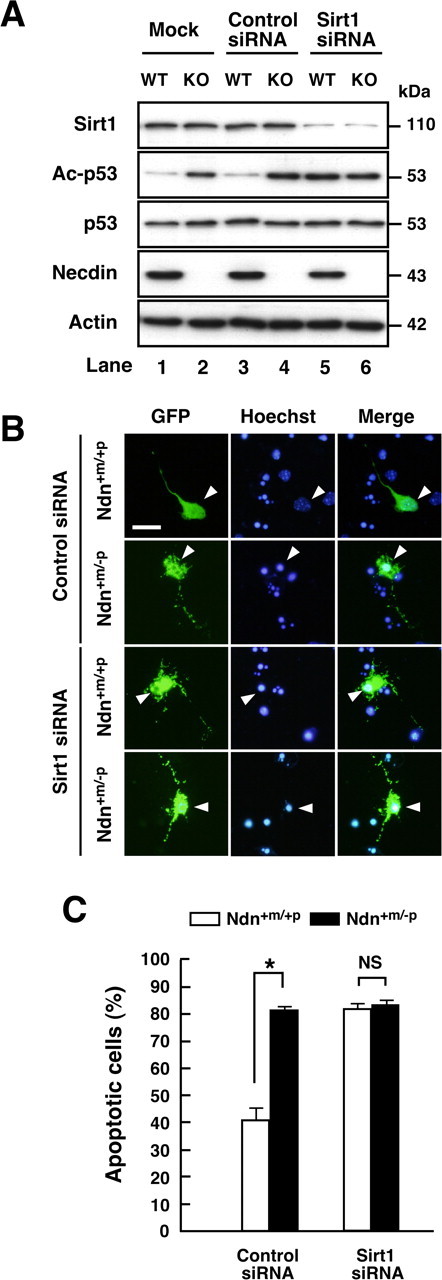
Knockdown of endogenous Sirt1 expression in neurons increases p53 acetylation and apoptosis. A, Western blot analysis. Cortical neurons were prepared from wild-type (Ndn+m/+p, WT) and necdin-deficient (Ndn+m/−p, KO) mouse forebrain and transfected with Sirt1 siRNA and negative control siRNA (Control siRNA). Neurons were harvested 24 h later, and the lysates were immunoblotted with antibodies to Sirt1, acetyl-p53 (Lys373) (Ac-p53), p53, necdin, and actin. B, Immunostaining. Cortical neurons transfected with negative control siRNA (Control siRNA) and Sirt1 siRNA were visualized by cotransfected GFP and immunostained with Hoechst 33342 (arrowheads). Scale bar, 10 μm. C, Quantification of apoptotic neurons. Neurons with condensed or fragmented nuclei among >40 GFP-positive cells were counted (mean ± SEM; n = 3). *p < 0.01. NS, Not significant (p > 0.05).
p53 colocalizes with necdin and Sirt1 in H2O2-treated neurons
Finally, we examined the colocalization of necdin and Sirt1 with p53 in vivo. We used organotypic slice cultures of E14.5 mouse forebrain for the detection of acetylated p53 immunoreactivity (Fig. 9A). The population of acetylated p53-positive cells in necdin-deficient was significantly increased (2.1 ± 0.08 × the wild-type level; n = 3; p <0.01) (Fig. 9B, top). Acetylated p53 was localized to the nucleus (Fig. 9B, bottom). These cells with acetylated p53-positive nuclei were located in the cortical plate, suggesting that acetylated p53-positive cells are postmitotic neurons. We then analyzed the distribution of necdin, Sirt1, and p53 in the slices treated with H2O2 by immunohistochemistry. The p53 immunoreactivity was very low in the forebrain slices under unstimulating conditions, but was localized predominantly to the nucleus when treated with H2O2 (Fig. 9C). Necdin, Sirt1, and p53 were distributed mainly in the nucleus stained with Hoechst 33342, although extranuclear staining of necdin and Sirt1 was also noted in these cells (Fig. 9D). Double immunostaining revealed that both necdin and Sirt2 were present in almost all p53-positive cells in slice cultures treated with H2O2 (Fig. 9E), suggesting that necdin, Sirt1, and p53 form a stable complex in neurons after H2O2 treatment.
Figure 9.
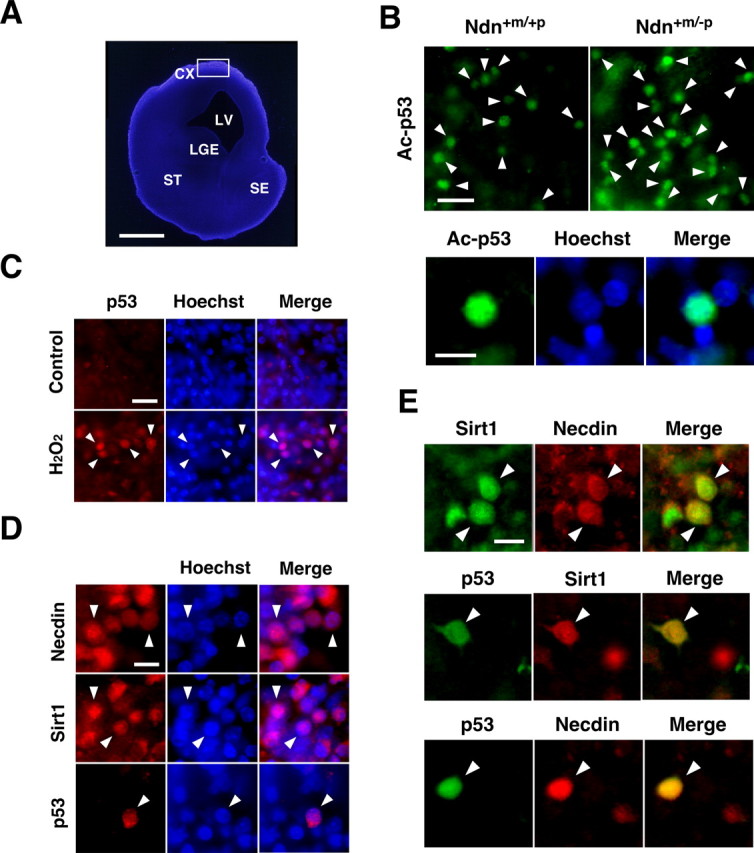
p53 colocalizes with necdin and Sirt1 in organotypic forebrain cultures after H2O2 treatment. A, DNA staining of slice culture of E14.5 mouse forebrain. Organotypic 150-μm-thick slices were prepared from E14.5 forebrain, cultured for 6 h, and stained with Hoechst 33342. CX, Cortex; LV, lateral ventricle; LGE, lateral ganglionic eminence; ST, striatum; SE, septum. B, Immunostaining for acetyl-p53 (Lys373). Slice cultures were fixed and immunostained for acetyl-p53 (Lys373) (Ac-p53). Acetylated p53-positive cells (arrowheads) in cortical regions (boxed region; A) of wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/−p) mouse forebrain are presented. Acetylated p53 is accumulated in the nucleus (bottom). C, D, E, Immunostaining for necdin, Sirt1, and p53. Slice cultures were treated with H2O2 (200 μm) for 6 h and immunostained with antibodies against p53, necdin, and Sirt1. Arrowheads point to representative immunopositive cells (C, D) and double-immunopositive cells (E). Scale bars: (in A) 500 μm; (in B) top, 20 μm; bottom, 10 μm; (in C) 20 μm; (in D, E) 10 μm.
Discussion
The present study has shown that necdin promotes p53 deacetylation by facilitating the interaction between Sirt1 and p53 to suppress p53-dependent neuronal apoptosis. Based on the present observations, we propose that necdin is a key component of the complex containing Sirt1 and p53 to confer the resistance to DNA damage on postmitotic neurons (Fig. 10). p53 plays a key role in apoptosis of postmitotic neurons (Slack et al., 1996; Xiang et al., 1996). In response to DNA damage, p53 first undergoes phosphorylation, which increases its ability to recruit CBP/p300 acetyltransferase (Lambert et al., 1998). p53 is then acetylated by CBP/p300 which enhances sequence-specific DNA binding of p53 to activate transcription of its downstream targets with proapoptotic properties (Gu and Roeder, 1997). The acetylated p53 level is downregulated by Sirt1, which prevents p53-dependent apoptosis (Luo et al., 2001; Vaziri et al., 2001). Furthermore, Sirt1-deficient cells exhibit p53 hyperacetylation and apoptosis in response to ionizing radiation (Cheng et al., 2003). Therefore, Sirt1 negatively regulates the acetylation status of p53 and prevents cell death caused by detrimental stresses. Necdin-deficient neurons, like Sirt1-deficient cells, have high levels of both p53 acetylation and p53-dependent apoptosis (Fig. 5). Necdin-deficient neurons contain higher levels of acetylated p53 than wild-type neurons at the initial stage (0–2 h) of CPT treatment, whereas wild-type neurons contain higher total p53 levels than necdin-deficient neurons 2–6 h after CPT treatment (Fig. 5C). Necdin increases total p53 levels and decreases acetylated p53 levels in H1299 cells when cotransfected with p53 and Sirt1 (Fig. 3C). In addition, siRNA-mediated Sirt1 knockdown increases both p53 acetylation and apoptosis in wild-type neurons but not in necdin-deficient neurons (Fig. 8). In organotypic cultures treated with H2O2, p53 colocalizes with necdin and Sirt1 in the nucleus (Fig. 9E). These data suggest that necdin forms a stable nuclear complex with deacetylated p53 and Sirt1, and this complex dampens the proapoptotic activity of p53 after DNA-damaging insults.
Figure 10.

A model for DNA damage-induced p53 acetylation regulated by necdin and Sirt1 in neurons. p53 is first phosphorylated in response to DNA damage and subsequently acetylated by CBP/p300 (CBP). Sirt1 inhibits neuronal apoptosis through p53 deacetylation. In wild-type neurons (Wild-type), necdin facilitates the association between Sirt1 and p53 to promote p53 deacetylation leading to survival. In paternal Ndn-deficient neurons (Necdin-null), increased p53 acetylation enhances apoptosis. For details, see Discussion. P, Phospho; Ac, acetyl.
p53 undergoes acetylation in response to a variety of insults (Ito et al., 2001). Two lysines of p53 at positions 320 (K320) and 373 (K373) are differentially acetylated to influence cell fate (Knights et al., 2006); acetylation of K320 favors cell survival, whereas acetylation of K373 promotes cell death. In addition, acetylation of K320 is required for neurite outgrowth and axonal regeneration (Di Giovanni et al., 2006). CBP/p300 acetylates C-terminal lysines including K373 and K382 of human p53 (Lambert et al., 1998), and the acetylation levels of these lysines are reduced by Sirt1 (Luo et al., 2001; Vaziri et al., 2001). Using the antibody against acetylated human p53 K373, we demonstrated that acetylation of mouse p53 K370 is increased in dispersed and organotypic cultures from necdin-deficient mice (Figs. 5C, 9B). Thus, the necdin-modulated acetylation of the C-terminal lysines including K370 may be crucial for neuronal fate. Although acetylated p53 K370 levels were high in necdin-null neurons even under unstimulating conditions, there was no difference in the apoptotic population between wild-type and necdin-null neurons (Fig. 5A,C). However, necdin-null neurons showed a high degree of CPT-induced apoptosis. Furthermore, enhanced apoptosis in necdin-null neurons was completely blocked by p53 siRNA and the p53 inhibitor pifithrin-α (Fig. 6). These findings suggest that p53 hyperacetylation is prerequisite to the high susceptibility of necdin-null neurons to DNA damage.
The activity of Sirt1 is modulated by several endogenous regulators such as FHL2 (four and a half LIM2) (Yang et al., 2005), the tumor suppressor HIC1 (hypermethylated in cancer 1) (Chen et al., 2005), the cell cycle regulator E2F1 (Wang et al., 2006), AROS (active regulator of SIRT1) (Kim et al., 2007b), and DBC1 (deleted in breast cancer 1) (Kim et al., 2008; Zhao et al., 2008). Among them, FHL2 and AROS are positive regulators of Sirt1. FHL2 is a tissue-specific coactivator of the androgen receptor expressed in the heart and prostate (Müller et al., 2000), whereas AROS is expressed in the heart, liver, small intestine, and lung (Kim et al., 2007b). Because necdin is expressed in almost all neurons throughout the nervous system (Aizawa et al., 1992; Uetsuki et al., 1996) (Fig. 1B), it is possible that necdin is a major positive regulator of Sirt1 in neurons.
RSV is a natural polyphenolic compound that significantly increases the Sirt1 activity through allosteric interaction, resulting in an increased Sirt1 affinity for both NAD and acetylated substrate (Howitz et al., 2003). It was unexpected that neither Sirt1-mediated p53 deacetylation nor neuronal survival was promoted by RSV in necdin-null neurons (Fig. 7). RSV is suggested to act on Sirt1 at an allosteric site N terminal to the catalytic domain (sirtuin homology domain) (amino acids 183–225) (Milne et al., 2007). We found that necdin interacts with the N-terminal domain (amino acids 1–235) comprising this region (Fig. 2D). Although the detailed mechanism underlying the RSV insensitiveness of necdin-deficient neurons remains to be elucidated, it seems likely that necdin affects the protein structure that is indispensable for RSV-mediated allosteric activation of Sirt1.
The human necdin gene NDN is located on chromosome 15q11.2-q12 (Nakada et al., 1998), a region deleted in the genomic imprinting-associated neurodevelopmental disorder Prader–Willi syndrome (PWS). NDN is maternally imprinted, expressed only from the paternal allele, and not expressed in individuals with PWS (Jay et al., 1997; MacDonald and Wevrick, 1997). Paternal Ndn-deficient mice lack necdin expression and display PWS-related phenotypes such as early postnatal lethality, impaired neuron development, and abnormal behaviors (Gérard et al., 1999; Muscatelli et al., 2000; Ren et al., 2003; Kuwako et al., 2005; Lee et al., 2005; Pagliardini et al., 2005). Recent three-dimensional magnetic resonance imaging studies have demonstrated a variety of morphological abnormalities in the brain affected by PWS (Yamada et al., 2006; Miller et al., 2007). Although there is currently no definitive proof of a relationship between PWS and abnormal cortical development at the cellular level, we assume that necdin deficiency increases neuronal propensity toward apoptosis via p53 pathway in PWS.
Necdin is the first identified protein among MAGE family proteins, all of which contain the MAGE homology domain (Maruyama et al., 1991). MAGE family proteins are divided into two groups based on their sequence similarities (Barker and Salehi, 2002). Type I MAGE proteins (MAGE-A, B, C subfamilies) are the precursors of tumor rejection antigens recognized by cytolytic T lymphocytes, but their functions are primarily unknown. In contrast, type II MAGE proteins such as necdin, MAGE-D1 (NRAGE, Dlxin-1) (Salehi et al., 2000), chicken MAGE (López-Sánchez et al., 2007), and Drosophila MAGE (Nishimura et al., 2008) are expressed in neurons and regulate neuronal apoptosis. MAGE-A1 acts as a potent transcriptional repressor through the interaction between SKIP (ski-interacting protein) and HDAC1 (Laduron et al., 2004). Furthermore, MAGE-A2 represses p53 functions through the recruitment with HDAC3 (Monte et al., 2006). The present study has shown that necdin interacts with Sirt1 histone deacetylase. Therefore, it is possible that MAGE proteins modulate acetylation levels of specific proteins by recruiting histone deacetylases. Deacetylation of p53 is mediated by HDAC in PC12 cells via the NGF-tropomyosin-related kinase A (TrkA) signaling pathway (Vaghefi and Neet, 2004). Moreover, HDAC inhibitor trichostatin A induces apoptosis of cultured neurons (Morrison et al., 2006). These findings suggest that HDAC promotes neuronal survival via p53 deacetylation. Necdin promotes differentiation and survival of sensory neurons in dorsal root ganglia by enhancing NGF-TrkA signaling pathway (Takazaki et al., 2002; Kuwako et al., 2005). Thus, necdin might also modulate p53 deacetylation via NGF-TrkA signaling pathway linked to HDAC. The present study focused on the modulatory effects of necdin on Sirt1-mediated p53 deacetylation in cortical neurons, because necdin preferentially interacts with Sirt1 (Fig. 2E). However, it is also possible that necdin interacts with HDACs other than Sirt1 to modulate protein acetylation in different types of neurons. We are currently investigating whether necdin regulates neuronal life and death through acetylation of necdin target proteins via Sirt1 and other HDACs.
It is tempting to speculate that necdin and Sirt1 are major hub proteins in a protein–protein interaction network involved in neuronal survival and lifespan extension of mammalian species. Expression of these two genes is coordinately regulated under specific conditions. For example, Sirt1 expression is increased in mice carrying the mutated SOD1 gene SOD1G37R, a mouse model of human amyotrophic lateral sclerosis, and RSV protects neurons against SOD1G93A toxicity (Kim et al., 2007a). Interestingly, the necdin level is increased in motor neurons of mice carrying the SOD1G93A gene (Ferraiuolo et al., 2007). Furthermore, calorie restriction, which elevates Sirt1 expression in mammalian cells (Cohen et al., 2004), also increases necdin expression in mouse skeletal muscle, whereas its expression decreases in aged mice (Lee et al., 1999). These findings suggest that necdin and Sirt1 cooperate to promote neuronal survival in response to various kinds of cellular and environmental stresses. Further studies on the interactions between necdin and Sirt1 will provide valuable insights into the regulation of protein acetylation-mediated fate decisions of neurons in various developmental and neuropathological conditions.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research B2 (18300122) from the Japan Society for the Promotion of Science and from the National Project on Protein Structure and Functional Analysis from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Drs. Y. Horio and M. Niinobe for research materials and Drs. M. Kurita, T. Kuwajima, K. Kuwako, I. Nishimura, and M. Niinobe for helpful discussions.
References
- Aizawa T, Maruyama K, Kondo H, Yoshikawa K. Expression of necdin, an embryonal carcinoma-derived nuclear protein, in developing mouse brain. Brain Res Dev Brain Res. 1992;68:265–274. doi: 10.1016/0165-3806(92)90069-9. [DOI] [PubMed] [Google Scholar]
- Andrieu D, Meziane H, Marly F, Angelats C, Fernandez PA, Muscatelli F. Sensory defects in Necdin deficient mice result from a loss of sensory neurons correlated within an increase of developmental programmed cell death. BMC Dev Biol. 2006;6:56. doi: 10.1186/1471-213X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PA, Salehi A. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res. 2002;67:705–712. doi: 10.1002/jnr.10160. [DOI] [PubMed] [Google Scholar]
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Knights CD, Rao M, Yakovlev A, Beers J, Catania J, Avantaggiati ML, Faden AI. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;25:4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27:9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard M, Hernandez L, Wevrick R, Stewart CL. Disruption of the mouse necdin gene results in early post-natal lethality. Nat Genet. 1999;23:199–202. doi: 10.1038/13828. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay P, Rougeulle C, Massacrier A, Moncla A, Mattei MG, Malzac P, Roëckel N, Taviaux S, Lefranc JL, Cau P, Berta P, Lalande M, Muscatelli F. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat Genet. 1997;17:357–361. doi: 10.1038/ng1197-357. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007a;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007b;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Taniura H, Yoshikawa K. Ectopic expression of necdin induces differentiation of mouse neuroblastoma cells. J Biol Chem. 2002;277:42128–42135. doi: 10.1074/jbc.M205024200. [DOI] [PubMed] [Google Scholar]
- Kurita M, Kuwajima T, Nishimura I, Yoshikawa K. Necdin downregulates cdc2 expression to attenuate neuronal apoptosis. J Neurosci. 2006;26:12003–12013. doi: 10.1523/JNEUROSCI.3002-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima T, Taniura H, Nishimura I, Yoshikawa K. Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J Biol Chem. 2004;279:40484–40493. doi: 10.1074/jbc.M404143200. [DOI] [PubMed] [Google Scholar]
- Kuwajima T, Nishimura I, Yoshikawa K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J Neurosci. 2006;26:5383–5392. doi: 10.1523/JNEUROSCI.1262-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwako K, Taniura H, Yoshikawa K. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J Biol Chem. 2004;279:1703–1712. doi: 10.1074/jbc.M308454200. [DOI] [PubMed] [Google Scholar]
- Kuwako K, Hosokawa A, Nishimura I, Uetsuki T, Yamada M, Nada S, Okada M, Yoshikawa K. Disruption of the paternal necdin gene diminishes TrkA signaling for sensory neuron survival. J Neurosci. 2005;25:7090–7099. doi: 10.1523/JNEUROSCI.2083-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laduron S, Deplus R, Zhou S, Kholmanskikh O, Godelaine D, De Smet C, Hayward SD, Fuks F, Boon T, De Plaen E. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 2004;32:4340–4350. doi: 10.1093/nar/gkh735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem. 1998;273:33048–33053. doi: 10.1074/jbc.273.49.33048. [DOI] [PubMed] [Google Scholar]
- Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O'Neill MA, Wevrick R. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum Mol Genet. 2005;14:627–637. doi: 10.1093/hmg/ddi059. [DOI] [PubMed] [Google Scholar]
- López-Sánchez N, González-Fernández Z, Niinobe M, Yoshikawa K, Frade JM. Single mage gene in the chicken genome encodes CMage, a protein with functional similarities to mammalian type II Mage proteins. Physiol Genomics. 2007;30:156–171. doi: 10.1152/physiolgenomics.00249.2006. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997;6:1873–1878. doi: 10.1093/hmg/6.11.1873. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Usami M, Aizawa T, Yoshikawa K. A novel brain-specific mRNA encoding nuclear protein (necdin) expressed in neurally differentiated embryonal carcinoma cells. Biochem Biophys Res Commun. 1991;178:291–296. doi: 10.1016/0006-291x(91)91812-q. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Taniura H, Uetsuki T, Yoshikawa K. Necdin acts as a transcriptional repressor that interacts with multiple guanosine clusters. Gene. 2001;272:173–179. doi: 10.1016/s0378-1119(01)00544-3. [DOI] [PubMed] [Google Scholar]
- McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JL, Couch JA, Schmalfuss I, He G, Liu Y, Driscoll DJ. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in Prader-Willi syndrome. Am J Med Genet A. 2007;143:476–483. doi: 10.1002/ajmg.a.31508. [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, Rodolfo M, Schneider C. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A. 2006;103:11160–11165. doi: 10.1073/pnas.0510834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D'Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller JM, Isele U, Metzger E, Rempel A, Moser M, Pscherer A, Breyer T, Holubarsch C, Buettner R, Schüle R. FHL2, a novel tissue-specific coactivator of the androgen receptor. EMBO J. 2000;19:359–369. doi: 10.1093/emboj/19.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P, Cremer H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet. 2000;9:3101–3110. doi: 10.1093/hmg/9.20.3101. [DOI] [PubMed] [Google Scholar]
- Nakada Y, Taniura H, Uetsuki T, Inazawa J, Yoshikawa K. The human chromosomal gene for necdin, a neuronal growth suppressor, in the Prader-Willi syndrome deletion region. Gene. 1998;213:65–72. doi: 10.1016/s0378-1119(98)00206-6. [DOI] [PubMed] [Google Scholar]
- Niinobe M, Koyama K, Yoshikawa K. Cellular and subcellular localization of necdin in fetal and adult mouse brain. Dev Neurosci. 2000;22:310–319. doi: 10.1159/000017455. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Sakoda JY, Yoshikawa K. Drosophila MAGE controls neural precursor proliferation in postembryonic neurogenesis. Neuroscience. 2008;154:572–581. doi: 10.1016/j.neuroscience.2008.03.075. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Ren J, Wevrick R, Greer JJ. Developmental abnormalities of neuronal structure and function in prenatal mice lacking the Prader-Willi syndrome gene necdin. Am J Pathol. 2005;167:175–191. doi: 10.1016/S0002-9440(10)62964-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Lee S, Pagliardini S, Gérard M, Stewart CL, Greer JJ, Wevrick R. Absence of Ndn, encoding the Prader-Willi syndrome-deleted gene necdin, results in congenital deficiency of central respiratory drive in neonatal mice. J Neurosci. 2003;23:1569–1573. doi: 10.1523/JNEUROSCI.23-05-01569.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto J, Miura T, Shimamoto K, Horio Y. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004;556:281–286. doi: 10.1016/s0014-5793(03)01444-3. [DOI] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL, Verdi JM, Barker PA. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–288. doi: 10.1016/s0896-6273(00)00036-2. [DOI] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmüller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazaki R, Nishimura I, Yoshikawa K. Necdin is required for terminal differentiation and survival of primary dorsal root ganglion neurons. Exp Cell Res. 2002;277:220–232. doi: 10.1006/excr.2002.5558. [DOI] [PubMed] [Google Scholar]
- Taniura H, Taniguchi N, Hara M, Yoshikawa K. Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J Biol Chem. 1998;273:720–728. doi: 10.1074/jbc.273.2.720. [DOI] [PubMed] [Google Scholar]
- Taniura H, Matsumoto K, Yoshikawa K. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem. 1999;274:16242–16248. doi: 10.1074/jbc.274.23.16242. [DOI] [PubMed] [Google Scholar]
- Taniura H, Kobayashi M, Yoshikawa K. Functional domains of necdin for protein-protein interaction, nuclear matrix targeting, and cell growth suppression. J Cell Biochem. 2005;94:804–815. doi: 10.1002/jcb.20345. [DOI] [PubMed] [Google Scholar]
- Tennese AA, Gee CB, Wevrick R. Loss of the Prader-Willi syndrome protein necdin causes defective migration, axonal outgrowth, and survival of embryonic sympathetic neurons. Dev Dyn. 2008;237:1935–1943. doi: 10.1002/dvdy.21615. [DOI] [PubMed] [Google Scholar]
- Uetsuki T, Takagi K, Sugiura H, Yoshikawa K. Structure and expression of the mouse necdin gene. Identification of a postmitotic neuron-restrictive core promoter. J Biol Chem. 1996;271:918–924. doi: 10.1074/jbc.271.2.918. [DOI] [PubMed] [Google Scholar]
- Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, Miura M, Yoshikawa K. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19:6955–6964. doi: 10.1523/JNEUROSCI.19-16-06955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaghefi H, Neet KE. Deacetylation of p53 after nerve growth factor treatment in PC12 cells as a post-translational modification mechanism of neurotrophin-induced tumor suppressor activation. Oncogene. 2004;23:8078–8087. doi: 10.1038/sj.onc.1207953. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, Chen J. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Matsuzawa H, Uchiyama M, Kwee IL, Nakada T. Brain developmental abnormalities in Prader-Willi syndrome detected by diffusion tensor imaging. Pediatrics. 2006;118:e442–e448. doi: 10.1542/peds.2006-0637. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]