

Abstract
Neurotrophic factors, including glial cell line-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF), promote survival of midbrain dopaminergic neurons, but the death pathways activated in the dopaminergic neurons by deprivation of these factors are poorly studied. We show here that deprivation of GDNF or BDNF triggers a novel mitochondria-independent death pathway in the cultured embryonic dopaminergic neurons: cytochrome c was not released from the mitochondria to cytosol, proapoptotic protein Bax was not activated, and overexpressed Bcl-xL did not block the death. Caspases were critically required, because the death was completely blocked by caspase inhibitor BAF [boc-aspartyl(OMe)-fluoromethylketone] and overexpression of dominant-negative mutants of caspase-9, -3, and -7 significantly blocked the death. Also, the death receptor pathway was involved, because blockage of caspase-8 or FADD (Fas-associated protein with death domain), an adapter required for caspase-8 activation, inhibited death induced by GDNF or BDNF deprivation. Ligation of Fas by agonistic anti-Fas antibody induced apoptosis in the GDNF- or BDNF-maintained neurons, and inhibition of Fas by Fas-Fc chimera blocked the death of GDNF- or BDNF-deprived neurons, whereas FAIML (long isoform of Fas apoptosis inhibitory molecule) could control the activity of Fas in the dopaminergic neurons.
Keywords: dopaminergic neurons; GDNF, BDNF; apoptosis; caspases; Fas
Introduction
Most neuronal populations undergo a period of ontogenetic death during which the initially overproduced neurons are reduced to the final number by target-derived neurotrophic factors (Barde, 1989; Oppenheim, 1991). The intrinsic death program of the neurons is suppressed by neurotrophic factors or, conversely, released in their absence (Johnson and Deckwerth, 1993; Chang et al., 2002). The molecular nature of this program is, however, poorly described for different neuronal types.
Survival of the midbrain dopaminergic (DA) neurons that degenerate in Parkinson's disease is potently promoted by glial cell line-derived neurotrophic factor (GDNF) in vitro (Lin et al., 1993; Burke et al., 1998), in vivo (Oo et al., 2003, 2005), and in several models of Parkinson's disease (Airaksinen and Saarma, 2002; Bespalov and Saarma, 2007; Lindholm et al., 2007). GDNF binds to coreceptor GFRα1, and this complex activates receptor tyrosine kinase Ret (Bespalov and Saarma, 2007). Genetic manipulations of Ret in the DA neurons (Granholm et al., 2000; Jain et al., 2006; Kramer et al., 2007; Mijatovic et al., 2007) have given controversial results whether and/or when Ret physiologically regulates survival/death of DA neurons. Treatment of Parkinson's patients with GDNF has also been contradictory, because some studies reported considerable improvement (Gill et al., 2003; Slevin et al., 2005), whereas others did not (Lang et al., 2006). These contradictions require further studies.
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, also promotes survival of DA neurons (Krieglstein et al., 1996; Baquet et al., 2005) but via a different receptor tyrosine kinase, TrkB. The physiological role of BDNF in the ontogenetic (Kramer et al., 2007) or pathological (Baquet et al., 2005; Sun et al., 2005) death/survival of DA neurons is poorly understood.
Classically, apoptosis occurs via either death receptor or mitochondrial pathway (Danial and Korsmeyer, 2004; Thorburn, 2004; Riedl and Salvesen, 2007). Ligated death receptors recruit and activate apical pro-caspase-8 via adapters as Fas-associated protein with death domain (FADD) (Peter and Krammer, 2003; Peter et al., 2007). In the mitochondrial pathway, activated proapoptotic proteins (e.g., Bax) release proteins (including cytochrome c) from the mitochondria to the cytosol, leading to the assembly of an apoptosome and activation of apical caspase-9. Apical caspases cleave and activate executionary caspase-3, -6, and -7, which ultimately demolish the cell (Shacka and Roth, 2006; Riedl and Salvesen, 2007). Some caspases (e.g., caspase-2) are activated via a different but poorly characterized mechanism (Troy et al., 2000; Baliga et al., 2004). Also, some nonclassical caspase-dependent apoptotic pathways have been described (Mehlen et al., 1998; Bredesen et al., 2006).
We recently showed that in the GDNF-deprived sympathetic neurons, caspase-2 and -7 are activated via a novel pathway without mitochondria and death receptors: Bax is not translocated to the mitochondria; cytochrome c is not released to the cytosol and caspase-9 and -3, but also caspase-8 and FADD are not involved (Yu et al., 2003). Here, we addressed the death pathways in GDNF- and BDNF-deprived DA neurons. In both cases, the neurons died via a nonclassical apoptotic pathway in which death receptors and caspases, but not mitochondria, were activated.
Materials and Methods
Cultures of 13-d-old mouse ventral mesencephalon and survival assays.
The midbrain floors were dissected from the ventral mesencephali of 13-d-old NMRI strain mouse embryos. Tissues were incubated with 0.5% trypsin (ICN Biomedical) in HBSS (Ca2+/Mg2+ free) (Invitrogen) for 15 min at 37°C, then mechanically dissociated using a large fire-polished Pasteur pipette. For survival assays, the cells were then plated onto the culture dishes coated with poly-l-ornithine (Sigma-Aldrich). To get the equal cell number for all experimental points, only the microisland areas of standard size (3 mm), scratched to the dishes, were coated. Equal volumes of cell suspension (4 μl, containing 1000–3000 neurons) were plated onto these areas. The cells were grown in DMEM/F12 medium (Invitrogen) containing N2 supplement (Invitrogen) for 5 d with GDNF (100 ng/ml) (Amgen) or BDNF (50 ng/ml) (R&D Systems). To deprive GDNF, the cultures were washed three times with normal medium, and function-blocking anti-GDNF antibodies (Amgen) were added. To remove BDNF, the cultures were gently washed three times with BDNF-free medium. The compounds of interest were then added and cultures grown for 3 additional days. To visualize the dopaminergic neurons, the cultures were stained with tyrosine hydroxylase (TH) antibodies (Millipore Bioscience Research Reagents), and the TH-positive neurons were microscopically counted by a “blind” experimenter not aware of the identity of the experimental groups. The following compounds were used: broad-range caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone (BAF) (Merck Biosciences) at 50 μm, staurosporine (Cell Signaling Technology) at 300 nm, mouse Fas/TNFRSF6/Fc Chimera (Fas-Fc) (R&D Systems) at 100 ng/ml, and anti-Fas antibody Jo2 (Cell Signaling Technology) at 100 ng/ml. Function-blocking anti-TrkC antibody (#AF1404; 100 ng/ml; R&D Systems) was used as a control for Jo2, because it does not contain toxic preservatives.
Transfections.
DA neurons were transfected as published previously (Yu and Arumäe, 2008). Briefly, GDNF- and BDNF-dependent cultures were transfected with calcium phosphate coprecipitation technique using the Ca-P kit (Invitrogen) according to the manufacturer's instructions. Plasmids of interest at 1 μg per well were cotransfected with reporter plasmid for enhanced green fluorescent protein (EGFP) (Clontech) at 0.2 μg per well. At that ratio, virtually all EGFP-expressing neurons coexpressed the cotransfected plasmid. The relevant empty vectors (pCR3.1, pMV2B, or pCDNA3) without the insert were always included as mock controls. The factors were deprived the next day after transfection. Fluorescent (EGFP-expressing) neurons were “blindly” counted from each dish immediately after factor deprivation (initial) and at the third day (final). The results were expressed as a percentage of initial fluorescent neurons. To exclude EGFP-expressing non-DA neurons, the cultures were stained with TH antibodies at the end of experiment, and the number of EGFP-positive, TH-negative neurons was subtracted from the initial and final number. All experiments were repeated at least three times on the independent cultures.
The expression plasmids for Bcl-xL, Ku70, dominant-negative mutants of caspase-2, -3, -7, -8, and -9, and FADD have been described previously (Yu et al., 2003). Plasmid for mitochondrially targeted yellow fluorescent protein (mtYFP) was a kind gift from Dr. A. Miyawaki (The Institute of Physical and Chemical Research, Hirosawa, Wako-city, Japan). Fas and long isoform of Fas apoptosis inhibitory molecule (FAIML) plasmids were kindly provided by Prof J. Comella (Universitat de Lleida-Hospital Universitari Arnau de Vilanova, Lleida, Spain)
Immunocytochemistry.
The neurons were grown on glass coverslips, fixed with 4% paraformaldehyde in PBS, permeabilized with 0.3% Triton X-100 (Sigma-Fluka), and blocked with 10% horse serum (Invitrogen) in TBS containing 0.3% Triton X-100. The following antibodies were used: cytochrome c (#556432; BD Bioscience); TH (Millipore Bioscience Research Reagents); Bax (#554082; BD Pharmingen), Pitx3 (Zymed), Fas and its immunizing peptide (M-20; Santa Cruz Biotechnology), FasL and its immunizing peptide (N-20; Santa Cruz Biotechnology), and cyanine 3 (Cy3)- or Alexa-488-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). The specimens were mounted in Vectashield (Vector Laboratories). The images were captured at room temperature with Leica TCS SP2 Spectral confocal and multiphoton system (Leica): an inverted microscope (model DM IRE2); HCX PL APO [objective, 63×; numerical aperture, 1.4–0.6 (oil)]; and TCS SP2 scan unit and interactive LCS software. Individual images were identically processed with Photoshop CS3 (Adobe Systems).
Reverse transcription-PCR.
Dopaminergic neurons were cultured for 5 d in vitro (DIV) and deprived or not deprived of the factors for 24 h with or without BAF. Total RNA from dopaminergic neurons, 3T3 [positive control, expressing both Fas and Fas ligand (FasL)] and Chinese hamster ovary (negative control) cells was isolated by Micro Scale RNA Isolation kit (Ambion) including DNase I treatment, according to the instruction manual. Total RNA (200–500 ng) were reverse transcribed using the Transcriptor First Strand cDNA Synthesis Kit (Roche), and the cDNA was used directly for PCR. Two microliters of cDNA of each sample were amplified by PCR with the Expand High Fidelity PCR System (Roche). The following primers were used. Fas: forward, 5′-GTGTTCGCTGCGCCTC-3′; reverse, 5′-GGTTCTGCGACATTCGGC-3′ (Lesné et al., 2002); Fas ligand: forward, 5′-TTTCATGGTTCTGGTGGCTCTGGT-3′; reverse, 5′-AGCGGTTCCATATGTGTCTTCCCA-3′. PCR was performed after an initial denaturation for 5 min at 95°C, followed by 36 cycles of 30 s denaturation at 95°C, 30 s annealing at 60°C, and 30 s extension at 72°C. As the negative controls, reverse transcription (RT) reaction was omitted from the 3T3 sample, or the whole procedure was performed in the water without added cDNA.
Immunoblot and coimmunoprecipitation.
For the immunoblotting, midbrain neurons were grown with GDNF or BDNF for 5 DIV, and lysed in 2% SDS in 125 mm Tris-HCl, pH 6.8. Lysates from the 3T3 cells or cortical cultures of embryonic mice were prepared in the same way. The proteins were separated on the SDS-PAGE and transferred to the filter by standard procedures. The filters were probed with antibodies to FAIML kindly provided by Prof. J. Comella (Segura et al., 2007) and reprobed with antibodies to α-actin (Sigma).
For the coimmunoprecipitation experiments, midbrain neurons were cultured for 5 DIV with GDNF or BDNF and transfected with expression plasmid for mouse Fas together with EGFP as published previously (Yu and Arumäe, 2008). After 2 d, the cells were lysed as published by Segura et al. (2007). Fas was precipitated by biotin-coupled anti-Fas antibody (BD Biosciences) or control biotin-coupled anti-GFRα2 antibody (BD Biosciences), and the immunocomplexes were collected with NeutrAvidin (Thermo Fisher Scientific). SDS-PAGE was performed right after immunoprecipitation.
Results
Embryonic dopaminergic neurons die in culture because of GDNF or BDNF deprivation
Because the death of GDNF- or BDNF-deprived DA neurons had not been described previously, we first characterized this process. Deprivation of 5-d-old GDNF- or BDNF-dependent 13-d-old midbrain floor cultures of the respective factors for 3 d leads to the loss of approximately one-half of the TH-positive neurons in both cases, as normalized to corresponding factor-maintained cultures (Fig. 1 A). The remaining TH-positive neurons survived without neurotrophic support, because their number did not change in the cultures deprived of GDNF or BDNF up to 7 d (data not shown). The number of TH-positive neurons did not change in the factor-maintained cultures (data not shown); thus, the cultures did not deteriorate during the experiments. The death was completely blocked by caspase inhibitor BAF (Fig. 1 A). Trophic factor deprivation may downregulate the TH that we used as a marker to count the results. We therefore immunostained the cultures with the antibodies to paired-like homeodomain transcription factor-3 (Pitx3), another marker for the DA neurons (Nunes et al., 2003). Again, approximately one-half of the Pitx3-expressing neurons were lost in the GDNF- or BDNF-deprived cultures (Fig. 1 B). Thus, two independent markers showed the same reduction in the number of DA neurons. Moreover, this reduction was blocked by caspase inhibitor. It is not probable that caspase inhibition blocks only the downregulation of TH but not the death of DA neurons. We conclude that GDNF or BDNF deprivation led to death of the DA neurons rather than loss of the markers in neurons that did not die. GDNF- and BDNF-dependent neurons were analyzed in parallel throughout the study and gained the identical treatment.
Figure 1.

Dopaminergic neurons die caspase dependently by GDNF or BDNF deprivation. Thirteen-day-old mouse dopaminergic neurons were plated at similar density and grown with 100 ng/ml GDNF or 50 ng/ml BDNF for 5 d. Neurotrophic factors were then removed (GDNF−, BDNF−) or not removed (GDNF+, BDNF+) for 3 d in the presence or absence of caspase inhibitor BAF (50 μm). A, B, The number of neurons positive for TH (A) or Pitx3 (B) from the factor-deprived cultures is expressed as a percentage of corresponding factor-maintained cultures. The means ± SEM of three independent experiments are shown. Statistical significance of the differences was estimated by Student's t test. The null hypothesis was rejected at p < 0.05.
Mitochondrial death pathway is not activated in GDNF- and BDNF-deprived dopaminergic neurons
To study the involvement of mitochondrial apoptotic machinery, we first tried to visualize the mitochondria of the DA neurons in our cultures. Because several antibodies to mitochondrial markers did not give satisfactory results (data not shown), we turned to chimeric yellow fluorescent protein (mtYFP) engineered to be specifically targeted to mitochondria (Fig. 2 A). The factor-maintained cells were transfected with mtYFP and immunolabeled the next day with antibodies to cytochrome c. In the double-fluorescent neurons, distribution of the two labels was indistinguishable, showing that cytochrome c antibody indeed stained mitochondria in the healthy neurons. Because such staining labels all the cells in the culture, we also stained mtYFP-transfected cultures with TH antibodies to recognize the pattern of mitochondria in the DA neurons (Fig. 2 A). We then deprived the cultures of GDNF or BDNF for 48 h and double stained the cultures with antibodies to cytochrome c and TH. In these experiments, death of the neurons was blocked by caspase inhibitor BAF to facilitate the assay. In the vast majority of TH-positive neurons of both GDNF- and BDNF-deprived cultures, cytochrome c localization was punctate, as in the neurons maintained with these factors (Fig. 2 B, Table 1). Similar results were obtained at 72 h after neurotrophic factor deprivation (data not shown). As a positive control for cytochrome c release, we treated the cultures with staurosporine (Meuer et al., 2007). Staurosporine dramatically reduced the number of the neurons with punctate mitochondrial localization. Instead, most of the staurosporine-treated neurons showed diffuse cytochrome c staining characteristic of its cytosolic localization (Fig. 2 C). Thus, although the DA neurons can release cytochrome c to the cytosol, this did not happen by withdrawal of GDNF or BDNF.
Figure 2.
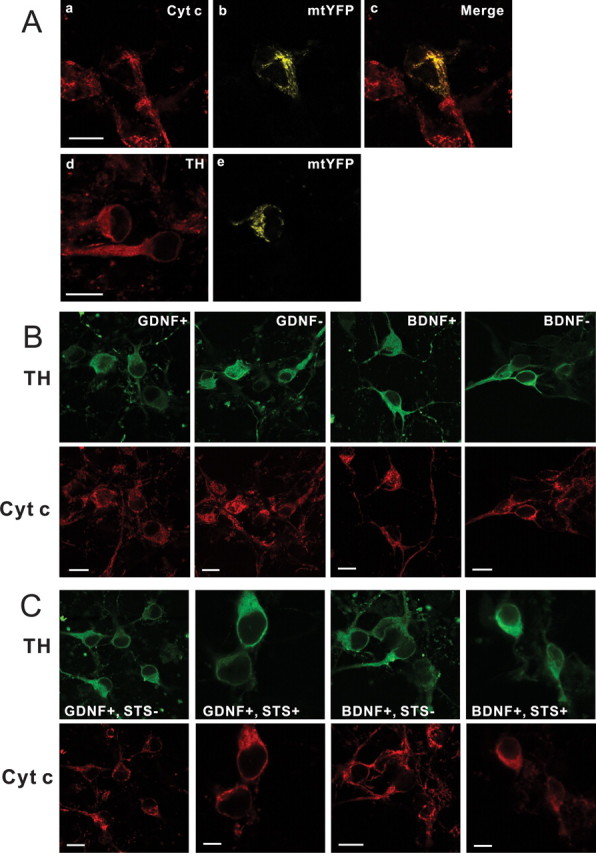
Cytochrome c is not released from the mitochondria of GDNF- or BDNF-deprived dopaminergic neurons. A, Confocal microscopic images of representative GDNF-maintained neurons stained with cytochrome c (Cyt c) antibodies (a) or expressing mitochondrial marker mtYFP (b). c, Colocalization of cytochrome c and mtYFP in the mitochondria. d, e, Mitochondria in the TH-positive neurons (d) are revealed by mtYFP (e). B, Bottom, Representative cytochrome c immunostaining patterns of GDNF- or BDNF-maintained or -deprived neurons. Top, Corresponding TH immunostaining images. The GDNF- or BDNF-maintained (GDNF+, BDNF+) or -deprived (GDNF−, BDNF−) neurons all show strongly punctate immunostaining (mitochondrial localization). C, Typical cytochrome c immunostaining patterns of GDNF- or BDNF-maintained neurons treated or not treated with staurosporine (STS). Note the diffuse cytosolic localization of cytochrome c in the staurosporine-treated cultures, whereas it shows punctate mitochondrial localization in the factor-maintained neurons. Scale bars, 10 μm.
Table 1.
Quantitation of the TH-positive neurons with punctate pattern of cytochrome c in the cultures maintained with or deprived of GDNF or BDNF for 48 h
| Factor | % Punctate Cyt c |
|---|---|
| GDNF+ | 99.8 ± 0.1 (1367 neurons) |
| GDNF− | 98.1 ± 0.6 (1146 neurons) |
| BDNF+ | 99.7 ± 0.1 (1173 neurons) |
| BDNF− | 97.7 ± 0.3 (795 neurons) |
The number of TH-positive neurons with a punctate pattern of cytochrome c (Cyt c) is expressed as the percentage of all TH-positive neurons in a given culture. The results are the means ± SEM of four independent experiments, each in three parallels. Collective absolute numbers are shown in the parentheses.
To study the involvement of Bax, we overexpressed Ku70, a protein that binds the N terminus of Bax, thereby inhibiting its translocation to mitochondria (Sawada et al., 2003a,b; Yu et al., 2003). Ku70 had no effect on the death of GDNF- or BDNF-deprived neurons (Fig. 3). Immunostaining of the GDNF- or BDNF-deprived, BAF-saved cultures with antibodies to Bax also revealed its cytosolic, not punctate, localization (data not shown). Overexpressed antiapoptotic Bcl-2 family member Bcl-xL, which efficiently blocks the mitochondrial death pathway (Yu et al., 2003), did not block the death of GDNF- or BDNF-deprived DA neurons. Collectively, these data show that mitochondrial pathway is not activated in the GDNF- or BDNF-deprived DA neurons.
Figure 3.

Overexpression of antiapoptotic proteins in the GDNF- or BDNF-deprived dopaminergic neurons. GDNF- or BDNF-maintained neurons were transfected with expression plasmids for Ku70, Bcl-xL, or empty pMV2B plasmid (vector) (together with the plasmid for EGFP) and maintained with (GDNF+, BDNF+) or without (GDNF−, BDNF−) neurotrophic factors for 3 d. The numbers of fluorescent neurons counted at the end of the experiment were normalized to those counted at the factor deprivation. Transfected non-TH neurons, identified by post hoc TH immunostaining, were subtracted from the counts. The mean ± SEM of four independent experiments is shown.
Caspases are required for the death of GDNF- or BDNF-deprived dopaminergic neurons
As shown on Figure 1, caspase inhibitor BAF completely blocked the death of GDNF- or BDNF-deprived DA neurons. To study which caspases are required, we overexpressed dominantly blocking isoforms of caspase-2, -3, -7, and -9 in the GDNF- or BDNF-deprived, but also the factor-maintained, neurons. As shown on Figure 4, blocking of caspase-3, caspase-7, and caspase-9 significantly inhibited the death of GDNF- or BDNF-deprived neurons, whereas blocking of caspase-2 had no statistically significant effect. The constructs did not affect the survival of neurotrophic factor-maintained neurons (Fig. 4).
Figure 4.

Inhibition of caspases in the GDNF- or BDNF-deprived dopaminergic neurons. GDNF- or BDNF-deprived (GDNF−, BDNF−) or -maintained (GDNF+, BDNF+) neurons were transfected with dominant-negative (DN) mutants of indicated caspases (together with the plasmid for EGFP) and maintained with or without neurotrophic factors for 3 d. The numbers of fluorescent neurons counted at the end of the experiment were normalized to those counted at the factor deprivation. Transfected non-TH neurons, identified by post hoc TH immunostaining, were subtracted from the counts. The means ± SEM of four independent experiments are shown. Data of each DN caspase were compared with control plasmid pCDNA3 (vector) by one-way ANOVA and post hoc Dunnett's t test. The null hypothesis was rejected at p < 0.05.
The death receptor pathway is activated in GDNF- and BDNF-deprived dopaminergic neurons
The above data show that death of GDNF- and BDNF-deprived DA neurons requires caspase activation that occurs independently of mitochondria. To check the involvement of death receptors, a second major apoptotic pathway, we first studied the Fas receptor and FasL expression in our cultures. By RT-PCR from the whole cultures, the fragments specific for both Fas and FasL were clearly observed (Fig. 5 B). However, the levels of these fragments did not change significantly by factor deprivation for 24 h (Fig. 5 B). Antibodies to Fas or FasL stained virtually all neurons in the culture, including the TH-positive DA neurons (Fig. 5 A). In both cases, neuronal somata as well as neurites were immunolabeled. In the control stainings, the immunoreactivity for Fas or FasL was strongly blocked by preincubation of the antibodies with cognate-blocking peptides, but not with noncognate peptides (Fig. 5 A). To further verify the specificity of the antibodies, we transfected the 3T3 cells with expression plasmids for Fas or FasL together with the plasmid for EGFP, and stained these cultures with respective antibodies. Anti-Fas antibodies stained the EGFP-positive Fas-transfected cells, and anti-FasL antibodies stained the EGFP-positive FasL-transfected cells, but not vice versa. We conclude that the DA neurons (and also other neurons) in our cultures express both Fas and FasL.
Figure 5.
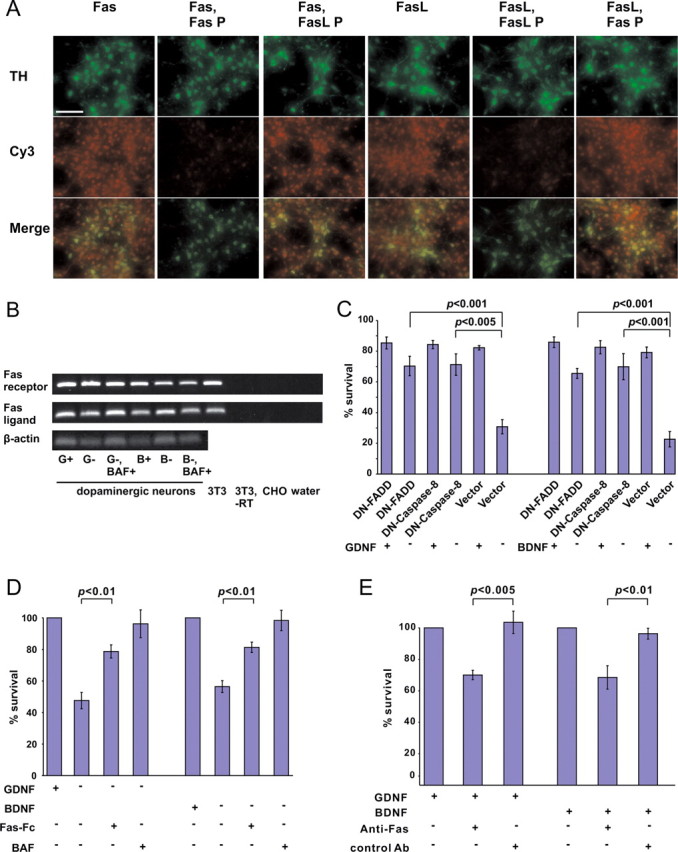
Activation of Fas-like receptor is required for the death of GDNF- or BDNF-deprived neurons. A, Virtually all neurons, including the TH-positive ones, express both Fas and FasL. GDNF-maintained midbrain cultures (5 DIV) were costained with antibodies to TH and either Fas or FasL (both revealed by Cy3-conjugated secondary antibodies). Specificity of immunostaining was checked by preincubation of the antibodies with the blocking peptide to either Fas (Fas P) or FasL (FasL P). Scale bar, 10 μm. B, RT-PCR analysis of Fas and FasL transcripts in the midbrain cultures. Midbrain cultures (5 DIV) were deprived or not deprived of GDNF (G) or BDNF (B) for 24 h in the presence or absence of caspase inhibitor BAF. Shown are 479 bp fragments of Fas and 247 bp fragments of FasL. 3T3 cells expressing both Fas and FasL served as a positive control. No fragments were obtained from the Chinese hamster ovary (CHO) cells. The fragments were also not obtained when RT reaction was omitted from the 3T3 samples (−RT), or when the reactions were performed without added cDNAs (water). C, GDNF- or BDNF-dependent neurons were transfected with expression plasmids for dominant-negative (DN) caspase-8, FADD, or empty pCR3.1 plasmid (vector) (together with the plasmid for EGFP) and maintained with or without neurotrophic factors for 3 d. The numbers of fluorescent neurons counted at the end of the experiment were normalized to those counted at the factor deprivation. Transfected non-TH neurons, identified by post hoc TH immunostaining, were subtracted from the counts. Statistical comparison of the means between expression plasmids and vector was performed by one-way ANOVA and post hoc Dunnett's t test. The null hypothesis was rejected at p < 0.05. D, Dopaminergic neurons were selected with GDNF or BDNF for 5 d, then cultured without neurotrophic factors in the presence or absence of Fas-Fc for an additional 3 d. Factor-deprived cultures in the presence of caspase inhibitor BAF served as positive controls. The cultures were then stained with TH antibodies, and TH-positive neurons were counted and expressed as a percentage of corresponding factor-maintained neurons. The mean ± SEM of three independent experiments is shown. Statistical significance of the differences was estimated by one-way ANOVA and post hoc Tukey's honestly significant difference test. The null hypothesis was rejected at p < 0.05. E, Dopaminergic neurons were selected with GDNF or BDNF for 5 d, then treated with agonistic Jo2 anti-Fas antibody or control anti-TrkC antibody for an additional 3 d. The cultures were then stained with TH antibodies. TH-positive neurons were counted and expressed as a percentage of corresponding factor-maintained neurons. The mean ± SEM of four independent experiments is shown. Statistical significance of the difference between groups treated with anti-Fas antibody and those treated with control antibody was estimated by Student's t test. The null hypothesis was rejected at p < 0.05.
We then transfected the cultures with the plasmid for dominantly blocking mutant of caspase-8. Dominant-negative mutants of FADD, an adapter that links pro-caspase-8 to most of the death receptors (Peter and Krammer, 2003; Peter et al., 2007), were transfected as well. Both constructs significantly blocked the death of GDNF- or BDNF-deprived DA neurons (Fig. 5 B), suggesting an activation of Fas-like receptor. Fas-Fc, a chimeric protein that prevents interaction of Fas and FasL in motoneurons (Raoul et al., 2006), significantly blocked the death of both GDNF- and BDNF-deprived neurons (Fig. 5 C). Moreover, ligation of Fas by agonistic Jo2 anti-Fas antibody (but not the control antibody) killed a significant portion of GDNF- or BDNF-maintained DA neurons (Fig. 5 D), suggesting the presence of functional Fas-like system. Interestingly, Jo2 antibody was reported not to kill the NGF-maintained sympathetic neurons (Putcha et al., 2002). Thus, the Fas-like death receptors are activated in the GDNF- or BDNF-deprived DA neurons.
Recently it was shown that FAIML associates with Fas and prevents the apoptotic activity of Fas in the neurons (Segura et al., 2007). FAIML is also expressed in our midbrain cultures as shown by immunoblotting (Fig. 6 A), but its expression in the DA neurons among other neurons in the mixed cultures could not be demonstrated, because the antibodies did not work in the immunocytochemistry. Repeated immunoblot experiments with the GDNF- or BDNF-deprived cultures did not reveal significant changes in the level of FAIML. This is, however, expected because the putative changes of FAIML in the minor fraction of apoptotic neurons could be overwhelmed by FAIML in the large number of nonresponsive neurons. Endogenous FAIML was coprecipitated with transfected Fas from the GDNF- or BDNF-maintained midbrain cultures (Fig. 6 B) but not from 3T3 cells (data not shown), showing physical association of these proteins. Overexpressed Fas killed significant number of GDNF- or BDNF-maintained DA neurons, and this was blocked by cotransfected FAIML (Fig. 6 C). In turn, overexpressed FAIML significantly blocked the death of GDNF- or BDNF-deprived DA neurons, and this was abolished by cotransfected Fas (Fig. 6 C). Thus, FAIML and Fas can functionally interact with each other in the DA neurons.
Figure 6.

FAIML associates with Fas and inhibits its apoptotic activity. A, Immunoblot of the midbrain cultures maintained with GDNF or BDNF for 5 DIV. Lysates of mouse 3T3 cells or mouse cortex were included as negative or positive controls, respectively. Reprobing of the filter with antibodies to actin shows the total protein loading. B, Coimmunoprecipitation of FAIML with Fas. GDNF- or BDNF-maintained midbrain cultures (5 DIV) were transfected with expression plasmid for Fas for 2 d and immunoprecipitated with biotin-anti-Fas antibody or control antibody (biotin-GFRα2). The filter was probed for coprecipitated endogenous FAIML (∼23 kDa) and reprobed for Fas (about 45 kDa). IP, Immunoprecipitation. C, FAIML and Fas interact functionally in the DA neurons. GDNF- or BDNF-maintained neurons were transfected with expression plasmids for FAIML, Fas, FAIML and Fas together (FAIML/Fas), or empty pcDNA3 plasmid (vector) (all together with the plasmid for EGFP) and maintained with (GDNF+, BDNF+) or without (GDNF−, BDNF−) neurotrophic factors for 3 d. The numbers of fluorescent neurons counted at the end of the experiment were normalized to those counted at the factor deprivation. Transfected non-TH neurons, identified by post hoc TH immunostaining, were subtracted from the counts. The mean ± SEM of four independent experiments is shown. Statistical significance of the differences was estimated by one-way ANOVA and post hoc Tukey's honestly significant difference test. The null hypothesis was rejected at p < 0.05.
Discussion
We have previously shown that GDNF-deprived sympathetic neurons activate caspases via a novel mitochondria-independent death pathway (Yu et al., 2003). Here we show that also the embryonic DA neurons do not activate mitochondrial death pathway by deprivation of GDNF (but also BDNF): Bax is not translocated to the mitochondria and Bax inhibition does not block the death, cytochrome c is not released to the cytosol, and antiapoptotic mitochondrial protein Bcl-xL does not block the death. In the immunostaining experiments, we had to prevent cell death by caspase inhibition, because the changes in the localization of Bax and cytochrome c occur only briefly before death and were almost impossible to detect. The apoptotic mitochondrial changes are believed to occur without caspase involvement (Chang et al., 2002; Gogvadze et al., 2006), but caspase-2 is reported to be activated upstream of mitochondria (Guo et al., 2002; Lassus et al., 2002; Robertson et al., 2004). We cannot exclude that caspase inhibition affected the movement of Bax and cytochrome c in our factor-deprived neurons. However, caspase-2 was not critical for the death of DA neurons. Moreover, experiments with Ku70 or Bcl-xL were performed without caspase inhibitors. We therefore conclude that mitochondria were not activated in the neurotrophic factor-deprived DA neurons.
Caspases were still absolutely required for the death of GDNF- and BDNF-deprived DA neurons. When compared with the GDNF-deprived sympathetic neurons, the involved caspases were completely different: caspase-2 was critical for the death of GDNF-deprived sympathetic neurons but not GDNF-deprived DA neurons. Instead, the DA neurons died via caspase-9, -3, and -7, which were not essential in the sympathetic neurons. How caspase-9 is activated without cytosolic cytochrome c remains an open question. Caspase-9 is activated at the apoptosome by dimerization (Boatright et al., 2003; Pop et al., 2006), but it is also cleaved in the apoptotic cells, which enhances its catalytic activity (Bao and Shi, 2007; Riedl and Salvesen, 2007). We speculate that caspase-8, activated at the death receptors, could cleave and activate caspase-9 in our neurons, as recently shown in other systems (McDonnell et al., 2003; Gyrd-Hansen et al., 2006). Indeed, blockage of caspase-8 prevented the death of the neurons, suggesting that caspase-9 is activated downstream of caspase-8. Our attempts to visualize cleavage products of the caspases by Western blotting failed, most probably because of the small amount of the material available. Immunostaining of the cultures with antibodies to activated caspases also did not give consistent results in our hands. Thus, further studies are required to understand caspase-9 activation in our model.
Death receptors were activated in the GDNF- and BDNF-deprived DA neurons, because the death was significantly blocked by inhibition of caspase-8 or FADD, but also with overexpression of Fas inhibitor FAIML. Fas agonists and antagonists as well as the PCR results strongly suggest the existence of functional Fas-like receptor on the surface of the DA neurons, and its activation by GDNF or BDNF deprivation. Most probably the Fas and FasL interact via neuritic contacts that are extensive in our dense cultures, as also suggested for trophic factor-deprived motoneurons (Raoul et al., 1999; Ugolini et al., 2003; Raoul et al., 2006). Fas and FasL were constitutively present in the midbrain cultures, suggesting that an inhibitory system should prevent the unwanted activation of Fas. Indeed, we found that a neuron-specific inhibitory protein FAIML (Segura et al., 2007) was expressed in the midbrain cultures. FAIML interacts with Fas, both physically, as revealed by coimmunoprecipitation, and functionally, as revealed by transfection of DA neurons. Thus, the findings of Segura et al. (2007) were repeated in our model. It is tempting to speculate that in the healthy DA neurons, Fas could perform some nonapoptotic functions (Peter et al., 2007) with its apoptotic activity blocked by FAIML, whereas FAIML could be degraded or removed from Fas by apoptotic stimulus such as neurotrophic factor deprivation.
We found that de-ligation of Ret activates different death pathways in sympathetic and DA neurons, whereas the same pathways were activated in the DA neurons by de-ligation of Ret and TrkB. We included BDNF for comparison to GDNF expecting different death pathways, because it was in the sympathetic neurons deprived of NGF (de-ligation of TrkA that is homologous to TrkB) (Yu et al., 2003). This was, however, not the case. The neurons can indeed sense the lack of neurotrophic support in many ways, depending on the neuronal type and/or the deprived factor.
Thus, we show that the DA neurons have their own way to respond to neurotrophic factor deprivation, in which caspases are activated via death receptors and without mitochondria. Whether and when this pathway is activated in vivo remains to be studied. It is controversial whether GDNF actually controls the number of DA neurons during development (Janec and Burke, 1993; Jackson-Lewis et al., 2000; Krieglstein, 2004). Indeed, function-blocking antibodies to GDNF reduced the death of neonatal DA neurons in vivo (Oo et al., 2003, 2005), but genetic manipulations of Ret affected adult rather than neonatal DA neurons (Granholm et al., 2000; Jain et al., 2006; Kramer et al., 2007; Mijatovic et al., 2007). More studies, including mice with conditional deletion of GDNF in the striatum, are required to solve the issue. Also, the activation of Fas in the DA neurons during ontogenetic death is not yet studied in vivo. However, death receptors are activated in Parkinson's disease. Indeed, human postmortem studies have revealed increased number of DA neurons positive for activated caspase-8 (Hartmann et al., 2001), as well as selective loss of FADD-positive DA neurons (Hartmann et al., 2002). Caspase-8 also cleaves and inactivates parkin, a gene associated with several familial forms of the disease (Kahns et al., 2003). Our data showing activation of death receptors by GDNF (and BDNF) removal suggest that suppression of death receptors could be one mechanism by which GDNF improves the symptoms of Parkinson's disease (Gill et al., 2003) (but see Lang et al., 2006) and could also be a potential target for the treatment of this disease.
Footnotes
This work was supported by the Academy of Finland, Finnish Centre of Excellence Programme 44896, an Academy of Finland research fellow grant (U.A.), and a Sigrid Jusélius Foundation grant (M.S.). We thank Prof. Klaus Unsicker and Jutta Fey for teaching the technique of midbrain cultures. We acknowledge Amgen for providing research materials. We acknowledge Jaan-Olle Andressoo for useful inventions in the culture technique and Maili Jakobson for providing help with the RT-PCR. We are greatly thankful to Prof. Joan Comella for kindly providing research tools for FAIML and for useful discussions. We acknowledge Dr. Juha Partanen for help with Pitx3 antibodies.
References
- Airaksinen and Saarma, 2002.Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Baliga et al., 2004.Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004;11:1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- Bao and Shi, 2007.Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2007;14:56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- Baquet et al., 2005.Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde, 1989.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- Bespalov and Saarma, 2007.Bespalov MM, Saarma M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. 2007;28:68–74. doi: 10.1016/j.tips.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Boatright et al., 2003.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Bredesen et al., 2006.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke et al., 1998.Burke RE, Antonelli M, Sulzer D. Glial cell line-derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture. J Neurochem. 1998;71:517–525. doi: 10.1046/j.1471-4159.1998.71020517.x. [DOI] [PubMed] [Google Scholar]
- Chang et al., 2002.Chang LK, Putcha GV, Deshmukh M, Johnson EM., Jr Mitochondrial involvement in the point of no return in neuronal apoptosis. Biochimie. 2002;84:223–231. doi: 10.1016/s0300-9084(02)01372-x. [DOI] [PubMed] [Google Scholar]
- Danial and Korsmeyer, 2004.Danial NN, Korsmeyer SJ. Cell death. Critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Gill et al., 2003.Gill SS, Patel NK, Hotton GR, O'Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- Gogvadze et al., 2006.Gogvadze V, Orrenius S, Zhivotovsky B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim Biophys Acta. 2006;1757:639–647. doi: 10.1016/j.bbabio.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Granholm et al., 2000.Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L, Westphal H, Hoffer B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J Neurosci. 2000;20:3182–3190. doi: 10.1523/JNEUROSCI.20-09-03182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo et al., 2002.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- Gyrd-Hansen et al., 2006.Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Høyer-Hansen M, Elling F, Wallach D, Flavell R, Kroemer G, Nylandsted J, Jäättelä M. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol. 2006;26:7880–7891. doi: 10.1128/MCB.00716-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann et al., 2001.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–2255. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann et al., 2002.Hartmann A, Mouatt-Prigent A, Faucheux BA, Agid Y, Hirsch EC. FADD: a link between TNF family receptors and caspases in Parkinson's disease. Neurology. 2002;58:308–310. doi: 10.1212/wnl.58.2.308. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis et al., 2000.Jackson-Lewis V, Vila M, Djaldetti R, Guegan C, Liberatore G, Liu J, O'Malley KL, Burke RE, Przedborski S. Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J Comp Neurol. 2000;424:476–488. doi: 10.1002/1096-9861(20000828)424:3<476::aid-cne6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Jain et al., 2006.Jain S, Golden JP, Wozniak D, Pehek E, Johnson EM, Jr, Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci. 2006;26:11230–11238. doi: 10.1523/JNEUROSCI.1876-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janec and Burke, 1993.Janec E, Burke RE. Naturally occurring cell death during postnatal development of the substantia nigra pars compacta of rat. Mol Cell Neurosci. 1993;4:30–35. doi: 10.1006/mcne.1993.1004. [DOI] [PubMed] [Google Scholar]
- Johnson and Deckwerth, 1993.Johnson EM, Jr, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- Kahns et al., 2003.Kahns S, Kalai M, Jakobsen LD, Clark BFC, Vandenabeele P, Jensen PH. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem. 2003;278:23376–23380. doi: 10.1074/jbc.M300495200. [DOI] [PubMed] [Google Scholar]
- Kramer et al., 2007.Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein, 2004.Krieglstein K. Factors promoting survival of mesencephalic dopaminergic neurons. Cell Tissue Res. 2004;318:73–80. doi: 10.1007/s00441-004-0920-8. [DOI] [PubMed] [Google Scholar]
- Krieglstein et al., 1996.Krieglstein K, Maysinger D, Unsicker K. The survival response of mesencephalic dopaminergic neurons to the neurotrophins BDNF and NT-4 requires priming with serum: comparison with members of the TGF-beta superfamily and characterization of the serum-free culture system. J Neural Transm Suppl. 1996;47:247–258. doi: 10.1007/978-3-7091-6892-9_17. [DOI] [PubMed] [Google Scholar]
- Lang et al., 2006.Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, Brooks DJ, Hotton G, Moro E, Heywood P, Brodsky MA, Burchiel K, Kelly P, Dalvi A, Scott B, Stacy M, Turner D, Wooten VG, Elias WJ, Laws ER, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- Lassus et al., 2002.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- Lesné et al., 2002.Lesné S, Blanchet S, Docagne F, Liot G, Plawinski L, MacKenzie ET, Auffray C, Buisson A, Piétu G, Vivien D. Transforming growth factor-beta1-modulated cerebral gene expression. J Cereb Blood Flow Metab. 2002;22:1114–1123. doi: 10.1097/00004647-200209000-00009. [DOI] [PubMed] [Google Scholar]
- Lin et al., 1993.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Lindholm et al., 2007.Lindholm P, Voutilainen MH, Laurén J, Peränen J, Leppänen VM, Andressoo JO, Lindahl M, Janhunen S, Kalkkinen N, Timmusk T, Tuominen RK, Saarma M. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo . Nature. 2007;448:73–77. doi: 10.1038/nature05957. [DOI] [PubMed] [Google Scholar]
- McDonnell et al., 2003.McDonnell MA, Wang D, Khan SM, Vander Heiden MG, Kelekar A. Caspase-9 is activated in a cytochrome c-independent manner early during TNFalpha-induced apoptosis in murine cells. Cell Death Differ. 2003;10:1005–1015. doi: 10.1038/sj.cdd.4401271. [DOI] [PubMed] [Google Scholar]
- Mehlen et al., 1998.Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, Bredesen DE. The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature. 1998;395:801–804. doi: 10.1038/27441. [DOI] [PubMed] [Google Scholar]
- Meuer et al., 2007.Meuer K, Suppanz IE, Lingor P, Planchamp V, Göricke B, Fichtner L, Braus GH, Dietz GPH, Jakobs S, Bähr M, Weishaupt JH. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 2007;14:651–661. doi: 10.1038/sj.cdd.4402087. [DOI] [PubMed] [Google Scholar]
- Mijatovic et al., 2007.Mijatovic J, Airavaara M, Planken A, Auvinen P, Raasmaja A, Piepponen TP, Costantini F, Ahtee L, Saarma M. Constitutive Ret activity in knock-in multiple endocrine neoplasia type b mice induces profound elevation of brain dopamine concentration via enhanced synthesis and increases the number of TH-positive cells in the substantia nigra. J Neurosci. 2007;27:4799–4809. doi: 10.1523/JNEUROSCI.5647-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes et al., 2003.Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A. 2003;100:4245–4250. doi: 10.1073/pnas.0230529100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo et al., 2003.Oo TF, Kholodilov N, Burke RE. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal glial cell line-derived neurotrophic factor in vivo . J Neurosci. 2003;23:5141–5148. doi: 10.1523/JNEUROSCI.23-12-05141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo et al., 2005.Oo TF, Ries V, Cho J, Kholodilov N, Burke RE. Anatomical basis of glial cell line-derived neurotrophic factor expression in the striatum and related basal ganglia during postnatal development of the rat. J Comp Neurol. 2005;484:57–67. doi: 10.1002/cne.20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim, 1991.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Peter and Krammer, 2003.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Peter et al., 2007.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, Wallach D, Wiltrout RH, Zörnig M, Lynch DH. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Pop et al., 2006.Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22:269–275. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Putcha et al., 2002.Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM., Jr Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–453. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul et al., 1999.Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the fas death receptor. J Cell Biol. 1999;147:1049–1062. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul et al., 2006.Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B, Henderson CE, Haase G. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci U S A. 2006;103:6007–6012. doi: 10.1073/pnas.0508774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl and Salvesen, 2007.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Robertson et al., 2004.Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S. Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep. 2004;6:643–648. doi: 10.1038/sj.embor.7400153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada et al., 2003a.Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003a;5:320–329. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- Sawada et al., 2003b.Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol. 2003b;5:352–357. doi: 10.1038/ncb955. [DOI] [PubMed] [Google Scholar]
- Segura et al., 2007.Segura MF, Sole C, Pascual M, Moubarak RS, Perez-Garcia MJ, Gozzelino R, Iglesias V, Badiola N, Bayascas JR, Llecha N, Rodriguez-Alvarez J, Soriano E, Yuste VJ, Comella JX. The long form of Fas apoptotic inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J Neurosci. 2007;27:11228–11241. doi: 10.1523/JNEUROSCI.3462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka and Roth, 2006.Shacka JJ, Roth KA. Bcl-2 family and the central nervous system: from rheostat to real complex. Cell Death Differ. 2006;13:1299–1304. doi: 10.1038/sj.cdd.4401974. [DOI] [PubMed] [Google Scholar]
- Slevin et al., 2005.Slevin JT, Gerhardt GA, Smith CD, Gash DM, Kryscio R, Young B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg. 2005;102:216–222. doi: 10.3171/jns.2005.102.2.0216. [DOI] [PubMed] [Google Scholar]
- Sun et al., 2005.Sun M, Kong L, Wang X, Lu XG, Gao Q, Geller AI. Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson's disease. Brain Res. 2005;1052:119–129. doi: 10.1016/j.brainres.2005.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn, 2004.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Troy et al., 2000.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini et al., 2003.Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaün D, Henderson CE, Kato AC, Pettmann B, Hueber AO. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo . J Neurosci. 2003;23:8526–8531. doi: 10.1523/JNEUROSCI.23-24-08526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu and Arumäe, 2008.Yu LY, Arumäe U. Survival assay of transiently transfected dopaminergic neurons. J Neurosci Methods. 2008;169:8–15. doi: 10.1016/j.jneumeth.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Yu et al., 2003.Yu LY, Jokitalo E, Sun YF, Mehlen P, Lindholm D, Saarma M, Arumäe U. GDNF-deprived sympathetic neurons die via a novel nonmitochondrial pathway. J Cell Biol. 2003;163:987–997. doi: 10.1083/jcb.200305083. [DOI] [PMC free article] [PubMed] [Google Scholar]