

Abstract
Exploring mechanisms that govern neuronal responses to metabolic stress is essential for the development of therapeutic strategies aimed at treatment of neuronal injury and disease. AMP-activated protein kinase (AMPK) is a key enzyme regulating cellular energy homeostasis that responds to changes in cellular energy levels by promoting energy-restorative and inhibiting energy-consumptive processes. Recent studies have suggested that AMPK might have a neuroprotective function. However, the existing evidence is contradictory and almost exclusively derived from in vitro studies based on drug treatments and metabolic stress models. To tackle these issues in vivo, we used the Drosophila visual system. In this report, we describe a novel Drosophila mutant, alicorn (alc), encoding the single β regulatory subunit of AMPK. Loss of alc using the eyFlp system causes severe early-onset progressive nonapoptotic neurodegeneration in the retina, the optic lobe, and the antennae, as well as behavioral and neurophysiological defects. Retinal degeneration occurs immediately after normal neuronal differentiation, can be enhanced by exposure to light, and can be prevented by blocking photoreceptor excitation. Furthermore, AMPK is required for proper viability of differentiated photoreceptors by mechanisms unrelated to polarity events that AMPK controls in epithelial tissues. In conclusion, AMPK does not affect photoreceptor development but is crucial to maintaining integrity of mature neurons under conditions of increased activity and provides protection from excitotoxicity.
Keywords: AMPK, Drosophila, excitotoxicity, neurodegeneration, visual system, polarity
Introduction
AMP-activated protein kinase (AMPK) is an evolutionarily highly conserved key metabolic sensor of the cell. In all eukaryotic systems, it exists as a heterotrimer, composed of the catalytic α subunit and regulatory β and γ subunits, with all three being essential for the formation of an active, stable complex (Dyck et al., 1996; Woods et al., 1996). AMPK is activated through phosphorylation by at least two upstream kinases: the tumor suppressor LKB1 complex and calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) (Hong et al., 2003; Woods et al., 2003, 2005; Hawley et al., 2005) but also allosterically with binding of AMP (Scott et al., 2004; Sanders et al., 2007). Energetic stress, reflected by the rise of AMP/ATP ratio, various pathological stresses (including hypoxia, ischemia, oxidative damage, and glucose deprivation), as well as exercise and dietary hormones, activate AMPK, which responds by regulating a wide variety of cellular metabolic processes. This ultimately results in increased ATP production and decreased ATP consumption (Hardie et al., 2003).
Neurons are particularly sensitive to fluctuations in energy levels, for two reasons: first, they are highly metabolically active cells executing a number of energy-demanding processes (e.g., maintaining ion gradients across membranes and producing action potentials), and thus they account for a high proportion of total body energy turnover. Second, neurons have a rather inflexible metabolism in that they show poor capacity to store nutrients (Hardie and Frenguelli, 2007). High neuronal metabolic activity suggests that AMPK could play a pivotal role in neuronal maintenance. It is therefore not surprising that AMPK is highly expressed in the CNS (Turnley et al., 1999; Culmsee et al., 2001). However, the wider role of AMPK in nervous system development and function remains essentially elusive, and data available on the subject are contradictory and scarce (Hardie and Frenguelli, 2007).
Namely, there is emerging evidence that AMPK may have a neuroprotective role: AMPK is activated in the brain in response to metabolic insults such as ischemia, hypoxia, or glucose deprivation (Culmsee et al., 2001; Gadalla et al., 2004; McCullough et al., 2005), but it remains unclear whether the activation is beneficial. A study by Culmsee et al., 2001) using isolated hippocampal neurons reported that AMPK activation under energy-stress conditions promotes neuronal survival (Culmsee et al., 2001). Additional evidence comes from Drosophila in which mutation of the AMPK γ subunit resulted in a progressive neurodegenerative phenotype (Tschape et al., 2002). McCullough et al., 2005) reported that, in an in vivo CNS injury model (i.e., cerebral ischemia) as well as in an in vitro stroke model (hippocampal tissue slices subjected to oxygen-glucose deprivation), there is a global activation of AMPK. Paradoxically, pharmacological activators of AMPK had a detrimental and inhibitors a beneficial effect in these model systems.
Understanding normal physiological functions of AMPK will provide insight into mechanisms of protection against metabolic stress and neurodegeneration. Here we provide the first in vivo evidence conclusively demonstrating the involvement of AMPK in protection of mature neurons from increased metabolic activity.
Materials and Methods
Drosophila strains and culture conditions.
l(2)45Ad2 and Df(2R)Np5 lines were obtained from the Bloomington Stock Center (Bloomington, IN). βAMPK alleles were recombined with FRT{ry+t7.2 = neoFRT}42D for mitotic recombination. The chromosome with the l(2)45Ad2 allele was recombined with a wild-type Canton S chromosome to remove contaminating mutations distally to 45A11 on chromosome arm 2R. Clones were induced using ey–Gal4 UAS–Flp (EGUF) and eyFlp for the optic system and hsFlp for follicle cell clones. EGUF clones were made with an FRT42D chromosome carrying either a GMR–hid construct (referred to as “EGUF–hid” in text), resulting in development of an entirely mutant eye (Stowers and Schwarz, 1999), or a ubi–GFP marker (referred to as “EGUF–GFP” in text), resulting in homozygous mutant clones and wild-type twin spots (green). Rescue constructs were driven by ey–Gal4, da–Gal4, and tub–Gal4. norpAP24 mutant was obtained from G. Zhai (University of Miami, Miami, FL). Flies were reared under controlled temperature conditions of 25°C and, when specified, under 12 h light/dark cycle or in constant darkness from embryonic stage onward. Pupal staging was performed under the assumption that 1% pupal development (p.d.) corresponds to 1 h at 25°C. For energetic stress experiments, flies were cultured on a starvation medium (1% yeast extract, 3.5% wheat flour, and 0.8% agar) compared with the control group raised on 5% glucose, 5% yeast extract, 3.5% wheat flour, and 0.8% agar (Mirouse et al., 2007). For other experiments, flies were kept on standard fly medium. For induction of follicle cell clones, eclosed females were put on starvation or control medium for 2 d during eclosion, heat-shocked for 2 consecutive days thereafter (2 h at 37°C each), and dissected 2 d after the last heat shock (Mirouse et al., 2007).
Construction of βAMPK transgene.
Sequencing of cDNA clone RE12077 corresponding to a βAMPK–RA transcript (Drosophila Genomics Resource Center, Bloomington, IN) revealed three single nucleotide changes when compared with the wild-type βAMPK sequence, at positions +283, +398, and +333, the last of which was a predicted silent mutation. The other two nucleotide substitutions (T283C and A398G), resulting in predicted amino acid changes T95A and V133A, were reverted in a single PCR reaction using QuickChange Multi Site-Directed Mutagenesis kit (Stratagene) and the following mutagenized primers: 5′-tggctccgctcctGccgctgcctcctc-3′ and 5′-cagttccagttcccgTgtagtacgggtcgtc-3′. After sequence verification, the cDNA was cloned into pUAST and pUASP vectors (Brand and Perrimon, 1993) as EcoRI/KpnI and NotI/XbaI fragments, respectively. The transgenes were introduced into a yw1118 stock by P-element-mediated transformation.
mRNA quantification analysis.
Total RNA was isolated from first-instar larvae using RNeasy Micro kit (Qiagen). Quantitative reverse transcription-PCR analysis was performed by using SyBr-Green methodology on ABI Prism 7000 Sequence Detection System (Applied Biosystems) with the following primers: for βAMPK, 5′-ggatacaccgctatcgtgtgaa-3′ and 5′-gggcgtacaagtggttaagca-3′; for CG8788, 5′-atggatagcgtcaagtcgttcttc-3′ and 5′-cacattggcctgcctgttg-3′.
Scanning electron microscopy.
For scanning EM, whole flies were fixed in 50% ethanol, 5% neutral buffered Formalin, and 2% paraformaldehyde overnight at 4°C. Subsequently, samples were dehydrated through a series of 10 min ethanol and acetone washes and finally treated with hexamethyldisilazane for 30 min at room temperature. Pictures were taken using a field emission scanning electron microscope JSM-7401F (JEOL) at the Katholieke Universiteit Leuven Electron Microscopy Core Facility of the Department of Human Genetics (Leuven, Belgium).
Behavioral analysis and electroretinogram measurements.
Phototaxis and negative geotaxis tests were performed as previously described (Benzer, 1973). For electroretinogram (ERG) recordings, flies were immobilized with nail polish. A reference electrode was inserted in the thorax, whereas the recording electrode, filled with 3 m NaCl, was placed on the eye surface (Alawi and Pak, 1971). Light flashes of 1 s were delivered using a halogen lamp. Data were digitized with pClamp (Molecular Devices) and analyzed with Clampfit software (Molecular Devices) and Microsoft Excel.
Tissue sections and histological staining.
For paraffin sections of fly heads, aged flies were fixed in Carnoy's fixative (60% ethanol, 30% chloroform, and 10% glacial acetic acid) for 4 h at room temperature. After four washes in ethanol (30 min each), flies were kept in methylbenzoate overnight and subsequently transferred to a mixture of methylbenzoate and paraffin at 60°C. Finally, heads were embedded in paraffin, and sections of 10 μm thickness were made using a Hacker-Bright 5040 microtome (Hacker Instruments and Industries). Sections were stained with hematoxylin/eosin as follows: slides were deparaffinated in xylol and rehydrated through washes in series of decreasing concentrations of ethanol; next, they were stained in 0.1% hematoxylin (Merck), 0.02% KIO3, 5% KAl(SO4)2·12H2O for 2.5 min, and eosin (Romil) for 10 s. Subsequently, slides were dehydrated and rinsed in xylol and finally mounted in DPX (Romil). For semithin plastic sections of the retina, aged flies were dissected and fixed in 2% glutaraldehyde, 1% OsO4, 50 mm sodium cacodylate, and 4.2 mm HCl for 30 min on ice and postfixed in 2% OsO4 for 2 h on ice. Subsequently, fly heads were dehydrated through acetone series and embedded with Spurr Low Viscosity Embedding kit (Polysciences). The 100-nm-thin sections were stained with 1% Azure II (Fluka), 1% Methylene Blue (Fluka), and 1% disodiumtetraborate. Images were obtained using an Olympus microscope BX61 and CellD̂ 2.6 imaging software, processed with Adobe PhotoshopCS2 (Adobe Systems), and assembled in CorelDrawX3 (Corel Corporation).
Immunocytochemistry.
Immunofluorescence on ovaries and pupal eye discs was performed using standard procedures (Walther and Pichaud, 2006). Primary antibodies were as follows: rabbit anti-atypical protein kinase C (aPKC) (1:500; Santa Cruz Biotechnology), mouse anti-Armadillo (Arm) (1:50, N2 7A1; Developmental Studies Hybridoma Bank), rabbit anti-Bazooka (Baz) [1:1000, a gift from A. Wodarz, Georg-August University, Göttingen, Germany (Wodarz et al., 1999)], rabbit anti- Discs lost (Dlt) (1:1000), rabbit anti-Pon (Partner of Numb) [1:1000, received from Y. Jan, Howard Hughes Medical Institute, San Francisco, CA (Lu et al., 1998)], rabbit anti-green fluorescent protein (GFP) (1:1000; Invitrogen), and mouse anti-GFP (1:500; Abcam). Actin staining was performed with rhodamine-conjugated phalloidin (Invitrogen). Secondary antibodies coupled with cyanine 3 or fluorescein (anti-mouse and anti-rabbit, 1:200; Jackson ImmunoResearch) were used. Images were taken on a confocal microscope (Leica DM RXA) using Leica Confocal Software version 2.5, processed with Adobe PhotoshopCS2, and assembled in CorelDrawX3.
Results
Drosophila alicorn encodes the β subunit of AMP-activated protein kinase
A P-element insertion line (P{lArB}K12) associated with a subtle bristle number defect was found to target CG8057 [alicorn (alc)], the single Drosophila homolog of the β subunit of AMPK (Lyman et al., 1996; Norga et al., 2003). alc has two predicted transcripts arising from two different transcription initiation sites (Fig. 1 A) whose existence was confirmed with Northern blot (data not shown). Inverse PCR revealed that the insertion site of P{lArB}K12 was in the 5′ untranslated region (UTR) of the larger alc transcript, at position −283, 591 bp away from the nearest neighboring gene, CG8788. A lethal excision Δ12.125 failed to complement the deficiency covering the entire genomic region [Df(2R)Np5] (Fig. 1 C). Southern blot and PCR analysis of Δ12.125 (data not shown) demonstrated a deletion of 1718 bp of the genome surrounding the P-element insertion site, including the entire first exon of both alc transcripts and 39 bp of the predicted 5′ UTR of the neighboring CG8788. Quantitative real-time PCR results on the lethal excision line showed that the expression levels of CG8788 remained unaltered, whereas alc expression was entirely absent (Fig. 1 B). Lack of alc expression was confirmed with whole-mount in situ hybridization of mutant embryos (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Furthermore, we found that l(2)45Ad2, an ethyl methanesulfonate (EMS)-induced mutant from the same genomic region (Dockendorff et al., 2000), failed to complement the lethality of the excision line Δ12.125 (Fig. 1 C). Sequencing revealed a nonsense point mutation in the second exon of the gene (Fig. 1 D), replacing codon 233/112 (in transcripts RA/RB, respectively) for Arg with a premature stop signal. The resulting truncated peptide lacks the C-terminal region, which is very highly conserved between evolutionarily distant species, and contains a predicted complex-interacting region. Therefore, we predict alcAd2 to be a functional null of βAMPK.
Figure 1.

alc encodes the β subunit of AMPK. A, Genomic organization and position of the alc gene. The alc locus encodes two predicted transcripts (RA and RB) having different transcription initiation sites (UTRs marked in yellow). Inverse PCR mapped the P{lArB}K12 to the 5′ UTR of the larger transcript (marked by the P-element symbol; for details, see Results). The cDNA recue construct, corresponding to the alc RA transcript, is marked in green. The extent of the imprecise excision of the P element is marked with brackets (for details, see Results). B, Quantitative real-time PCR results on animals homozygous for the imprecise excision (Δ/Δ) or transheterozygous over a deficiency from the region (Δ/DfNp5) show lack of alc expression (blue bars) and unaltered expression levels of the neighboring gene CG8788 (green bars) compared with the wild type (WT); values were corrected for expression of a housekeeping gene (actin 5C). Error bars indicate SD. C, Complementation analysis of P-element insertion (P{lArB}K12), lethal excision (Δ12.125), an EMS mutant (l(2)45Ad2), and Df(2R)Np5 indicates allelism; +/− indicates presence/absence of adult escapers. D, Amino acid sequence of the highly conserved C-terminal region of βAMPK orthologs in evolutionarily distant species. Position of the EMS-induced nonsense mutation is indicated. The putative complex-interacting region is underlined in red.
Both alc mutations result in lethality over a range of developmental stages, from first larval instar to pupal stage, possibly at least partially attributable to a strong maternal contribution. Lethal phase was identical in transheterozygous and hemizygous mutant combinations, further supporting the fact that alcAd2 and alc Δ12.125 are in fact null alleles of the same gene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Lethality of the homozygous excision allele and transheterozygous mutant forms was rescued to viable adulthood with overexpression of wild-type alc from a cDNA rescue construct (Fig. 1 A), using ubiquitous daughterless- and tubulin–Gal4 driver lines. In situ hybridization of wild-type embryos using a probe generated against both alc transcripts revealed a strong maternal contribution and a broad, ubiquitous expression pattern during embryonic development (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Loss of βAMPK causes early-onset progressive neurodegeneration in the entire eyeless expression domain
To circumvent organismal lethality and look at the effect of the loss of AMPK in neuronal tissue, we performed clonal analysis of alc mutants using the ey–Gal4 UAS–Flp (EGUF) system combined with a GMR–hid construct. This results in the development of an entirely mutant eye (Stowers and Schwarz, 1999). Aside from a strong expression in larval eye-antennal imaginal discs, the eyeless enhancer is also expressed in parts of the brain, among others, the optic lobe (Hauck et al., 1999; Newsome et al., 2000). Although external eye morphology was grossly normal (Fig. 2 A), alc EGUF–hid mutants had a striking external phenotype (Fig. 2 B,C): although normal at eclosion, the antennae progressively accumulated an uncharacterized semitransparent organic deposit (hence, the name of the mutant), which appeared to originate from the culture media. The exact cause of this phenomenon is unclear at present, but this is likely a reflection of internal degenerative defects that are already present at day 1 (Fig. 2 D). Furthermore, aged mutant animals displayed behavioral changes. The antennae have been linked previously to gravitaxis in Drosophila and other insects (Horn and Kessler, 1975; Armstrong et al., 2006). Indeed, when a negative geotaxis test was performed, it showed a severe impairment of the response in mutants already at day 1 and almost complete lack of response by day 7, whereas the response of control heterozygous flies was comparable with wild type (Fig. 2 E). This behavioral defect could successfully be rescued by expressing the wild-type βAMPK protein.
Figure 2.

Loss of alc causes severe early-onset neurodegeneration in the entire eyeless expression domain. A, Normal external eye morphology of alc eyFlp mutants (shading in the eye is attributable to color differences between red/heterozygous and white/mutant tissue). B, Deposit formed on the antennae of 35-d-old alc EGUF–hid mutant (arrows point to bristle loss). C, Antennal degeneration and head bristle loss (arrows) in 30-d-old eyFlp mutants. D, Plastic semithin section of the antennae, showing severe degeneration at 1 d of age; arrowheads point to second (II) and third (III) antennal segments. E, Negative geotaxis test performed on two groups of 7–12 flies per genotype (mean value of 10 tests per group). Error bars indicate SEM. eyFlp mutants geotax poorly already at day 1 and almost completely lack the response by day 7. F, Progressive neurodegeneration in the optic lobes of alc EGUF–hid mutants. Inset at day 1, Magnification of the right optic lobe. Similar results were obtained with the lethal excision allele (alc Δ12.125) (data not shown). Expression of viral apoptosis inhibitor p35 does not rescue degeneration. All defects can be rescued by expression of wild-type βAMPK.
To investigate the consequences of the loss of AMPK in the rest of the eyeless expression domain, we first examined the morphology of the brain. Paraffin brain sections of adult alc clonal mutants revealed vacuolization and neuronal degeneration in the optic lobes (Fig. 2 F). This neurodegeneration had an early onset, because it was apparent already at 1 d of age. To the contrary, brains of pharate clonal mutant adults had a normal morphology (data not shown). The neurodegeneration was clearly progressing, because the optic lobes of 14-d-old adults were profoundly altered. The neurodegenerative phenotype could also be rescued with wild-type alc expression.
Next, we explored the mechanism of neuronal death. AMPK has been linked previously to apoptosis, paradoxically to both its activation and suppression (Ido et al., 2002; Russell et al., 2004; Shaw et al., 2004; Okoshi et al., 2008). To determine whether neuronal death in the brain is apoptotic, we attempted a rescue by the viral apoptosis inhibitor p35 (Hay et al., 1994). We did not observe any difference between p35-expressing and control mutant brains (Fig. 2 F); thus, we concluded that the observed neuronal death is independent of the p35-mediated apoptotic pathway.
In conclusion, alc clonal mutants appeared to show a general early-onset progressive neurodegenerative phenotype in the entire expression domain of eyeless.
βAMPK does not affect photoreceptor cell polarity and eye development
Recent studies on the AMPK α subunit in Drosophila demonstrated that AMPK is a key mediator of apico-basal epithelial cell polarity (Lee et al., 2007; Mirouse et al., 2007). In addition, LKB1, an upstream activator of AMPK, has been shown previously to function during establishment of neuronal cell polarity (Asada et al., 2007; Barnes et al., 2007; Shelly et al., 2007). However, the role of AMPK in neuronal polarity is unknown (Williams and Brenman, 2008). To determine the effect of the loss of AMPK in the eye, we first examined the establishment and maintenance of apico-basal cell polarity during the development of retinal photoreceptor neurons.
The Drosophila compound eye consists of ∼800 clusters called ommatidia, each comprising eight photoreceptor cells (PRCs) that are closely connected by adherens junctions (AJs). These clusters are generated in the eye disc epithelium during the third-instar larval stage, but it is during pupal development that the apical domain of differentiating PRCs undergoes a remarkable cell shape change (Longley and Ready, 1995) (Fig. 3 A). This results in the formation of rhabdomeres, stacks of microvilli packed with photopigment rhodopsin, on the apical surface of the cell. During pupal morphogenesis of PRCs, two protein complexes that determine apico-basal cell polarity play a decisive role (Nam and Choi, 2003): the Crumbs complex, consisting of Crumbs–Stardust–Dpatj (Drosophila PALS-1-associated tight junction protein), and the Par-6 complex, comprising Par-6–aPKC–Baz. Each of them is characterized by a specific subcellular distribution (Fig. 3 A).
Figure 3.
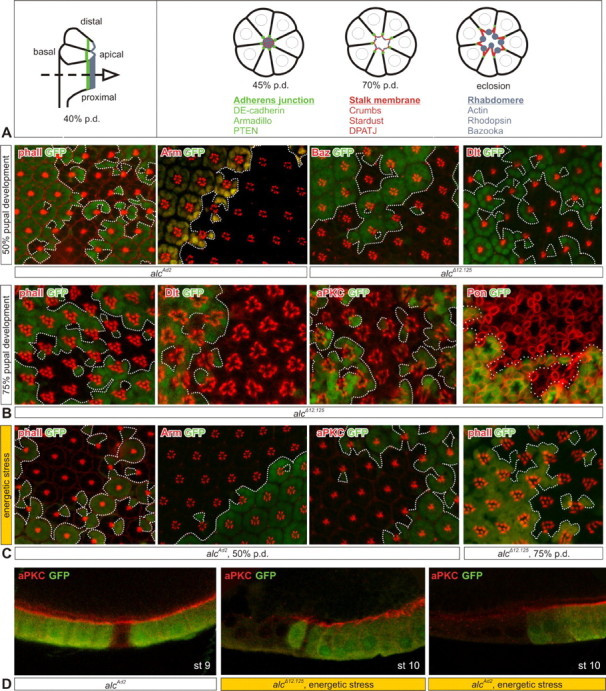
AMPK does not affect photoreceptor cell polarity and development. A, Development of the Drosophila compound eye and subcellular distribution of various markers. Photoreceptor cells presented schematically, at 45 and 70% p.d. and in the adult. Green, Adherens junctions; red, subapical stalk membrane; blue, apical rhabdomeres; dashed arrow at 40% p.d. indicates tangential view of the drawings on the right side of the panel and micrographs on B and C. B, Distribution of various subcellular markers (indicated in the top left corner) in alc EGUF–GFP clones at 50% (top row) and 75% pupal development (bottom row). GFP marks twin spots and any remaining heterozygous cells, whereas mutant cells are GFP negative; clone borders are delineated with a dotted line. C, Energetic stress does not induce a change in marker distribution (also for Dpatj and Baz) (data not shown). D, alc follicle cell clones under normal and energetic stress conditions (for details, see Results); mutant cells are marked by the absence of GFP (green). Oocyte stage (st) is indicated in the bottom right corner, and markers are indicated in the top left. Percentage of pupal development is approximate and varies within 4%.
To investigate photoreceptor development, we produced alc EGUF–GFP clones, which are flanked by wild-type twin spots (marked by GFP expression). We stained developing pupal retinas for actin (which labels the rhabdomeres and outlines photoreceptor morphology), as well as different polarity and AJ markers. At 50% p.d., actin staining revealed no morphological defects in mutant photoreceptors (Fig. 3 B). In addition, when staining for polarity and AJ markers, Arm, Baz, Dlt, and others [Dlg (Discs large), DE-cadherin, and Par-6] (data not shown), we could not detect any differences between heterozygous and homozygous mutant tissue. Photoreceptor subtype specification and differentiation are mostly completed by 50% p.d.; therefore, we conclude that mutant photoreceptors develop normally. To look at maintenance of polarity, we examined pupal eyes at 75% p.d. Also at these later stages, ∼24 h before eclosion, mutant photoreceptors have a normal general morphology and distribution of polarity determinants (Fig. 3 B).
To test the possibility that the effect of AMPK on PRC polarity would only become apparent under sensitizing conditions of starvation, as proposed by Mirouse et al., 2007), we allowed larvae with alc mutant eye clones to develop on a starvation medium, containing no sugar and five times less yeast than in normal diet (Mirouse et al., 2007). Although this treatment, predictably, delayed pupariation by >90 h, it did not induce changes in polarity or development of mutant photoreceptors, as witnessed by staining for actin, Arm, and aPKC, as well as Dpatj and Baz (data not shown) at 50% and 75% p.d. (Fig. 3 C).
This prompted us to examine whether alc follicle cell mutants would exhibit the same kind of defects that were described for α subunit mutants by Mirouse et al. For this purpose, we compared follicle epithelia of starved and nonstarved adult females. While under nonstarved conditions, βAMPK mutant follicle cells displayed no polarity defects as demonstrated by the normal distribution of polarity markers (Fig. 3 D, left panel), these defects did appear when applying energy stress; apical markers such as aPKC completely lost their cortical localization and are downregulated (shown for the two alc mutants on the middle and right panels of Fig. 3 D). This is in agreement with the previously reported findings on the α subunit (Mirouse et al., 2007).
In conclusion, although AMPK is an important factor determining polarity of epithelial cells, we show that it does not affect photoreceptor cell polarity, even under conditions of energetic stress.
alc adult mutants show rapid and severe photoreceptor degeneration
Further extending our findings, we examined the adult eye for consequences of loss of alc. First, we tested adult alc ey–Flp mutants in a phototaxis assay in which they showed a severe impairment of phototaxis response already at 1 d of age and almost complete nonresponsiveness by day 7 (Fig. 4 A). As is the case with negative geotaxis, alc heterozygotes show an impairment in behavior compared with wild-type controls. This semidominance is most likely caused by haploinsufficiency. Such dosage-sensitive genetic characteristics of behavioral phenotypes have been reported previously (Anholt and Mackay, 2004; Jordan et al., 2006; Rollmann et al., 2007). This behavioral defect could effectively be rescued to wild-type levels by alc expression. Subsequently, we subjected the mutant flies to ERG analysis, taking extracellular recordings that measure the response of photoreceptor neurons to a light stimulus. ERG recordings were strikingly different compared with heterozygous controls (Fig. 4 B) and revealed a reduction of “on-transients” and “off-transients,” which suggests a possible neurotransmitter release defect at the synapse between R1–R6 photoreceptors and second-order neurons in the lamina. Furthermore, decreased amplitude of depolarization for the stimulus duration indicated phototransduction defects. These ERG defects could also be reverted with a UAS–alc construct.
Figure 4.

Retinal degeneration in alc mutant adult clones. A, Phototaxis test performed on two groups of 7–10 flies per genotype (10 tests per group). Error bars indicate SEM. eyFlp mutants phototax poorly already at day 1 and almost completely lack the response by day 7. B, ERG recordings taken from aged alc EGUF–hid mutants show striking differences in on- and off-transients and photoreceptor depolarization when compared with heterozygous controls; average recording of 10 flies per genotype, 5 recordings per fly. Scale bar, 500 ms. C, Progressive retinal degeneration in EGUF–hid mutants, as documented by semithin retinal sections. Extensive vacuolization (black arrows), rhabdomere loss, ommatidial disorganization, and presence of large vesicular structures (arrowheads) can be observed. The phenotype has an early onset and is progressing with age. In pharate adults, the retinas already show an occasional photoreceptor loss (white arrows). The EMS (alcAd2) mutants have similar defects (data not shown). All mutant phenotypes can be rescued by wild-type alc expression.
To further explore the retinal morphology of adult alc mutants, we made semithin sections of the retina (Fig. 4 C). Retinas homozygous for either of the alc alleles were profoundly altered, revealing extensive photoreceptor degeneration. Similar to the brain, retinal degeneration had an early onset, with elements of the phenotype present already in pharate adults, showing occasional photoreceptor (rhabdomere) loss (white arrows in Fig. 4 C). Interestingly, the phenotype was rapidly progressing and was significantly worse as early as day 1, with photoreceptor loss being more frequent and the appearance of vacuoles (black arrows in Fig. 4 C). Vacuolization of tissue and photoreceptor degeneration were further progressing with age, eventually resulting in general disorganization of ommatidial architecture at day 14. Additionally, large vesicular structures were apparent in the pigment cells (arrowheads in Fig. 4 C). Again, the retinal phenotype was rescued by expressing wild-type βAMPK.
In conclusion, alc clonal mutants showed phototaxis and neurophysiological defects and extensive age-dependent retinal degeneration with an early onset. Therefore, AMPK is required for viability of fully differentiated photoreceptors by mechanisms unrelated to photoreceptor development and polarity.
Retinal degeneration in alc mutants is activity dependent
We further wanted to dissect the mechanism underlying neurodegeneration of mutant photoreceptors. The sudden onset of degeneration after eclosion and its rapid progression, together with AMPK being a cellular metabolic sensor, prompted us to postulate that the degenerative phenotype is a consequence of neuronal activity. To explore this possibility, we first tested whether photoreceptor degeneration is light dependent (Fig. 5 A). To this end, we raised and kept one group of alc clonal mutants in conditions of constant darkness, whereas another group was kept under controlled 12 h light/dark cycle. Notably, the phenotypes of alc dark- and light-reared EGUF–hid mutants showed striking differences: dark-reared flies at day 1 had an entirely preserved ommatidial organization. Although at 1 week of age, some signs of neurodegeneration in the dark-reared flies were obvious, the phenotype was present to a far lesser extent compared with flies kept on a light/dark cycle; vacuolization was barely present, and ommatidial architecture was grossly preserved.
Figure 5.
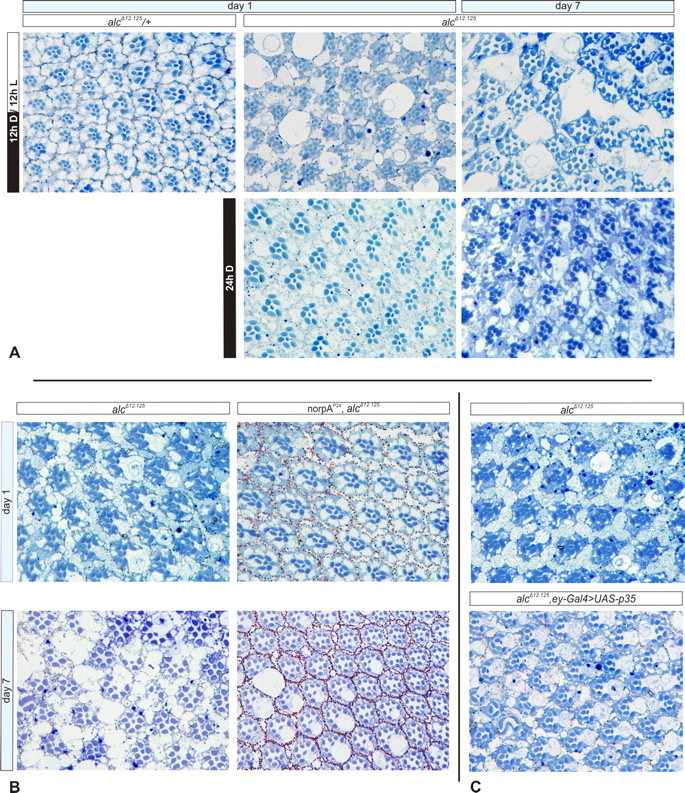
Retinal degeneration is activity-dependent and nonapoptotic. A, Retinal sections of flies kept under a controlled 12 h light/dark cycle compared with mutants reared in constant darkness reveals striking differences in severity of the phenotype, indicating that light enhances retinal degeneration in alc EGUF–hid mutants. B, norpAP24 (encoding a phospholipase C) mutation blocks the phototransduction cascade, renders photoreceptors unresponsive to light, and rescues alc neurodegeneration; at day 7, mild vacuolization is apparent, but there are no signs of photoreceptor loss. C, Expression of viral apoptosis inhibitor p35 does not ameliorate the mutant phenotype at day 1.
To further establish whether neuronal activity is mediating photoreceptor degeneration, we investigated whether blocking the phototransduction cascade could lead to amelioration of the phenotype. For this purpose, we used a mutant allele of the norpA (no receptor potential A) gene encoding a phospholipase C necessary for phototransduction and light perception (Bloomquist et al., 1988). In norpAP24 mutants, phototransduction is blocked, and the flies do not react to light and show no response to light pulse in ERG recordings (McKay et al., 1995; Shieh et al., 1997). Remarkably, in a norpAP24 mutant background, photoreceptors of alc EGUF mutants retained normal morphology, and neurodegeneration was fully suppressed at day 1 (Fig. 5 B). At 1 week of age, although vacuolization was present, there were no signs of photoreceptor (rhabdomere) loss and the ommatidial architecture was almost entirely preserved. Thus, we have shown that the retinal neurodegenerative phenotype of alc mutants can be rescued by blocking neuronal excitation.
Next, we wanted to explore whether apoptosis is responsible for the observed degeneration and loss of photoreceptors. To this end, we used a transgenic construct carrying a viral apoptosis-inhibiting factor p35 to try and rescue neurodegenerative defects (Hay et al., 1994). With this treatment, we found no suppression of the phenotype (Fig. 5 C), both vacuolization and rhabdomere loss were still apparent, and retinas were virtually indistinguishable from mutant controls lacking the p35 transgenic construct. In a separate control experiment, expression of p35 rescued the GMR–hid-induced gross morphological eye defects resulting from apoptotic cell death triggered by the hid gene (data not shown). Therefore, we conclude that the p35-mediated apoptotic pathway is not responsible for retinal defects of alc mutants, as shown previously for optic lobe degeneration (Fig. 2 F).
Discussion
AMP-activated protein kinase is an evolutionarily conserved heterotrimeric enzyme whose primary role involves maintenance of energy balance in the eukaryotic cell. The structure and function of each of the subunits is conserved through evolution; however, although there are two or three genes encoding each subunit in the mammalian systems, there is but a single gene for each of them in Drosophila, making it an attractive model system to study the functions of AMPK in vivo (Pan and Hardie, 2002). Although it has been the focus of much research, little is known about the precise mechanisms underlying the regulatory functions of the β and γ subunits: β acts as a scaffold via its C-terminal domain and contains a carbohydrate-binding domain that associates the mammalian enzyme complex with glycogen, whereas the gamma subunit contains the so-called Bateman domains, responsible for binding of AMP and therefore allosteric activation of AMPK (Hardie, 2007). Finally, all three subunits are essential for the formation of an active, stable complex: a knock-out of a single subunit produces a functional knock-out of the entire enzyme (Dyck et al., 1996; Woods et al., 1996; Pan and Hardie, 2002).
Recent research suggests that AMPK participates in physiological functions beyond those associated with responding to energy status. The fact that stimuli like dietary restriction and exercise, which have a beneficial effect on different neurodegeneration animal and cell culture models (Bruce-Keller et al., 1999; Zhu et al., 1999; Endres et al., 2003; Adlard et al., 2005), also lead to AMPK activation (Hardie et al., 2003; Dasgupta and Milbrandt, 2007) suggests possible involvement of this enzyme in neuroprotective processes. Furthermore, in the nervous system, as in most other tissues, normal aging is associated with increased amounts of oxidative stress and perturbed cellular energy metabolism involving impaired efficiency of mitochondrial ATP production (Floyd and Hensley, 2002; Navarro, 2004). Therefore, one can imagine that the need for a functional metabolic sensor increases during aging. Up to date, the available information on the possible role of AMPK in neuroprotection is mostly derived from in vitro studies based on drug treatments and metabolic stress models and is contradictory.
In this study, we used the Drosophila visual system to address these issues and modeled changes in cellular metabolic status by influencing normal physiological activity of photoreceptors. Knock-out of AMPK using the ey–Flp system results in a rapid and severe neurodegeneration in the entire eyeless expression domain immediately on completion of normal neuronal differentiation and development. Just before eclosion, the eye-antennal system of mutant flies appears to be structurally intact, externally as well as internally, with only occasional photoreceptor loss already present. Strikingly, however, immediately after eclosion, and progressively with age, severe behavioral (phototaxis and negative geotaxis), and neurophysiological defects, optic lobe and antennal degeneration follow. It thus appears that the entire sensory system, along with parts of the brain, all deriving from the eyeless-driven AMPK knock-out succumbs to neurodegeneration with the start of neuronal activity. Although the eyeless enhancer used in the ey–Flp construct has been shown to drive expression in parts of the brain, as well as the eyes (Hauck et al., 1999; Newsome et al., 2000), there still is a possibility that the observed brain degeneration is (partly) a secondary, non-cell-autonomous effect. However, this remains to be proven.
One of the striking features of AMPK clonal mutants is the extensive retinal degeneration, which can be markedly reduced by rearing flies in the dark and almost completely prevented by blocking photoreceptor excitation. Therefore, although AMPK does not seem to be required for photoreceptor development, it is crucial for maintenance and integrity of mature neurons. The fact that the mutant photoreceptors develop normally and maintain their structural integrity until they start to activate in response to light suggests that this is precisely the time when the absence of AMPK appears to have the gravest consequences. This is in agreement with the fact that activation (excitation) of neuronal cells very efficiently leads to energy depletion (Hardie and Frenguelli, 2007). The key factor that is responsible for bringing the energy levels back to normal in this situation is missing. Among different proposed genetic mechanisms of neurodegeneration, the AMPK phenotype illustrates how loss-of-function mutations can lead to neurodegeneration.
In support of this paradigm, Kuramoto et al. (2007) have reported recently that, as a result of increased neuronal activity, AMPK is activated and suppresses neuronal excitation by activating GABAB receptors on postsynaptic neurons. Altogether, these data reveal that neurons protect themselves against excitotoxicity and that failure of such a system causes neurodegeneration. Potentially, this could be exploited to protect neurons from degeneration under adverse conditions.
In addition, we show that, although AMPK regulates apico-basal cell polarity in epithelial tissues, it does not do the same in photoreceptors. Namely, mutant photoreceptors develop normally, despite their need for a precisely regulated distribution and function of cellular polarity determinants. Moreover, changes in photoreceptor polarity cannot be evoked even when subjecting the mutants to energetic stress. This is the first indication of a tissue-specific function of AMPK in this process and suggests that AMPK may not be as functionally universal in this regard as previously thought. It has been postulated before that AMPK activation in general may have cell- or tissue-specific outcomes (Ramamurthy and Ronnett, 2006). This could be attributable to differential expression and/or activity of its upstream regulators, as well as its downstream effectors mediating such responses. This raises intriguing questions. How is AMPK differentially regulated in cells or tissues to provide regulation of cellular polarity only in certain instances? Also, what are the mediators of the effect of AMPK on cellular polarity, and is their expression and/or activity regulated in a cell- or tissue-specific manner?
Finally, the exact mechanism of progressive neuronal death observed as a consequence of loss of AMPK remains unresolved. Dying by apoptosis requires a lot of energy in the form of ATP (Edinger and Thompson, 2004). Therefore, apoptotic death would indeed not be expected in the situation in which cells are deprived of energy and are in addition lacking a key energy sensor. In accordance with this, both we and Tschape et al. (2002) in a study with the γ subunit mutant were unable to demonstrate any role of apoptosis in neurodegeneration of AMPK mutants. Conversely, autophagy (a catabolic program in which cellular constituents are degraded for energy production) is a process activated during periods of nutrient starvation and ATP depletion (Edinger and Thompson, 2004). It has been shown that, under metabolic stress conditions, AMPK induces autophagy rather than apoptosis to ensure survival of cells (Liang et al., 2007). In addition, recent studies have indicated that autophagy is protective against neurodegeneration (Levine and Kroemer, 2008). In additional support of this model, Lippai et al., 2008) have shown recently that a Drosophila P-element insertion mutant of the AMPK γ subunit results in autophagic defects during hormone-induced metamorphosis in third-instar larval fat body (Lippai et al., 2008). It will therefore be interesting to investigate the possible role of autophagy in neuronal loss observed in alc mutants.
Footnotes
This work was supported by Flanders Institute for Biotechnology (VIB) (P.C.), Interuniversity Attraction Poles Grant P6/43 (P.C.), and Fonds voor Wetenschappelijk Onderzoek (FWO-Flanders) Grants G.0397.06N (K.K.N.) and G.0285.05 and G024508N (P.C.). M.R.S. is a recipient of a doctoral fellowship of the Katholieke Universiteit Leuven (OE/05/31). K.K.N. is a Senior Clinical Investigator of the FWO-Flanders. We thank T. Ooms, I. Bosmans, P. Lauwers, and A. Schellens for technical assistance, and R. Habets and P. Verstreken for help with ERG measurements. We are also grateful to G. Zhai, A. Wodarz, D. Montell, and Y. Jan for generously sending fly stocks and reagents, as well as to C. Dotti, G. Zhai, B. Dermaut, and B. Hassan for critical reading of this manuscript. We also thank The Bloomington Drosophila Stock Center and The Developmental Studies Hybridoma Bank for providing flies and antibodies.
References
- Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawi AA, Pak WL. On-transient of insect electroretinogram: its cellular origin. Science. 1971;172:1055–1057. doi: 10.1126/science.172.3987.1055. [DOI] [PubMed] [Google Scholar]
- Anholt RR, Mackay TF. Quantitative genetic analyses of complex behaviours in Drosophila . Nat Rev Genet. 2004;5:838–849. doi: 10.1038/nrg1472. [DOI] [PubMed] [Google Scholar]
- Armstrong JD, Texada MJ, Munjaal R, Baker DA, Beckingham KM. Gravitaxis in Drosophila melanogaster: a forward genetic screen. Genes Brain Behav. 2006;5:222–239. doi: 10.1111/j.1601-183X.2005.00154.x. [DOI] [PubMed] [Google Scholar]
- Asada N, Sanada K, Fukada Y. LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J Neurosci. 2007;27:11769–11775. doi: 10.1523/JNEUROSCI.1938-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Benzer S. Genetic dissection of behavior. Sci Am. 1973;229:24–37. doi: 10.1038/scientificamerican1273-24. [DOI] [PubMed] [Google Scholar]
- Bloomquist BT, Shortridge RD, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak WL. Isolation of a putative phospholipase C gene of Drosophila, norpA, and its role in phototransduction. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff TC, Robertson SE, Faulkner DL, Jongens TA. Genetic characterization of the 44D–45B region of the Drosophila melanogaster genome based on an F2 lethal screen. Mol Gen Genet. 2000;263:137–143. doi: 10.1007/s004380050040. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Gao G, Widmer J, Stapleton D, Fernandez CS, Kemp BE, Witters LA. Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. J Biol Chem. 1996;271:17798–17803. doi: 10.1074/jbc.271.30.17798. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schrock H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U. Mechanisms of stroke protection by physical activity. Ann Neurol. 2003;54:582–590. doi: 10.1002/ana.10722. [DOI] [PubMed] [Google Scholar]
- Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- Hardie, 2007.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hardie and Frenguelli, 2007.Hardie DG, Frenguelli BG. A neural protection racket: AMPK and the GABAB receptor. Neuron. 2007;53:159–162. doi: 10.1016/j.neuron.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Hardie et al., 2003.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- Hauck et al., 1999.Hauck B, Gehring WJ, Walldorf U. Functional analysis of an eye specific enhancer of the eyeless gene in Drosophila . Proc Natl Acad Sci USA. 1999;96:564–569. doi: 10.1073/pnas.96.2.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley et al., 2005.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hay et al., 1994.Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila . Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hong et al., 2003.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn and Kessler, 1975.Horn E, Kessler W. The control of antennae lift movements and its importance on the gravity reception in the walking blowfly, Calliphora erythrocephala . J Comp Physiol. 1975;97:189–203. [Google Scholar]
- Ido et al., 2002.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–167. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- Jordan et al., 2006.Jordan KW, Morgan TJ, Mackay TF. Quantitative trait loci for locomotor behavior in Drosophila melanogaster . Genetics. 2006;174:271–284. doi: 10.1534/genetics.106.058099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto et al., 2007.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee et al., 2007.Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, Chung J. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Levine and Kroemer, 2008.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang et al., 2007.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Lippai et al., 2008.Lippai M, Csikos G, Maroy P, Lukacsovich T, Juhasz G, Sass M. SNF4Agamma, the Drosophila AMPK gamma subunit is required for regulation of developmental and stress-induced autophagy. Autophagy. 2008;4:476–486. doi: 10.4161/auto.5719. [DOI] [PubMed] [Google Scholar]
- Longley and Ready, 1995.Longley RL, Jr, Ready DF. Integrins and the development of three-dimensional structure in the Drosophila compound eye. Dev Biol. 1995;171:415–433. doi: 10.1006/dbio.1995.1292. [DOI] [PubMed] [Google Scholar]
- Lu et al., 1998.Lu B, Rothenberg M, Jan LY, Jan YN. Partner of Numb colocalizes with Numb during mitosis and directs Numb asymmetric localization in Drosophila neural and muscle progenitors. Cell. 1998;95:225–235. doi: 10.1016/s0092-8674(00)81753-5. [DOI] [PubMed] [Google Scholar]
- Lyman et al., 1996.Lyman RF, Lawrence F, Nuzhdin SV, Mackay TF. Effects of single P-element insertions on bristle number and viability in Drosophila melanogaster . Genetics. 1996;143:277–292. doi: 10.1093/genetics/143.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough et al., 2005.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- McKay et al., 1995.McKay RR, Chen DM, Miller K, Kim S, Stark WS, Shortridge RD. Phospholipase C rescues visual defect in norpA mutant of Drosophila melanogaster . J Biol Chem. 1995;270:13271–13276. doi: 10.1074/jbc.270.22.13271. [DOI] [PubMed] [Google Scholar]
- Mirouse et al., 2007.Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol. 2007;177:387–392. doi: 10.1083/jcb.200702053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nam and Choi, 2003.Nam SC, Choi KW. Interaction of Par-6 and Crumbs complexes is essential for photoreceptor morphogenesis in Drosophila . Development. 2003;130:4363–4372. doi: 10.1242/dev.00648. [DOI] [PubMed] [Google Scholar]
- Navarro, 2004.Navarro A. Mitochondrial enzyme activities as biochemical markers of aging. Mol Aspects Med. 2004;25:37–48. doi: 10.1016/j.mam.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Newsome et al., 2000.Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Norga et al., 2003.Norga KK, Gurganus MC, Dilda CL, Yamamoto A, Lyman RF, Patel PH, Rubin GM, Hoskins RA, Mackay TF, Bellen HJ. Quantitative analysis of bristle number in Drosophila mutants identifies genes involved in neural development. Curr Biol. 2003;13:1388–1396. doi: 10.1016/s0960-9822(03)00546-3. [DOI] [PubMed] [Google Scholar]
- Okoshi et al., 2008.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- Pan and Hardie, 2002.Pan DA, Hardie DG. A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. Biochem J. 2002;367:179–186. doi: 10.1042/BJ20020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy and Ronnett, 2006.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol (Lond) 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollmann et al., 2007.Rollmann SM, Yamamoto A, Goossens T, Zwarts L, Callaerts-Vegh Z, Callaerts P, Norga K, Mackay TF, Anholt RR. The early developmental gene Semaphorin 5c contributes to olfactory behavior in adult Drosophila . Genetics. 2007;176:947–956. doi: 10.1534/genetics.106.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell et al., 2004.Russell RR, III, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders et al., 2007.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott et al., 2004.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw et al., 2004.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly et al., 2007.Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shieh et al., 1997.Shieh BH, Zhu MY, Lee JK, Kelly IM, Bahiraei F. Association of INAD with NORPA is essential for controlled activation and deactivation of Drosophila phototransduction in vivo. Proc Natl Acad Sci USA. 1997;94:12682–12687. doi: 10.1073/pnas.94.23.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers and Schwarz, 1999.Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschape et al., 2002.Tschape JA, Hammerschmied C, Muhlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. EMBO J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnley et al., 1999.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Walther and Pichaud, 2006.Walther RF, Pichaud F. Immunofluorescent staining and imaging of the pupal and adult Drosophila visual system. Nat Protoc. 2006;1:2635–2642. doi: 10.1038/nprot.2006.379. [DOI] [PubMed] [Google Scholar]
- Williams and Brenman, 2008.Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–198. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Wodarz et al., 1999.Wodarz A, Ramrath A, Kuchinke U, Knust E. Bazooka provides an apical cue for Inscuteable localization in Drosophila neuroblasts. Nature. 1999;402:544–547. doi: 10.1038/990128. [DOI] [PubMed] [Google Scholar]
- Woods et al., 1996.Woods A, Salt I, Scott J, Hardie DG, Carling D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996;397:347–351. doi: 10.1016/s0014-5793(96)01209-4. [DOI] [PubMed] [Google Scholar]
- Woods et al., 2003.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Woods et al., 2005.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Zhu et al., 1999.Zhu H, Guo Q, Mattson MP. Dietary restriction protects hippocampal neurons against the death-promoting action of a presenilin-1 mutation. Brain Res. 1999;842:224–229. doi: 10.1016/s0006-8993(99)01827-2. [DOI] [PubMed] [Google Scholar]