

Abstract
The ATP-gated P2X7 receptor (P2X7R) is a highly unusual calcium-permeable cationic channel in that within seconds of its activation, dramatic and reversible cytoskeletal rearrangements with prominent membrane blebbing occurs. Agonist-induced membrane currents at hyperpolarized potentials show pronounced facilitation during the initial 30–100 s of receptor activation but mechanisms responsible have not been elucidated. We measured facilitation of ATP-gated currents in HEK cells expressing rat P2X7R and delineated distinct calcium-dependent and independent processes. The calcium-dependent facilitation was composed of an instantaneous (millisecond time domain) and slowly developing (time constant, 20 s with maximum agonist stimulation) component. Both components were prevented when recording with a highly specific calmodulin (CaM) inhibitory peptide but only the instantaneous component was reduced by expression of the dominant-negative EF-handless CaM mutant. Coimmunoprecipitation assays detected low levels of CaM binding to unstimulated P2X7R, and this increased by 50% during 45 s stimulation of the receptor. We identified a novel 1-5-16 Ca2+-dependent CaM binding motif in the intracellular C terminus of P2X7R; mutations in this domain resulted in the absence of calcium-dependent facilitation and binding of CaM to unstimulated or stimulated receptor. Blockade of CaM binding also delayed membrane blebbing by threefold. Our results demonstrate that CaM binds constitutively to closed P2X7R channels and dynamically during channel activation to significantly enhance and prolong calcium entry. This is the first example of CaM deregulating, rather than tightly controlling, calcium entry through an ion channel.
Keywords: ionotropic receptor, patch clamp, mutagenesis, facilitation, calmodulin, inflammation
Introduction
There are seven members of the ATP-gated P2X purinergic receptor family (P2X1–7Rs). They all form cation-selective homomeric and/or heteromeric channels with variable but significant calcium permeability (North, 2002; Burnstock, 2007). P2X1–6Rs are found throughout the central and peripheral nervous system in which they act as excitatory neurotransmitter receptors (North, 2002; Burnstock, 2007). In contrast, P2X7Rs are prominent in activated macrophage and microglia at sites of inflammation or injury in which they act as the main physiological stimulus for rapid release of the proinflammatory cytokines, interleukin-1β (IL-1β) and IL-18 (Ferrari et al., 2006; Di Virgilio, 2007). Studies using mice in which the P2X7R has been deleted and recent studies with highly selective P2X7R antagonists have further supported a significant role for this receptor in chronic pain associated with inflammatory diseases (Labasi et al., 2002; Chessell et al., 2005).
The properties of the P2X7R ion channel are remarkably different from other P2X receptors, and highly unusual relative to any other ligand-gated ion channel in several aspects (North, 2002), but of most relevance is that agonist-evoked currents dramatically change their onset and offset kinetics with repeated applications of the same agonists, and activation also rapidly engages a series of cytoskeletal and mitochondrial alterations that have not been directly associated with brief activation of any other ion channel. These include actin/α-tubulin rearrangements, phosphatidylserine translocation, mitochondrial swelling and loss of mitochondrial membrane potential, and membrane blebbing. All of these events occur within 2–30 s of maximum receptor activation, are fully reversible, and do not lead to cell death unless receptor stimulation is prolonged, after which cell death is inevitable (Mackenzie et al., 2001, 2005; Verhoef et al., 2003; Pfeiffer et al., 2004; Adinolfi et al., 2005; Di Virgilio, 2007).
Repeated activation of P2X7R evokes one of two diametrically opposite types of current response depending on the metabolic state of the cell. When protein tyrosine phosphatase activity is high, currents desensitize completely during agonist applications and peak amplitude of currents run down permanently by >90% with successive agonist applications (Kim et al., 2001). However, the more common response is a slow onset, non-desensitizing current that shows strong run-up, or facilitation, of both onset kinetics and peak current amplitude with repeated applications (Surprenant et al., 1996; Chessell et al., 1997; Rassendren et al., 1997). Mechanisms underlying the facilitation of P2X7R currents is, up to now, unknown, but facilitating currents are associated with, and appear to be responsible for, many of the downstream events, such as membrane blebbing, that occur on P2X7R stimulation.
We sought to determine how facilitation of P2X7R currents occur by using mutagenesis-based strategies, coimmunoprecipitation (co-IP) assays, and whole-cell recordings from HEK cells expressing P2X7R. We found the facilitation was composed of a calcium-dependent and a calcium-independent process with the calcium-dependent facilitation resulting from calmodulin (CaM) binding to an unusual CaM binding domain in the C terminus of P2X7R. Data obtained from the present study present a unique example of CaM deregulating (via facilitated calcium entry), rather than tightly controlling, calcium homeostasis of ion channel function.
Materials and Methods
Chemicals, antibodies, and peptides.
Culture media, sera, and other cell culture reagents were obtained from Invitrogen. ATP, Triton X-100, salts and chemicals for the solutions, calmodulin inhibitory binding peptide (CIP) (LKKFNARRKLKGAILTTMLA), and Kodak films were obtained from Sigma-Aldrich, and KN-62 from Calbiochem. The control peptide (LKCFNARCKLKGAILCTMLA) was generated by Alta Bioscience. Anti-Glu–Glu and rabbit IgG antibodies came from Bethyl Laboratories, polyclonal rabbit anti-C-terminal rat P2X7 receptor primary antibodies from Alomone Labs (epitope from residue 576–595; Alomone Labs), monoclonal mouse anti-calmodulin primary antibodies from Millipore, goat anti-rabbit and rabbit anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies from Dako UK, and ECL-plus kit from GE Healthcare.
Cell culture, transfection, and site-directed mutagenesis.
Human embryonic kidney (HEK293) cells were maintained in Dulbecco's modified Eagle's: F12 medium, supplemented with 10% heat-inactivated fetal calf serum and 2 mm l-glutamine at 37°C in a humidified 5% CO2 incubator. Cells were grown to 80–90% confluence and transiently transfected with 1 μg of cDNA encoding rat P2X7 receptors (Surprenant et al., 1996) and green fluorescent protein (GFP) expression plasmid (0.1 μg; for fluorescent detection of transfected cells) using Lipofectamine 2000 (Invitrogen). They were then plated either into 75 cm2 tissue culture flasks (for coimmunoprecipitation experiments) or onto 13 mm glass coverslips (for electrophysiological studies). Point mutations were introduced into rat P2X7 receptor constructs in pcDNA3 (Invitrogen) using the PCR overlap extension method and Accuzyme proof-reading DNA polymerase (Bioline). Nucleotide changes in the coding sequence were responsible for I541 to T541 and S552 to C552 amino acid mutations into the rat P2X7 protein. The primers used for these site-directed mutagenesis experiments were for the I541 to T541 mutation: forward, 5′-GGAGAGGCCACCAACAGCAAG-3′; reverse, 5′-CTTGCTGTTGGTGGCCTCTCC-3′, and for the S552 to C552 mutation: forward, 5′-GTGCGTACAGGTGCTATGCCACC-3′; reverse, 5′-GGTGGCATAGCACCTGTACGCAC-3′. Wild-type (WT) and mutated rP2X7 receptors bore C-terminal Glu–Glu (EE) epitope tags (EYMPME). The full coding regions of the constructs used in this study were verified by DNA sequencing.
In some cases, HEK293 cells were also cotransfected with the calmodulin dominant-negative mutant CaM1234 YFP (yellow fluorescent protein)-tagged (CaMmut) (Xia et al., 1998) kindly provided by Dr. Elizabeth Seward (University of Sheffield, Sheffield, UK). When CaMmut was coexpressed with P2X7R, GFP was not used.
Electrophysiological, dye uptake, and video recordings.
Electrophysiological recordings were performed 24–48 h after transfection by the patch-clamp technique. Patch pipettes were pulled from borosilicate glass (World Precision Instruments) to a resistance of 4–6 MΩ. Currents were recorded in the whole-cell configuration, under voltage-clamp mode at room temperature using a MultiClamp 700A patch-clamp amplifier (Molecular Devices), and analog signals were filtered at 10 kHz and digitized using a 1322A Digidata converter. Cell capacitance and series resistances were electronically compensated. Membrane potential was held at −60 mV except where indicated otherwise. Agonist (ATP) was applied using a RSC 200 fast-flow delivery system (Bio-Logic Science Instruments). Facilitation curves were built by measuring the maximal current at each time point from initiation of ATP application (prolonged applications), or measuring instantaneous current during the initial 10 s application of ATP and calling this “0,” and current amplitude at end of each 10 s pulse (at 40 s intervals) was measured and plotted as a function of total duration of ATP application. A facilitation index was calculated using the latter protocol by integrating the total currents of the eight first 10-s-long applications of ATP, and expressing this as a ratio of I8/I0. Concentration–response curves to ATP were realized by an initial application of maximum ATP (10 mm), and then by applying decreasing concentrations of agonist, before or after full P2X7R facilitation. Concentration–responses curves were plotted using Origin Microcal 6.0 software (OriginLab) and EC50 defined using the sigmoidal equation provided by this software. Time to initial onset of membrane blebs was measured in parallel experiments using standard CCD video microscopy (Mackenzie et al., 2001, 2005); we previously showed that time course of membrane blebbing in a cell during whole-cell recording (using standard intracellular solutions) does not differ significantly from uncompromised cells (Mackenzie et al., 2001). Ethidium bromide (20 μm present throughout) fluorescence was measured as detailed previously (Pelegrin and Surprenant, 2006) using 20× objective under a Nikon confocal microscope; 30 cells were imaged per coverslip and averaged, and the slope of the initial fluorescent signal was used as the most accurate and consistent measurement for comparisons.
Solutions.
The external physiological saline solution (PSS) had the following composition (in mm): 147 NaCl, 10 HEPES, 13 d-glucose, 2 KCl, 2 CaCl2, and 1 MgCl2. Agonist solutions were prepared in this PSS at the concentration indicated in the figure legends. Control intracellular solution had the following composition (in mm): 147 NaCl, 10 HEPES, 10 EGTA, 2 KCl. EGTA concentration was varied as indicated in the text, or BAPTA (5 or 10 mm) was used in place of EGTA. WebMaxC (http://www.stanford.edu/∼cpatton/webmaxc/webmaxcE.htm) was used to calculate intracellular Ca2+ concentrations in presence of EGTA and BAPTA; we used 3 mm ([Ca2+]i, 320 nm) and 4 mm ([Ca2+]i, 720 nm) CaCl2 with 5 mm BAPTA in the experiments described in Figure 3A. Osmolarity and pH values were 295–315 mOsm and 7.3, respectively. Ten minutes was allowed after achieving whole-cell conditions before beginning experimental protocols to obtain complete dialysis of intracellular solutions, although in a smaller number of experiments we noted no differences when we waited only 5 min after cell break-in.
Figure 3.
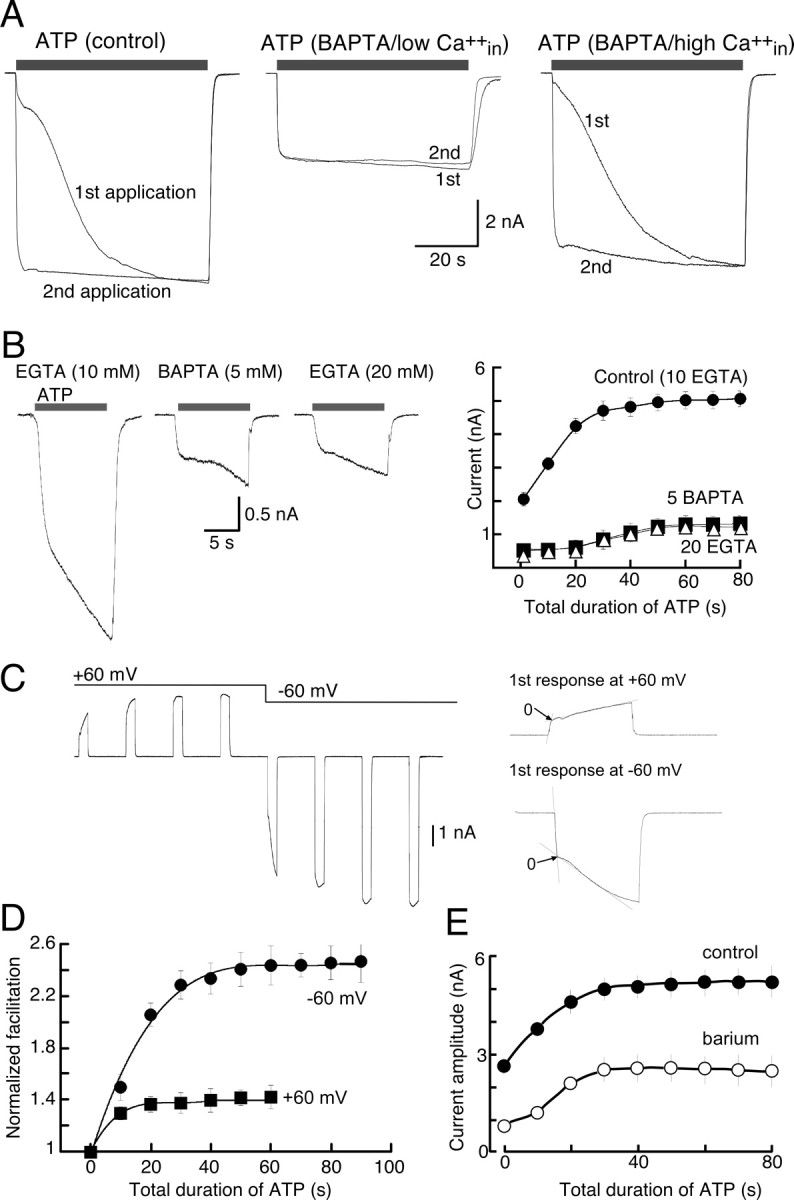
Calcium-dependent and calcium-independent facilitation of P2X7R currents. A, Representative traces of currents in response to sustained (60 s) ATP application (protocol shown in Fig. 1A) with different concentrations of intracellular calcium buffers; each set shows currents in response to first and second agonist application. Membrane capacitance was approximately the same for each cell shown (14, 14.8, and 14.2 pF, respectively) and thus can be directly compared. B, Representative traces of initial currents in response to brief (10 s) applications of ATP recorded with different concentrations of intracellular calcium buffers as indicated, and summary of all experiments (protocol as per Fig. 1C) (n = 7–9 for each point). C, Example of currents recorded in response to ATP (5 mm, 10 s applications at 40 s intervals) at +60 and −60 mV. A rapidly facilitating current occurred at +60 mV (zero calcium influx), but when the potential was taken to −60 mV the subsequent facilitation was the same as that observed in protocols described in Figure 1C. D, Summary of all experiments as shown in B; time constant of facilitation was 19.5 ± 0.8 s at −60 mV and 9.2 ± 0.6 s at +60 mV (n = 6). E, Results from experiments performed using protocol described in Figure 1C in control solution or in barium solution as indicated (n = 8 for control and 16 for barium). Error bars indicate SEM.
Coimmunoprecipitation assays.
Two days after transfection, HEK293 cells previously plated in 75 cm2 tissue culture flasks were washed twice with 15 ml of PSS, and then either vehicle buffer or ATP (5 mm) was added for 45 s. Solution was immediately removed and the cells were scraped and lysed in presence of 1 ml of radioimmunoprecipitation assay lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm EDTA), containing 1% Triton X-100 and protease inhibitors (Complete; Roche) for 1 h at 4°C. Cell lysates were centrifuged at 16,000 × g for 10 min to remove insoluble debris. Total protein samples were removed and assayed for protein content using the Bio-Rad Protein Assay Kit (Bio-Rad). Co-IP experiments were performed using the ExactaCruz F (Santa Cruz Biotechnology). The supernatants obtained from the cell lysate centrifugation (1 ml) were precleared with 40 μl of rabbit IP matrix beads for 1 h. Two types of immunoprecipitation (IP) antibody–IP matrix complexes were prepared and incubated overnight at 4°C. Each complex contained 500 μl of PBS solution, 40 μl of IP matrix beads, and 2 μg of antibodies. In one complex, the antibodies used were rabbit polyclonal anti-EE to pull down rP2X7 receptors Glu–Glu tagged, and the other complex was done using rabbit IgG as a negative control. Matrixes were centrifuged, and pellet matrixes were washed two times in PBS before adding the samples. Samples were separated in two volumes (500 μl) and incubated with each of the two IP antibody–IP matrix complexes overnight at 4°C. After incubation, samples were centrifuged and pellet washed five times with PBS. Pellets were resuspended in 35 μl of 2× SDS-reducing electrophoresis buffer and boiled for 3 min. IP samples were loaded, as well as total cell lysates in 10% polyacrylamide gels (5 μg of total protein for P2X7 receptor identification and 2 μg for calmodulin identification) and transferred to polyvinylidene difluoride membranes. Western blotting was performed according to standard protocols. P2X7 proteins were visualized using polyclonal rabbit anti-rat P2X7 C terminus primary antibody and anti-rabbit ExactaCruz F-HRP-conjugated secondary antibody (both at 1/2000 dilution), and calmodulin proteins were visualized using monoclonal mouse anti-calmodulin primary antibody (1/1000 dilution) and rabbit anti-mouse HRP-conjugated secondary antibody (1/2000 dilution), followed by detection using the ECL-plus kit and Kodak Bio-Max MS film. Gels were analyzed by densitometry measurements using GeneSnap/GeneTools software (Syngene). Relative CaM binding to WT and I541T, S552C double-mutant P2X7 receptors was expressed in diagrams from densitometric measurements. To compare protein expression levels, semiquantitative densitometric analyses were performed on Western blots using β-actin to normalize band densities. For each IP experiment, the CaM band value was divided by the correspondent light chain IgG band value and normalized to the results obtained for the WT P2X7 receptor in absence of ATP stimulation.
Data analysis and statistics.
Average results are expressed as the mean ± SEM from the number of cells or assays indicated in the figure legends. Statistics were performed using SigmaStat 3.0.1a (Systat Software). One-way ANOVA on ranks followed by a Student–Newman–Keuls test were used to compare the facilitation index in the different condition tested, as well as relative CaM bindings to WT and mutant P2X7 receptors.
Results
Facilitation of P2X7R currents
Fast-flow, sustained application of a maximum concentration of ATP at −60 mV evoked an initial rapid onset of inward current, typical of ligand-gated ion channels (e.g., millisecond time domain); this was followed by an approximately threefold increase in current amplitude over the subsequent 20 s (Fig. 1A,D). As described previously (Mackenzie et al., 2001, 2005; Verhoef et al., 2003; Adinolfi et al., 2005), reversible membrane blebbing occurred within 40–60 s of sustained ATP application (Fig. 1B) (see below). Because these blebs often resulted in patch recording instability (∼40% of recordings), we quantified ATP-mediated facilitation using the protocol illustrated in Figure 1C, which did not result in membrane blebbing. Ten second duration applications of ATP (5 mm) were applied at 40 s intervals, the instantaneous current (termed “0”) evoked by the first agonist application and the current at the end of each 10 s application (termed “1, 2, n”) were measured (Fig. 1C). Plots of current amplitude as a function of total duration of ATP using either protocol were not significantly different (Fig. 1D); in either case, the increase in current was well fit by a single exponential function with time constants of 18.2 ± 0.5 s (n = 12) and 17.2 ± 0.4 s (n = 41) for prolonged application and repetitive application, respectively. Facilitation was measured similarly for lower concentrations of ATP, with facilitation time constants of >2 min observed with 1 mm ATP (Fig. 1E). ATP concentration response curves were determined for initial currents and after complete facilitation; EC50 value was shifted ∼2.5-fold to the left after facilitation (Fig. 2A,B). Currents remained fully facilitated for the duration of recordings (up to 2 h).
Figure 1.

Current facilitation at rat P2X7R. Two protocols to measure ATP-induced current facilitation were prolonged application for 1 or 2 min (A) or 10 s duration applications at 40 s intervals (C); ATP concentration was 5 mm for all experiments in A–D. Membrane blebbing (indicated at arrows in A and asterisks in B) is illustrated by photomicrographs in B which are frames extracted at times indicated from video recording of two P2X7R-expressing cells in parallel experiment (without electrophysiological recording) to that illustrated in A. D, Time course of facilitation measured directly as current amplitude at 1 s and thereafter 10 s intervals during prolonged application of ATP (open symbols; n = 11) or as initial “instantaneous” current and then sustained current at end of each 10 s duration application (filled circles; n = 41). The lines are best-fit single exponential functions; time constants were 18.2 ± 0.5 and 17.2 ± 0.4 s, respectively, and were not significantly different. E, Concentration dependence to time course of ATP-mediated facilitation of P2X7R currents; histograms show time constant of facilitation obtained from experiments as in C (n = 11, 12, and 41, respectively). Holding potential was −60 mV. Error bars indicate SEM.
Figure 2.

Facilitation of P2X7R currents increases agonist affinity. A, Currents recorded in response to increasing concentrations of ATP applied for 10 s duration before and after currents had fully facilitated (by repeated applications of maximum ATP concentration, as per protocol shown in Fig. 1C). B, Concentration–response curves for initial and fully facilitated ATP-evoked currents; half-maximal (EC50) values are 4.9 ± 0.5 and 2.0 ± 0.2 mm, respectively (n = 17). Error bars indicate SEM.
Distinct calcium-dependent and calcium-independent components to facilitation
We asked whether there was a calcium-dependent process involved in the facilitation of ATP-induced currents in three ways. First, currents were elicited while recording with varying concentrations of EGTA or BAPTA for intracellular chelation of calcium (Fig. 3A,B). High concentrations of EGTA (20 mm) or BAPTA (5 mm) with low intracellular calcium (calculated <5 nm) decreased both instantaneous and sustained current by 60–80%, but no significant differences were observed when these same chelators were used with high intracellular calcium (calculated as 720 nm by inclusion of 4 mm Ca2+ in intracellular solution) (Fig. 3A). Lower concentration of intracellular calcium (nominal 320 nm) yielded currents that showed only 16 ± 7% less facilitation (range, 0–45%; n = 6) than control (nominal 5 nm) intracellular calcium (data not shown). Additionally, no significant differences in initial or facilitated currents were observed when using either 1 or 10 mm EGTA (Figs. 3B, 5). Second, ATP-evoked currents (10 s duration/40 s intervals) were elicited at +60 mV (zero calcium influx). Current amplitude increased during the first ATP application, it reached maximum by the second or third application (Fig. 3C), and full facilitation was ∼60% less than that elicited at −60 mV (Fig. 3D). After steady-state facilitation was achieved while holding the membrane potential at +60 mV, the membrane potential was returned to −60 mV and ATP applications continued. The degree of facilitation at −60 mV was not significantly altered by the previous ATP applications (at +60 mV) (Fig. 3C,D). This smaller and more rapid (calcium-independent) facilitation of currents at +60 mV was not significantly different when recording with intracellular BAPTA solution (data not shown) (n = 3). These results suggest that distinct mechanisms underlie facilitation at +60 and −60 mV, or that the calcium-independent facilitation observed at +60 mV also occurs at −60 mV but that it has already been activated and fully saturated. We have not explored further the calcium-independent facilitation observed at +60 mV. Third, we substituted barium for calcium in the external solution and found that both instantaneous and sustained currents were decreased by 50–75% (Figs. 3E, 5). These results clearly differentiate both a calcium-dependent and a calcium-independent component to the agonist-evoked current at P2X7R. The kinetics of the facilitation observed at +60 mV, with high intracellular EGTA or BAPTA at −60 mV, or with barium replacing extracellular calcium (−60 mV), were all similar and were of a more rapid time course than the agonist-evoked current at −60 mV when recording with ≤10 mm intracellular EGTA (8 ± 0.6 s at +60 mV vs 18 s at −60 mV) (Figs. 1, 3, 5). The calcium-independent component of facilitation constituted ∼25–30% of the total facilitation recorded at −60 mV (Figs. 3, 5).
Figure 5.

Summary of facilitation of P2X7R currents under experimental conditions used in this study. A, Representative current traces recorded under the conditions indicated; each set of traces (top and bottom) are from same cell at I0 (first application of ATP) and I8 (eighth application). B, Facilitation index was calculated (as detailed in Materials and Methods) as the integrated current at I0/integrated current at I8; single asterisks indicate significantly different at p < 0.02, double asterisks indicate p < 0.005, and n values are shown in parentheses above each bar. Error bars indicate SEM.
Calmodulin is responsible for the calcium-dependent component of facilitation
Because the amino acid sequence of P2X7R lacks EF hand motifs, we thought it unlikely that direct binding of calcium to the P2X7R could account for the facilitation. We asked whether CaM may be involved by including the well characterized and highly specific inhibitor of CaM, CIP (30 μm) in the patch pipette (Zhang et al., 1998). Intracellular dialysis with CIP inhibited the instantaneous current to the same degree as BAPTA chelation and was nearly as effective in reducing the sustained current and full facilitation (Figs. 4A, 5). No differences were observed when recordings were made with the control, CaM-inactive, peptide (Figs. 4A, 5). We then coexpressed P2X7R with the dominant-negative CaM mutant lacking all four EF hands (Xia et al., 1998; DeMaria et al., 2001). The instantaneous current was reduced to the same degree as we observed when recording with intracellular BAPTA or CIP (∼80% inhibition), but the sustained current and degree of facilitation were inhibited by only 25–40% (Figs. 4B, 5). Sustained currents and full facilitation were further reduced when BAPTA was included in the intracellular solution (Figs. 4B, 5). These inhibitory effects could not be attributed to activation of calmodulin kinase II (CaMKII), because inclusion of the CaMKII inhibitor KN-62 (10 μm) into the patch pipette had no effect on ATP-induced current facilitation (Fig. 5).
Figure 4.

Calmodulin modulates calcium-dependent facilitation of P2X7R currents. A, Representative traces of initial currents in control (10 mm EGTA intracellular), with CIP (30 μm), or with control peptide (30 μm) in pipette, and summary of all experiments (n = 11 or 12 for each point). B, Representative traces of initial current from control, CaMmut-expressing, and CaMmut plus intracellular BAPTA, and summary of all experiments (n = 8–14 for each point). The graphs for intracellular BAPTA are the same in A and B for comparison. Error bars indicate SEM.
Using the CaM Target Database (http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html), we identified a 17 residue putative CaM binding sequence in the P2X7R C terminus (Fig. 6A). This sequence was no longer identified as a possible CaM binding domain when we replaced two of these residues (isoleucine with threonine at residue 541 and serine with cysteine at residue 552). Therefore, we made this double mutation in the P2X7R and found that ATP-evoked current facilitation no longer showed any calcium dependence nor was it altered by intracellular dialysis with CIP (Fig. 6B) or by recording with high intracellular calcium (nominal 720 nm; n = 5). The (calcium-independent) fully facilitated currents at the mutant P2X7R were 50–60% less than at the WT P2X7R [2.9 ± 0.2 nA at P2X7R(I541T/S552C) vs 5.8 ± 0.4 nA at WT P2X7R; n = 8]. These differences could not be explained by lower protein expression because protein expression was significantly higher for the mutant P2X7R (data not shown). In addition to the classical “IQ” Ca2+-independent CaM-binding motif (which binds apocalmodulin), there are now dozens of other motifs on ion channels that have been identified as CaM binding targets, and even more target sequences that have been implicated as contributing to, or influencing, CaM binding (http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html) (Bhattacharya et al., 2004; O'Day and Myre, 2004; Yamniuk and Vogel, 2004). Some of these sequences are called Ca2+-dependent and only bind CaM under its 4 Ca2+-associated form. The CaM binding motif we identified here (Fig. 6A) does not conform to the relatively common 1-10 or 1-14 motifs delineated at other ion channels (Yamniuk and Vogel, 2004; Abriel and Kass, 2005; Halling et al., 2006), but it does fit within the Ca2+-dependent 1-16 motif family (sequences whose key bulky amino acids are spaced 14 residues apart); thus, our sequence is I-x(14)-W. However, there is an additional bulky residue in the middle of this sequence [I-x(3)-L-x(10)-W], making this sequence a 1-5-16 motif, which has not been reported as a subfamily within the 1-16 motif. Indeed, there are no reported subfamilies within the 1-16 family because this has been considered to be unique to CaMKK (Ca2+/CaM-dependent protein kinase kinase) binding (Osawa et al., 1999).
Figure 6.

Identification of a CaM interacting domain in C terminus of P2X7R. A, Amino acid sequence of intracellular C-terminal residues of rat P2X7R (N356 to Y595). The boxed residues indicate putative CaM binding site according to Calmodulin Target Database (http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html); this sequence is expanded below to show the two sites we mutated. B, Summary of facilitation recorded from P2X7R(I541T/S552C) mutant under control (10 mm EGTA intracellular), BAPTA (5 mm intracellular), or CaM inhibitory peptide (30 μm CIP intracellular) (n = 18, 16, and 12, respectively). C, ATP concentration–response curves obtained for initial and fully facilitated currents from P2X7R(I541T/S552C) mutant expressing cells (n = 6). ATP EC50 values were 3.2 ± 0.3 and 1.5 ± 0.1 mm, respectively, and these were not significantly different from values obtained at wild-type receptor. D, Time to initial onset of membrane blebbing measured under conditions indicated; numbers in parentheses are n values, and asterisks indicate significantly different at p < 0.005. Error bars indicate SEM.
The ATP concentration response curve showed the same twofold leftward shift after full facilitation at P2X7R(I541T/S552C) (Fig. 6C), indicating that the increased agonist potency is not calmodulin dependent.
CaM-dependent membrane blebbing and CaM-independent dye uptake
Dramatic but reversible membrane blebbing is a signature feature of both ectopic and endogenously expressed P2X7R, and previous experiments have demonstrated a prominent, although partial, dependence on calcium (Mackenzie et al., 2001, 2005; Verhoef et al., 2003; Pfeiffer et al., 2004; Adinolfi et al., 2005). We investigated whether calmodulin may be involved in ATP-induced blebbing by measuring time to initial onset of membrane blebs as a reliable and quantifiable assay (Mackenzie et al., 2005). In wild-type P2X7R-expressing cells, initial blebs appeared at ∼30–40 s of ATP application, whereas initiation of blebs was delayed by twofold to threefold in cells preincubated with BAPTA-AM (Fig. 6D). These values are similar to those observed in previous studies (Mackenzie et al., 2005). Coexpression of CaMmut resulted in a small, but not significant, delay in membrane blebbing, although when cells expressing P2X7R and CaMmut were incubated in BAPTA-AM the delay to bleb onset was again increased by 2.5-fold (Fig. 6D). In contrast, time to membrane blebbing was threefold longer in cells expressing P2X7R(I541T/S552C) than in cells expressing wild-type P2X7R (Fig. 6D).
The best known feature of P2X7R function is that activation of the cationic ion channel is followed within seconds by a nonselective permeability to molecules up to 900 Da, referred to as the “large pore” or “dye uptake” pathway. This dye uptake pathway had previously been thought to result from a time-dependent dilatation of the P2X7R ion channel itself (North, 2002; Di Virgilio, 2007), but recent work has identified an associated protein, pannexin-1, as (or responsible for) the “large pore” opening (Pelegrin and Surprenant, 2006). Neither P2X7R-mediated dye uptake, nor pannexin-1 channel activity itself, is calcium dependent (Surprenant et al., 1996; Chessell et al., 1997; Virginio et al., 1999; Pelegrin and Surprenant, 2006). In the present study, there were no significant differences in ethidium uptake in response to stimulation with ATP (3 mm) at wild-type P2X7R or mutant P2X7R(I541T/S552C); 180 cells from six preparations were recorded in each case and values did not differ significantly (p = 0.26).
Calmodulin binds constitutively and dynamically to P2X7R
We used co-IP to assay direct protein–protein interaction between ectopically expressed P2X7R and endogenous calmodulin by immunoprecipitating epitope-tagged P2X7R and blotting for CaM. CaM clearly associated with P2X7R in the absence of stimulation with ATP (Fig. 7A, lane 2) and this association was significantly greater when co-IP was performed on cells harvested at 45 s in the presence of ATP (Fig. 7A, lane 5). Semiquantitative densitometric analysis showed a 50% increase in CaM binding to P2X7R during ATP stimulation when immunoprecipitated protein was normalized to IgG bands (Fig. 7C). In contrast, we did not detect any CaM binding when we performed similar experiments on the P2X7R(I541T/S552C) mutant, although the lysates obtained from cells expressing the mutant P2X7R showed much greater CaM protein concentration than did lysates from the wild-type P2X7R-expressing cells (Fig. 7B,C). These results provide direct biochemical support for our conclusion that a significant amount of CaM binds to wild-type, but not P2X7R(I541T/S552C) mutant receptor.
Figure 7.

CaM shows both constitutive and dynamic physical interaction with P2X7R. A, B, Co-IP experiments on cells expressing wild type or P2X7R(I541T/S552C) mutant, as indicated. Under basal unstimulated conditions, CaM coimmunoprecipitates with wild-type P2X7R (A, lane 2) and the amount of CaM protein associated with P2X7R is enhanced by 45 s stimulation with ATP (A, lane 5). No CaM binding was detected from immunoprecipitates of mutant P2X7R under basal or stimulated conditions (B, lanes 2 and 5). C, Semiquantitative densitometric measurements of CaM binding from all co-IP experiments as illustrated (n = 4); protein levels were normalized to the corresponding IgG band level for wild-type P2X7R in absence or presence of ATP as indicated. No CaM binding was detected even with prolonged film exposure from co-IPs on P2X7R(I541T/S552C)-expressing cells (n = 4). Numbers on left are molecular weight markers in kilodaltons. Error bars indicate SEM.
Discussion
Calmodulin is the most abundant calcium sensor in eukaryotic cells, and the most intensively studied. It is able to assume myriad conformations specified both by distinct target interaction sites and its own state(s) of bound calcium (Saimi and Kung, 1994; Levitan, 1999; Bhattacharya et al., 2004; Yamniuk and Vogel, 2004). These structural features allow CaM to regulate the function of diverse ion channels: from the best known example of calcium-dependent inactivation of voltage-gated calcium channels (Halling et al., 2006) through calcium-dependent activation, and calcium-independent trafficking, of SK potassium channels (Maylie et al., 2003) to more recent examples of both calcium-dependent and -independent inactivation of TRP channel family members (Kiselyov et al., 2005; Zhu, 2005; Derler et al., 2006). But, regardless of the specific ion channel or underlying molecular mechanism, the end result is to prevent calcium overload by tightly regulating global or microdomain calcium entry and ion channel activity. Here, we show CaM binds both constitutively and dynamically to a novel CaM binding motif in the C terminus of the P2X7 receptor. This results in dramatic facilitation of the membrane current with saturating levels of calcium entry and mitochondrial calcium release (Mackenzie et al., 2005) over many seconds to minutes, coincident with rapid cytoskeletal rearrangements and zeiotic membrane blebbing. This presents a unique example of CaM-mediated deregulation of calcium entry through an ion channel.
We delineated distinct calcium-dependent and calcium-independent facilitation processes and found that only the calcium-dependent process was CaM dependent. Our results also suggest CaM binds both constitutively and dynamically to the P2X7R. That is, strong chelation of intracellular Ca2+ with BAPTA or high EGTA, substitution of barium for calcium, addition of the CaM inhibitory peptide, or transfection with the dominant-negative CaMmut all reduced the instantaneous current by ∼80%, and co-IP revealed basal CaM association with unactivated P2X7R protein. These results, and the finding that intracellular application of the CaMKII inhibitor, KN62, had no effect on P2X7R currents provide fairly conclusive evidence that CaM is in a constitutive association with the unactivated P2X7R. Because the instantaneous current was decreased by inhibition of CaM or CaM binding to P2X7R, we hypothesize the constitutively bound CaM regulates initial channel gating on receptor activation. In contrast, the slow time course of facilitation of currents at −60 mV (from >3 min at low ATP to 18 s with maximum ATP concentration) was only minimally inhibited by the dominant-negative CaMmut. This result suggests that CaM also associates in a dynamic manner during prolonged or repeated receptor activation, a conclusion further supported by our biochemical results which demonstrated significantly increased co-IP of CaM with P2X7R when cells were stimulated with ATP for 45 s. Our findings that barium replacement or intracellular dialysis with 20 mm EGTA or 5 mm BAPTA inhibited P2X7R facilitation by 70–80% provide the evidence for our conclusion that this fraction of the facilitation is, indeed, calcium dependent. This conclusion is further supported by the finding that facilitation was not altered compared with control when we increased intracellular calcium to >500 nm by including 4 mm Ca2+ in the BAPTA-containing solution. However, we saw no difference in the instantaneous current, or the fully facilitated current, when using either 1 or 10 mm intracellular EGTA, whereas most studies on voltage-gated calcium channels or neurotransmitter release processes find 10 mm intracellular EGTA to be as efficient as 5 mm BAPTA (Kits and Mansvelder, 2000). The most likely explanation is that the massive calcium entry associated with the large (several nanoamperes), non-desensitizing P2X7R current over many seconds rapidly saturates EGTA at <20 mm, as has been found for synaptic transmission at the squid giant synapse (Adler et al., 1991) and for calcium-dependent inactivation of voltage-gated calcium channels by several endogenous calcium buffering proteins (Kreiner and Lee, 2006). It has also been shown that mitochondrial stores of Ca2+ are released in the millisecond to second time domain on P2X7R stimulation (Adinolfi et al., 2005; Mackenzie et al., 2005), thus providing an additional calcium surge that may be expected to saturate these buffers.
We also delineated a calcium-independent component to the ATP-induced facilitation P2X7R currents, but we have not obtained any evidence for its underlying mechanism. This component was threefold more rapid than the CaM/calcium-dependent facilitation. The calcium-independent facilitation was not altered by inhibiting CaM with the CaM inhibitory peptide, nor was it altered in the P2X7R(I541T/S552C) mutant, making it unlikely that indirect actions of CaM are involved. The 2.5-fold increase in agonist affinity, indicated by the leftward shift in the agonist concentration–response curve consequent to full facilitation, appears to result solely from this calcium-independent facilitation process because it was unaltered in the P2X7R(I541T/S552C) mutant receptor. The P2X7R(I541T/S552C) mutant should prove a valuable tool in future studies to elucidate possible mechanisms because it will allow us to investigate this distinct facilitation process without interference from the calcium/CaM-dependent current facilitation.
Rapid cytoskeletal rearrangements leading to prominent membrane blebbing are key cellular events downstream of P2X7R activation (Pfeiffer et al., 2004; Adinolfi et al., 2005; Mackenzie et al. 2005). We, and others, have demonstrated that P2X7R-induced membrane blebbing in microglia and other immune cells, and in mammalian cells ectopically expressing P2X7R, show both calcium-dependent and calcium-independent mechanisms (Pfeiffer et al., 2004; Adinolfi et al., 2005; Mackenzie et al., 2005) When intracellular calcium is strongly chelated using BAPTA-AM, the time to onset of membrane blebbing is significantly increased (Fig. 6D) and the delayed-onset blebs that do form are fewer but larger than when calcium is present (Mackenzie et al., 2005). Two key results from the present study indicate the calcium-dependent membrane blebbing results from the dynamic association of CaM to P2X7R during sustained, or repetitive, ATP applications. First, the dominant-negative CaMmut did not alter the time course of membrane blebbing, nor their morphology, and second, time to onset of membrane blebbing was delayed in the P2X7R(I541T/S552C) mutant to the same extent as when BAPTA-AM was present in wild-type-expressing P2X7R. Proteomic and co-IP analyses have previously identified α-actinin and the F-actin binding protein, supervillin, as physically associating with P2X7R in a functional complex (Kim et al., 2001). Calcium-dependent inactivation of the glutamate-gated NMDA receptor involves the competitive association of CaM and α-actinin with specific residues (in a 1-5-8-14 CaM consensus motif) of the intracellular C terminus of the NR1 subunit (Zhang et al., 1998; Krupp et al., 1999; Xia and Storm, 2005). Thus, it will be important to address whether a similarly intimate structural and functional relationship exists for CaM, α-actinin, and P2X7R using a similar strategy to that used in studies of the NMDA receptor (Zhang et al., 1998; Krupp et al., 1999), by examining ATP-induced current facilitation, membrane blebbing, and CaM/α-actinin/P2X7R co-IP in cells overexpressing wild-type and mutated α-actinin with wild type and P2X7R(I541T/S552C) mutants.
CaM modulation of ion channels has previously always been found to limit calcium entry, by inactivation of voltage-dependent and voltage-independent cation channels and/or by activation of potassium channels (Maylie et al., 2003; Abriel and Kass, 2005; Xia and Storm 2005; Zhu, 2005; Halling et al., 2006). Here, we find CaM dramatically increases calcium entry by a time-dependent facilitation of the calcium-permeable P2X7R cation channel. The P2X7R (previously termed P2Z receptor) has long been known as the “cytolytic P2X receptor” because prolonged agonist application leads to inevitable cell death via tissue-specific apoptosis and/or necrosis (North, 2002; Ferrari et al., 2006; Burnstock, 2007; Di Virgilio, 2007). It is likely CaM binding plays a role to enhance, or activate, the cell death process during long-term activation of P2X7R under conditions of chronic inflammatory disease. However, the reversible, noncytolytic, sequelae in response to brief ATP application of the type used in the present study have become a more recent focus of attention, particularly in regard to early mechanisms underlying neuroinflammatory insults, such as hypoxia and stroke (Di Virgilio, 2007; Simi et al., 2007). In resting, unactivated microglia or peripheral macrophage, P2X7R-induced currents and downstream signaling (cytoskeletal rearrangements and migratory rate, release of cytokines) are minimal, but alterations in extracellular sodium and potassium concentrations are known to significantly increase P2X7R currents and cellular signaling within seconds (Michel et al., 1999; Verhoef et al., 2005), a time course too rapid to be accounted for by transcriptional upregulation. Molecular mechanisms that may explain such rapid sensitization of P2X7R responses have not been previously elucidated. Our results provide a likely molecular mechanism and allow us to propose the following scenario: The most immediate extracellular consequences of acute neuronal injury or neuroinflammatory insult are rapid alterations in sodium/potassium concentrations concomitant with release of high levels of intracellular ATP into the localized milieu surrounding the insult (Burnstock, 2007; Simi et al., 2007). These conditions will sensitize microglial P2X7Rs, prolong their activation, and increase calcium entry for the several seconds required for dynamic CaM binding to the CaM binding domain we identified on the P2X7R C terminus. CaM binding during receptor activation will result in prolonged current facilitation with continuous calcium influx and increased cytoskeletal alterations, thus promoting rapid microglial migration into the damaged area. During initial neuroinflammatory insult, CaM deregulation (via facilitation) of calcium entry through this highly unusual ion channel would prove beneficial, not detrimental, to neuronal survival.
Footnotes
This work was supported by AstraZeneca Charnwood and the Biotechnology and Biological Sciences Research Council. We thank Elizabeth Martin, Isabel Hui, and Weihong Ma for support with cell culture and transfections.
References
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel NaV1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, Di Virgilio F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels and promotes serum-independent growth. Mol Biol Cell. 2005;16:3260–3272. doi: 10.1091/mbc.E04-11-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Bunick CG, Chazin WJ. Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta. 2004;1742:69–79. doi: 10.1016/j.bbamcr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- Chessell IP, Michel AD, Humphrey PP. Properties of the pore-forming P2X7 purinoceptor in mouse NTW8 microglial cells. Br J Pharmacol. 1997;121:1429–1437. doi: 10.1038/sj.bjp.0701278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GB. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Derler I, Hofbauer M, Kahr H, Fritsch R, Muik M, Kepplinger K, Hack ME, Moritz S, Schindl R, Groschner K, Romanin C. Dynamic but not constitutive association of calmodulin with rat TRPV6 channels enables fine tuning of Ca2+-dependent inactivation. J Physiol (Lond) 2006;577:31–44. doi: 10.1113/jphysiol.2006.118661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F. Liaisons dangereuses: P2X7 and the inflammasome. Trends Pharmacol Sci. 2007;28:465–472. doi: 10.1016/j.tips.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Halling DB, Aracena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE. 2006;re15:1–11. doi: 10.1126/stke.3182006er1. [DOI] [PubMed] [Google Scholar]
- Kim M, Jiang LH, Wilson HL, North RA, Surprenant A. Proteomic and functional evidence for a P2X7 receptor signalling complex. EMBO J. 2001;20:6347–6358. doi: 10.1093/emboj/20.22.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Kim JY, Zeng W, Muallem S. Protein-protein interaction and function of TRPC channels. Pflügers Arch. 2005;451:116–124. doi: 10.1007/s00424-005-1442-2. [DOI] [PubMed] [Google Scholar]
- Kits KS, Mansvelder HD. Regulation of exocytosis in neuroendocrine cells: spatial organization of channels and vesicles, stimulus-secretion coupling, calcium buffers and modulation. Brain Res Rev. 2000;33:78–94. doi: 10.1016/s0165-0173(00)00023-0. [DOI] [PubMed] [Google Scholar]
- Kreiner L, Lee A. Endogenous and exogenous Ca2+ buffers differentially modulate Ca2+-dependent inactivation of CaV2.1 calcium channels. J Biol Chem. 2006;281:4691–4698. doi: 10.1074/jbc.M511971200. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas C, Heinemann SF, Westbrook GL. Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labasi JM, Petrushova N, Donovan C, McCurdy S, Lira P, Payette MM, Brissette W, Wicks JR, Audoly L, Gabel CA. Absence of the P2X7 receptor alters leukocyte function and attenuates an inflammatory response. J Immunol. 2002;168:6436–6445. doi: 10.4049/jimmunol.168.12.6436. [DOI] [PubMed] [Google Scholar]
- Levitan IB. It is Calmodulin after all. Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- Mackenzie A, Wilson HL, Koss-Toth E, Dower S, North RA, Surprenant A. Rapid secretion of interleukin 1β by microvesicle shedding. Immunity. 2001;8:825–835. doi: 10.1016/s1074-7613(01)00229-1. [DOI] [PubMed] [Google Scholar]
- Mackenzie A, Young MT, Adinolfi E, Surprenant A. Pseudoapoptosis induced by brief activation of ATP-gated P2X7 receptor. J Biol Chem. 2005;280:33968–33976. doi: 10.1074/jbc.M502705200. [DOI] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol (Lond) 2003;554:255–261. doi: 10.1113/jphysiol.2003.049072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Chessell IP, Humphrey PP. Ionic effects on human recombinant P2X7 receptor function. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:102–109. doi: 10.1007/pl00005328. [DOI] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- O'Day DH, Myre MA. Calmodulin-binding domains in Alzheimer's disease proteins: extending the calcium hypothesis. Biochem Biophys Res Comm. 2004;320:1051–1054. doi: 10.1016/j.bbrc.2004.06.070. [DOI] [PubMed] [Google Scholar]
- Osawa M, Tokumitsu H, Swindells MB, Kurihara H, Orita M, Shibanuma T, Furuya T, Ikura M. A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase. Nat Struct Biol. 1999;6:819–824. doi: 10.1038/12271. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ. The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophage via p38 MAP kinase and Rho. J Leukoc Biol. 2004;75:1173–1182. doi: 10.1189/jlb.1203648. [DOI] [PubMed] [Google Scholar]
- Rassendren F, Buell G, Newbolt A, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor (P2X7): cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- Saimi Y, Kung C. Ion channel regulation by calmodulin binding. FEBS Lett. 1994;350:155–158. doi: 10.1016/0014-5793(94)00782-9. [DOI] [PubMed] [Google Scholar]
- Simi A, Lerouet D, Pinteaux E, Brough D. Mechanisms of regulation for interleukin-1beta in neurodegenerative disease. Neuropharmacology. 2007;52:1563–1569. doi: 10.1016/j.neuropharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Surprenant A, Rassendren F, Kawashim E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- Verhoef PA, Estacion M, Schilling W, Dubyak GR. P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases and IL-1beta release. J Immunol. 2003;170:5728–5738. doi: 10.4049/jimmunol.170.11.5728. [DOI] [PubMed] [Google Scholar]
- Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion and cytolysis by the P2X7 receptor. J Immunol. 2005;175:7623–7634. doi: 10.4049/jimmunol.175.11.7623. [DOI] [PubMed] [Google Scholar]
- Virginio C, MacKenzie A, North RA, Surprenant A. Kinetics of cell lysis, dye uptake and permeability changes in cells expressing the rat P2X7 receptor. J Physiol (Lond) 1999;519:3. doi: 10.1111/j.1469-7793.1999.0335m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Yamniuk AP, Vogel HJ. Calmodulin's flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol Biotechnol. 2004;27:33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]
- Zhu MX. Multiple roles of calmodulin and other Ca2+ binding proteins in the functional regulation of TRP channels. Pflügers Arch. 2005;451:105–115. doi: 10.1007/s00424-005-1427-1. [DOI] [PubMed] [Google Scholar]