

Abstract
It is established that increases in neuropeptide Y (NPY) expression are associated with hyperphagia and obesity. These effects can be reversed by estrogen, a recognized anorexigen. We found that 17β-estradiol (E2) regulates biphasic NPY gene expression in a clonal, immortalized hypothalamic cell line, N-38, through estrogen receptor (ER) action at the level of the NPY promoter. However, rapid, nongenomic actions of estrogen, linked to the phosphatidylinositol 3-kinase (PI3-K)/Akt and ERK1/2 mitogen-activated protein kinase (MAPK) pathways, may also play a role. We therefore examined the changes in the phosphorylation status of Akt, ERK1/2, and cAMP response element-binding protein (CREB) after treatment with 10 nm E2 in the N-38 neurons and found activation of these signaling proteins within 5–30 min. We also demonstrated possible cross talk between the estrogen-activated PI3-K/Akt and MAPK/extracellular signal-regulated kinase pathways using pharmacological inhibitors. We find that only ERα is involved in the early signaling events using the ERα agonist 4,4′,4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol and the ERβ agonist 2,3-bis(4-hydroxyphenyl)-propionitrile. Furthermore, we can detect colocalization of ERα and caveolin-1, a membrane-associated signaling protein. Remarkably, we find that the membrane-mediated events are critical for the long-term estrogen-mediated repression of NPY gene expression that can be mapped to within −97 bp of the NPY promoter. To link the early signaling events to downstream effectors, we detected induction of c-fos and inactivation of MSK-1 by estrogen and binding of CREB to this minimal promoter region. These observations suggest that rapid estrogen-mediated signaling is mediated by ERα, and the signal transduction events potentiate the genomic actions of estrogen on NPY gene expression in the N-38 NPY neurons.
Keywords: neuropeptide Y, membrane signaling, neurons, hypothalamic cell lines, estrogen receptor, transcription
Introduction
The genomic actions of estrogen are primarily mediated by two nuclear steroid receptors, estrogen receptor (ER) α and ERβ (Enmark and Gustafsson, 1999). The existence of nongenomic actions has led to the notion of alternative pathways that mediate acute estrogen responses via cell-surface ERs integrated or associated with the plasma membrane and activation of signal transduction cascades (Kelly and Levin, 2001; Vasudevan and Pfaff, 2007), including the phosphatidylinositol 3-kinase (PI3-K)/Akt and mitogen-activated protein kinase (MAPK) pathways (Björnström and Sjöberg, 2005; Vasudevan and Pfaff, 2007). Although nongenomic and genomic estrogenic effects have often been viewed as distinct modes of estrogen action, this may not necessarily hold true because inhibition of one can limit the effectiveness of the other downstream (Björnström and Sjöberg, 2005; Levin, 2005).
The most potent estrogen is 17β-estradiol (E2), and its main role is to regulate the neuronal network that controls reproductive function (Herbison, 1998). In addition to this role, estrogen is also critical in the regulation of appetite, energy expenditure, cell proliferation, and vascular protection (Shimizu et al., 1996, 1997; Mendelsohn and Karas, 1999; Hill and Levine, 2003). Therefore, it is evident that the physiological mechanisms that control reproduction are linked to other important regulatory systems, such as those that control energy balance (Acosta-Martinez et al., 2007). Neuropeptide Y (NPY) neurons are implicated in feeding behavior, energy use, and reproductive function (Xu et al., 2000). Because estrogen receptors are present on NPY neurons in vivo (Estrada et al., 2003), and in vitro (Belsham et al., 2004; Titolo et al., 2006), the feedback effects of estrogen on gonadotropin-releasing hormone (GnRH) neurons may be mediated via the NPY neuronal framework. Evidence also indicates that estrogen may play a role in the regulation of NPY synthesis (Shimizu et al., 1996; Ainslie et al., 2001; Pelletier et al., 2007). In an attempt to elucidate how estrogen may regulate the NPY neuron, we used a clonal, immortalized hypothalamic neuronal cell line expressing NPY, the N-38 neurons (Belsham et al., 2004). We reported recently that E2 mediates the biphasic control of NPY gene expression (Titolo et al., 2006). We linked this biphasic regulation of the NPY gene to the changing levels or ratio of the ER subtypes in the N-38 NPY neurons (Titolo et al., 2006). Thus, E2-mediated signaling and its control of the NPY neuron may be dynamic and crucial to maintaining diverse homeostatic functions (Acosta-Martinez et al., 2007).
Our findings demonstrate rapid E2 signaling in the N-38 neurons and that activation of these signaling cascades are essential for the long-term estrogen-mediated control of NPY gene expression. In addition, using ER-specific agonists, these studies provide evidence that ERα is involved in nongenomic estrogen signaling in our NPY-expressing cell model. We then link the E2-mediated membrane signaling to downstream effectors, including c-fos activation, cAMP response element-binding protein (CREB) binding to the NPY promoter, and MSK-1 inactivation.
Materials and Methods
Cell culture and reagents.
N-38 neurons were cultured in monolayer in DMEM (Invitrogen) and supplemented with 5% fetal bovine serum (HyClone Laboratories), 4.5 mg/ml glucose, and penicillin/streptomycin and maintained at 37°C in an atmosphere of 5% CO2 as described previously (Belsham et al., 2004). Information regarding the phenotypic profile of the N-38 cell line can be found on the website http://cellutionsbiosystems.com or in previous manuscripts (Belsham et al., 2004; Titolo et al., 2006). During steroid treatment, cells were serum starved for 12–16 h in phenol red-free DMEM, and then treatments were performed in phenol red-free medium supplemented with charcoal-stripped serum (Titolo et al., 2006). All of the experiments were performed using E2 and, unless noted otherwise, is the estrogen stated in the text. E2 was obtained from Sigma-Aldrich. ERα-selective agonist 4,4′,4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT) and ERβ-selective agonist 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN) were obtained from Tocris Bioscience. Final concentration of estradiol and ER-selective agonists in the treatments was 10 nm,whereas control cells were treated with comparable concentration of vehicle (ethanol). Although there is evidence that DPN has a 10-fold lower Kd for ERβ than E2 or PPT has for ERα, PPT and DPN dose curves did not demonstrate significant differences in their effective concentrations; therefore, 10 nm was chosen for both. The PI3-K inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-1-benzopyran-4-one-hydrochloride] and the MAPK inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] were obtained from Cell Signaling Technology and dissolved in dimethylsulfoxide (Sigma-Aldrich). Cells were treated with inhibitors 45–60 min before estradiol treatment and had final concentrations of 25 μm.
SDS-PAGE and Western blot analysis.
Confluent N-38 cells were washed with ice-cold PBS and lysed in high-salt buffer (0.4 m NaCl, 20 mm HEPES, pH 8.0, 1 mm MgCl2, 0.5 mm EDTA, 0.5 mm DTT, 1 mm PMSF, 0.1% NP-40, 1% protease inhibitor cocktail, and 1% phosphatase inhibitor cocktail) for 40 min on ice. Lysates were cleared at 12,000 × g for 20 min at 4°C and then treated for 4 min at 96°C with SDS-PAGE sample buffer containing β-mercaptoethanol. Protein concentration was determined by the BCA protein assay kit (Pierce). Lysate protein (45 μg) was resolved on SDS-PAGE gels and blotted onto Hybond-C nitrocellulose membranes (GE Healthcare). The resulting blot was blocked with 5% skim milk in PBS containing 0.2% Tween 20, and incubated with primary antibody overnight at 4°C. For studies of Akt, ERK1/2, CREB, and MSK-1 phosphorylation, the blots were first probed with phospho-specific antibodies to detect phospho-Akt (Ser473, 1:1000; Cell Signaling Technology), phospho-ERK1/2 (Thr202, Tyr204, 1:200; Santa Cruz Biotechnology), phospho-CREB (Ser133, 1:1000; Cell Signaling Technology), or phospho-MSK-1 (Thr581, 1:1000; Cell Signaling Technology). The same blot was reprobed for total (nonphosphorylated) Akt, ERK1/2, CREB, or MSK-1 protein (1:1000) to verify equal loading. Immunoreactive bands were visualized with horseradish peroxidase-labeled secondary sheep anti-mouse antisera at a 1:5000 dilution and enhanced chemiluminescence (ECL kit; GE Healthcare) as described by the manufacturer.
Reverse-transcriptase PCR.
N-38 neurons were incubated in the presence of 17β-estradiol (10 nm) or vehicle (ethanol) over a 48 h time course. Cells were then harvested at the indicated times. Total RNA was isolated using the guanidium-thiocyanate phenol chloroform extraction method (Chomczynski and Sacchi, 1987) and quantified. Real-time reverse transcriptase (RT)-PCR was performed with SYBR green PCR master mix (Applied Biosystems), according to the instructions of the manufacturer, and run on the Applied Biosystems Prism 7000 real-time PCR machine. Approximately 50 ng of template was used for the PCR, and the primer sequences for the NPY transcript are as follows: NPY-248-SYBR sense, 5′CAG AAA ACG CCC CCA GAA; and NPY-324-SYBR antisense, 5′AAA AGT CGG GAG AAC AAG TTT CAT T. Real-time RT-PCR values were calculated by the ΔΔCT method and normalized to γ-actin mRNA levels at the corresponding time points, as described previously (Titolo et al., 2006).
Transient transfections.
The full-length human −1078 NPY 5′ flanking gene plasmid was generated by PCR as described previously (Mayer et al., 2003). Deletion constructs were generated using PCR, and the −1078 NPY plasmid was used as the template. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. The cells were incubated with deletion mutant constructs (−1078, −936, −876, −769, −721, −597, −448, −278, and −97) of the human NPY 5′ flanking region in the pGL2–enh plasmid or the pGL2–enh plasmid alone. After 6 h transfection, the cells were treated with 10 nm estradiol or vehicle and incubated for an additional 24 h before harvesting. Relative luciferase units were normalized by total cellular protein, and protein concentrations were determined using the bicinchoninic acid protein assay reagent. Luciferase assays were performed as described previously (Titolo et al., 2006).
Immunocytochemistry.
N-38 cells were plated on eight-well chamber slides (BD Biosciences) in DMEM overnight. During steroid treatment, cells were serum starved for 12–16 h in phenol red-free DMEM and treated with 10 nm E2 for 30 min before fixation in 2% paraformaldehyde. Cells were blocked with 1% BSA–PBS and incubated with primary antibody overnight at 4°C. No antibody wells served as controls. The primary ERα antibody was used at a 1:50 1% BSA–PBS dilution of mouse monoclonal anti-human for ERα (DakoCytomation). For colocalization, primary antibody for rabbit polyclonal caveolin-1 (Santa Cruz Biotechnology) was used at a 1:50 dilution. Cells were washed with PBS and incubated with a FITC-conjugated AffiniPure goat anti-mouse secondary antibody (Jackson ImmunoResearch) in a 1:100 dilution at room temperature for 90 min. Cells were then washed with PBS and incubated with rhodamine-labeled (Texas Red) biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories) in a 1:100 dilution at room temperature for 30 min. After washing cells with PBS, gaskets were removed from the chamber slides and mounted with DakoCytomation fluorescent mounting media. Fixed cells were then visualized with a confocal laser scanning microscope at a magnification of 400× (LSM 510; Carl Zeiss). FITC fluorescence was excited by the 488 nm argon laser line, whereas rhodamine was excited by the 543 nm helium–neon laser line.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays were performed using previously described methodology (Cui et al., 2005). Cells were cross-linked with 1% formaldehyde for 10 min at room temperature and then washed with ice-cold PBS. The cell pellet was resuspended in cell lysis buffer [5 mm PIPES (KOH), pH 8.0, 85 mm KCl, and 0.5% NP-40] containing protease inhibitors and incubated for 10 min on ice. The nuclei were lysed in the nuclear lysis buffer (50 mm Tris, pH 8.1, 10 mm EDTA, and 1% SDS) containing protease inhibitors and sonicated three times for 20 s each on ice. Supernatants were then recovered by centrifugation at 20,000 rpm for 10 min at 4°C and diluted five times in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris, pH 8.1, and 167 mm NaCl) plus protease inhibitors. The sample was precleared with 80 μl of a 50% salmon sperm DNA/protein G agarose slurry (Upstate) for 30 min at 4°C. Ten percent of the lysate was used as the input control (50-fold dilution). The rest of the supernatant was incubated with 2 μl of anti-CREB antibody (Cell Signaling Technology) at 4°C overnight, and then 60 ml of a 50% salmon sperm DNA/protein G agarose slurry (beads) was further added for 1 h at 4°C. Immunoprecipitates were washed sequentially for 5 min each in low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris, pH 8.1, and 150 mm NaCl), high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris, pH 8.1, and 500 mm NaCl), and LiCl wash buffer (0.25 m LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, and 10 mm Tris, pH 8.0). Bead precipitates were then washed twice with Tris–EDTA buffer and eluted two times with elution buffer (1% SDS and 0.1 m NaHCO3). The eluates were combined and incubated at 65°C for 5 h to reverse the formaldehyde cross-linking. The DNA was precipitated and dissolved in water and treated with proteinase K at 45°C for 2 h. DNA was purified using Qiaquick spin columns (Qiagen) and eluted in 50 ml of water. For PCR, 2 ml of DNA was amplified for 35 cycles with annealing temperature of 60°C. The following primers were used: 5′GAA GTG GCT GTG GGA GTC AC3′ (sense) and 5′CTC TGG GTG CCT CTG TGA G3′ (antisense) from −118 to +22 bp of the mouse NPY promoter (containing the −97 bp region). The controls indicated were “beads alone” [no antibody or DNA (“bead”)] or the “input DNA from the lysate” [no immunoprecipitation (“input”)].
Statistical analysis.
Data were analyzed using one-way or two-way ANOVA by Prism software (GraphPad Software), and statistical significance of the results was determined using Tukey's multiple comparison test with p < 0.05.
Results
E2 rapidly phosphorylates Akt, ERK1/2, and CREB in N-38 neurons
Using Western blot analysis and phospho-specific antibodies, we determined whether estrogen activates signal transduction cascades in the N-38 neurons. Specifically looking at the PI3-K and MAP kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) MAPK pathways, we demonstrate that 10 nm 17β-estradiol induces the phosphorylation of Akt (Fig. 1A), ERK1/2 (Fig. 1B), and CREB (Fig. 1C) in the N-38 neurons. Phosphorylation was maximal at 30 min after estrogen treatment, and this activation dropped to basal levels by 60 min after being normalized to total protein. Original Western blots for each of the estradiol treatments versus vehicle can be seen in the following figures (see Figs. 2–4).
Figure 1.
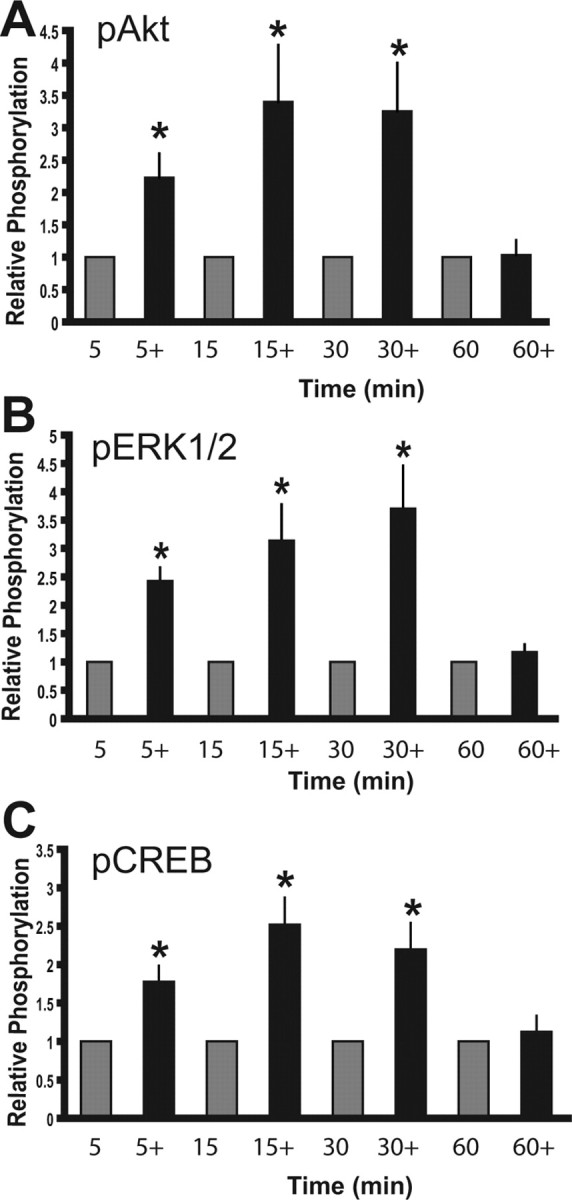
E2 activates signal transduction events in N-38 neurons. N-38 neurons were serum starved for 12–16 h before treatment with 10 nm estradiol (+) or vehicle alone over a 60 min time course. At the indicated time points, cell lysates from N-38 neurons were isolated and subjected to SDS-PAGE. A–C, Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against Akt (A), ERK1/2 (B), and CREB (C). All results shown are normalized to total protein relative to corresponding control protein levels (set to 1.0) at each time point and are expressed as mean ± SEM (n = 3 independent experiments). *p < 0.05, by two-way ANOVA followed by Tukey's post hoc test.
Figure 2.
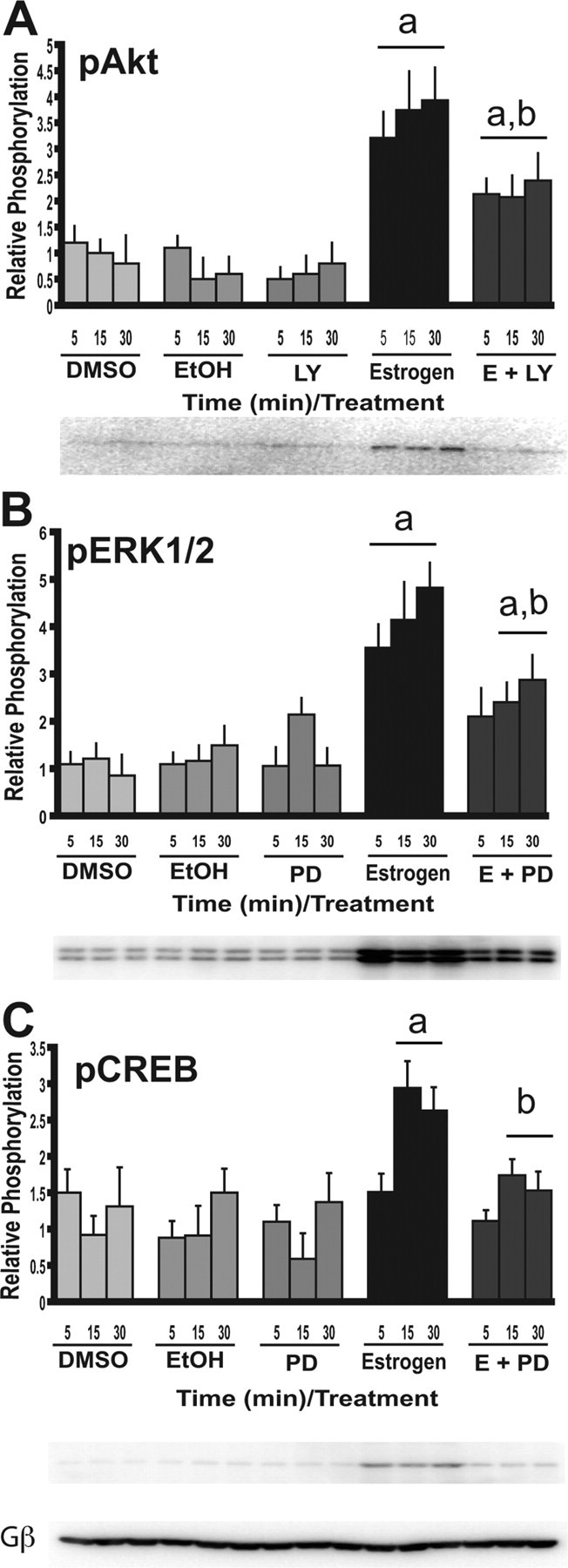
PI3-K and MEK inhibitors reduce the activation of the signal transduction events induced by E2. N-38 neurons were serum starved for 12–16 h before treatment with 10 nm estradiol or vehicle alone over a 30 min time course. Cells were treated the PI3-K inhibitor [LY294002 (LY)] and MAPK inhibitor [PD98059 (PD)] for 45–60 min at a final concentration of 25 μm before estradiol exposure. At the indicated time points, cell lysates from N-38 neurons were isolated and subjected to SDS-PAGE. A–C, Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against Akt (A), ERK1/2 (B), and CREB (C). All results shown are normalized to total protein relative to corresponding control protein levels at each time point and are expressed as mean ± SEM (n = 3 independent experiments). ap < 0.05 compared with vehicle controls; bp < 0.05 compared with E2 treatment by two-way ANOVA, followed by Tukey's post hoc test. Representative Western blots are shown. Gβ, G-protein β subunit.
Figure 3.
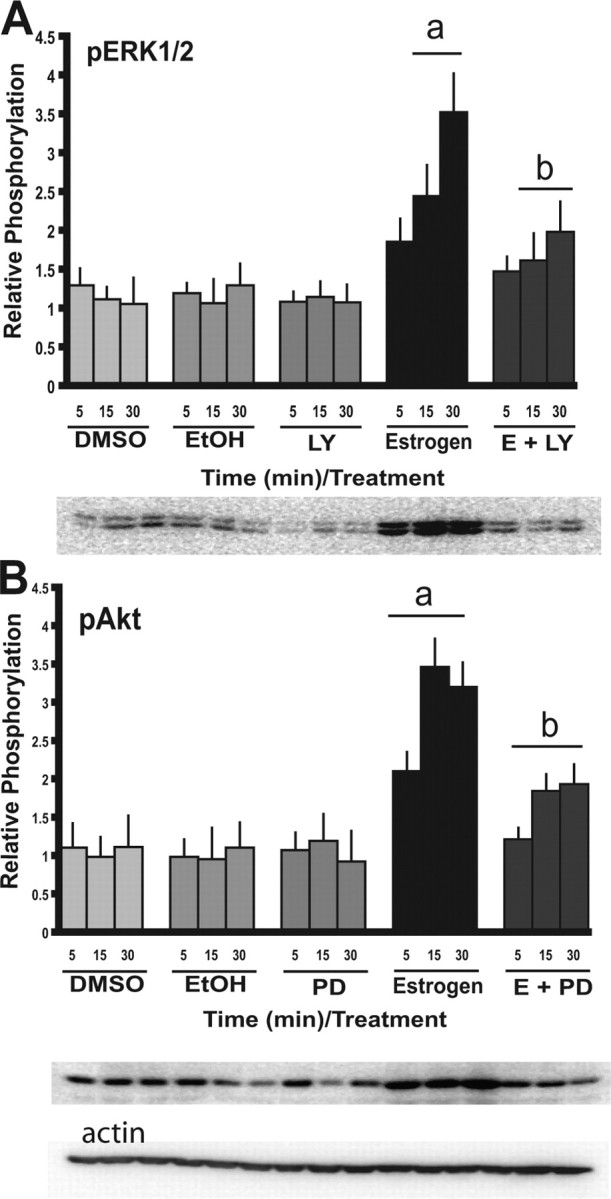
Cross talk between the PI3-K and MAPK signal transduction cascades. N-38 neurons were serum starved for 12–16 h before treatment with 10 nm estradiol or vehicle alone over a 30 min time course. Cells were treated the PI3-K inhibitor [LY294002 (LY)] and MAPK inhibitor [PD98059 (PD)] for 45–60 min at a final concentration of 25 μm before estradiol exposure. At the indicated time points, cell lysates from N-38 neurons were isolated and subjected to SDS-PAGE. A, B, Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against Akt (A) and ERK1/2 (B). All results shown are normalized to total protein relative to corresponding control protein levels at each time point and are expressed as mean ± SEM (n = 3 independent experiments). ap < 0.05 compared with vehicle controls; bp < 0.05 compared with E2 treatment by two-way ANOVA, followed by Tukey's post hoc test. Representative Western blots are shown.
Figure 4.
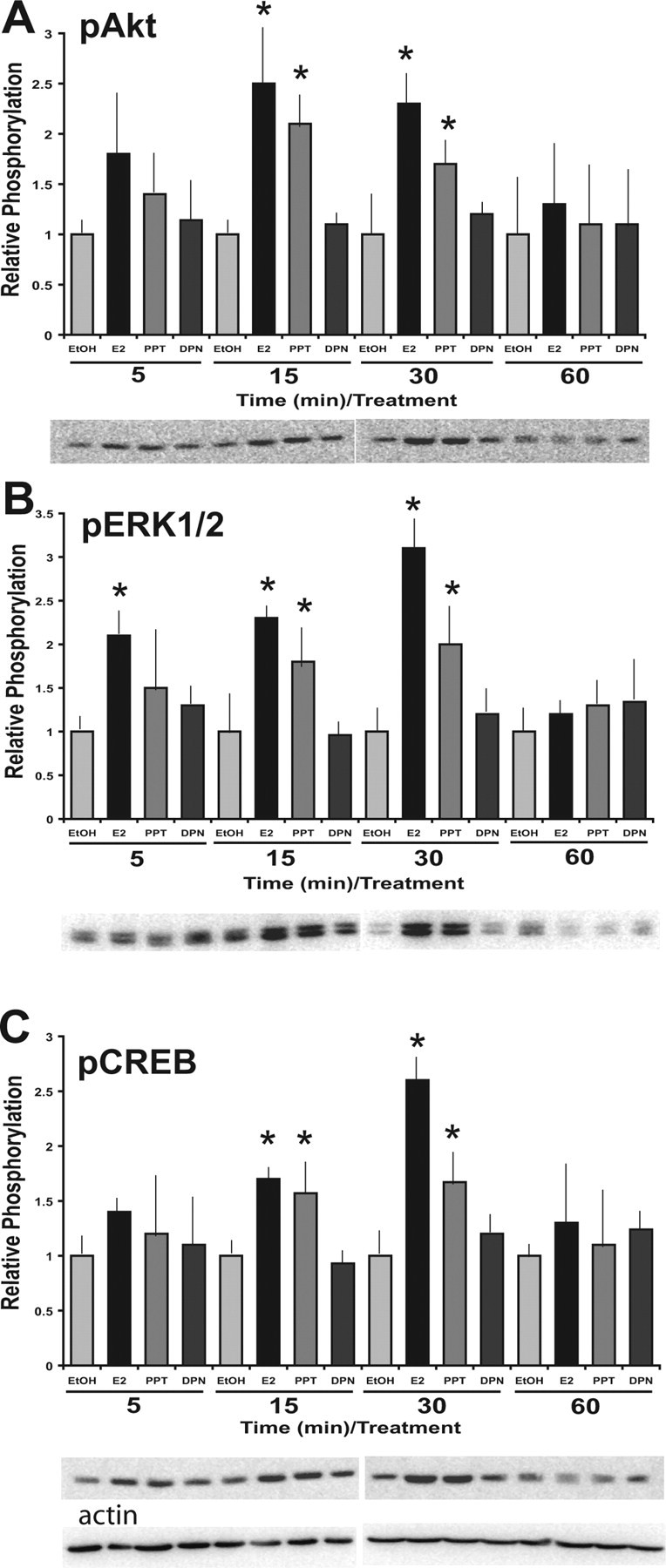
ERα-selective agonist, PPT, induces signal transduction events in the N-38 neurons. N-38 neurons were serum starved for 12–16 h before treatment with 10 nm estradiol PPT, DPN, or vehicle alone over a 60 min time course. At the indicated time points, cell lysates from N-38 neurons were isolated and subjected to SDS-PAGE. A–C, Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against Akt (A), ERK1/2 (B), and CREB (C). All results shown are normalized to total protein relative to corresponding control protein levels at each time point and are expressed as mean ± SEM (n = 3 independent experiments). *p < 0.05, by two-way ANOVA, followed by Tukey's post hoc test. Representative Western blots are shown.
PI3-K and MEK inhibitors reduce the E2-mediated phosphorylation of Akt, ERK1/2, and CREB in N-38 neurons
To determine whether or not the phosphorylation of Akt and ERK1/2 by E2 is mediated by the PI3-K and MEK/ERK MAPK pathways, respectively, we used pharmacological inhibitors of these signaling pathways. We demonstrate that the PI3-K inhibitor LY294002 reduces the level of phosphorylation of Akt by E2 from 5–30 min exposure (Fig. 2A). Similarly, the MEK inhibitor PD98059 attenuated the level of phosphorylation of ERK1/2 by E2 from 5–30 min exposure (Fig. 2B). These results show that the estrogen-mediated activation of Akt and ERK1/2 is via the PI3-K and MAPK pathways, respectively.
Because we have shown that estrogen activates CREB in the N-38 neurons, it was of interest to determine the signaling cascade that mediates this activation in response to E2. It has been shown previously that the MEK/ERK MAPK pathway has been linked to the activation of CREB and subsequent gene regulation at cAMP-responsive elements (CRE) (Wade and Dorsa, 2003; Hennessy et al., 2005). To elucidate the possibility that the E2-mediated phosphorylation of CREB occurs via the MAPK pathway, we pretreated N-38 neurons with PD98059 and observed its affect on the activation of CREB after E2 treatment. In the N-38 neurons, we have shown that the MEK inhibitor reduces the level of phosphorylation of CREB when compared with E2 treatment alone (Fig. 2C). This result suggests that E2 induces CREB phosphorylation in the N-38 neurons via the MEK/ERK MAPK pathway.
Cross talk between the PI3-K and MAPK signal transduction pathways after E2 treatment in the N-38 neurons
Because the PI3-K and MEK inhibitors did not completely abolish the E2-mediated induction of Akt and ERK1/2, respectively, it is possible that there is cross talk between the two pathways. The notion of cross talk between the PI3-K and MAPK signaling cascades during the transmission of estrogen signals has already been suggested (Bar-Sagi and Hall, 2000; Lehman and Gomez-Cambronero, 2002). In an attempt to determine possible cross talk in the N-38 cells, we used the PI3-K inhibitor and observed its effects on E2-mediated ERK1/2 phosphorylation, and used the MEK inhibitor and observed its effects on the activation of Akt by E2. We demonstrate that LY294002 significantly reduced the level of phosphorylation of ERK1/2 in the N-38 neurons (Fig. 3A), whereas PD98059 also diminished the activity of Akt (Fig. 3B). These data suggest that both the PI3-K and MAPK pathways are necessary for full activation of ERK1/2 and Akt. However, using both inhibitors in concert (data not shown) did not reduce the signal further; therefore, there are likely other factors that may be involved in the signaling.
E2-mediated activation of the PI3-K and MAPK pathways is mediated by ERα in the N-38 neurons
Rapid physiological changes can be caused by more than one membrane-associated protein that interacts with E2. However, many of the observed nongenomic estrogenic effects may be attributable to classical nuclear ERs conforming to the demands of a membrane receptor. To further elucidate the possible role of ERα and/or ERβ in mediating the rapid effects of E2 in N-38 neurons, we used ER-selective agonists and observed their effect on the activation of ERK1/2 and Akt. Using the ERα and ERβ-selective ligands PPT and DPN, respectively, we have shown that 10 nm PPT induces Akt (Fig. 4A), ERK1/2 (Fig. 4B), and CREB (Fig. 4C) phosphorylation from 5 to 30 min after E2 exposure. DPN, conversely, had no considerable effects, even at higher concentrations (data not shown). These findings suggest that the activation of Akt, ERK1/2, and CREB by E2 may be mediated by nuclear ERα in N-38 neurons. However, because the phosphorylation by PPT was not quite as effective as E2 alone, even at higher concentrations, we might speculate that there may be another factor involved, such as a nonclassical ER; thus, we decided to look for membrane localization of the classic ERα.
There is some evidence that the classic nuclear ERα can localize to the cell membrane (Pappas et al., 1995; Razandi et al., 1999). If ERα is responsible for the membrane signaling in the N-38 NPY neurons, we should be able to detect colocalization with another membrane protein, such as caveolin (Razandi et al., 2002). Using immunocytochemistry with ERα and caveolin-1 antibodies, we found that ERα can be localized to the membrane (Fig. 5A). After a 30 min exposure to 10 nm E2, there was an increase in the signal at the cell membrane (Fig. 5B), indicating that ERα may translocate to the membrane during exposure to estrogen.
Figure 5.

ERα can be colocalized to the cell membrane in N-38 NPY neurons. Confocal laser scanning microscopy of N-38 cells labeled with ERα–FITC or caveolin-1–rhodamine (Cav-Rhod). A, B, N-38 cells were serum starved for 12–16 h before treatment with vehicle (A) or 10 nm E2 (B) before being fixed in 2% paraformaldehyde and exposed to primary antibody directed against ERα. Colocalization of the ERα–FITC (green) and caveolin–rhodamine (red) fluorescence was observed in the merge (yellow) of the two images. C, Negative control cells were in the absence of primary antibody (Ab) and resulted in minimal staining. Cells were visualized with a confocal laser-scanning microscope at a magnification of 400×. FITC fluorescence was excited by the 488 nm argon laser line, whereas rhodamine was excited by the 543 nm helium–neon laser line. Representative images are shown from n = 3.
Inhibition of the PI3-K and MAPK signaling pathways affects the E2-mediated regulation of the NPY gene
We have reported previously that E2 biphasically regulates NPY gene expression in the N-38 neurons (Titolo et al., 2006). We determined that E2 initially downregulates NPY mRNA levels from 4 to 10 h, followed by a steady induction that peaks at 48 h, after which NPY mRNA levels begin to return to basal levels. To determine whether or not the regulation of NPY gene expression by E2 is dependent on rapid signaling cascades, we used inhibitors of the PI3-K and MAPK pathways and observed the effect on NPY mRNA levels. We specifically looked at five specific time points that prove the biphasic regulation of NPY mRNA by estrogen: 2, 8, 12, 24, and 48 h time points. Interestingly, we observed that, when the N-38 neurons were cotreated with E2 and the PI3-K or MEK inhibitors, not only is the induction of NPY mRNA attenuated but the repression seen at 8 h E2 exposure is also blocked (Fig. 6). Although the MEK inhibitor may take longer to produce its long-term effects, we believe that it is also involved because of the trend at 24 h and the significant effect at 48 h. The inhibitors alone had no effect on NPY gene expression. Therefore, it is evident that both the PI3-K and MAPK pathways are critical for mediating the effects of E2 on NPY gene expression. Together, these data suggest that early membrane effects of estrogen are critical for the subsequent genomic events occurring at the level of NPY gene expression in N-38 neurons. These findings suggest that there is a link between membrane signaling and gene expression in these neurons.
Figure 6.

PI3-K and MAPK inhibitors effect the E2-mediated regulation of NPY mRNA in N-38 neurons. N-38 neurons were serum starved for 12–16 h before treatment with 10 nm estradiol or vehicle alone over a 48 h time course. Cells were treated the PI3-K inhibitor [LY294002 (LY)] and MAPK inhibitor [PD98059 (PD)] for 45–60 min at a final concentration of 25 μm before estradiol exposure. At the indicated time points, total RNA was extracted and used as a template for real-time RT-PCR with primers specifically designed to amplify NPY. Real-time RT-PCR products were amplified on an Applied Biosystems Prism 7000. NPY mRNA levels were quantified using the ΔΔCT method and normalized to the internal control (actin). All results shown are relative to corresponding control mRNA levels (set to 1.0) at each time point and are expressed as mean ± SEM (n = 3 independent experiments). ap < 0.05 compared with vehicle controls; bp < 0.05 compared with estrogen treatment, by Student's t test.
Downstream effectors connect the membrane signaling to the NPY promoter
Previously, we localized the estrogen-mediated repression of the NPY gene to within −1078 of the human NPY 5′ regulatory region (Titolo et al., 2006). To assess the cis-regulatory sequences necessary for the regulated expression of the NPY gene in the N-38 cells, we transiently transfected reporter plasmids representing sequential deletions of the human −1078 NPY 5′ regulatory region. A significant decrease in reporter gene activity occurred with 10 nm E2 treatment over 24 h, and an estrogen-responsive region can be mapped to within −97 bp of the NPY 5′ regulatory region (Fig. 7A). This allows us to speculate that the repressive effects of estrogen on NPY gene expression may be a transcriptional event involving the binding of transcription factors to the NPY promoter. With analysis of this region using transcription factor binding database TRANSFAC, we found that there are a number of transcription factor binding sites with in this region, including SP-1, AP-1, AP-2, and a CRE half-site (Fig. 8). This is consistent with previous analysis of the human NPY gene promoter between −87 and −36, in which SP-1 and AP-2 sites were found to be functionally important for basal NPY transcription (Minth and Dixon, 1990; Minth-Worby, 1994) and also with the CREB activation after E2 exposure in the N-38 neurons.
Figure 7.

E2-mediated repression of NPY transcription can be mapped to within −97 bp of the human NPY 5′ regulatory region and CREB can bind to this region. A, N-38 neurons were transfected with pGL2–enh luciferase expression vectors containing sequential deletions of the human NPY 5′ regulatory region. After transfection, cells were treated with 10 nm E2 or vehicle for 24 h. Luciferase activity was normalized to protein content. All results shown are relative to control luciferase levels for each deletion mutant and are expressed as mean ± SEM from three experiments, each done in triplicate. B, C, N-38 neurons were serum starved for 12–16 h before treatment with 10 nm E2 (+) or vehicle alone over a 90 min time course. At the indicated time points, total RNA or cell lysates from N-38 neurons were isolated and subjected to real-time PCR or SDS-PAGE. B, Transcriptional analysis of c-fos gene expression in N-38 neurons. C, Western blot analysis was performed with enhanced chemiluminescence using specific antibodies directed against phospho-MSK-1. All results shown are normalized to histone gene expression or β-actin protein relative to corresponding control protein levels at each time point and are expressed as mean ± SEM (n = 3–7 independent experiments; representative Western blots are shown). *p < 0.05, by two-way ANOVA, followed by Tukey's post hoc test. D, ChIP assays demonstrate that CREB binds to the NPY promoter region. Formaldehyde cross-linked, chromatin-associated DNA from N-38 neurons were immunoprecipitated with an antibody to CREB. DNA fragments were subjected to PCR amplification using primers flanking the −118 to +22 bp region of the NPY promoter. A 141 bp PCR product was observed and sequenced. Negative controls included a no-antibody immunoprecipitation with the protein G agarose beads (No Ab), whereas the positive control consisted of 10% of the total chromatin in the absence of immunoprecipitation (Input; 50-fold dilution). Representative gel is shown; n = 3.
Figure 8.

Schematic diagram of signal transduction pathways induced by estradiol in the clonal, hypothalamic NPY cell model N-38. In the hypothalamic cell model N-38, we provide evidence for the activation of two specific signal transduction pathways by E2. The ERK1/2 MAPK and PI3-K/Akt pathways are induced by E2 activation, likely through ERα localized to the membrane. CREB is also involved in the E2-mediated regulation of NPY gene expression, as we have described its binding to the mouse NPY promoter region. Within the minimal promoter region, we detect a number of transcription factor binding sites, including SP-1, AP-1, AP-2, and a CRE half-site. These findings implicate a circuit from E2-mediated membrane activation to control of NPY gene expression in the nucleus, which is dependent on rapid membrane signaling events.
Because the early membrane signaling events are linked to the transcriptional response, we attempted to study some of the key effector proteins that may be involved in this process. Because the NPY promoter has been shown to contain multiple AP-1 sites, c-fos was our initial target. During exposure to 10 nm E2, we found that c-fos gene expression was induced twofold in the N-38 NPY neurons (Fig. 7B). Recent evidence from progesterone receptor analysis indicates that MSK-1 is phosphorylated by progestins (Vicent et al., 2006). MSK-1 has been shown to phosphorylate histone H3, facilitating changes in transcriptional activity (Thomson et al., 1999). To determine whether this may be a common mechanism in steroid regulation of gene expression, we analyzed MSK-1 activity using a phospho-specific MSK-1 antibody. We found that the MSK-1 protein is dephosphorylated or inactivated during E2 exposure at 30 and 60 min (Fig. 7C). ChIP assays were used to determine whether the CREB cis-elements present within the −118 to +22 bp fragment of the mouse NPY promoter were active and whether E2 would affect CREB binding to this region. A proven antibody to CREB was used to immunoprecipitate the regions bound by the CREB transcription factor. The ChIP assay demonstrated that CREB binds to the −118 to +22 bp region as detected by PCR, using specific primers to this region of the DNA (Fig. 7D). Compared with control, the estrogen treatment displayed a weaker association of CREB to this region. PCR analysis of the immunoprecipitation control lacking antibody did not display any amplification of the fragment (beads), whereas the preimmunoprecipitation control showed equal amplification of the fragment (input). Although additional analysis of this complex promoter region must now be undertaken, we can start to formulate a picture of the cellular events responsible for the regulation of the NPY gene by estrogen during the initiation of membrane signaling (Fig. 8).
Discussion
The concept of nongenomic estrogen signaling was first proposed in the late 1970s when it was discovered that estrogen can bind receptor proteins located in the cell membrane and initiate a rapid generation of cAMP in endometrial cells (Pietras and Szego, 1977). Since then, nongenomic estrogen signaling has been linked to other aspects of neuroendocrinology, including GnRH secretion, electrophysiological responses in neurons, and reproductive behavior, including lordosis (for review, see Vasudevan and Pfaff, 2007). Importantly, we determined that the E2-mediated regulation of the NPY gene is also dependent on rapid signal transduction cascades activated by estrogen.
Akt is the principle downstream effector of PI3-K, triggering several of its cellular effects, such as cell growth and survival, or apoptosis. Akt mediates not only cell survival but also appears to mediate the neuroprotective effects of estrogen in neurons (Ivanova et al., 2002). Estrogen-mediated MAPK signaling can acutely affect the cell by the activation of signal transduction cascades that can contribute to rapid cell proliferation (Migliaccio et al., 1998). In addition to demonstrating that estrogen activates both the MAPK and PI3-K pathways in N-38 neurons, we provide evidence for possible interactions between the two signaling pathways with the use of pharmacological inhibitors. The notion of possible cross talk between the MAPK and PI3-K signaling cascades during the transmission of estrogen signals has been suggested (Bar-Sagi and Hall, 2000; Lehman and Gomez-Cambronero, 2002). PI3-K can activate ras, which is an upstream effector of the MEK/ERK MAPK pathway. The ras protein can interact with the catalytic unit of the PI3-K, p110, and then converge on the MEK/ERK MAPK pathway by activating ERK1/2 (Bar-Sagi and Hall, 2000). However, efforts must continue on this theory to demonstrate how these two signaling cascades may regulate each other and contribute to estrogen signaling and to define the intermediate steps involved.
Recent evidence has shown that nuclear ERs can mediate many of the nongenomic effects of estrogen (Björnström and Sjöberg, 2005; Vasudevan and Pfaff, 2007). These studies have revealed that classical nuclear ERs are located at the plasma membrane of a variety of cell types, including endothelial cells, spermatozoa, and neurons in the brain (Pappas et al., 1995; Clarke et al., 2000), hence proving that membrane estrogen signaling through nuclear receptors is a more common phenomenon than originally thought. However, an issue that should be addressed is which of the nuclear ERs is responsible for the rapid events of membrane signaling and how it is connected to E2-mediated signal transduction systems. Studies have suggested that ERα can localize to the plasma membrane and functionally regulate signal transduction cascades (Razandi et al., 1999; Levin, 2001). These signaling cascades have been linked to diverse estrogen-related processes such as neurovascular and vascular protection (Migliaccio et al., 1998; Mendelsohn and Karas, 1999). The notion of ERα being associated with nongenomic estrogen action was also shown in vivo in mouse astrocytes (Pawlak and Beyer, 2005), wherein astrocytes collected from ERα knock-out mice displayed a marked decrease in estrogen binding at the plasma membrane, in addition to a decrease in the level of activity of the MEK/ERK MAPK and PI3-K pathways.
The mechanisms through which ERα is linked to these signal transduction cascades remain to be determined. Recently, a newly identified scaffold protein, termed the modulator of nongenomic activity of ER (MNAR), has been speculated to bridge ERα activation and MAPK phosphorylation (Wong et al., 2002; Barletta et al., 2004; Pawlak and Beyer, 2005; Pawlak et al., 2005; Song et al., 2005). MNAR is found in a variety of estrogen-responsive tissues, including the brain (Pawlak and Beyer, 2005), embryonic neurons, and astrocytes (Pawlak and Beyer, 2005), as well as the N-38 neurons (data not shown). The high level of expression of MNAR in the prenatal hypothalamus in addition to the prolonged expression of MNAR in the cortex coincides well with the fact that these are estrogen-sensitive periods in these regions of the brain (Carrer and Cambiasso, 2002). In addition, MNAR contains protein motifs that can interact with the activation function 2 domain of ERα and the Src homology 2 domain of Src, thus creating an ERα–MNAR–Src complex (Pawlak et al., 2005). The activation of Src can then lead to the activation of the MEK/ERK MAPK pathway, ultimately regulating cell proliferation, neuroprotection, and the maturation of neurons.
Although genomic and nongenomic mechanisms have been viewed as distinct modes of steroid hormone action, signal transduction pathways may possibly connect the two pathways and have a unique or synergistic effect on the cell. This would also allow for a distinct mechanism by which estrogen can regulate transcription at alternate response elements. In this study, we used selective PI3-K and MEK inhibitors to ultimately link early membrane estrogenic events to the full expression of long-term genomic actions in the N-38 neurons. Integrating both rapid and prolonged signaling in different sites of the cell can provide plasticity in the response of the cell to the steroid. In breast cancer cells, estrogen binding to the putative membrane ER will activate the insulin-like growth factor receptor, resulting in the activation of downstream signaling to MAPK and PI3-K (Levin, 2005). The activation of MEK/ERK MAPK and PI3-K signaling pathways can then upregulate the transcriptional activity of endogenous nuclear ERs (Kato et al., 1995; Martin et al., 2000). In addition, activation of PI3-K from the membrane can also cause the repression of downstream inhibitory kinases (Levin, 2005). For instance, the phosphorylation, and thus inactivation, of GSK-3β, a downstream effector of the PI3-K/Akt pathway, increases the transcriptional activity of nuclear ERs. In nerve cells, genomic and nongenomic modes of estrogen action have been shown to synergize to initiate transcription (Vasudevan et al., 2001). These data further demonstrate that rapid nongenomic actions of estrogen are critical for later genomic events and full transcriptional potency. For instance, in our study, we have demonstrated inactivation of MSK-1 by estrogen. MSK-1 has been linked to chromatin remodeling and the initiation of transcription, through its phosphorylation of histone H3 (Thomson et al., 1999). Stimulation of the MEK/ERK MAPK pathway can regulate MSK-1 and in turn has been associated with immediate early gene induction in fibroblasts (Chadee et al., 1999; Thomson et al., 1999). In our case, we can propose an association between inactivation of MSK-1 and the initial repression of the NPY gene by E2. We find that c-fos is induced by E2 and speculate that MSK-1 likely plays some role in this process, completing the circuit between the membrane and gene regulatory events.
Estrogen can also rapidly affect a number of other downstream effector molecules such as CREB (Wade and Dorsa, 2003). CREB has been shown recently to contribute to the functional regulation in specific regions of the brain, such as the POA, hippocampus, and hypothalamus (Abrahám and Herbison, 2005; Rønnekleiv and Kelly, 2005). CREB is phosphorylated after the MAPK pathway has been activated by estrogen and can then lead to the regulation of genes that contain CREs (Zhou et al., 1996; Kousteni et al., 2003). It is of interest that estrogen induces c-fos gene expression by ER tethering to Sp-1 at GC-rich binding sites (Duan et al., 1998) but may also regulate the gene through a CRE located in a cis-inducible element in the c-fos promoter. We show that CREB activation is mediated by the MEK/ERK MAPK pathway because the MEK pathway inhibitor PD98059 reduced the E2-induced phosphorylation of CREB in N-38 neurons. Furthermore, we find that CREB directly binds to the −118 to +22 bp region of the NPY promoter, although there is only a half-CRE site in this region. We also find that both ERs bind to the NPY promoter, without a classic ER element (data not shown). We speculate that the ERs may tether to Sp-1 GC-rich sites in the NPY promoter, although this will require additional investigation into the protein interactions within this complex region of the NPY gene.
The precise mechanism by which estrogen signals in the brain remains to be elucidated. However, it is conclusive that the effects of estrogen include complex cellular mechanisms ranging from nuclear to membrane-mediated action. In this way, a cell can use rapid modifications of cellular proteins via membrane ERs or a more prolonged modification of proteins via nuclear ERs according to cellular needs. In our case, we found that estrogen activates both the PI3-K/Akt and ERK1/2 MAPK pathways to initiate a cascade of downstream events ultimately coupled to the control of the NPY gene. We detect the E2-mediated phosphorylation of CREB, which may induce c-fos, an AP-1 site binding protein. One or both of these transcription factors may act directly at the level the NPY promoter, and we detect direct binding of CREB and the ERs within 100 bp of the transcriptional start site. We also find that estrogen inactivates MSK-1, a histone H3 kinase, linked to chromatin remodeling and the initiation of transcription. Hence, we can link E2-mediated signaling events at the membrane to the long-term repression of NPY gene expression in the N-38 NPY neurons.
Footnotes
This work was supported by Canadian Institutes of Health Research, Canadian Diabetes Association, Canada Foundation for Innovation, and the Canada Research Chairs Program (D.D.B.). We thank the Belsham laboratory for critical reading of this manuscript. Thanks to Dr. Kuiru Wei and Laura Fick for technical assistance.
References
- Abrahám IM, Herbison AE. Major sex differences in non-genomic estrogen actions on intracellular signaling in mouse brain in vivo. Neuroscience. 2005;131:945–951. doi: 10.1016/j.neuroscience.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Acosta-Martinez M, Horton T, Levine JE. Estrogen receptors in neuropeptide Y neurons: at the crossroads of feeding and reproduction. Trends Endocrinol Metab. 2007;18:48–50. doi: 10.1016/j.tem.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Ainslie DA, Morris MJ, Wittert G, Turnbull H, Proietto J, Thorburn AW. Estrogen deficiency causes central leptin insensitivity and increased hypothalamic neuropeptide Y. Int J Obes Relat Metab Disord. 2001;25:1680–1688. doi: 10.1038/sj.ijo.0801806. [DOI] [PubMed] [Google Scholar]
- Barletta F, Wong CW, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol Endocrinol. 2004;18:1096–1108. doi: 10.1210/me.2003-0335. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–238. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- Belsham DD, Cai F, Cui H, Smukler SR, Salapatek AM, Shkreta L. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology. 2004;145:393–400. doi: 10.1210/en.2003-0946. [DOI] [PubMed] [Google Scholar]
- Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- Carrer HF, Cambiasso MJ. Sexual differentiation of the brain: genes, estrogen, and neurotrophic factors. Cell Mol Neurobiol. 2002;22:479–500. doi: 10.1023/A:1021825317546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, Davie JR. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999;274:24914–24920. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Clarke CH, Norfleet A, Clarke M, Watson C, Cunningham K, Thomas M. Perimembrane localization of the estrogen receptor alpha protein in neuronal processes of cultured hippocampal neurons. Neuroendocrinology. 2000;71:34–42. doi: 10.1159/000054518. [DOI] [PubMed] [Google Scholar]
- Cui H, Cai F, Belsham DD. Anorexigenic hormones leptin, insulin, and alpha-melanocyte-stimulating hormone directly induce neurotensin (NT) gene expression in novel NT-expressing cell models. J Neurosci. 2005;25:9497–9506. doi: 10.1523/JNEUROSCI.2269-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan R, Porter W, Safe S. Estrogen-induced c-fos protooncogene expression in MCF-7 human breast cancer cells: role of estrogen receptor Sp1 complex formation. Endocrinology. 1998;139:1981–1990. doi: 10.1210/endo.139.4.5870. [DOI] [PubMed] [Google Scholar]
- Enmark E, Gustafsson JA. Oestrogen receptors—an overview. J Intern Med. 1999;246:133–138. doi: 10.1046/j.1365-2796.1999.00545.x. [DOI] [PubMed] [Google Scholar]
- Estrada KM, Pompolo S, Morris MJ, Tilbrook AJ, Clarke IJ. Neuropeptide Y (NPY) delays the oestrogen-induced luteinizing hormone (LH) surge in the ovariectomized ewe: further evidence that NPY has a predominant negative effect on LH secretion in the ewe. J Neuroendocrinol. 2003;15:1011–1020. doi: 10.1046/j.1365-2826.2003.01087.x. [DOI] [PubMed] [Google Scholar]
- Hennessy BA, Harvey BJ, Healy V. 17beta-Estradiol rapidly stimulates c-fos expression via the MAPK pathway in T84 cells. Mol Cell Endocrinol. 2005;229:39–47. doi: 10.1016/j.mce.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons. Endocr Rev. 1998;19:302–330. doi: 10.1210/edrv.19.3.0332. [DOI] [PubMed] [Google Scholar]
- Hill JW, Levine JE. Abnormal response of the neuropeptide Y-deficient mouse reproductive axis to food deprivation but not lactation. Endocrinology. 2003;144:1780–1786. doi: 10.1210/en.2002-221024. [DOI] [PubMed] [Google Scholar]
- Ivanova T, Mendez P, Garcia-Segura LM, Beyer C. Rapid stimulation of the PI3-kinase/Akt signalling pathway in developing midbrain neurones by oestrogen. J Neuroendocrinol. 2002;14:73–79. doi: 10.1046/j.0007-1331.2001.00742.x. [DOI] [PubMed] [Google Scholar]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JA, Gomez-Cambronero J. Molecular crosstalk between p70S6k and MAPK cell signaling pathways. Biochem Biophys Res Commun. 2002;293:463–469. doi: 10.1016/S0006-291X(02)00238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE, Stoica A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–4511. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- Mayer CM, Cai F, Cui H, Gillespie JM, MacMillan M, Belsham DD. Analysis of a repressor region in the human neuropeptide Y gene that binds Oct-1 and Pbx-1 in GT1–7 neurons. Biochem Biophys Res Commun. 2003;307:847–854. doi: 10.1016/s0006-291x(03)01289-0. [DOI] [PubMed] [Google Scholar]
- Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17:2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minth CD, Dixon JE. Expression of the human neuropeptide Y gene. J Biol Chem. 1990;265:12933–12939. [PubMed] [Google Scholar]
- Minth-Worby CA. Transcriptional regulation of the human neuropeptide Y gene by nerve growth factor. J Biol Chem. 1994;269:15460–15468. [PubMed] [Google Scholar]
- Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- Pawlak J, Beyer C. Developmental expression of MNAR mRNA in the mouse brain. Cell Tissue Res. 2005;320:545–549. doi: 10.1007/s00441-005-1090-z. [DOI] [PubMed] [Google Scholar]
- Pawlak J, Karolczak M, Krust A, Chambon P, Beyer C. Estrogen receptor-alpha is associated with the plasma membrane of astrocytes and coupled to the MAP/Src-kinase pathway. Glia. 2005;50:270–275. doi: 10.1002/glia.20162. [DOI] [PubMed] [Google Scholar]
- Pelletier G, Li S, Luu-The V, Labrie F. Oestrogenic regulation of pro-opiomelanocortin, neuropeptide Y and corticotrophin-releasing hormone mRNAs in mouse hypothalamus. J Neuroendocrinol. 2007;19:426–431. doi: 10.1111/j.1365-2826.2007.01548.x. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265:69–72. doi: 10.1038/265069a0. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in CHO cells. Mol Endocrinol. 1999;13:307–319. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100–115. doi: 10.1210/mend.16.1.0757. [DOI] [PubMed] [Google Scholar]
- Rønnekleiv OK, Kelly MJ. Diversity of ovarian steroid signaling in the hypothalamus. Front Neuroendocrinol. 2005;26:65–84. doi: 10.1016/j.yfrne.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Ohtani K, Kato Y, Tanaka Y, Mori M. Withdrawal of [corrected] estrogen increases hypothalamic neuropeptide Y (NPY) mRNA expression in ovariectomized obese rat. Neurosci Lett. 1996;204:81–84. doi: 10.1016/0304-3940(96)12322-3. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Shimomura Y, Nakanishi Y, Futawatari T, Ohtani K, Sato N, Mori M. Estrogen increases in vivo leptin production in rats and human subjects. J Endocrinol. 1997;154:285–292. doi: 10.1677/joe.0.1540285. [DOI] [PubMed] [Google Scholar]
- Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–353. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Thomson S, Mahadevan LC, Clayton AL. MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin Cell Dev Biol. 1999;10:205–214. doi: 10.1006/scdb.1999.0302. [DOI] [PubMed] [Google Scholar]
- Titolo D, Cai F, Belsham DD. Coordinate regulation of neuropeptide Y and agouti-related peptide gene expression by estrogen depends on the ratio of estrogen receptor (ER) alpha to ERbeta in clonal hypothalamic neurons. Mol Endocrinol. 2006;20:2080–2092. doi: 10.1210/me.2006-0027. [DOI] [PubMed] [Google Scholar]
- Vasudevan N, Pfaff DW. Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr Rev. 2007;28:1–19. doi: 10.1210/er.2005-0021. [DOI] [PubMed] [Google Scholar]
- Vasudevan N, Kow LM, Pfaff DW. Early membrane estrogenic effects required for full expression of slower genomic actions in a nerve cell line. Proc Natl Acad Sci USA. 2001;98:12267–12271. doi: 10.1073/pnas.221449798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent GP, Ballaré C, Nacht AS, Clausell J, Subtil-Rodríguez A, Quiles I, Jordan A, Beato M. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–381. doi: 10.1016/j.molcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Wade CB, Dorsa DM. Estrogen activation of cyclic adenosine 5′-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology. 2003;144:832–838. doi: 10.1210/en.2002-220899. [DOI] [PubMed] [Google Scholar]
- Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. 2002;99:14783–14788. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu M, Hill JW, Levine JE. Attenuation of luteinizing hormone surges in neuropeptide Y knockout mice. Neuroendocrinology. 2000;72:263–271. doi: 10.1159/000054595. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Watters JJ, Dorsa DM. Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology. 1996;137:2163–2166. doi: 10.1210/endo.137.5.8612562. [DOI] [PubMed] [Google Scholar]