

Abstract
Channelopathies are often linked to defective protein folding and trafficking. Among them, the calcium channelopathy episodic ataxia type-2 (EA2) is an autosomal dominant disorder related to mutations in the pore-forming Cav2.1 subunit of P/Q-type calcium channels. Although EA2 is linked to loss of Cav2.1 channel activity, the molecular mechanism underlying dominant inheritance remains unclear. Here, we show that EA2 mutants as well as a truncated form (DI-II) of the Cav3.2 subunit of T-type calcium channel are misfolded, retained in the endoplasmic reticulum, and subject to proteasomal degradation. Pulse-chase experiments revealed that misfolded mutants bind to nascent wild-type Cav subunits and induce their subsequent degradation, thereby abolishing channel activity. We conclude that this destructive interaction mechanism promoted by Cav mutants is likely to occur in EA2 and in other inherited dominant channelopathies.
Keywords: voltage-gated calcium channel, P/Q-type, T-type, dominant-negative activity, episodic ataxia type 2, misfolding, endoplasmic reticulum, proteasome
Introduction
Episodic ataxia type 2 (EA2) [on-line mendelian inheritance in man (OMIM) #108500] is a prototypical episodic neurological syndrome linked to mutations in the pore-forming Cav2.1 (α1A) subunit of P/Q-type Ca2+ channels (Ophoff et al., 1996; Pietrobon, 2002). These channels play a major role in neurotransmitter release (Urbano et al., 2003) throughout the nervous system, particularly in the cerebellar Purkinje cells. It is believed that P/Q-type mutations associated with EA2 reduce the precision of intrinsic Purkinje cell pace making, which likely accounts for the spontaneous attacks of ataxia in EA2 patients (Donato et al., 2006; Walter et al., 2006). The affected gene, CACNA1A, is localized on human chromosome 19p13 (Diriong et al., 1995). CACNA1A is also linked to familial hemiplegic migraine type 1 (FHM1) (OMIM #141500) and spinocerebellar ataxia type 6 (OMIM #183086) (Ophoff et al., 1996; Pietrobon, 2002). These allelic diseases are autosomal dominant disorders, yet the molecular mechanisms involved are unknown. Regarding EA2, >20 CACNA1A nonsense or missense mutations have been identified, and the general consensus is that these mutations, in contrast to FHM1, result in loss of channel activity (Ophoff et al., 1996; Jen, 2000; Guida et al., 2001; Wappl et al., 2002; Pietrobon, 2005). Haploinsufficiency has been suggested as a mechanism for EA2 (Guida et al., 2001; Wappl et al., 2002). This should cause a ∼50% reduction of Cav2.1 activity in patients with completely inactive EA2 mutations. However, analysis of Cav2.1(+/−) heterozygous mice, which exhibit a ∼50% inhibition of the P/Q current, revealed no apparent neurological abnormalities (Jun et al., 1999; Fletcher et al., 2001). Additionally, it has also been shown that some EA2 mutants exert a dominant-negative effect (Jouvenceau et al., 2001; Page et al., 2004; Jeng et al., 2006). Several mechanisms have been proposed for this effect, including competition of mutant channels for auxiliary subunits and/or the cell trafficking machinery (Arikkath et al., 2002; Cao et al., 2004; Wan et al., 2005; Raike et al., 2007; Jeng et al., 2008) and translational arrest (Page et al., 2004).
In this study, we explore the folding, trafficking, and stability of Cav channels in the presence of mutants. Pulse-chase analysis reveals that wild-type Cav2.1 channels are quickly degraded in the presence of EA2 mutants and similarly for Cav3.2 channels in the presence of truncated mutant (DI-II). Altogether, endoplasmic reticulum (ER) retention and proteasomal degradation of Cav mutants appear to be the basis of the dominant-negative activity. From this study, a model emerges where misfolded mutants interact with wild-type channels and promote their premature proteasomal degradation. This destructive interaction mechanism (DIM) provides a likely framework to decipher the principle of dominance in EA2 as well as other calcium channelopathies that involve misfolded mutants.
Materials and Methods
Molecular biology.
Green fluorescent protein (GFP) N-terminally fused Cav2.1 (GFP-Cav2.1) was constructed by cloning human Cav2.1 cDNA in pbA-eGFP expression vector. Hemagglutinin-tagged Cav2.1 (Cav2.1-HA) was constructed by replacing amino acids 1726–1733 in the domain IV pore loop of Cav2.1 in pbA-eGFP expression vectors (AF004883) (Mullner et al., 2004) by the HA epitope tag sequence YPYDVPDYA using standard splice overlap extension PCR. The construct was confirmed by sequencing. GFP-Cav2.1 and Cav2.1-HA cDNA were transferred in pCIneo expression vector (Promega, Madison, WI). R1279X, G293R, and AY1593/94D Cav2.1 mutant constructs have been described previously (Wappl et al., 2002). CD4 constructs have also been described previously (Zerangue et al., 2001). The generation of the HA-tagged Cav3.2 subunit has been described previously (Dubel et al., 2004; Vitko et al., 2007). Truncated isoforms of the Cav3.2 subunit were created using PCR techniques, automated DNA sequencing, and cloning into the pEGFP-C1 expression vector (BD Biosciences, Franklin Lakes, NJ).
Antibodies and reagents.
The 12CA5 (mouse) and 3F10 (rat) monoclonal antibodies were used to detect HA-tagged proteins (Roche Applied Science, Indianapolis, IN). The murine anti-CD4 antibody was obtained from Beckman Coulter (Fullerton, CA). Anti-GFP monoclonal and polyclonal antibodies were obtained from Clontech (Mountain View, CA). Anti-Cav2.1 polyclonal antibodies were purchased from Alomone Labs (Jerusalem, Israel). Secondary HRP-conjugated antibodies were obtained from The Jackson Laboratory (Bar Harbor, ME). Alexa-Fluor-conjugated antibodies and cholera toxin (CT) were obtained from (Invitrogen, Carlsbad, CA). MG-132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), leupeptin, and NH4Cl were obtained from Calbiochem (San Diego, CA).
Cell cultures and transfections.
Human embryonic kidney 293 (HEK293) and NG108-15 cells were cultured as described previously (Chemin et al., 2002; Vitko et al., 2007). For optimal transfection, cells were plated at 50–70% confluence. Cell lines were transfected using the JetPEI transfection reagent (QBiogene, Irvine, CA) according to the manufacturer protocol. The Cav2.1 constructs were cotransfected with α2/δ1 and β1b subunits (1:2:2 molar ratio), unless otherwise indicated.
Surface expression measurement.
Luminometry-ELISA analysis was performed as described previously (Vitko et al., 2007). Briefly, cells were transfected in 24-well plates. Forty hours after transfection, the cells were fixed for 5 min in 4% paraformaldehyde followed by two washes in PBS. Four wells (105 cells per well) for each condition were incubated for 30 min in blocking solution (PBS plus 1% fetal bovine serum). The expression of HA-tagged channels was measured using a rat anti-HA (3F10, 1/1000) and secondary goat anti-rat coupled to horseradish peroxidase (1/5000). After extensive washes in PBS, SuperSignal substrate (femto; Pierce Chemical, Rockford, IL) was added, and luminescence was measured (Victor 2 luminometer). The surface expression was measured in nonpermeabilized cells, and total expression was measured after Triton X-100 (0.1%) permeabilization. The percentage of surface expression corresponds to the ratio of surface/total relative light unit values.
Immunocytochemistry.
NG108-15 cells were seeded on poly-ornithine-coated coverslips the day before transfection. Two days after transfection, cells were washed in PBS, fixed for 5 min in 4% paraformaldehyde at room temperature, and permeabilized with 0.5 mg/ml saponin in 10 mm glycine PBS. Cells were then incubated with blocking solution [5% fetal calf serum (FCS)] for 30 min. Incubation of the primary antibody was performed at room temperature for 1 h. After washes in PBS, incubation of the secondary antibody was performed at room temperature for 30 min. Coverslips were next mounted onto a microscope slide and were observed on Leica (Wetzlar, Germany) SP2 confocal microscope using a 63× oil immersion objective.
Western blotting.
Forty-eight hours after transfection, cells cultured in 35 mm dishes were lysed on ice for 20 min with NP-40 buffer containing 10 mm Tris-HCl, pH 7.4, 120 mm NaCl, 1% NP-40, and mixture inhibitors (Roche Applied Science). Cell lysates were spun at 10,000 × g for 30 min at 4°C. Protein content of the supernatant was determined using the BCA Protein Assay kit (Pierce Chemical). Sixty micrograms of proteins were mixed with a 4× loading buffer and then loaded on SDS-PAGE. Proteins were then transferred onto nitrocellulose membranes and blocked with 5% powdered nonfat milk. Primary antibody was incubated for 1 h at room temperature in PBS-T (Tween 0.05%). After two washes in PBS-T, secondary HRP-coupled antibodies were incubated for 1 h in PBS-T. The signal was detected using the Super Signal West Pico Chemiluminescent system (Pierce Chemical).
Pulse-chase and immunoprecipitation.
Pulse-chase analyses were performed as described previously (Mezghrani et al., 2001). Briefly, cells were labeled with 250 μCi/ml promix (GE Healthcare Bio-Sciences, Little Chalfont, Buckinghamshire, UK) for 20 min in a free methionine/cysteine medium. Next, cells were washed two times with complete medium and chased in the presence of a fivefold excess of methionine/cysteine for the indicated time. At the end of the chase, cells were lysed in NP-40 buffer (1% NP-40, 150 mm NaCl, 10 mm Tris-HCl, pH 7,6, and mixture inhibitors; Roche Applied Science) supplemented with 10 mm N-ethyl maleimide for 30 min on ice. Lysates were centrifuged at 10,000 × g for 20 min, precleared for 1 h with FCS-Sepharose beads, and incubated overnight with specific antibodies and protein-A Sepharose beads (GE Healthcare Bio-Sciences). Beads were washed three times with lysis buffer, and bead-bound proteins were resolved by standard SDS-PAGE and quantified by autoradiography using automated densitometric scanning (GE Healthcare). Individual bands were quantified by the ImageQuant software.
Electrophysiology.
Whole-cell Ca2+ currents were recorded 2–4 d after transfection. Cav2.1 and Cav3.2 currents were elicited by test pulses (TPs) to 0 and −30 mV, respectively. The current density was calculated according to the capacitance of the cell and expressed in pA/pF. Extracellular solution contained the following (in mm): 2 CaCl2, 160 tetraethylammonium (TEA)Cl, and 10 HEPES (pH to 7.4 with TEAOH). Pipettes (2–3 MÙ) were filled with a solution containing the following (in mm): 110 CsCl, 10 EGTA, 10 HEPES, 3 Mg-ATP, and 0.6 GTP (pH to 7.2 with CsOH). Detailed acquisition and analysis procedures can be found in a previous study (Chemin et al., 2002).
Results
The EA2 mutant (R1279X) exerts a specific dominant-negative effect on the Cav2.1 protein
Several EA2 mutations result in truncated forms of the Cav2.1 subunit of P/Q-type channels, such as the R1279X mutant (Fig. 1A) described previously (Wappl et al., 2002). To investigate the properties of R1279X in coexpression experiments with the wild-type Cav2.1 subunit, various epitope-tagged constructs were engineered, especially an R1279X mutant N-terminally fused to GFP (R1279X) (Fig. 1A). Functional expression of Cav2.1-HA was obtained both in HEK293 cells (Fig. 1B) and in undifferentiated NG108-15 cells (Fig. 1C) that do not express high-voltage-activated (HVA) calcium currents (Chemin et al., 2002). Typical HVA P/Q-type currents (Fig. 1, insets) were recorded in the presence of 2 mm extracellular Ca2+. In these two heterologous expression systems, the coexpression of R1279X with Cav2.1-HA (1:1) resulted in a strong reduction of the Ca2+ current density (>80% in HEK293 cells and ∼70% in NG108-15 cells) (Fig. 1B,C, black bars). The remaining current (Fig. 1B,C, insets) displayed no significant electrophysiological difference in terms of steady state and kinetic properties (data not shown). Similar results were obtained using untagged Cav2.1 and the R1279X mutant, excluding a role of the different tags used in the described effects (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Whether this R1279X mutant exhibits inhibitory activity onto other types of Ca2+ channels was evaluated in differentiated NG108-15 cells, which display T-type and HVA currents, essentially L- and N-types (Lukyanetz, 1998; Chemin et al., 2002). Figure 1D shows that transfection of R1279X induces no change in the T-type current density both in proliferative and in differentiated NG108-15 cells (white bars), as well as no significant change in native HVA current density (black bars). In addition, coexpression in undifferentiated NG108-15 cells of R1279X with human Cav2.2 that codes for N-type channels produced no significant change in N-type current (10.3 ± 4.7 pA/pF, n = 11 for Cav2.2 alone; 11.7 ± 2.5 pA/pF, n = 10 for Cav2.2 in the presence of R1279X).
Figure 1.

Inhibition of Cav2.1 Ca2+ currents by the R1279X EA2-truncated form of Cav2.1 protein. A, Schematic cartoon representing the main Cav2.1 constructs used in this study. B, The Ca2+ current density in HEK293 cells expressing the Cav2.1 channel protein. The cells were cotransfected with plasmids encoding the human Cav2.1 subunit, the β1b subunit, and the α2/δ1 subunit. Mean ± SEM values of the current density for cells expressing the Cav2.1 subunit alone (white bar; n = 18) or in the presence of the R1279X mutant (+R1279X) (black bar; n = 22) are presented (***p < 0.001; Student's t test). The inset shows representative current traces at TP −40 and 0 mV [holding potential (HP), −80 mV]. C, Same experiments as in B performed in the neuroblastoma cell line NG108-15. The HP was −50 mV (***p < 0.001; Student's t test). Cav2.1 subunit alone (white bar; n = 15) or in the presence of the R1279X mutant (+R1279X) (black bar; n = 22). D, Measurements of native T-type and HVA Ca2+ current densities in R1279X-transfected NG108-15 cells. The T-current density (left) was measured in proliferative NG108-15 cells (Prolif.; 2–3 d after transfection; n = 9) and in differentiated NG108-15 cells (Diff.; 6 d after transfection; 3–4 d after switching to differentiation medium; n = 11) using HP −100 mV and TP −30 mV. The HVA current density, mainly corresponding to L- and N-type channel activities, was measured using HP −50 mV and TP 0 mV (right).
The R1279X mutant impairs trafficking of the wild-type Cav2.1 protein
This dominant-negative effect could be caused by either a functional inhibition of wild-type channels at the plasma membrane or by an alteration of Cav2.1 trafficking. To visualize the pool of wild-type Cav2.1 at the plasma membrane in the absence or presence of R1279X (+GFP and +R1279X, respectively), we performed immunofluorescence studies in HEK293 cells coexpressing wild-type Cav2.1 (Cav2.1-HA) and the auxiliary β1b and α2/δ1 subunits (Fig. 2A). With coexpressed GFP (control), wild-type Cav2.1 can be detected at the surface of the cells (Fig. 2A, top). However, after coexpression of R1279X, no HA staining is detected in nonpermeabilized cells (Fig. 2A, middle). The absence of Cav2.1 at the cell surface suggests that R1279X alters trafficking of the channel complex. Immunofluorescence in permeabilized cells revealed a preferential distribution of wild-type Cav2.1 in intracellular compartments (Fig. 2A, bottom). Interestingly, despite the low expression of the wild-type Cav2.1 protein, it appeared to strongly colocalize with the R1279X mutant (Fig. 2A). A similar pattern of expression was observed in NG108-15 cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). A more prominent punctuate labeling can bee seen in NG108-15 cells (supplemental Fig. 2, top, available at www.jneurosci.org as supplemental material), that likely reflects specific membrane domains and/or channel clustering.
Figure 2.
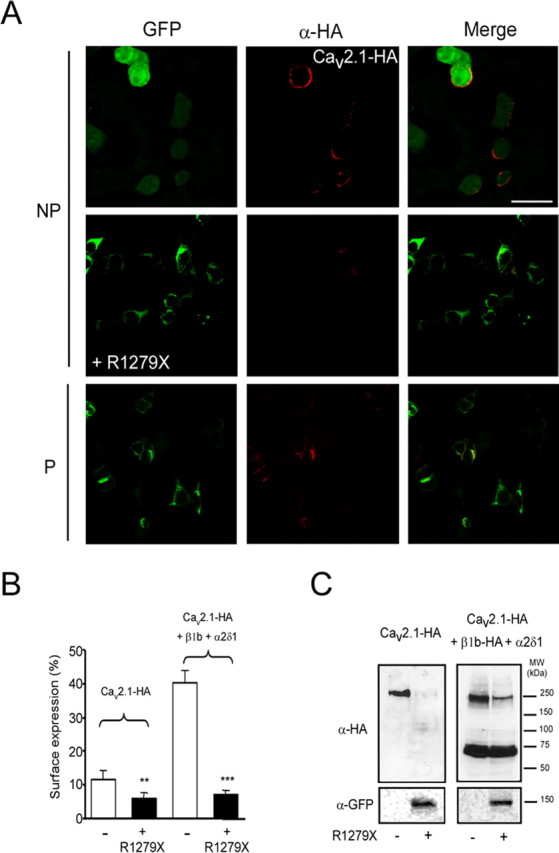
Effect of R1279X mutant on surface expression of Cav2.1 channel. A, HEK293 cells were cotransfected with the HA-tagged wild-type Cav2.1 subunit alone (with free GFP) or together with the R1279X mutant (fused to GFP). Left column, GFP fluorescence. Middle column, Staining of cells with monoclonal rat anti-HA antibody (primary antibody) and Alexa594 (secondary antibody). NP, Nonpermeabilized; P, permeabilized. Images were acquired using a Leica SP2 confocal microscope, with a 63× oil immersion objective. B, Luminometric ELISA assays to quantify surface expression of the HA-tagged Cav2.1 in the presence of the R1279X mutant and auxiliary subunits (**p < 0.01, ***p < 0.001; Student's t test). C, Western blots performed on HEK293 cells expressing Cav2.1-HA alone (−) or in the presence of R1279X (+) without (left) or with (right) auxiliary subunits. β1b-HA was used in these experiments (right).
We next performed luminometric assays in HEK293 cells to quantify Cav2.1 surface expression in the presence/absence of the R1279X mutant and the presence/absence of auxiliary subunits (Fig. 2B,C). Approximately 12% of total Cav2.1-HA was present at the cell surface when expressed alone, whereas in the presence of the auxiliary subunits, Cav2.1 surface expression reached ∼40% (Fig. 2B). R1279X significantly interfered with surface expression under both conditions (Fig. 2B). The remaining Cav2.1 surface expression in the presence of R1279X (∼5–7%) was not further modulated by the presence of auxiliary subunits.
Total expression was also strongly reduced (∼50%) in the presence of the R1279X mutant, again regardless of the presence of auxiliary subunits (data not shown). The ability of R1279X to downregulate Cav2.1 expression was verified in Western blot experiments with Cav2.1-HA (Fig. 2C). Again, we observed a strong inhibition of Cav2.1 expression, regardless of the presence of auxiliary subunits. In these experiments, we used an HA-tagged version of the β1b subunit that allowed us to visualize both Cav2.1 and β1b subunits. The β1b subunit expression was unchanged in the presence of R1279X, indicating that the observed downregulation of the Cav2.1 subunit is not attributable to a decrease in β subunit expression (Fig. 2C) and that the dominant-negative effect of R1279X acts specifically on Cav2.1. We also verified by Western blot that the untagged R1279X mutant downregulated Cav2.1 expression (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). To further address that this effect was not caused by the saturation of the transcription/translation machinery, we coexpressed the Cav2.1 subunit with the β2-adrenergic receptor and a CD4 protein with an ER retention site (KKXX) (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). No detectable downregulation with unrelated membrane proteins was observed supporting the specificity of this dominant-negative effect. In addition, we report that R1279X is able to slightly downregulate (∼20%) Cav2.2 channel expression (supplemental Fig. 3C,D, available at www.jneurosci.org as supplemental material). In agreement with previous reports (Page et al., 2004), we found that coexpression of R1279X with the Cav2.2 subunit in HEK293 cells resulted in a small but not significant reduction of N-type current (16.2 ± 6.1, n = 10 and 12.8 ± 5.0, n = 12 for Cav2.2 alone and Cav2.2 plus R1279X, respectively). Together, these results reveal that R1279X induces a preferential downregulation of Cav2.1 channel.
The presence of R1279X mutant induces Cav2.1 protein instability
The lower protein amount could result from a decrease in protein synthesis and/or from an increase in protein degradation/instability. We therefore designed pulse-chase experiments to study Cav2.1 channel stability in the presence of R1279X (Fig. 3A,B). These experiments were performed using in vivo labeling of cells with a [35S]methionine-cysteine mixture (see Materials and Methods), followed by wash out for an indicated time (1–4 h) and subsequent immunoprecipitation. In the absence of R1279X, the Cav2.1 protein decreases slightly (in the range of 25–35%) during 4 h chase (Fig. 3A, left, C, open squares). In contrast, in the presence of R1279X, the Cav2.1 signal decreased rapidly and significantly during the chase (down to 10% after 4 h) (Fig. 3A, right, C, filled squares). Notably, a 140 kDa protein coimmunoprecipitated with wild-type Cav2.1 (Fig. 3A, right, arrow). This protein, which was only present after R1279X coexpression, most likely corresponds to the R1279X construct. Similar experiments were performed in the presence of auxiliary subunits (Fig. 3B,C, circles). In the absence of R1279X, a ∼60 kDa band was observed, which most likely corresponds to the β1b subunit, because it is known to coimmunoprecipitate with Cav2.1 (Fig. 3B, arrow), and it is absent in transfections without auxiliary subunits (Fig. 3A). In the presence of R1279X, Cav2.1 was rapidly degraded (Fig. 3B, right, C), regardless of the presence of auxiliary subunits. Quantification revealed a large and marked change in channel stability as evident from the reduction of the half-life of the Cav2.1 subunit from >4 h to 1 h (Fig. 3C). This destabilization cannot be rescued by coexpression of the auxiliary subunits (Fig. 3C). An alternative possibility is that R1279X expression could affect cellular homeostasis, leading to a nonspecific dominant effect. To test this hypothesis, we analyzed the effect of R1279X expression on CD4 receptor stability by pulse chase (Fig. 3D). No significant change of CD4 stability was observed after 4 h of chase (Fig. 3D, quantifications), suggesting that Cav2.1 protein stability is selectively affected.
Figure 3.

Channel misfolding and instability induced by the R1279X mutant. A, B, Pulse-chase experiments were performed on HEK293 cells 48 h after transfection (see Materials and Methods). Cells were labeled with 35S-methionine-cysteine for 20 min. The chase was performed for 0, 1, 2, or 4 h as indicated. After lysis, the Cav2.1 subunit was immunoprecipitated with an anti-HA antibody. The arrow indicates the band corresponding to the EA2 mutant that coimmunoprecipitates with the wild-type Cav2.1 subunit. In B (left), the β1b is detected (arrow). C, Quantification of three independent experiments using GE Healthcare software. D, Pulse-chase analyses performed on HEK293 cells transfected with CD4-AAXX alone or in the presence of R1279X. Quantification was performed as described above. E, Immunoprecipitations performed on cells coexpressing various subunit arrangements (as indicated) in the absence or presence of an untagged R1279X mutant (right) using standard SDS-PAGE (6%). Western blots (WBs) were performed with the indicated antibodies. F, Immunoprecipitations were performed between β1b-HA and GFP-R1279X as described in E. MW, Molecular weight.
In the presence of R1279X, the loss of β subunit interaction in pulse-chase experiments (Fig. 3B, right) suggests that its binding to Cav2.1 protein is disabled by R1279X. To further support this observation, we next performed coimmunoprecipitation assays using cells coexpressing β1b-HA and GFP-Cav2.1 (Fig. 3E). As already observed in pulse-chase experiments, β1b-HA coimmunoprecipitated with GFP-Cav2.1 (Fig. 3E, lane 3), but no GFP-Cav2.1 coimmunoprecipitated with β1b-HA (Fig. 3E, lane 4). As predicted from the pulse-chase experiments, no β1b-HA coimmunoprecipitated with GFP-Cav2.1 in the presence of R1279X (Fig. 3E, lane 6), providing further support that R1279X expression prevents Cav2.1-β1b interaction. Next, we tested for a possible interaction between R1279X and the β1b subunit (Fig. 3F). No detectable interaction between β1b-HA and R1279X was detected by coimmunoprecipitation (Fig. 3F, lanes 2, 3). Collectively, these data demonstrate that R1279X destabilizes the wild-type Cav2.1 channel and interferes with its association with the β1b subunit. R1279X coimmunoprecipitates with wild-type Cav2.1, suggesting that the destabilization process requires a direct interaction between the R1279X and wild-type Cav2.1 proteins.
Dominant-negative effect with a similar truncated form of Cav3.2
To investigate whether this mechanism also applies to other Cav channels, notably T-type Ca2+ channels that do not depend critically on β and α2/δ subunits for correct functionality (Perez-Reyes, 2003), we constructed two truncated forms, the two first domains (termed DI-II) and the two last (termed DIII-IV) (Fig. 4A) of the Cav3.2 channel. Immunofluorescence experiments were performed in NG108-15 cells using an HA-tagged Cav3.2 construct (Cav3.2-HA) described previously (Dubel et al., 2004). Similar to that observed with R1279X, DI-II suppressed surface expression of wild-type Cav3.2 (Fig. 4B, middle). In contrast, DIII-IV did not alter Cav3.2 surface expression (Fig. 4B, bottom). Furthermore, wild-type Cav3.2 and DIII-IV colocalized at the plasma membrane.
Figure 4.
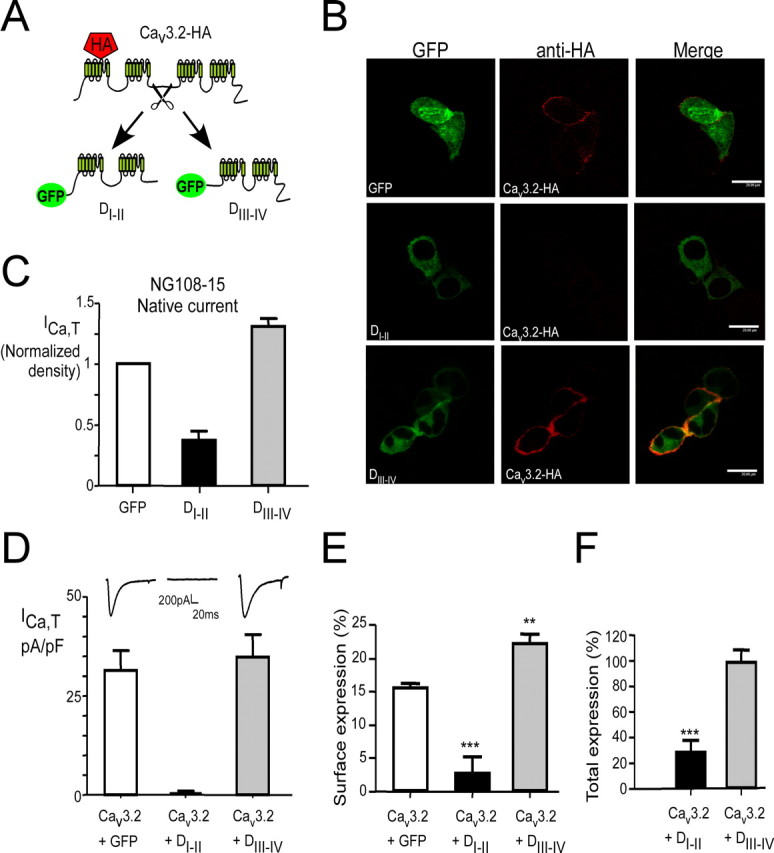
Opposite effects of truncated Cav3.2 forms on recombinant and endogenous Cav3.2 channels. A, Cartoon representing the main Cav3.2 constructs used in this study. B, Immunofluorescence was performed on nonpermeabilized NG108-15 cells expressing Cav3.2-HA in the presence of truncated forms (DI-II or DIII-IV) with a monoclonal rat anti-HA (3F10) antibody and an Alexa-594 secondary antibody. Images were acquired using a Leica SP2 confocal microscope with a 63× oil immersion objective. C, Measurements of the T-type Ca2+ current density (ICa,T) in NG108-15 cells (white bars) transfected with DI-II (n = 19; black bar) or DIII-IV (n = 13; gray bar) Cav3.2 constructs. Note that expression of DI-II, but not DIII-IV, significantly downregulated the endogenous T-type current density. D, Effect on the T-current density measured in HEK293 cells of the coexpression of DI-II (n = 15; black bar) or DIII-IV (n = 7; gray bar) with the wild-type Cav3.2 protein. Note that coexpression of the DI-II construct with the wild-type Cav3.2 protein completely downregulated the T-type current density (see the corresponding insets). E, F, Luminometry assay was performed to quantify surface (E) and total expression (F) of the HA-tagged Cav3.2 in the presence of DI-II and DIII-IV mutants. The data correspond to the average of three independent experiments (**p < 0.01, ***p < 0.001; Student's t test).
Proliferative NG108-15 cells endogenously express T-type channels related to the Cav3.2 subunit (Chemin et al., 2002). We performed patch-clamp recordings of the native T-type current (ICa,T) in NG108-15 cells after transfection of either DI-II or DIII-IV constructs (Fig. 4C). These experiments reveal that overexpression of DI-II, but not of DIII-IV, significantly affects the density of ICa,T in NG108-15 cells. In HEK293 cells, coexpression of DI-II with wild-type Cav3.2 caused abolishment of ICa,T (Fig. 4D, black bar). No change in the T-current density was observed after cotransfection of DIII-IV with wild-type Cav3.2 (Fig. 4D, gray bar). In agreement with the electrophysiological data, luminometry analysis confirmed the inhibition of Cav3.2 surface expression (Fig. 4E) and revealed a strong inhibition of total expression (Fig. 4F) in the presence of DI-II. Note that coexpression of the wild-type Cav3.2 subunit with DIII-IV actually results in a slight increase of Cav3.2 surface expression (Fig. 4E). This is in contrast with the dominant activity that has been observed for the DIII-IV form of Cav2.2 (Raghib et al., 2001).
The DI-II truncated form induces Cav3.2 channel instability
We next performed pulse-chase analysis to study whether the loss of Cav3.2 channel expression in the presence of DI-II relates to protein instability. When expressed alone, Cav3.2-HA remained stable (Fig. 5A, left). In accordance with the observations with R1279X, DI-II drastically destabilized wild-type Cav3.2 with only 10% remaining after 4 h (Fig. 5A, middle, B). A band corresponding to DI-II (120 kDa) coimmunoprecipitated with Cav3.2-HA and disappeared during the chase. Anti-GFP immunoprecipitation from the same fractions identified this band as DI-II (Fig. 5A, right, arrowhead).
Figure 5.

Misfolding induction of DI-II truncated form by direct interaction with the Cav3.2 channel. A, Pulse-chase experiments were performed on HEK293 cells transfected with Cav3.2 alone (left) and in the presence of DI-II (middle and right) for the indicated time periods. After lysis, Cav3.2-HA subunits were immunoprecipitated with anti-HA antibody. The arrow (right) indicates the DI-II-truncated form that coimmunoprecipitates with the wild-type channel. B, Quantification of three independent experiments as shown in A was performed using GE Healthcare software. C, Coimmunoprecipitations were performed on cells expressing the Cav3.2-HA subunit in the presence of the two DI-II and DIII-IV truncated forms. Immunoprecipitated samples (IP) were resolved on 6% SDS-PAGE, and Western blots (WB) were performed with indicated antibodies on the same membranes. MW, Molecular weight.
Coimmunoprecipitation experiments were then designed to identify whether the two truncated forms of Cav3.2 interact with the wild-type channel (Fig. 5C). DI-II could be coimmunoprecipitated with Cav3.2 and conversely Cav3.2 coimmunoprecipitated with DI-II (Fig. 5C, lanes 3 and 4, respectively). DIII-IV did not coimmunoprecipitate with Cav3.2 (Fig. 5C, compare lanes 9 and 10). These data extend our analysis of the dominant-negative effects of R1279X by showing that a truncated Cav3.2 protein suppresses functional expression of its wild-type channel counterpart. Notably, the dominant-negative effect appeared to be specific for constructs comprising domains I and II, because DIII-IV did not affect channel stability.
EA2 missense mutants exert a dominant-negative effect through induction of protein misfolding and instability
Missense mutations also exist in EA2 and progressive ataxia (Pietrobon, 2005). We therefore analyzed the effects of two of these mutants, G293R and AY1593/94D (Yue et al., 1997; Wappl et al., 2002). When expressed in NG108-15 cells, only the untagged and tagged G293R mutants produced a detectable current (in 5 of 33 cells) (Fig. 6A) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material), in agreement with data reported previously (Wappl et al., 2002). The coexpression of these missense mutants with Cav2.1-HA (1:1) resulted in a marked loss of the channel activity (Fig. 6A), whereas T-type channel activity was not affected (Fig. 6B).
Figure 6.

EA2 missense mutants act in a dominant-negative manner by misfolding and instability induction. A, Histograms of the mean Ca2+ current density (± SEM) obtained in a representative batch of NG108-15 cells expressing wild-type subunit alone (Cav2.1-HA, n = 18); the EA2 mutants alone (Cav2.1-HA-G293R, n = 33, 5 with detectable current; Cav2.1-HA-AY1593/94D, n = 6); and the wild-type and EA2 mutants together (Cav2.1-HA+Cav2.1-HA-G293R, n = 23, 3 with detectable current; Cav2.1-HA+Cav2.1-HA-AY1593/94D, n = 10). These experiments were conducted in the presence of the auxiliary β1b and α2/δ1 subunits. B, Normalized native T-type Ca2+ current densities (mean ± SEM) in the NG108-15 cells analyzed in A. The differences are not statistically significant between the various conditions tested (Student's t test). C, Western blots with the indicated antibodies (WB) were performed on HEK293 cells coexpressing Cav2.1-HA alone or in the presence of G293R and AY1593/94D mutants together with auxiliary subunits. The bottom panel shows the Western blot from cells expressing missense mutants alone. D, Pulse-chase analyses were performed on HEK293 cells transfected with wild-type Cav2.1 alone or in the presence of G293R. Quantification of three independents experiment was performed as described above. MW, Molecular weight.
The ability of missense EA2 mutants to downregulate Cav2.1 protein expression was investigated by Western blot (Fig. 6C). We observed a strong inhibition of the Cav2.1 expression, independent of the presence of auxiliary subunits (Fig. 6C, top). Mutant expression was verified by Western blot with anti-Cav2.1 polyclonal antibodies (Fig. 6C, bottom). Next, we performed a pulse-chase analysis to study whether the loss of Cav2.1 expression in the presence of missense channels was also related to the induction of protein instability (Fig. 6D). As obtained above with truncated mutants, G293R drastically destabilized wild-type Cav2.1, reducing its half-life to <1 h (Fig. 6D). Altogether, these data indicate that the G293R mutant acts in a dominant-negative manner, similar to the truncated Cav mutants.
Dominant activity implies ER retention and proteasomal degradation
The destabilization mechanism depicted above is likely to depend on the intrinsic instability of the Cav mutants and their ER localization (Gelman and Kopito, 2003). Therefore, we analyzed the subcellular localization of the EA2 mutants and the truncated Cav3.2 constructs in NG108-15 by confocal microscopy (Fig. 7A). R1279X, G293R, and DI-II were not detected at the cell surface, because no colocalization with CT (a plasma membrane marker) was observed (Fig. 7A, magnification). The finding of a trafficking defect of the G293R mutant is in good agreement with that observed previously (Wan et al., 2005) and was further validated by luminometry experiments (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material).
Figure 7.
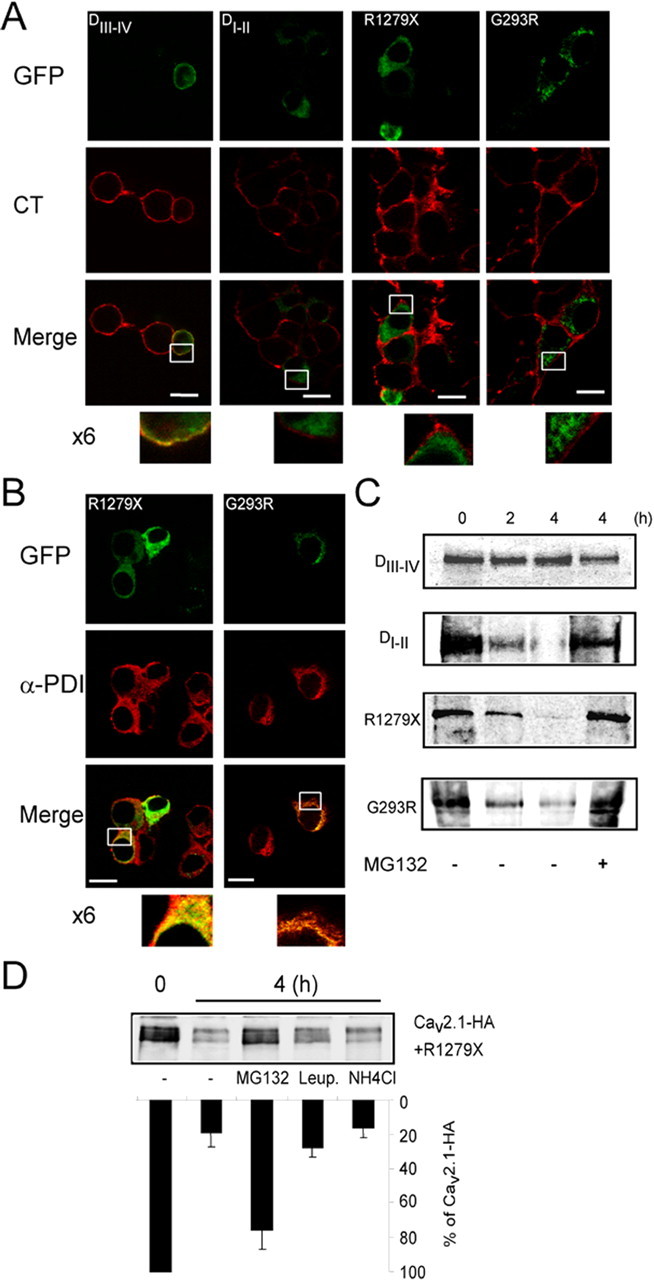
Endoplasmic reticulum retention and proteasomal degradation of the dominant-negative Cav mutants. A, Confocal images of nonpermeabilized NG108-15 cells expressing EA2 mutants and truncated Cav3.2 subunits. Alexa 594-coupled CT was used as plasma membrane marker (0.5 μg/ml). B, Confocal images of immunofluorescence staining performed on permeabilized NG108-15 cells expressing indicated EA2 mutants and truncated Cav3.2. Polyclonal anti-protein disulfide isomerase (Assay Designs, Ann Arbor, MI) was used as ER marker. C, Pulse-chase experiments were performed as in Figure 3 on cells transfected with EA2 mutants (R1279X and G293R) and Cav3.2 truncated forms. Chase was done for the indicated time (hours) with or without MG-132 proteasome inhibitor (50 μm). After lysis, the truncated Cav channels were immunoprecipitated with anti-GFP antibody. D, Representative pulse chase performed on HEK293 cells transfected with Cav2.1 and the R1279X mutant and the corresponding quantification (n = 3). During the chase, cells were treated with MG-132 (50 μm), leupeptin (20 μm), and NH4Cl (10 mm).
In contrast, DIII-IV colocalized with CT (Fig. 7A, left column). In permeabilized cells (Fig. 7B), staining of R1279X and G293R, as well as DI-II staining, appeared predominantly in the perinuclear region (i.e., the ER). Figure 7B shows that both R1279X and G293R mutants fully colocalized with an ER resident protein, protein disulfide isomerase. The same pattern was obtained with DI-II (data not shown). These data establish a correlation between the dominant-negative effect, exerted by R1279X, G293R, and DI-II and intracellular retention.
Because most misfolded proteins retained in the ER are degraded by the proteasome, a mechanism called endoplasmic reticulum-associated degradation (ERAD) (for review, see Gelman and Kopito, 2003), we performed pulse-chase experiments to evaluate the proteasome-dependent stability of the EA2 mutants and truncated Cav3.2 constructs. Fig. 7C shows that R1279X, G293R, and DI-II were degraded rapidly (for quantification, see supplemental Fig. 4A, available at www.jneurosci.org as supplemental material) and protected from degradation by the proteasome inhibitor MG-132 (50 μm) when added during the chase (Fig. 7C). Such stabilization was not obtained in the presence of lysosomal degradation inhibitors (data not shown). In contrast with EA2 mutants and DI-II, DIII-IV remained stable during the chase (Fig. 7C) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). These results show that the truncated forms with dominant-negative activity are recognized as misfolded proteins and become degraded by the proteasome.
Next, we investigated whether misfolded mutant forms induce the proteasomal destruction of the wild-type channels (Fig. 7D). Pulse-chase experiments on cells coexpressing wild-type channels (Cav2.1-HA and Cav3.2-HA) and truncated forms revealed that MG-132 treatment, but neither leupeptin nor NH4Cl (lysosomal protease inhibitors), stabilizes wild-type channels after 4 h (Fig. 7D) (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). We observed a similar effect of the proteasome inhibitor on Cav2.1 wild-type channel with the G256R missense EA2 mutant (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). Altogether, these data indicate that misfolded forms of different Cav isoforms promote misfolding-induced ERAD of the wild-type Cav subunits and their degradation by the proteasome. This mechanism critically relies on the instability of the Cav mutants themselves, which pull their wild-type protein counterparts into degradation pathways through a direct interaction.
Discussion
We provide evidence that the dominant-negative effect induced by Cav mutants associated with EA2 and progressive ataxia relies on a DIM of these mutants with wild-type Cav2.1 culminating in the degradation of the latter. This dominant-negative behavior appears as a general destructive mechanism for voltage-gated Ca2+ channels, because a construct made of the first two domains of the Cav3.2 subunit acts similarly to promote Cav3.2 degradation. From these data, we propose a model in which the newly synthesized mutant channels physically interact with wild-type Cav subunits to promote their degradation. The hypothesis of an interaction between wild-type and mutant channels was proposed in previous studies (Page et al., 2004; Jeng et al., 2006). Here, we show that these incorrectly folded mutant channels are subject to ERAD. These misfolded mutants are able to drive wild-type Cav proteins toward degradation (i.e., DIM), which results in a powerful dominant-negative mechanism. DIM requires concomitant translation of the mutant and wild-type proteins, interaction of these two proteins, and induction of instability and proteasomal degradation. Because DIM critically involves the ER retention machinery, it is expected that the dominant activity of EA2 mutants may depend on the expression system/experimental temperature used. Indeed, studies in Xenopus oocytes, for which the ER retention machinery is leaky for misfolded mammalian proteins (Denning et al., 1992), have revealed no (Wappl et al., 2002) or modest (Jeng et al., 2006) dominant activity effect of EA2 mutants. Importantly, ER retention of misfolded proteins can be overcome by lowering culture temperature in mammalian cells (Denning et al., 1992; Morello et al., 2000), as recently reported for EA2 mutants (Jeng et al., 2008). Other data suggest that Cav2.1 subunit splice variants might be differentially affected by DIM (Jeng et al., 2006) and that auxiliary subunit expression and stoichiometry may modulate DIM (Arikkath et al., 2002; Raike et al., 2007). Altogether, our data reconcile the apparently controversial data on EA2 mutant dominant activity that appear to be principally attributable to variation in the ER retention and cellular degradative capacity.
Another striking finding of this study is that the presence of both misfolded and wild-type Cav in the ER enables their physical interaction. Pulse-chase and immunoprecipitation experiments validate this interaction (Figs. 3, 5). Our data suggest that the interaction between misfolded and wild-type Cav proteins depends primarily on determinants distributed along the domain I-II region of the Cav subunits, rather than the region from domain III to the C terminus, because DIII-IV does not immunoprecipitate with wild-type Cav3.2. Interestingly, it has been shown that DI-II and DIII-IV hemichannels of the Cav2.2 are able to form functional channels, suggesting their ability to associate with one another (Raghib et al., 2001). Altogether, these data, which indicate that a Cav–Cav interaction may be possible because of intramolecular as well as intermolecular interactions, open new questions regarding assembly and stochiometry of Cav proteins in a Ca2+ channel complex.
P/Q-type Ca2+ channels are heteromultimeric complexes comprising a pore-forming Cav2.1 subunit and auxiliary β and α2/δ subunits. The β subunit binds to the I-II loop of the Cav2.1 subunit [α1 interacting domain (AID)] and promotes its sorting from the ER (Isom et al., 1994; Gurnett and Campbell, 1996; Bichet et al., 2000). Interestingly, R1279X is retained in the ER and rapidly degraded, even in the presence of the β subunit. Coexpression of the β subunit does not attenuate DIM, indicating that the interaction of wild-type Cav2.1 with β subunit is lost in the presence of R1279X. Importantly, immunoprecipitation experiments indicate that the β subunit does not bind to the EA2 mutant, suggesting a conformational defect of the AID site. Again, the lack of β subunit binding can be interpreted as misfolding of the EA2 mutant. This indicates that interaction with EA2 mutants strongly affects the folding of nascent wild-type Cav2.1 subunits and interferes with β subunit interaction. Our data are in accordance with those of Page et al. (2004), who reported that dominant-negative suppression of Cav2.2 does not involve sequestration of the β subunit and is not prevented by expression of an increased amount of β subunit (Page et al., 2004).
Coexpression of EA2 mutants with the wild-type Cav2.1 subunit induces ER aggregation and rapid Cav2.1 degradation, which suggests induction of protein misfolding. At this point, Cav misfolding should be considered as a disease-related phenomenon and, reciprocally, EA2 may be considered as a misfolding protein disorder, as are many other neurodegenerative diseases. Defective protein trafficking has emerged as an important mechanism in many inheritable diseases (Kim and Arvan, 1998), and this applies to various channelopathies, including cystic fibrosis (Gelman and Kopito, 2003) and long QT2 syndrome (Delisle et al., 2004), as well as calcium channelopathies (Wan et al., 2005; Jeng et al., 2008). Within the Cav2.1 subunit, the ER quality control machinery may recognize many different regions (i.e., transmembrane domains and loops). At first glance, because the I-II loop contains multiple ER retention motifs (Bichet et al., 2000), it is tempting to speculate that a misfolded I-II loop may be responsible for EA2 mutant retention.
Once identified as misfolded proteins, the cellular machinery can activate several mechanisms of protein degradation, such as ATP-dependent ubiquitination and proteasomal degradation, or autophagic destruction by lysosomes (Schwartz and Ciechanover, 1999; Kostova and Wolf, 2003). To the best of our knowledge, the degradation process of misfolded Cav subunits is unknown. In this study, we provide evidence that the proteasome is critically involved in the degradation of both mutant and wild-type Cav subunit proteins. Page et al. (2004) suggested that accumulation of wild-type and truncated Cav proteins triggers a translation inhibition mechanism that is part of the unfolded protein response (UPR). This UPR activation would rely on pancreatic ER-resident, RNA-dependent protein kinase (PERK), because the dominant-negative effect was reduced partially after PERK inhibition (Page et al., 2004). UPR is composed of at least the PERK, inositol-requiring enzyme 1 (IRE1), and activating transcription factor-6 (ATF6) pathways (Harding et al., 2002). These proteins act as sensors of binding immunoglobulin protein (BiP), which is the major chaperone for protein folding. When BiP availability drops in the ER, the following UPR pathways are activated: PERK inhibits protein synthesis (Harding et al., 1999), whereas IRE1 and ATF6 increase ER chaperone expression that, in turn, increases ER exit and ERAD (Shamu and Walter, 1996; Yoshida et al., 2001). Persistent activation of PERK and IRE1 are well known to induce apoptosis (Schroder and Kaufman, 2006). Although we did not evaluate the level of persistent PERK activation, pulse-chase experiments that provide measurements of de novo Cav protein synthesis reveal no drastic reduction of both Cav2.1 (Fig. 3, compare line 0 in A and B) and Cav3.2 (Fig. 5, compare lane 0 in A, left and middle) translation in the presence of EA2 mutants and DI-II, respectively. Second, because PERK is an ER stress-sensing component and mediates broad translational suppression (Ron, 2002), one would expect low-voltage-activated and HVA Ca2+ currents to be significantly reduced in NG108-15 cells transfected with EA2 mutants, which we did not observe. Finally, R1279X does not affect expression of an unrelated membrane protein, CD4, and modestly the Cav2.2 channel. Similarly, Jouvenceau et al. (2001) reported no difference in potassium current density in cells cotransfected either with Kv1.1/Cav2.1 (wild-type) or Kv1.1/Cav2.1 (R1820X). Altogether, we propose that ERAD is a critical mechanism for dominant suppression of Ca2+ channel activity in EA2, which could be reinforced by UPR activation (Page et al., 2004).
This destructive interaction is likely to occur in EA2 and other channelopathies that are caused by improper folding of mutant proteins. For example, misassembly of mutant and wild-type channels resulting in targeted degradation was described for human ether-a-go-go-related gene (HERG) potassium channels in long QT2 syndrome (Kagan et al., 2000). HERG subunits assemble as tetramers in the ER to form functional potassium channels. Trafficking-defective mutants of HERG subunits have dominant-negative effects that suppress surface expression of wild-type subunits (Delisle et al., 2004). Another striking example is the human immunodeficiency virus 1 (HIV1) accessory protein Vpu, which shares a significant homology with the N-terminal region of TWIK-related acid-sensitive K+ channel 1 (TASK-1) channel and inhibits potassium current through a destructive interaction with the TASK-1 subunit (Hsu et al., 2004). These authors demonstrated nicely that bidirectional destruction occurred as a consequence of TASK-Vpu oligomerization: on one hand, HIV1 Vpu inhibits TASK current, although in contrast, TASK expression restrains the Vpu-mediated virus release (Hsu et al., 2004). The Cav channel pore subunit is a single entity of four transmembrane domains, and there is no biochemical evidence for oligomerization of mature Cav proteins. Precise analysis of Cav subunit folding in the ER by trapping conformational intermediates and eventual transient oligomerization would be necessary to further elucidate the mechanism of EA2 mutant misfolding induction. As a perspective for pharmacological therapy, the search for drugs able to alleviate this destructive interaction, such as small molecule chemical chaperones (Ulloa-Aguirre et al., 2004; Yam et al., 2005) may offer new therapeutic strategies for the treatment of EA2 and other channelopathies.
Footnotes
This work was supported by Agence Nationale pour la Recherche Grant 06-NEURO-035-01 and Austrian Science Fund Grant P17109 and by grants from the Fondation pour la Recherche sur le Cerveau and the Association Française contre les Myopathies. We are indebted to F. A. Rassendren, F. Bertaso, G. Barbara, and E. Kupfer for technical help and to the members of the Physiology Department at Institut de Génomique Fonctionelle for helpful discussions and suggestions. We thank S. Dubel for providing the β1b-HA construct, M. Hosey for β2-adrenergic receptor, and B. Schwappach for CD4 constructs.
References
- Arikkath J, Felix R, Ahern C, Chen CC, Mori Y, Song I, Shin HS, Coronado R, Campbell KP. Molecular characterization of a two-domain form of the neuronal voltage-gated P/Q-type calcium channel alpha(1)2.1 subunit. FEBS Lett. 2002;532:300–308. doi: 10.1016/s0014-5793(02)03693-1. [DOI] [PubMed] [Google Scholar]
- Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- Cao YQ, Piedras-Renteria ES, Smith GB, Chen G, Harata NC, Tsien RW. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron. 2004;43:387–400. doi: 10.1016/j.neuron.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Chemin J, Nargeot J, Lory P. Neuronal T-type α 1H calcium channels induce neuritogenesis and expression of high-voltage-activated calcium channels in the NG108–15 cell line. J Neurosci. 2002;22:6856–6862. doi: 10.1523/JNEUROSCI.22-16-06856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiac arrhythmias: ion channel protein trafficking. Circ Res. 2004;94:1418–1428. doi: 10.1161/01.RES.0000128561.28701.ea. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Diriong S, Lory P, Williams ME, Ellis SB, Harpold MM, Taviaux S. Chromosomal localization of the human genes for alpha 1A, alpha 1B, and alpha 1E voltage-dependent Ca2+ channel subunits. Genomics. 1995;30:605–609. doi: 10.1006/geno.1995.1284. [DOI] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto-Rostro M, Foucault I, Davies A, Wilkinson T, Rees M, Edwards FA, Dolphin AC. The ducky(2J) mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci. 2006;26:12576–12586. doi: 10.1523/JNEUROSCI.3080-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E, Nargeot J. Plasma membrane expression of T-type calcium channel alpha(1) subunits is modulated by high voltage-activated auxiliary subunits. J Biol Chem. 2004;279:29263–29269. doi: 10.1074/jbc.M313450200. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, Hosford DA, Tessarollo L, McEnery MW, Pietrobon D, Copeland NG, Jenkins NA. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–1290. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- Gelman MS, Kopito RR. Cystic fibrosis: premature degradation of mutant proteins as a molecular disease mechanism. Methods Mol Biol. 2003;232:27–37. doi: 10.1385/1-59259-394-1:27. [DOI] [PubMed] [Google Scholar]
- Guida S, Trettel F, Pagnutti S, Mantuano E, Tottene A, Veneziano L, Fellin T, Spadaro M, Stauderman K, Williams M, Volsen S, Ophoff R, Frants R, Jodice C, Frontali M, Pietrobon D. Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J Hum Genet. 2001;68:759–764. doi: 10.1086/318804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett CA, Campbell KP. Transmembrane auxiliary subunits of voltage-dependent ion channels. J Biol Chem. 1996;271:27975–27978. doi: 10.1074/jbc.271.45.27975. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell. 2004;14:259–267. doi: 10.1016/s1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- Jen J. Familial episodic ataxias and related ion channel disorders. Curr Treat Options Neurol. 2000;2:429–431. doi: 10.1007/s11940-000-0041-y. [DOI] [PubMed] [Google Scholar]
- Jeng CJ, Chen YT, Chen YW, Tang CY. Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am J Physiol Cell Physiol. 2006;290:C1209–C1220. doi: 10.1152/ajpcell.00247.2005. [DOI] [PubMed] [Google Scholar]
- Jeng CJ, Sun MC, Chen YW, Tang CY. Dominant-negative effects of episodic ataxia type 2 mutations involve disruption of membrane trafficking of human P/Q-type Ca2+ channels. J Cell Physiol. 2008;214:422–433. doi: 10.1002/jcp.21216. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, Hanna MG. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358:801–807. doi: 10.1016/S0140-6736(01)05971-2. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci USA. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan A, Yu Z, Fishman GI, McDonald TV. The dominant negative LQT2 mutation A561V reduces wild-type HERG expression. J Biol Chem. 2000;275:11241–11248. doi: 10.1074/jbc.275.15.11241. [DOI] [PubMed] [Google Scholar]
- Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev. 1998;19:173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanetz EA. Diversity and properties of calcium channel types in NG108–15 hybrid cells. Neuroscience. 1998;87:265–274. doi: 10.1016/s0306-4522(98)00057-8. [DOI] [PubMed] [Google Scholar]
- Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001;20:6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello JP, Petaja-Repo UE, Bichet DG, Bouvier M. Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci. 2000;21:466–469. doi: 10.1016/s0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- Mullner C, Broos LA, van den Maagdenberg AM, Striessnig J. Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter Cav2.1 Ca2+ channel gating: evidence for beta-subunit isoform-specific effects. J Biol Chem. 2004;279:51844–51850. doi: 10.1074/jbc.M408756200. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Page KM, Heblich F, Davies A, Butcher AJ, Leroy J, Bertaso F, Pratt WS, Dolphin AC. Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J Neurosci. 2004;24:5400–5409. doi: 10.1523/JNEUROSCI.0553-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr Opin Neurobiol. 2005;15:257–265. doi: 10.1016/j.conb.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, Dolphin AC. Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J Neurosci. 2001;21:8495–8504. doi: 10.1523/JNEUROSCI.21-21-08495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raike RS, Kordasiewicz HB, Thompson RM, Gomez CM. Dominant-negative suppression of Cav2.1 currents by alpha(1)2.1 truncations requires the conserved interaction domain for beta subunits. Mol Cell Neurosci. 2007;34:168–177. doi: 10.1016/j.mcn.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15:3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci USA. 2003;100:3491–3496. doi: 10.1073/pnas.0437991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko I, Bidaud I, Arias JM, Mezghrani A, Lory P, Perez-Reyes E. The I-II loop controls plasma membrane expression and gating of Cav3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J Neurosci. 2007;27:322–330. doi: 10.1523/JNEUROSCI.1817-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wan J, Khanna R, Sandusky M, Papazian DM, Jen JC, Baloh RW. CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics. Neurology. 2005;64:2090–2097. doi: 10.1212/01.WNL.0000167409.59089.C0. [DOI] [PubMed] [Google Scholar]
- Wappl E, Koschak A, Poteser M, Sinnegger MJ, Walter D, Eberhart A, Groschner K, Glossmann H, Kraus RL, Grabner M, Striessnig J. Functional consequences of P/Q-type Ca2+ channel Cav2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J Biol Chem. 2002;277:6960–6966. doi: 10.1074/jbc.M110948200. [DOI] [PubMed] [Google Scholar]
- Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005;19:12–18. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yue Q, Jen JC, Nelson SF, Baloh RW. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am J Hum Genet. 1997;61:1078–1087. doi: 10.1086/301613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci USA. 2001;98:2431–2436. doi: 10.1073/pnas.051630198. [DOI] [PMC free article] [PubMed] [Google Scholar]