

Abstract
The fragile X syndrome is caused by the lack of fragile X mental retardation protein (FMRP) attributable to silencing of the FMR1 gene. The metabotropic glutamate receptors (mGluRs) in the CNS contribute to different brain functions, including learning/memory, mental disorders, drug addiction, and persistent pain. Most of the previous studies have been focused on downstream targets of FMRP in hippocampal neurons, and fewer studies have been reported for the second-messenger signaling pathways between group I mGluRs and FMRP. Furthermore, no molecular study has been performed in the anterior cingulate cortex (ACC), a key region involved in high brain cognitive and executive functions. In this study, we demonstrate that activation of group I mGluR upregulated FMRP in ACC neurons of adult mice through the Ca2+-dependent signaling pathways. Using genetic approaches, we found that Ca2+/calmodulin-stimulated adenylyl cyclase 1 (AC1) and calcium/calmodulin-dependent kinase IV (CaMKIV) contribute to the upregulation of FMRP induced by stimulating group I mGluRs. The upregulation of FMRP occurs at the transcriptional level. The cAMP-dependent protein kinase is activated by stimulating group I mGluRs through AC1 in ACC neurons. Both AC1 and CaMKIV contribute to the regulation of FMRP by group I mGluRs probably through cAMP response element-binding protein activation. Our study has provided the first evidence for a molecular link between group I mGluRs and FMRP in ACC neurons and may help us to understand the pathogenesis of fragile X syndrome.
Keywords: adenylyl cyclase 1, CaMKIV, CREB, FMRP, group I mGluRs, cingulate cortex
Introduction
Fragile X syndrome, the most common inherited form of human mental retardation and an identified cause for autism, is caused by mutations of the FMR1 gene that encodes the fragile X mental retardation protein (FMRP) (Feng et al., 1995; Jin and Warren, 2003; Belmonte and Bourgeron, 2006; Huber, 2007). FMRP, an mRNA binding protein, is believed to play an important role in activity-dependent synaptic plasticity through biochemical regulation of local protein synthesis at synapses (Greenough et al., 2001; Huber et al., 2002; Bagni and Greenough, 2005). The abnormal functions of group I metabotropic glutamate receptor (mGluR)-dependent synaptic plasticity have been observed in hippocampus of Fmr1 knock-out (KO) mice (Huber et al., 2002; Bear et al., 2004; Hou et al., 2006; Nakamoto et al., 2007). The function of group I mGluR activation require the translation of preexisting mRNA near active synapses. FMRP normally functions as a repressor of translation of specific mRNAs (Greenough et al., 2001; Bear et al., 2004; Bagni and Greenough, 2005; Grossman et al., 2006). It is possible that the protein synthesis-dependent functions of mGluR activation are exaggerated because of the lack of FMRP in fragile X syndrome (Bear et al., 2004; Hou et al., 2006).
Recent studies from animal models reveal that the anterior cingulate cortex (ACC) plays an important role in cognitive learning, fear memory, and persistent pain (Frankland et al., 2001, 2004; Han et al., 2003; Zhao et al., 2005b; Zhuo, 2006, 2008). Trace fear memory is sensitive to attention-distracting stimulation and requires the activity of ACC (Han et al., 2003). Attention control difficulties have been observed in fragile X patients (Scerif et al., 2007). Interestingly, trace fear memory is impaired in Fmr1 KO mice, accompanied by alterations in synaptic plasticity in ACC (Zhao et al., 2005a; Hayashi et al., 2007). These findings suggest that the dysfunction of ACC attributable to lack of FMRP may be responsible for certain types of mental disorders in fragile X syndrome. Electrophysiological and behavioral studies in animals found that the mGluRs in ACC may contribute to activity-dependent synaptic plasticity and behavioral fear memory (Wei et al., 1999; Tang et al., 2005). It is conceivable that mGluRs may regulate the expression of FMRP in ACC neurons, and the loss of this signaling pathway may contribute to the pathogenesis of fragile X syndrome. However, previous studies of the regulation of FMRP by mGluRs are mostly performed in hippocampus, and no study has been reported for the signaling pathway linking mGluRs and FMRP in ACC.
In the present study, we have demonstrated that activation of group I mGluRs increased FMRP in neurons from adult mouse cingulate slices. Using gene KO mice lacking adenylyl cyclase 1 (AC1) or calcium/calmodulin-dependent kinase IV (CaMKIV), we found that both AC1 and CaMKIV contribute to the upregulation of FMRP by (RS)-3,5-dihydroxyphenylglycine (DHPG), an agonist of group I mGluRs. By measuring the activation of cAMP-dependent protein kinase (PKA) and cAMP response element-binding protein (CREB), we provide evidence that PKA is activated by stimulating group I mGluRs through AC1; DHPG application also activated CREB, and both AC1 and CaMKIV are involved in the activation of CREB. Because the CREB pathway is not affected in Fmr1 KO mice, we propose that activation of group I mGluRs may upregulate FMRP through the Ca2+-stimulated CREB signaling pathway.
Materials and Methods
Animals.
Adult male C57BL/6 mice were used in most of experiments. Mutant male mice lacking AC1, AC8, or CaMKIV KO mice were derived as described previously and bred for several generations (F12–F16) on C57BL/6 background (Wei et al., 2002a,b, 2006). Wild-type (WT) mice from littermates of the mutant mice were also used as additional control mice. Fmr1 WT and KO mice of the FVB.129P2-Fmr1tm1Cgr strain were generously provided by Dr. W. T. Greenough (University of Illinois at Urbana-Champaign, Urbana, IL), the mice were generated and maintained as reported previously (Weiler et al., 2004; Zhao et al., 2005a). All mice were housed under a 12 light/dark cycle with food and water provided ad libitum. All mouse protocols are in accordance with National Institutes of Health guidelines and approved by the Animal Care and Use Committee of University of Toronto.
Drugs and antibodies.
DHPG, (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate [(2R,4R)-APDC], (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid (ACPT-I), 6 methoxy-N-(4-methoxyphenyl)-4-quinazolinamine hydrochloride (MPMQ), 3-amino-6-chloro-5-dimethylamino-N-2-pyridinylpyrazinec arboxamide hydrochloride (ACDPP), dl-AP-3, and KT5720 [(9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo-[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester] were purchased from Tocris Bioscience (Ellisville, MO). Actinomycin D, cyclopiazonic acid (CPA), nifedipine (Nif), phosphatase inhibitor cocktail, and phosphatase inhibitor cocktail 1 and 2 were purchased from Sigma (St. Louis, MO). The anti-FMRP antibody, horseradish peroxidase-linked goat anti-mouse IgG, and goat anti-rabbit IgG for Western blot were purchased from Millipore Bioscience Research Reagents (Temecula, CA). The anti-CREB antibody and anti-phosphorylated-CREB (pCREB) antibody were purchased from Cell Signaling Technology (Danvers, MA). The anti-actin antibody was from Sigma. The anti-CaMKIV antibody was from BD Biosciences (San Jose, CA).
Brain slice preparations.
Mice were anesthetized with 2% halothane, and brain slices (300 μm) containing ACC were cut at 4°C using a vibratome, in oxygenated artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2 KCl, 26 NaHCO3, 2 CaCl2, 2 MgSO4, 1 NaH2PO4, and 10 d-glucose, pH 7.4. The slices were slowly brought to final temperature of 30°C in ACSF gassed with 95% O2/5% CO2 and incubated for at least 1 h before experiments. Slices then were exposed to different compounds of interest for the indicated times and snap frozen over dry ice. For biochemical experiments, the ACC regions were microdissected and sonicated in ice-cold homogenization buffer containing phosphatase and protease inhibitors. For electrophysiology and calcium imaging, brain slices were transferred to a submerged recovery chamber with oxygenated ACSF at room temperature.
Immunohistochemistry.
The ACC sections from the control and DHPG treatment group were processed simultaneously to allow the same condition and time for DAB staining. Cryostat-cut brain sections (30 μm) were processed with mouse anti-FMRP (1:500) and rabbit anti-pCREB antibody (1:500). The avidin–biotin protocol was used as described previously (Wei et al., 2002b). Images were collected on an Olympus (Tokyo, Japan) BX 60 microscope and analyzed using NIH Image J software (Scion, Frederick, MD).
Western blot analysis.
Western blot was conducted as described previously (Wang et al., 2007). The brain tissues were dissected and homogenized in lysis buffer containing 10 mm Tris-HCl, pH 7.4, 2 mm EDTA, 1% SDS, 1× protease inhibitor cocktail, and 1× phosphatase inhibitor cocktail 1 and 2. Protein was quantified by Bradford assay. Electrophoresis of equal amounts of total protein was performed on SDS-polyacrylamide gels. Separated proteins were transferred to polyvinylidene fluoride membranes at 4°C for analysis. Membranes were probed with 1:3000 dilution of anti-FMRP, 1:1000 dilution of anti-pCREB (Ser133) and anti-CREB, or 1:2000 dilution of anti-CaMKIV antibodies. The membranes were incubated in the appropriate horseradish peroxidase-coupled secondary antibody diluted 1:3000 for 1 h followed by enhanced chemiluminescence detection of the proteins with Western lightning chemiluminescence reagent plus according to the instructions of the manufacturer. To verify equal loading, membranes were also probed with 1:3000 dilution of anti-actin antibody. The density of immunoblots was measured using NIH ImageJ program.
Reverse transcription-PCR.
The total RNA from the ACC was isolated using RNAspin Mini kit (GE Healthcare, Little Chalfont, UK). Reverse transcription (RT)-PCR was performed using SuperScript One-Step RT-PCR System with Platinum TaqDNA Polymerase. A 25 μl PCR containing 0.5 μg of total RNA, 5 μl of 5× RT-PCR buffer, 400 μm dNTP, 0.5 μm of each primer, and 1 μl of RT-PCR enzyme. PCR conditions were adjusted to be in a linear range of amplification. The PCR cycles consisted of initial incubation at 94°C for 1 min; denaturation at 94°C for 45 s; annealing at 56°C for 45 s; and extension at 72°C for 1 min, for 30 cycles, and final extension at 72°C for 10 min. The primers for Fmr1 used in this experiment were as follows: sense, 5′-CCGAACAGATAATCGTCCACG-3′; antisense, 5′-ACGCTGTCTGGCTTTTCC TTC-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as an internal control by using the following primer sets: sense, 5-AACGACCCCTTCATTGAC-3′; antisense 5′-TCCACGACATACTCAGCAC-3′. RT-PCR products were electrophoresed on 1.5% agarose gels and visualized under UV light by ethidium bromide staining. The relative density of bands was analyzed by the NIH ImageJ program.
cAMP assay.
After treatment with DHPG, the ACC tissues were harvested and lysed in 0.1 m HCl. Direct cAMP measurements were performed using the direct cAMP enzyme immunoassay kit (Assay Designs, Ann Arbor, MI) according to the protocol of the manufacturer. Phosphodiesterase was inhibited by the addition of 1 mm 3-isobutyl-1 methylxanthine (Sigma) in this process.
PKA activity assay.
The PKA activity was measured using the PKA kinase activity kit (Assay Designs). The relative PKA activity in the samples was calculated according to the following formula: relative kinase activity = [absorbance of samples − absorbance of blank]/quantity of protein used in per assay.
Whole-cell patch-clamp recording.
Brain slices were transferred to a recording chamber and perfused with oxygenated ACSF solution at 3–4 ml/min at room temperature. Whole-cell patch-clamp recordings were made on the soma of ACC pyramidal neurons. Recording electrodes (2–3 MΩ) contained an K+-based internal solution composed of the following (in mm): 120 K-gluconate, 5 NaCl, 1 MgCl2, 0.5 EGTA, 0.1 Na3GTP, 10 HEPES, and 0.4 Oregon green BAPTA-1 (OGB-1), pH 7.2 (280–300 mOsm). Unless otherwise stated, the membrane potential was held at −60 mV for neurons throughout all experiments. To record DHPG-induced current, tetrodotoxin (1 mm) was added in the ACSF. Data were amplified and filtered at 2 kHz by a patch-clamp amplifier (Axopatch 200B), digitalized (DIGIDATA 1322A), stored, and analyzed by pClamp (Molecular Devices, Union City, CA).
Calcium imaging.
OGB-1 (0.4 mm; Invitrogen, Carlsbad, CA) was dialyzed into ACC neurons by whole-cell patch pipette. After entering whole-cell mode, the cells were maintained for >10 min to allow for filling with OGB-1 before image acquisition. Fluorescent signals were imaged by confocal microscope (Fluoview FV 1000; Olympus). The laser with a wavelength of 488 nm was used for excitation, and fluorescence was recorded through a bandpass filter (500–550 nm). The images were acquired using 40× 0.8 numeric aperture water-immersion objectives every 5 s after a 188 ms exposure to 488 nm light. XYT image galleries were collected, and average fluorescence intensity in the soma was measured for the quantification. The intensity was expressed as F/F0, where F0 is the fluorescence intensity before DHPG treatment.
Data analysis. Statistical comparisons were made using the paired t test or one-way or two-way ANOVA (Student–Newman–Keuls test was used for post hoc comparison). All data were presented as the mean ± SEM. In all cases, p < 0.05 is considered statistically significant.
Results
Activation of group I mGluRs upregulates FMRP in the ACC neurons
Previous studies of the regulation of FMRP by mGluRs have mainly been performed in cultured cortical neurons and hippocampal slices (Weiler et al., 1997; Todd et al., 2003; Hou et al., 2006). In this study, we investigated the effects of mGluR activation on the expression of FMRP in ACC slices from adult mice. By immunohistochemistry, we found FMRP was well expressed in the ACC at the basal condition. Application of the group I mGluR agonist DHPG (100 μm) to ACC slices for 30 min caused a widespread increase of FMRP immunoreactivity from layers II–VI in the ACC (Fig. 1A). The increase of FMRP levels by DHPG in the ACC was also measured by Western blot (196 ± 11% of the control levels; p < 0.01 compared with control; n = 6) (Fig. 1B). In contrast, bath application of the group II mGluR agonist (2R,4R)-APDC (10 μm) or group III mGluR agonist ACPT-I (100 μm) for 30 min did not cause changes in FMRP levels in the ACC slices (p > 0.05 compared with control; n = 4) (Fig. 1C). These data indicate that activation of group I mGluRs upregulates FMRP in ACC neurons.
Figure 1.
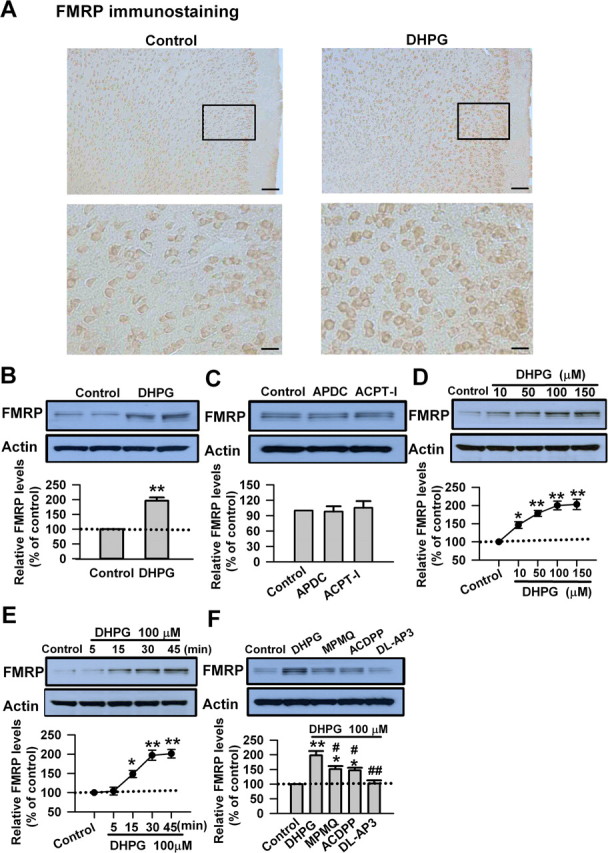
Activation of group I mGluRs upregulates FMRP in ACC neurons. A, Immunohistochemistry of FMRP in ACC. Compared with the control (left), the increase of the immunoreactivity of FMRP by DHPG (100 μm, 30 min; right) could be found in neurons from layers II–VI in ACC. A high-magnification image of the selected part showing the staining in layers II and III is provided at the bottom. Scale bars: top, 100 μm; bottom, 25 μm. B, Application of the group I mGluR agonist DHPG (100 μm) for 30 min increased the levels of FMRP in ACC, as measured by Western blot. C, Application of the group II mGluR agonist (2R,4R)-APDC (10 μm) or group III mGluR agonist ACPT-I (100 μm) for 30 min did not affect the levels of FMRP in ACC slices. D, The levels of FMRP were increased by DHPG (10–150 μm) in a dose-dependent manner. The highest level of FMRP was seen at 100 μm; DHPG at higher concentration (150 μm) did not cause additional increase of FMRP. E, DHPG (100 μm) increased FMRP in a time-dependent manner, the increase was observed at 15 min, and the highest increase was reached at 30 min. F, The increase of FMRP caused by DHPG was partially blocked by the selective mGluR1 antagonist MPMQ (10 μm) or a specific mGluR5 antagonist of ACDPP (10 μm); the presence of the group I mGluR antagonist dl-AP-3 (100 μm) completely blocked the increase of FMRP caused by DHPG in ACC slices. The antagonists were applied to slices 20 min before and during the DHPG treatment. Representative Western blot (top) and quantification data (bottom) of FMRP levels are shown for corresponding treatments from B–F. Data were normalized by the control values. *p < 0.05, **p < 0.01 compared with control; #p < 0.05, ##p < 0.01 compared with DHPG treatment; n = 3 mice for each group in A; n = 6 mice for each group in B; n = 4 mice for each group in C–F.
The effect of DHPG on FMRP expression was further characterized in ACC slices. We found that the FMRP levels were increased by DHPG (10–150 μm) in a dose-dependent manner. The highest level of FMRP was observed at 100 μm, whereas DHPG at 150 μm did not cause additional increase in FMRP level (p < 0.01 compared with control; n = 4) (Fig. 1D). In addition, DHPG (100 μm) increased FMRP expression in a time-dependent manner, the increase could be observed at 15 min, the highest level was reached after 30 min (p < 0.01 compared with control; n = 4) (Fig. 1E).
The group I mGluRs consist of two subtypes, mGluR1 and mGluR5 (Coutinho and Knopfel, 2002; Thuault et al., 2002; Moult et al., 2006). To identify which group I mGluR subtype(s) was responsible for the increase of FMRP caused by DHPG, we applied DHPG (100 μm) to ACC slices in the presence of either the selective mGluR1 antagonist MPMQ (10 μm) or a specific mGluR5 antagonist of ACDPP (10 μm). We found that the increase of FMRP caused by DHPG was only partially blocked by MPMQ or ACDPP (p < 0.05 compared with control or compared with DHPG treatment; n = 4) (Fig. 1F). However, the presence of group I mGluR antagonist dl-AP-3 (100 μm) completely blocked the increase of FMRP caused by DHPG in ACC slices (p < 0.01 compared with DHPG treatment; n = 4) (Fig. 1F). These data confirm that group I mGluRs, including mGluR1 and mGluR5, regulate FMRP in ACC neurons.
Calcium is critical for the regulation of FMRP by group I mGluRs
During group I mGluR activation, Ca2+ could be released from IP3-sensitive intracellular stores and acts as a second messenger (Rae et al., 2000; Fitzjohn et al., 2001; Coutinho and Knopfel, 2002; Heinke and Sandkuhler, 2007). The sarco/endoplasmic reticulum Ca2+/ATPase (SERCA) pump inhibitor cyclopiazonic acid depletes intracellular stores of Ca2+ by blocking Ca2+ reuptake into the stores (Rae et al., 2000; Heinke and Sandkuhler, 2007). To examine the role of Ca2+ release from intracellular stores in the upregulation of FMRP by DHPG, CPA (30 μm) was applied to ACC slices 20 min before and during the DHPG (100 μm, 30 min) treatment. We found that the upregulation of FMRP by DHPG was partially blocked by cyclopiazonic acid (193 ± 7 and 132 ± 9% of the control levels for DHPG and CPA treatment, respectively; p < 0.05 compared with DHPG treatment; n = 6) (Fig. 2A).
Figure 2.

Calcium mediates the regulation of FMRP by group I mGluRs in ACC neurons. A, The SERCA pump inhibitor CPA (50 μm) partially blocked the upregulation of FMRP by DHPG (100 μm, 30 min). Cyclopiazonic acid was applied to slices 20 min before and during DHPG treatment. B, L-type Ca2+ channels blocker Nif (25 μm) partially blocked the increase of FMRP caused by DHPG treatment. Nifedipine was applied to slices 20 min before and during DHPG treatment. C, Coapplication of cyclopiazonic acid with nifedipine almost completely blocked the upregulation of FMRP by DHPG treatment. CPA (50 μm) and Nif (25 μm) were applied to slices 20 min before and during DHPG treatment. D, Application of cyclopiazonic acid (50 μm), nifedipine (25 μm), or coapplication of cyclopiazonic acid with nifedipine for 30 min did not affect the basal levels of FMRP in ACC slices. Representative Western blot (top) and quantification data (bottom) of FMRP are shown for the corresponding treatments. Data were normalized by the control values. *p < 0.05, **p < 0.01 compared with control; #p < 0.05, ##p < 0.01 compared with DHPG treatment; n = 6 mice for each group in A–C; n = 4 mice for each group in D.
Activation of group I mGluRs can facilitate L-type voltage-dependent Ca2+ channels (L-VDCCs) in different cell types (Chavis et al., 1995, 1996; Heinke and Sandkuhler, 2005). DHPG treatment may induce Ca2+ influx through L-VDCCs in striatal neurons (Mao and Wang, 2003). Here, we found that application of L-VDCC blocker Nif (25 μm) 20 min before and during the DHPG treatment also partially blocked the increase of FMRP caused by DHPG (198 ± 12 and 146 ± 11% of the control levels for DHPG and nifedipine treatment, respectively; p < 0.05 compared with DHPG treatment; n = 6) (Fig. 2B). In addition, coapplication of cyclopiazonic acid with nifedipine almost completely blocked the upregulation of FMRP by DHPG treatment (190 ± 1% and 106 ± 11% of the control levels for DHPG and CPA plus nifedipine treatment, respectively; p < 0.01 compared with DHPG treatment; n = 6) (Fig. 2C). In contrast, cyclopiazonic acid, nifedipine, or coapplication of cyclopiazonic acid and nifedipine did not affect the basal levels of FMRP in ACC slices (p > 0.05 compared with control; n = 4) (Fig. 2D). These data indicate that both Ca2+ release from intracellular stores and Ca2+ influx through L-VDCCs are required for the regulation of FMRP by group I mGluRs in ACC neurons.
AC1 contributes to the upregulation of FMRP in the ACC neurons
The cAMP signaling pathway contributes to the activity-dependent synaptic plasticity in ACC neurons (Wei et al., 2002b, 2006; Liauw et al., 2005; Zhuo, 2008). The group I mGluRs have been shown to potentiate cAMP accumulation in cultured striatal neurons, striatum, and cerebral cortex (Cartmell et al., 1997, 1998; Schaffhauser et al., 1997). Among the cAMP signaling pathway, AC1 and AC8 are the two major Ca2+/calmodulin-stimulated AC isoforms (Cooper et al., 1998; Sunahara and Taussig, 2002; Wang and Storm, 2003; Cooper and Crossthwaite, 2006). No study has been reported for the involvement of AC1 and AC8 in the mGluR signaling pathway. Here we first used the AC1&8 double KO (DKO) mice to explore the role of Ca2+/calmodulin-stimulated ACs in the regulation of FMRP by group I mGluRs. We found that, although the basal FMRP levels were unaltered in ACC of AC1&8 DKO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 3A), the increase of FMRP caused by DHPG (100 μm, 30 min) was partially blocked in AC1&8 DKO mice compared with WT mice (p < 0.05 compared with WT mice; n = 4) (Fig. 3B). These findings indicate that Ca2+/calmodulin-stimulated ACs are involved in the regulation of FMRP by group I mGluRs in ACC neurons.
Figure 3.
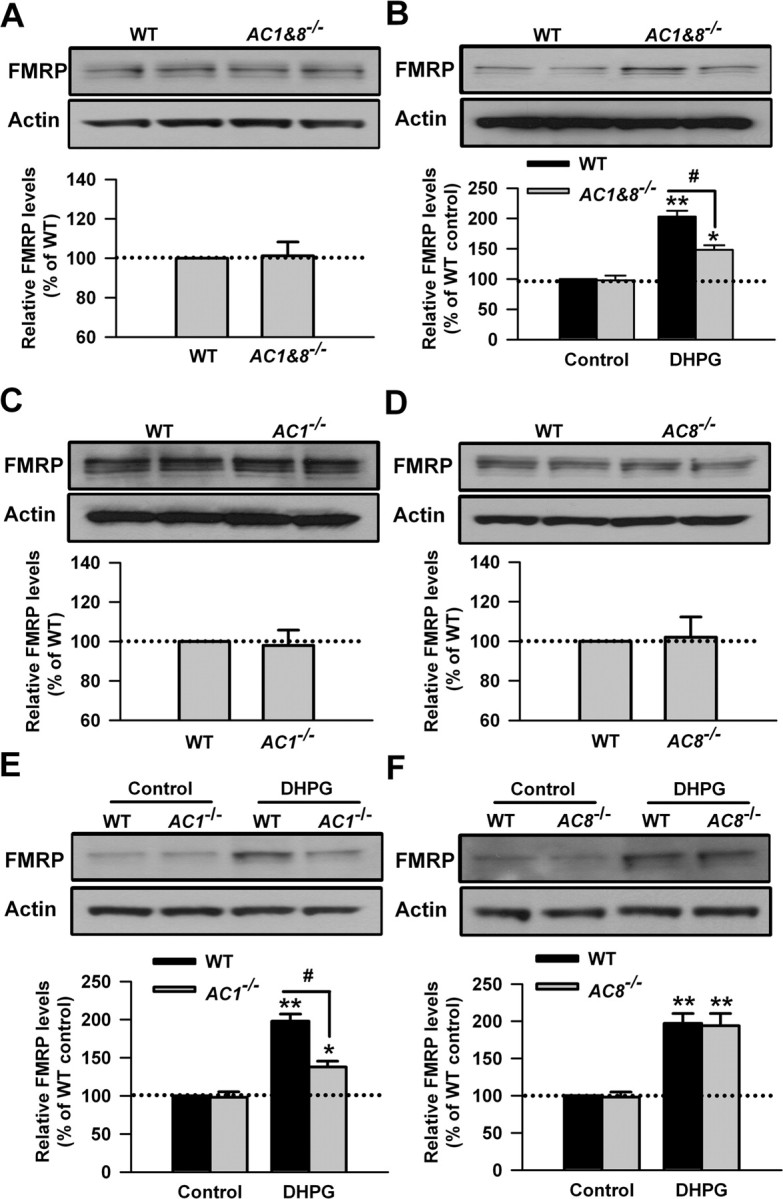
Upregulation of FMRP was partially blocked in ACC from mice lacking AC1. A, There was no difference in the basal levels of FMRP in ACC slices between WT and AC1&8 DKO mice, as shown by Western blot. B, The increase of FMRP in ACC slices treated by DHPG (100 μm, 30 min) was partially blocked in AC1&8 DKO mice compared with WT mice. C, D, There was no difference in the basal levels of FMRP in ACC slices between WT and AC1 KO (C) or AC8 KO (D) mice, as shown by Western blot. E, The increase of FMRP in ACC slices from AC1 KO mice was attenuated compared with WT mice. The slices were treated with DHPG (100 μm) for 30 min. F, The increase of FMRP attributable to DHPG treatment (100 μm, 30 min) in ACC slices from AC8 KO mice was not different from that in WT mice. Representative Western blot (top) and quantification data (bottom) of FMRP are shown for the corresponding treatments. Data were normalized by the WT control values. *p < 0.05, **p < 0.01 compared with control; #p < 0.05 compared with WT; n = 4 mice for each group in A and B; n = 6 mice for each group in C–F.
To address the specific roles of AC1 and AC8 in the upregulation of FMRP by stimulating group I mGluRs, we tested the effect of DHPG (100 μm, 30 min) in ACC slices from AC1 KO or AC8 KO mice, respectively. By Western blot, we found that there was no difference in the basal levels of FMRP in ACC slices between WT and AC1 KO or AC8 KO mice (p > 0.05 compared with WT mice; n = 6) (Fig. 3C,D). After ACC slices were treated with DHPG for 30 min, we found that the increase of FMRP in ACC slices from AC1 KO mice was attenuated compared with WT mice (198 ± 10 and 138 ± 9% of the WT control levels for WT and AC1 KO mice, respectively; p < 0.05 compared with WT mice; n = 6) (Fig. 3E). However, the increase of FMRP attributable to DHPG treatment was not changed in ACC slices from AC8 KO mice (197 ± 14 and 194 ± 17% of the WT control levels for WT and AC8 KO mice, respectively; p > 0.05 compared with WT mice; n = 6) (Fig. 3F). These results indicate that AC1, but not AC8, contributes to the regulation of FMRP by group I mGluRs in ACC neurons.
CaMKIV acts downstream of group I mGluRs in the ACC neurons
CaMKIV is a key effector in neuronal Ca2+ signaling and functions as a transcriptional activator (Bito et al., 1997; Ho et al., 2000; Hook and Means, 2001). It is expressed in both nuclei and cytosol of neurons in several brain regions, including cortex, cerebellum, hippocampus, and amygdala (Means et al., 1991; Sun et al., 1995; Ho et al., 2000). CaMKIV has been implicated in many aspects of neuronal Ca2+ signaling, including gene expression in response to excitatory neurotransmission (Ho et al., 2000; Hook and Means, 2001; Wei et al., 2002a). To explore the possible role of CaMKIV in the upregulation of FMRP by stimulating group I mGluRs, we tested the effect of DHPG (100 μm, 30 min) treatment in ACC slices from CaMKIV KO mice. We found that there was no difference in the basal levels of FMRP in ACC slices between WT and CaMKIV KO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 4A). However, after the treatment with DHPG for 30 min, we found that the increase of FMRP was partially blocked in ACC slices from CaMKIV KO mice compared with WT mice (202 ± 10 and 139 ± 11% of the WT control levels for WT and CaMKIV KO mice, respectively; p < 0.05 compared with WT mice; n = 4) (Fig. 4B). The data indicate that CaMKIV also acts as a downstream effector for group I mGluRs and contributes to the regulation of FMRP by group I mGluRs in ACC neurons.
Figure 4.
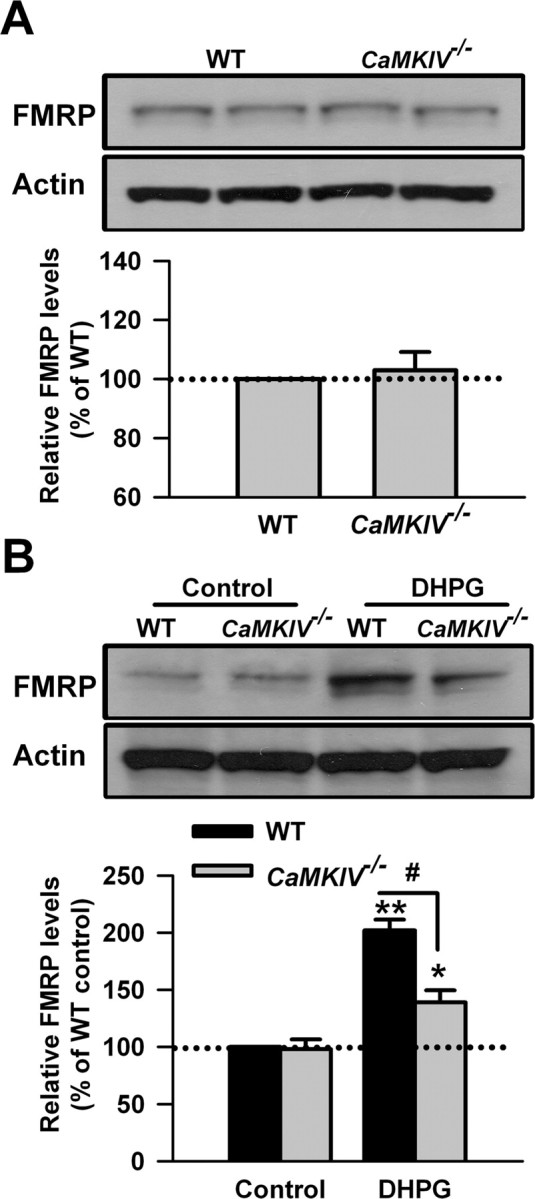
CaMKIV contributes to the upregulation of FMRP by group I mGluRs in ACC neurons. A, There was no difference in the basal levels of FMRP in ACC slices between WT and CaMKIV KO mice. B, The increase of FMRP after treatment with DHPG (100 μm) for 30 min was attenuated in ACC slices from CaMKIV KO mice compared with WT mice. Representative Western blot (top) and quantification data (bottom) of FMRP are shown for the corresponding treatments. Data were normalized by WT control values. *p < 0.05, **p < 0.01 compared with control; #p < 0.05 compared with WT; n = 4 mice for each group.
Upregulation of FMRP by group I mGluRs occurs at the transcription level
One previous study has shown that stimulation of group I mGluRs with DHPG induces the increase of FMRP in a protein synthesis-dependent manner in hippocampus CA1 area (Hou et al., 2006). To examine whether the upregulation of FMRP by stimulating group I mGluR occurs at the transcriptional or translational level in ACC neurons, we tested the effect of inhibiting gene transcription in ACC neurons. We found that pretreatment with a transcription inhibitor, actinomycin D (40 μm), 20 min before and during DHPG application (100 μm, 30 min) abolished the DHPG-induced increase of FMRP in ACC slice (187 ± 10 and 103 ± 13% of the control levels for DHPG and actinomycin D treatment, respectively; p < 0.01 compared with DHPG treatment; n = 6) (Fig. 5A). The data indicate that the regulation of FMRP by I mGluRs occurs at the transcriptional level.
Figure 5.

The upregulation of FMRP by group I mGluRs occurs at the transcriptional level. A, DNA transcription inhibitor actinomycin D (40 μm) abolished the DHPG-induced increase of FMRP in ACC slices, as shown by Western blot. Actinomycin D was applied to slices 20 min before and during DHPG (100 μm) treatment for 30 min. B, DHPG (100 μm, 15 min) treatment increased the levels of Fmr1 mRNA in ACC slices, as shown by RT-PCR. C, D, The increase of Fmr1 mRNA was attenuated in ACC slices from AC1 KO (C) or CaMKIV KO (D) mice compared with WT mice, as shown by RT-PCR. The size of PCR products is 141 and 191 bp for Fmr1 and GAPDH, respectively. Representative gels (top) and quantification data (bottom) of FMRP or Fmr1 mRNA are shown for the corresponding treatments. Data were normalized by WT control values. *p < 0.05, **p < 0.01 compared with control; #p < 0.05 compared with WT; n = 4 mice for each group.
Can Fmr1 mRNA be newly transcribed by stimulating I mGluRs in ACC neurons? To address this, we measured the levels of Fmr1 mRNA by RT-PCR. We found that DHPG (100 μm, 15 min) treatment could increase the levels of Fmr1 mRNA in ACC slices (220 ± 12% of the control levels; p < 0.01 compared with control; n = 4) (Fig. 5B). There was no difference in the basal levels of Fmr1 mRNA in ACC between WT and AC1 or CaMKIV KO mice (p > 0.05; n = 4) (Fig. 5C,D). However, the increase of Fmr1 mRNA caused by DHPG treatment was partially blocked in ACC slices from AC1 or CaMKIV KO mice (p < 0.05 compared with WT mice; n = 4) (Fig. 5C,D). These data further confirm that group I mGluR activation upregulates FMRP at the transcriptional level, and AC1 and CaMKIV are required for the transcriptional regulation of FMRP by group I mGluRs in ACC neurons.
PKA is activated by stimulating group I mGluRs
Once activated by Ca2+/calmodulin, the Ca2+/calmodulin-stimulated ACs convert ATP to cAMP (Sunahara and Taussig, 2002; Cooper, 2003; Wang and Storm, 2003). To further characterize the roles of Ca2+ and Ca2+/calmodulin-stimulated ACs in the regulation of FMRP by group I mGluRs, we measured the cAMP levels in ACC neurons. We found that application of DHPG (100 μm, 15 min) increased the cAMP levels in ACC slice. We then tested the effects of SERCA pump inhibitor cyclopiazonic acid and L-VDCC blocker Nif (25 μm). We found that application of CPA (30 μm) or Nif (25 μm) before and during DHPG treatment could partially block the increase of cAMP caused by DHPG (p < 0.05 compared with DHPG treatment; n = 4) (Fig. 6A), and the increase of cAMP was completely blocked by coapplication of cyclopiazonic acid and nifedipine (p < 0.01 compared with DHPG treatment; n = 4) (Fig. 6A). In contrast, cyclopiazonic acid, nifedipine, or coapplication of cyclopiazonic acid and nifedipine did not affect the basal levels of cAMP in ACC slices (p > 0.05 compared with control; n = 4) (Fig. 6B). We next measured the cAMP levels in ACC slices from AC1 KO or AC8 KO mice. We found that the increase of cAMP caused by DHPG treatment was blocked in AC1 KO mice (p < 0.05 compared with WT; n = 4) (Fig. 6C) but not in AC8 KO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 6C), whereas the basal levels of cAMP was not affected in ACC slices from AC1 KO or AC8 KO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 6C). These results demonstrate that both the intracellular calcium stores and external calcium influx through L-VDCC are required for the cAMP production after stimulation of group I mGluRs, and AC1 is specifically involved in this process.
Figure 6.
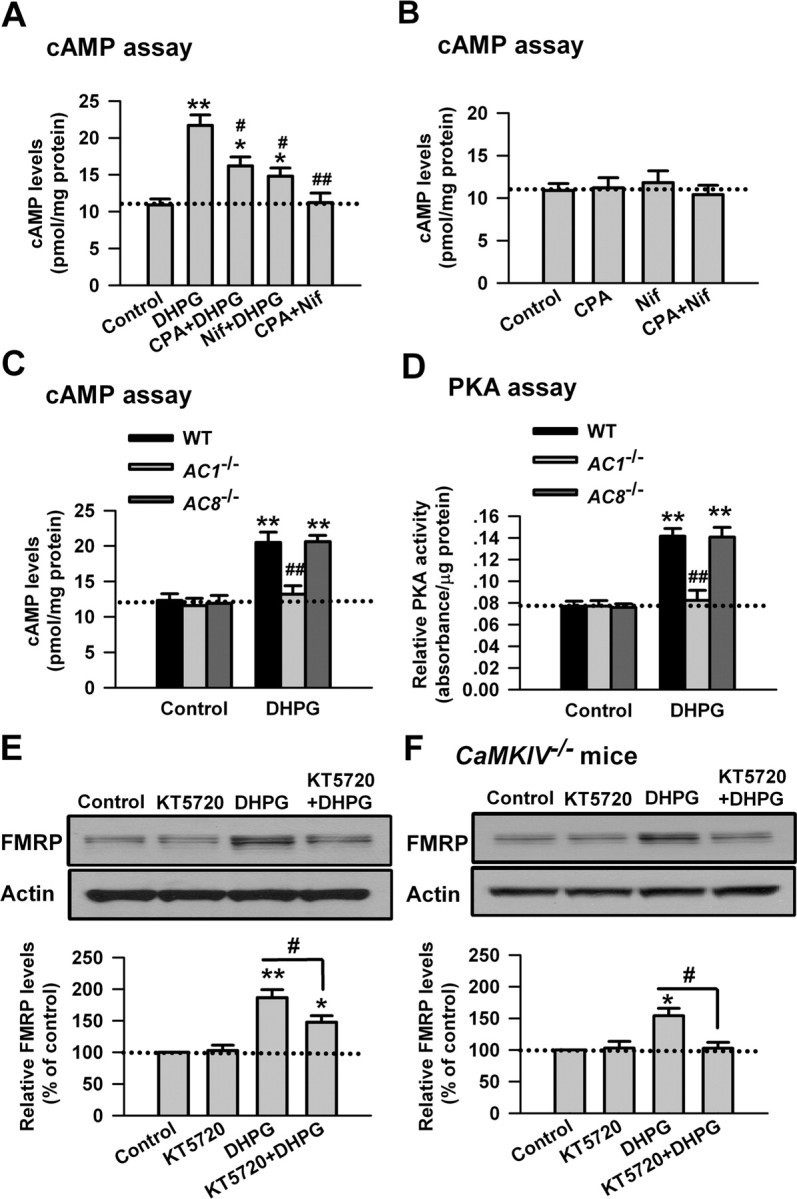
PKA is activated by stimulating group I mGluRs in ACC neurons. A, The cAMP levels was increased in ACC slices by DHPG (100 μm, 15 min) treatment as shown by cAMP assay. Both CPA (50 μm) and Nif (25 μm) partially blocked the increase of cAMP caused by DHPG. Coapplication of cyclopiazonic acid with nifedipine completely blocked the increase of cAMP caused by DHPG treatment. CPA (50 μm) or Nif (25 μm) was applied to slices 20 min before and during DHPG treatment. B, Application of cyclopiazonic acid (50 μm), nifedipine (25 μm), or coapplication of cyclopiazonic acid with nifedipine for 30 min did not affect the basal levels of cAMP in ACC slices. C, The increase of cAMP was abolished in AC1 KO but not in AC8 KO mice. D, The PKA activity was increased in ACC slices by DHPG (100 μm, 15 min) treatment, as shown by active PKA assay. The increase of the PKA activity was abolished in AC1 KO but not in AC8 KO mice. E, The PKA inhibitor KT5720 (10 μm) partially blocked the increase of FMRP caused by DHPG treatment. KT5720 was applied to slices 15 min before and during DHPG treatment. KT5720 (10 μm, 30 min) did affect the basal levels of FMRP in the ACC neurons. F, KT5720 (10 μm) abolished the increase of FMRP caused by DHPG treatment in ACC slices from CaMKIV KO mice. KT5720 was applied to slices 15 min before and during DHPG treatment. Representative Western blot (top) and quantification data (bottom) of FMRP are shown for the corresponding treatment in E and F. Data were normalized by the control values in E and F. *p < 0.05, **p < 0.01 compared with control in A–F; #p < 0.05, ##p < 0.01 compared with DHPG treatment in A, E, and F; ##p < 0.01 compared with WT in C and D; n = 4 mice for each group.
The cellular effects of cAMP are mediated by PKA, a major cAMP target, followed by the phosphorylation of relevant proteins (Sunahara and Taussig, 2002; Cooper, 2003; Wang and Storm, 2003). To investigate the role of PKA in the regulation of FMRP by group I mGluRs, we next measured the PKA activity in ACC slices. At the basal condition, there was no difference in the PKA activity in ACC slices between WT and AC1 KO or AC8 KO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 6A,B). DHPG (100 μm, 15 min) treatment could increase the PKA activity in ACC slices, as measured by the active PKA assay (p < 0.01 compared with DHPG treatment; n = 4) (Fig. 6D). However, the increase of PKA activity induced by DHPG was blocked in ACC slices from AC1 KO mice (p < 0.01 compared with WT; n = 4) (Fig. 6D) but not AC8 KO mice (p > 0.05 compared with WT mice; n = 4) (Fig. 6D). These results indicate that PKA is activated by stimulating group I mGluRs in ACC neurons, and AC1 plays a critical role in this activation.
To further confirm the role of PKA in the regulation of FMRP by group I mGluRs in ACC neurons, we then tested the effect of KT5720, a cell-permeable PKA inhibitor (Moore and Kennedy, 2006; Huang and Kandel, 2007). We found that application of KT5720 (10 μm) 15 min before and during DHPG (100 μm, 30 min) treatment could partially block the increase of pCREB caused by DHPG (187 ± 13 and 148 ± 10% of the control levels for DHPG and KT5720 treatment, respectively; p < 0.05 compared with DHPG treatment; n = 4) (Fig. 6E). Furthermore, application of KT5720 to ACC slices from CaMKIV KO mice could completely block the increase of FMRP caused by DHPG (154 ± 12 and 103 ± 9% of the control levels for DHPG and KT5720 treatment, respectively; p < 0.05 compared with DHPG treatment; n = 4) (Fig. 6F). However, application of KT5720 (10 μm, 15 min) did not affect the basal levels of FMRP in ACC slices from WT or CaMKIV KO mice (p > 0.05 compared with the control; n = 4) (Fig. 6E,F). These data further support the conclusion that the PKA signaling pathway is involved in the regulation of FMRP by group I mGluRs in ACC neurons.
CREB is activated by stimulating group I mGluRs
pCREB binds to the cAMP response element (CRE) site in the gene promoter and activates gene transcription (Bito et al., 1997; Lu et al., 1999; Shaywitz and Greenberg, 1999). Previous studies have shown that activation of group I mGluRs can induce the phosphorylation of CREB and subsequent expression of immediate early gene in cultured striatal neurons (Mao and Wang, 2003; Voulalas et al., 2005; Warwick et al., 2005). It has been reported that the FMR1 gene promoter contains the CRE site (Hwu et al., 1997; Garber et al., 2006), suggesting that CREB may be involved in the regulation of FMR1 gene transcription by group I mGluR activation.
Next, we proceeded to explore whether CREB could be activated by stimulating group I mGluRs in ACC neurons. We checked the phosphorylation of CREB at Ser133 residue, which is phosphorylated by PKA. By immunohistochemistry, we found that pCREB could be expressed at lower level in ACC at the basal condition. The immunoreactivity of pCREB was homogeneously increased by DHPG (100 μm, 15 min) treatment in the cells from layers II–VI of ACC slices (Fig. 7A). The increase of pCREB caused by DHPG in ACC slices was also measured by Western blot (221 ± 15% of the control levels; p < 0.01 compared with control; n = 6) (Fig. 7B). The data suggest that the regulation of FMRP by group I mGluRs in ACC neurons likely occurs through CREB activation.
Figure 7.
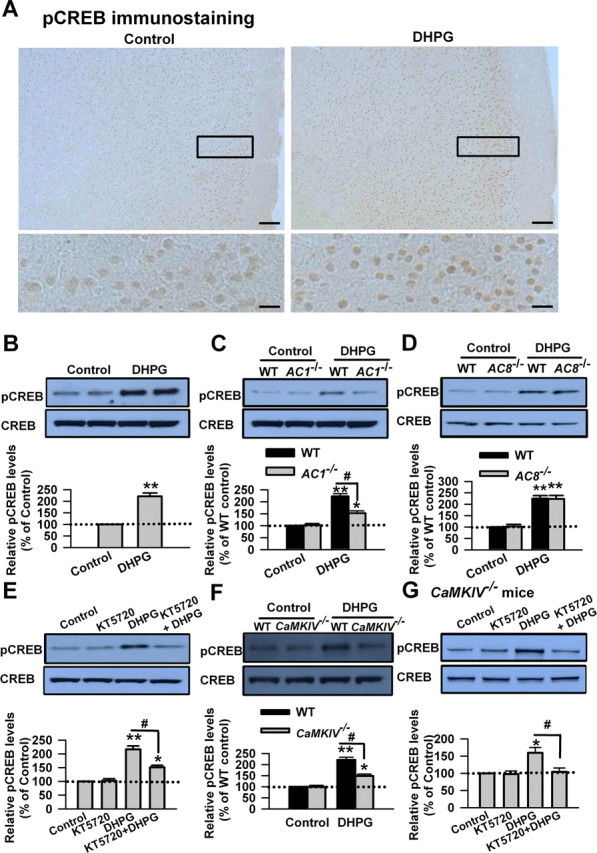
AC1 and CaMKIV contribute to the phosphorylation of CREB by group I mGluR activation. A, Immunohistochemistry of pCREB in ACC. Compared with the control (left), the increase of the immunoreactivity of pCREB caused by DHPG (100 μm, 15 min; right) could be found in neurons from layers II–VI in ACC. A high-magnification image of the selected region showing the staining in layers II–III is provided at the bottom. Scale bars: top, 100 μm; bottom, 20 μm. B, Activation of group I mGluRs by DHPG induced the phosphorylation of CREB in ACC slices. ACC slices were treated by DHPG (100 μm) for 15 min, and the phosphorylation of CREB at Ser133 residue was tested by Western blot. C, The phosphorylation of CREB induced by DHPG (100 μm, 15 min) treatment was significantly attenuated in ACC slices from AC1 KO mice compared with WT mice. The basal phosphorylation levels of CREB were not changed in ACC slices from AC1 KO mice. D, There is no difference in the phosphorylation levels of CREB after DHPG (100 μm, 15 min) treatment in ACC slices between AC8 KO and WT mice. The basal phosphorylation levels of CREB were not changed in ACC slices from AC8 KO mice. E, The PKA inhibitor KT5720 (10 μm) partially blocked the increase of pCREB caused by DHPG treatment. KT5720 was applied to slices 15 min before and during DHPG treatment. KT5720 (10 μm, 15 min) did affect the basal levels of pCREB in ACC neurons. F, The phosphorylation of CREB induced by DHPG (100 μm, 15 min) treatment was significantly attenuated in ACC slices from CaMKIV KO mice compared with WT mice. The basal phosphorylation levels of CREB were not changed in ACC slices from CaMKIV KO mice. G, KT5720 (10 μm) abolished the increase of pCREB caused by DHPG treatment in ACC slices from CaMKIV KO mice. KT5720 was applied to slices 15 min before and during DHPG treatment. Representative Western blot (top) and quantification data (bottom) of pCREB levels are shown for corresponding treatments. Data were normalized by the control values in B, E, and G and by the WT control values in C, D, and F; *p < 0.05, **p < 0.01 compared with control from B–G; #p < 0.05 compared with WT in C and F; #p < 0.05 compared with DHPG treatment in E and G; n = 3 mice for each group in experiments in A; n = 6 mice for each group in experiments in B; n = 4 mice for each group in experiments from C–G.
AC1 and CaMKIV are involved in CREB activation
Ca2+/calmodulin-dependent AC isoforms have been shown to be involved in the activation of PKA and the transcription factor CREB (Shaywitz and Greenberg, 1999; Kornhauser et al., 2002; Wei et al., 2002b; Wang et al., 2007). It is possible that AC1 or AC8 may contribute to the activation of CREB caused by stimulating group I mGluRs. We then tested the phosphorylation of CREB induced by DHPG (100 μm, 15 min) in ACC slices from AC1 KO or AC8 KO mice. We found that the phosphorylation of CREB induced by DHPG treatment in ACC slices from AC1 KO mice was significantly reduced compared with WT mice (223 ± 11 and 152 ± 10% of the WT control levels for WT and AC1 KO mice, respectively; p < 0.05 compared with WT mice; n = 6) (Fig. 7C). However, there is no difference in the phosphorylation of CREB after DHPG treatment in ACC slices between AC8 KO and WT mice (225 ± 13 and 227 ± 15% of the WT control levels for WT and AC8 KO mice, respectively; p > 0.05 compared with WT mice; n = 6) (Fig. 7D). These findings suggest that AC1 plays an isoform-specific role in the phosphorylation of CREB induced by stimulating group I mGluRs in ACC neurons.
To further confirm the role of PKA in the phosphorylation of CREB induced by stimulating group I mGluRs in the ACC neurons, we tested the effect of the PKA inhibitor KT5720. We found that KT5720 (10 μm, 15 min) did affect the basal levels of pCREB in ACC neurons (p > 0.05 compared with control; n = 4) (Fig. 7E). However, application of KT5720 (10 μm) 15 min before and during DHPG (100 μm, 15 min) treatment could partially block the increase of pCREB caused by DHPG (216 ± 12 and 151 ± 8% of the control levels for DHPG and KT5720 treatment, respectively; p < 0.05 compared with DHPG treatment; n = 4) (Fig. 7E). The data indicate that PKA is required for the activation of CREB by stimulating group I mGluRs in ACC neurons.
CaMKIV activates the transcription factor CREB by phosphorylating CREB on the regulatory Ser133 residue (Bito et al., 1997; Hook and Means, 2001). To investigate whether CaMKIV is involved in the phosphorylation of CREB caused by stimulating group I mGluRs, we then tested the phosphorylation of CREB induced by DHPG (100 μm, 15 min) in ACC slices from CaMKIV KO mice. We found that the basal levels of pCREB were not changed in ACC slices from CaMKIV KO mice (p > 0.05 compared with control; n = 4) (Fig. 7F). However, the phosphorylation of CREB induced by DHPG treatment was partially blocked in ACC slices from CaMKIV KO mice compared with WT mice (221 ± 12 and 149 ± 9% of the WT control levels for WT and CaMKIV KO mice, respectively; p < 0.05 compared with WT mice; n = 4) (Fig. 7F). These results indicate that CaMKIV contributes to the phosphorylation of CREB induced by stimulating group I mGluRs in ACC neurons. The application of KT5720 (10 μm) 15 min before and during DHPG (100 μm, 15 min) treatment completely blocked the increase of pCREB caused by DHPG in ACC slices from CaMKIV KO mice (159 ± 15 and 104 ± 11% from CaMKIV KO mice (159 ± 15 and 104 ± 11% of the control levels for DHPG and KT5720 treatment, respectively; p < 0.05 compared with DHPG treatment; n = 4) (Fig. 7G). The results further indicate that PKA and CaMKIV are the key molecules that phosphorylate CREB during group I mGluR activation in ACC neurons.
FMRP is downstream of group I mGluR–CREB signaling pathway
To further confirm that FMRP is downstream of group I mGluRs, we next characterized the function of group I mGluRs in ACC neurons of Fmr1 KO mice. By calcium imaging, we found that increase of Ca2+ signals caused by DHPG (100 μm) perfusion in ACC neurons of Fmr1 KO mice was similar to that of WT mice (p > 0.05 compared with WT; n = 6 and n = 8 for WT and Fmr1 KO mice, respectively) (Fig. 8A,B). By electrophysiological re-cording, we found that there was no difference in DHPG-induced currents in ACC neurons between WT and Fmr1 KO mice (p > 0.05 compared with WT; n = 11 and n = 8 for WT and Fmr1 KO mice, respectively) (Fig. 8C,D). These findings provide the direct evidence that genetic deletion of FMRP does not affect the function of group 1 mGluRs in ACC neurons.
Figure 8.
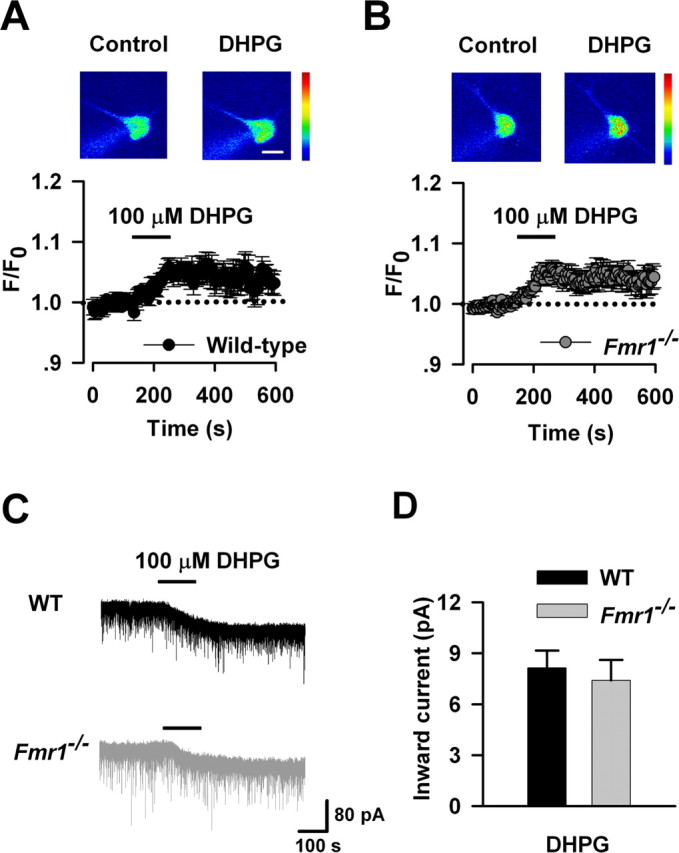
The function of group I mGluRs in ACC neurons is not affected in Fmr1 KO mice. A, Represent images (top) and time plot (bottom) showing that DHPG (100 μm) perfusion for 2 min increased the Ca2+ signals in ACC neurons from WT mice (n = 6 cells). Scale bar, 20 μm. B, Similar increase of Ca2+ signals were also found in ACC neurons from Fmr1 KO mice (n = 8 cells); p > 0.05 compared with that in A. C, Typical traces showing the inward current induced by perfusion of DHPG (100 μm) for 2 min. D, Pooled data showing that there is no difference in DHPG-induced currents between WT (n = 11 cells) and Fmr1−/− (n = 8 cells) mice; p > 0.05 compared with WT.
FMRP regulates the translation of specific mRNAs and functions as a dynamic regulator of synaptic plasticity (Bear et al., 2004; Garber et al., 2006; Nosyreva and Huber, 2006). FMRP is likely to be critically involved in the biochemical regulation of translation during mGluR activation (Weiler et al., 1997; Antar et al., 2004; Hou et al., 2006). To rule out the possibility that the changes in CREB might be caused by the upregulated FMRP attributable to group I mGluR activation, we then checked the levels of CREB in ACC slices from Fmr1 KO mice. By Western blot, we found that the basal levels of pCREB and CREB were not changed in ACC slices from Fmr1 KO mice compared with WT mice (p > 0.05; n = 6) (Fig. 9A). Similarly, the basal level of CaMKIV was not altered in ACC slices of Fmr1 KO mice (p > 0.05 compared with WT; n = 4) (Fig. 9A) In addition, the phosphorylation of CREB induced by DHPG (100 μm) treatment in ACC slices from Fmr1 KO mice was not affected compared with WT mice (223 ± 16 and 226 ± 19% of the WT control levels for WT and Fmr1 KO mice, respectively; p > 0.05 compared with WT mice; n = 6) (Fig. 9B). These data indicate that the expression and phosphorylation of CREB is not affected by the lack of FMRP and suggest that FMRP is downstream of the group I mGluR–CREB signaling pathway in ACC neurons.
Figure 9.
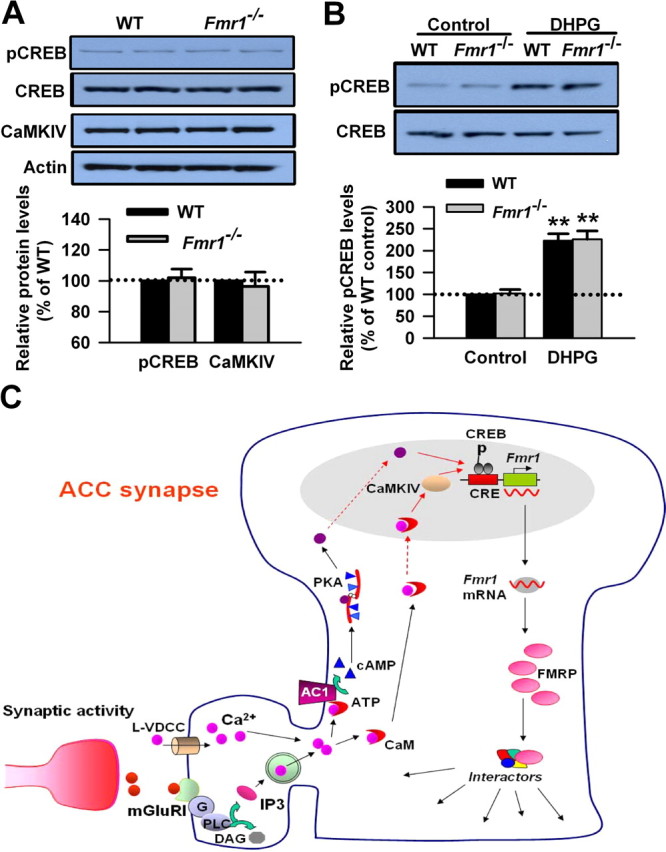
FMRP is downstream of the group I mGluR–CREB pathway in ACC neurons. A, There is no difference in basal levels of CREB in ACC between WT and Fmr1 KO mice. The basal levels of CaMKIV were not affected in ACC of Fmr1 KO mice compared with that of WT mice. B, The phosphorylation of CREB induced by DHPG treatment was not affected in Fmr1 KO mice compared with WT mice. The slices were treated with DHPG (100 μm) for 15 min. Representative Western blot (top) and quantification data (bottom) of pCREB levels are shown for corresponding treatments. Data were normalized by WT control values. **p < 0.01 compared with control; n = 4 mice for each group in A; n = 6 mice in B. C, The proposed signaling pathway for the regulation of FMRP by group I mGluRs in ACC neurons. Activation of group I mGluR triggers the Ca2+ release from intracellular calcium stores by IP3 and Ca2+ influx from L-VDCCs through membrane depolarization. Postsynaptic increases in Ca2+ leads to activation of Ca2+–calmodulin (CaM)-dependent pathways. Among them, Ca2+ and CaM-stimulated AC1 is activated, and this activation leads to the generation of the key second-messenger cAMP. Subsequently, cAMP activates the PKA. PKA then translocates to the nucleus and phosphorylates CREB. In addition to the cAMP–PKA pathway, rapid CaM translocation into the nucleus activates CaMKIV. CaMKIV then phosphorylates CREB. pCREB initiates the CREB-dependent transcription of Fmr1 gene and upregulates FMRP in the cytoplasm. FMRP may interact with several FRMP interactors and causes changes in neuronal functions in ACC.
Discussion
Our previous studies suggest that FMRP is required for the physiological function of ACC (Zhao et al., 2005a). Previous electrophysiological and behavioral studies suggest that the mGluRs in ACC may contribute to the activity-dependent synaptic plasticity and fear memory (Wei et al., 1999; Tang et al., 2005). Here we provide the direct biochemical evidence that activation of group I mGluRs upregulates FMRP in ACC neurons of adult mice. The upregulation of FMRP by group I mGluRs occurs at the transcriptional level. The activation of group I mGluRs induced the phosphorylation of CREB in ACC neurons. AC1 and CaMKIV, but not AC8, contribute to the upregulation of FMRP and the phosphorylation of CREB induced by stimulating group I mGluRs. The activation of group I mGluRs is linked to PKA through AC1. To our knowledge, this is the first time to show how the expression of FMRP is regulated in ACC neurons. Our results have provided novel evidence that FMRP acts as a key intracellular messenger downstream of Ca2+/calmodulin-dependent signaling pathways.
Ca2+ and the expression of FMRP during group I mGluR activation
Ca2+ signaling pathway plays a pivotal role in synaptic plasticity (Bito et al., 1997; Shaywitz and Greenberg, 1999; West et al., 2001). Ca2+ is released from IP3-sensitive intracellular stores during group I mGluR activation (Rae et al., 2000; Rae and Irving, 2004; Heinke and Sandkuhler, 2007). In this study, we found that application of sarco/endoplasmic reticulum Ca2+/ATPase pump inhibitor cyclopiazonic acid before and during group I mGluR activation partially blocked the upregulation of FMRP attributable to group I mGluR activation. Another possible source for intracellular Ca2+ is Ca2+ influx through L-VDCCs. Previous studies have shown that membrane depolarization by DHPG treatment can trigger the opening of L-VDCCs (Bianchi et al., 1999; Mao and Wang, 2003). We found that application of the L-VDCC blocker nifedipine also partially blocked the upregulation of FMRP attributable to group I mGluR activation. When the sarco/endoplasmic reticulum Ca2+/ATPase pump inhibitor cyclopiazonic acid was coapplied with nifedipine to ACC slices, the upregulation of FMRP was almost completely blocked. It indicates that both Ca2+ release from intracellular stores and Ca2+ influx from L-VDCCs are necessary for the upregulation of FMRP attributable to group I mGluR activation. The role of calcium in the regulation of FMRP by group I mGluRs suggests that the downstream Ca2+-dependent signaling cascades might be involved in this process.
AC1, CaMKIV, and the regulation of FMRP by group I mGluRs
Ca2+/calmodulin-stimulated ACs serve as the transducer of Ca2+ signaling in synaptic plasticity and neuronal survival (Wei et al., 2002b; Liauw et al., 2005; Wang et al., 2007; Zhuo, 2008). AC1 and AC8 are the two major AC isoforms stimulated by Ca2+/calmodulin in neurons (Cooper et al., 1998; Sunahara and Taussig, 2002; Watts, 2007). In this study, we found that genetic deletion of AC1, but not AC8, could partially block the increase of FMRP induced by group I mGluR activation. In addition, we showed that PKA is required for the upregulation of FMRP by group I mGluRs; AC1, rather than AC8, is involved in the activation of PKA by group I mGluRs. These results indicate that AC1, but not AC8, is critical for the upregulation of FMRP by stimulating group I mGluRs. The isoform-specific role of AC1 might be explained by the fact that AC1 is more sensitive to Ca2+ than AC8, and its calcium sensitivity is approximately fivefold lower than AC1 (Wang and Storm, 2003; Watts, 2007). Thus, in contrast with AC8, AC1 is the major downstream effector for group I mGluRs in ACC neurons. Genetic deletion of AC1 or inhibition of PKA cannot fully block the increase of FMRP induced by group I mGluR activation, suggesting that AC1–PKA is not the only signaling pathway involved in this process.
It is well known that CaMKIV transduces Ca2+ signaling and functions as a transcriptional activator in synaptic plasticity (Bito et al., 1997; Hook and Means, 2001; Wei et al., 2002a). It is likely that CaMKIV might be involved in the signaling pathway downstream of group I mGluRs. By using the CaMKIV KO mice, we confirmed that CaMKIV acts downstream of group I mGluRs and contributes to the regulation of FMRP by group I mGluRs in ACC neurons. We also found that inhibiting PKA could completely block the increase of FMRP induced by stimulating group I mGluRs in ACC slice from CaMKIV KO mice. This result further supports the conclusion that both AC1–PKA and CaMKIV are required for the upregulation of FMRP by the activation of group I mGluR in ACC neurons.
CREB and the regulation of FMRP by group I mGluRs
The CREB is a transcriptional factor that plays an important role in synaptic plasticity (Bito et al., 1997; Kornhauser et al., 2002; Gong et al., 2007). The activity of CREB is regulated by its phosphorylation; pCREB binds to the CRE site within the gene and activates the gene transcription (Bito et al., 1997; Shaywitz and Greenberg, 1999; Kornhauser et al., 2002). Previous studies have shown that there is the CRE site in FMR1 promoter and implicated CREB in the regulation of the FMR1 gene transcription in neural cells (Hwu et al., 1997; Smith et al., 2006). In this study, we found that the function of group I mGluRs and the phosphorylation of CREB attributable to group I mGluR activation in ACC neurons are not affected in Fmr1 KO mice, indicating that FMRP is downstream of the group I mGluR–CREB signaling pathway. This is the first time to provide direct evidence for the functions of mGluRs in ACC neurons of Fmr1 KO mice. Moreover, the group I mGluR-dependent regulation of FMRP in ACC neurons occurs at the transcriptional level, because new Fmr1 mRNA is transcribed, as shown by RT-PCR; the upregulation of FMRP is accompanied by the phosphorylation of CREB (Ser133). These results further support that CREB acts as a transcriptional factor for group I mGluR-dependent regulation of FMRP in the neurons.
There are several signaling pathways that lead to the activation of CREB (Fig. 9C). Ca2+ and cAMP are the principal second messengers that control the phosphorylation of CREB at its regulatory site (Ser133) (Bito et al., 1997; Shaywitz and Greenberg, 1999; Kornhauser et al., 2002). pCREB can be used as a marker for the activation of ACs in the CNS (Wei et al., 2002b; Wang et al., 2007). In this study, we found that pharmacological inhibition of PKA partially blocked the phosphorylation of CREB induced by DHPG in ACC slices, indicating that the cAMP–PKA signaling pathway in involved in this process. We then characterized the role of Ca2+/calmodulin-stimulated ACs in the phosphorylation of CREB induced by group I mGluR activation in ACC slices. We found that the phosphorylation of CREB induced by DHPG was attenuated in ACC slices from AC1 KO, but not AC8 KO mice, compared with WT mice. CaMKIV is implicated in various aspects of the neuronal Ca2+ signaling pathways, inducing the phosphorylation of CREB and the gene expression in response to excitatory neurotransmission (Bito et al., 1997; Ho et al., 2000). We found that the phosphorylation of CREB induced by DHPG was partially blocked in ACC slices from CaMKIV KO mice. Moreover, pharmacological inhibition of PKA could completely block the increase of pCREB induced by DHPG in ACC of CaMKIV KO mice. Together, these data indicate that AC1 and CaMKIV are critically involved in the activation of CREB induced by stimulating group I mGluRs, AC1 and CaMKIV contribute to the regulation of FMRP through CREB activation in ACC neurons.
A previous study showed that the DHPG-induced increase of FMRP is protein synthesis dependent and depends on the activation of mGluR5 rather than mGluR1 in hippocampus (Hou et al., 2006). In contrast, we found that the upregulation of FMRP by group I mGluRs occurs at the transcriptional level in cingulate cortex. In addition, the upregulation of FMRP depends on the activation of both mGluR1 and mGluR5 in ACC neurons. The differences between the findings from these two studies may be caused by the differences in the composition of neuronal types between hippocampus and cingulate cortex. Alternatively, the differences might reflect the possible functional diversity of group I mGluRs or FMRP in different brain areas. Future studies are definitely needed to study the mechanisms underlying the differences in the regulation of FMRP by group I mGluRs between hippocampus and cingulate cortex.
In summary, we have demonstrated that the activation of group I mGluRs upregulates FMRP in ACC neurons. Ca2+ is the key messenger for the regulation of FMRP by group I mGluRs; the regulation occurs at the transcriptional level. The activation of CREB is involved in this process. AC1 and CaMKIV, the major Ca2+ sensors, contribute to the upregulation of FMRP through the activation of CREB (for model, see Fig. 9C). Our study has provided strong evidence for the regulation of FMRP by group I mGluRs in ACC neurons and may help to further elucidate the cellular mechanisms underlying fragile X syndrome.
Footnotes
This work was supported by grants from the EJLB–Canadian Institutes of Health Research (CIHR) Michael Smith Chair in Neurosciences and Mental Health, Canada Research Chair, and NeuroCanada and CIHR Operating Grants CIHR66975 and CIHR84256. H.W. and L.-J.W. were supported by postdoctoral fellowships from The Fragile X Research Foundation of Canada. We thank Dr. Louis J. Muglia for providing the AC1 and AC8 KO mice and Dr. W. T. Greenough for the generous gift of Fmr1 WT and KO breeding pairs.
References
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Young SR, Wong RK. Group I mGluR activation causes voltage-dependent and -independent Ca2+ rises in hippocampal pyramidal cells. J Neurophysiol. 1999;81:2903–2913. doi: 10.1152/jn.1999.81.6.2903. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schaffhauser H, Wichmann J, Mutel V. mGluR-evoked augmentation of receptor-mediated cyclic AMP formation in neonatal and adult rat striatum. Br J Pharmacol. 1997;121:1263–1268. doi: 10.1038/sj.bjp.0701266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Goepfert F, Knoflach F, Pink JR, Bleuel Z, Richards JG, Schaffhauser H, Kemp JA, Wichmann J, Mutel V. Effect of metabotropic glutamate receptor activation on receptor-mediated cyclic AMP responses in primary cultures of rat striatal neurones. Brain Res. 1998;791:191–199. doi: 10.1016/s0006-8993(98)00094-8. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Bockaert J, Lansman JB. Modulation of calcium channels by metabotropic glutamate receptors in cerebellar granule cells. Neuropharmacology. 1995;34:929–937. doi: 10.1016/0028-3908(95)00082-h. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Cooper DM. Regulation and organization of adenylyl cyclases and cAMP. Biochem J. 2003;375:517–529. doi: 10.1042/BJ20031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Crossthwaite AJ. Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci. 2006;27:426–431. doi: 10.1016/j.tips.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Cooper DM, Karpen JW, Fagan KA, Mons NE. Ca2+-sensitive adenylyl cyclases. Adv Second Messenger Phosphoprotein Res. 1998;32:23–51. [PubMed] [Google Scholar]
- Coutinho V, Knopfel T. Metabotropic glutamate receptors: electrical and chemical signaling properties. The Neuroscientist. 2002;8:551–561. doi: 10.1177/1073858402238514. [DOI] [PubMed] [Google Scholar]
- Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol (Lond) 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, O'Brien C, Ohno M, Kirkwood A, Silva AJ. Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature. 2001;411:309–313. doi: 10.1038/35077089. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- Garber K, Smith KT, Reines D, Warren ST. Transcription, translation and fragile X syndrome. Curr Opin Genet Dev. 2006;16:270–275. doi: 10.1016/j.gde.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Gong B, Wang H, Gu S, Heximer SP, Zhuo M. Genetic evidence for the requirement of adenylyl cyclase 1 in synaptic scaling of forebrain cortical neurons. Eur J Neurosci. 2007;26:275–288. doi: 10.1111/j.1460-9568.2007.05669.x. [DOI] [PubMed] [Google Scholar]
- Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci USA. 2001;98:7101–7106. doi: 10.1073/pnas.141145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: fragile X syndrome and beyond. J Neurosci. 2006;26:7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CJ, O'Tuathaigh CM, van Trigt L, Quinn JJ, Fanselow MS, Mongeau R, Koch C, Anderson DJ. Trace but not delay fear conditioning requires attention and the anterior cingulate cortex. Proc Natl Acad Sci USA. 2003;100:13087–13092. doi: 10.1073/pnas.2132313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinke B, Sandkuhler J. Signal transduction pathways of Group I metabotropic glutamate receptor-induced long-term depression at sensory spinal synapses. Pain. 2005;118:145–154. doi: 10.1016/j.pain.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Heinke B, Sandkuhler J. Group I metabotropic glutamate receptor-induced Ca2+-gradients in rat superficial spinal dorsal horn neurons. Neuropharmacology. 2007;52:1015–1023. doi: 10.1016/j.neuropharm.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Means AR. Ca2+/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. 5-Hydroxytryptamine induces a protein kinase A/mitogen-activated protein kinase-mediated and macromolecular synthesis-dependent late phase of long-term potentiation in the amygdala. J Neurosci. 2007;27:3111–3119. doi: 10.1523/JNEUROSCI.3908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K. Fragile X syndrome: molecular mechanisms of cognitive dysfunction. Am J Psychiatry. 2007;164:556. doi: 10.1176/ajp.2007.164.4.556. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwu WL, Wang TR, Lee YM. FMR1 enhancer is regulated by cAMP through a cAMP-responsive element. DNA Cell Biol. 1997;16:449–453. doi: 10.1089/dna.1997.16.449. [DOI] [PubMed] [Google Scholar]
- Jin P, Warren ST. New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem Sci. 2003;28:152–158. doi: 10.1016/S0968-0004(03)00033-1. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221–233. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Liauw J, Wu LJ, Zhuo M. Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol. 2005;94:878–882. doi: 10.1152/jn.01205.2004. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Group I metabotropic glutamate receptor-mediated calcium signalling and immediate early gene expression in cultured rat striatal neurons. Eur J Neurosci. 2003;17:741–750. doi: 10.1046/j.1460-9568.2003.02495.x. [DOI] [PubMed] [Google Scholar]
- Means AR, Cruzalegui F, LeMagueresse B, Needleman DS, Slaughter GR, Ono T. A novel Ca2+/calmodulin-dependent protein kinase and a male germ cell-specific calmodulin-binding protein are derived from the same gene. Mol Cell Biol. 1991;11:3960–3971. doi: 10.1128/mcb.11.8.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Kennedy TE. Protein kinase A regulates the sensitivity of spinal commissural axon turning to netrin-1 but does not switch between chemoattraction and chemorepulsion. J Neurosci. 2006;26:2419–2423. doi: 10.1523/JNEUROSCI.5419-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci USA. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Rae MG, Irving AJ. Both mGluR1 and mGluR5 mediate Ca2+ release and inward currents in hippocampal CA1 pyramidal neurons. Neuropharmacology. 2004;46:1057–1069. doi: 10.1016/j.neuropharm.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Rae MG, Martin DJ, Collingridge GL, Irving AJ. Role of Ca2+ stores in metabotropic l-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J Neurosci. 2000;20:8628–8636. doi: 10.1523/JNEUROSCI.20-23-08628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scerif G, Cornish K, Wilding J, Driver J, Karmiloff-Smith A. Delineation of early attentional control difficulties in fragile X syndrome: focus on neurocomputational changes. Neuropsychologia. 2007;45:1889–1898. doi: 10.1016/j.neuropsychologia.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffhauser H, de Barry J, Muller H, Heitz MP, Gombos G, Mutel V. Involvement of a cyclic-AMP pathway in Group I metabotropic glutamate receptor responses in neonatal rat cortex. Eur J Pharmacol. 1997;334:289–297. doi: 10.1016/s0014-2999(97)01192-8. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Smith KT, Nicholls RD, Reines D. The gene encoding the fragile X RNA-binding protein is controlled by nuclear respiratory factor 2 and the CREB family of transcription factors. Nucleic Acids Res. 2006;34:1205–1215. doi: 10.1093/nar/gkj521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Means RL, LeMagueresse B, Means AR. Organization and analysis of the complete rat calmodulin-dependent protein kinase IV gene. J Biol Chem. 1995;270:29507–29514. doi: 10.1074/jbc.270.49.29507. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- Tang J, Ko S, Ding HK, Qiu CS, Calejesan AA, Zhuo M. Pavlovian fear memory induced by activation in the anterior cingulate cortex. Mol Pain. 2005;1:6. doi: 10.1186/1744-8069-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault SJ, Davies CH, Randall AD, Collingridge GL. Group I mGluRs modulate the pattern of non-synaptic epileptiform activity in the hippocampus. Neuropharmacology. 2002;43:141–146. doi: 10.1016/s0028-3908(02)00095-3. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci USA. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulalas PJ, Holtzclaw L, Wolstenholme J, Russell JT, Hyman SE. Metabotropic glutamate receptors and dopamine receptors cooperate to enhance extracellular signal-regulated kinase phosphorylation in striatal neurons. J Neurosci. 2005;25:3763–3773. doi: 10.1523/JNEUROSCI.4574-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Wang H, Gong B, Vadakkan KI, Toyoda H, Kaang BK, Zhuo M. Genetic evidence for adenylyl cyclase 1 as a target for preventing neuronal excitotoxicity mediated by N-methyl-d-aspartate receptors. J Biol Chem. 2007;282:1507–1517. doi: 10.1074/jbc.M607291200. [DOI] [PubMed] [Google Scholar]
- Warwick HK, Nahorski SR, Challiss RA. Group I metabotropic glutamate receptors, mGlu1a and mGlu5a, couple to cyclic AMP response element binding protein (CREB) through a common Ca2+- and protein kinase C-dependent pathway. J Neurochem. 2005;93:232–245. doi: 10.1111/j.1471-4159.2005.03012.x. [DOI] [PubMed] [Google Scholar]
- Watts VJ. Adenylyl cyclase isoforms as novel therapeutic targets: an exciting example of excitotoxicity neuroprotection. Mol Interv. 2007;7:70–73. doi: 10.1124/mi.7.2.6. [DOI] [PubMed] [Google Scholar]
- Wei F, Li P, Zhuo M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J Neurosci. 1999;19:9346–9354. doi: 10.1523/JNEUROSCI.19-21-09346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002a;5:573–579. doi: 10.1038/nn0602-855. [DOI] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron. 2002b;36:713–726. doi: 10.1016/s0896-6273(02)01019-x. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005a;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005b;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhuo M. Molecular mechanisms of pain in the anterior cingulate cortex. J Neurosci Res. 2006;84:927–933. doi: 10.1002/jnr.21003. [DOI] [PubMed] [Google Scholar]
- Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31:199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]