

Abstract
In addition to parenchymal amyloid-β (Aβ) plaques, Alzheimer's disease (AD) is characterized by Aβ in the cerebral vasculature [cerebral amyloid angiopathy (CAA)] in the majority of patients. Recent studies investigating vascular Aβ (VAβ) in amyloid precursor protein transgenic mice have suggested that passive immunization with anti-Aβ antibodies may clear parenchymal amyloid but increase VAβ and the incidence of microhemorrhage. However, the influences of antibody specificity and exposure levels on VAβ and microhemorrhage rates have not been well established, nor has any clear causal relationship been identified. This report examines the effects of chronic, passive immunization on VAβ and microhemorrhage in PDAPP mice by comparing antibodies with different Aβ epitopes (3D6, Aβ1–5; 266, Aβ16–23) and performing a 3D6 dose–response study. VAβ and microhemorrhage were assessed using concomitant Aβ immunohistochemistry and hemosiderin detection. 3D6 prevented or cleared VAβ in a dose-dependent manner, whereas 266 was without effect. Essentially complete absence of VAβ was observed at the highest 3D6 dose, whereas altered morphology suggestive of ongoing clearance was seen at lower doses. The incidence of microhemorrhage was increased in the high-dose 3D6 group and limited to focal, perivascular sites. These colocalized with Aβ deposits having altered morphology and apparent clearance in the lower-dose 3D6 group. Our results suggest that passive immunization can reduce VAβ levels, and modulating antibody dose can significantly mitigate the incidence of microhemorrhage while still preventing or reducing VAβ. These observations raise the possibility that Aβ immunotherapy can potentially slow or halt the course of CAA development in AD that is implicated in vascular dysfunction.
Keywords: Aβ-peptide, Alzheimer's disease, antibody, chronic, transgenic, vascular
Introduction
Overexpression of mutant human amyloid precursor protein (APP) in various transgenic mice leads to several Alzheimer's disease (AD)-type pathologies [for review, see Gotz et al. (2004) and Games et al. (2006)]. These include the development of parenchymal amyloid-β (Aβ) plaques, neuritic pathology, synaptic loss, and gliosis. A number of reports have shown that active (Schenk et al., 1999; Dickstein et al., 2006) and passive (Bard et al., 2000; Wilcock et al., 2004; Buttini et al., 2005) Aβ immunotherapeutic approaches are effective in reducing or eliminating these pathologies in preclinical studies (Janus et al., 2000; Morgan et al., 2000; Wilcock et al., 2004). In addition, other studies have shown improvement in various cognitive tests (Janus et al., 2000; Morgan et al., 2000; Wilcock et al., 2004). Importantly, these findings are supported by mounting correlative findings from both cognitive testing (Hock et al., 2003) and neuropathological examination of brains of patients who were enrolled in clinical trials of Aβ immunotherapy [AN1792 (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005)].
Recently, another common aspect of AD and cerebral amyloid angiopathy (CAA) pathology, vascular Aβ (VAβ), has been the subject of scrutiny in preclinical animal studies. In particular, it has been reported that passive immunization was associated with an increase in VAβ and microhemorrhage (Wilcock et al., 2004; Racke et al., 2005). However, predictive clinical implications remain unclear, especially in light of favorable behavioral outcomes in some of these same studies (Wilcock et al., 2004), the lack of ultrastructural differences in vascular morphology of hemosiderin-positive vessels in untreated and treated transgenic mice (Burbach et al., 2007), and, notably, the lack of evidence for significant hemorrhage or stroke-related consequences in ongoing clinical trials. Furthermore, little is known about the degree to which VAβ is ultimately affected by Aβ immunotherapeutic approaches. For instance, it is unknown whether reported increases in VAβ represent a transient phenomenon associated with Aβ clearance, whereas longer treatment might actually prevent or reverse vascular amyloid.
In view of these considerations, we sought to examine the effects of passive immunization in two chronic, 6 month treatment paradigms using PDAPP mice (Games et al., 1995), a model that favorably mimics the morphological and biochemical composition of amyloid deposits typical of sporadic AD (Johnson-Wood et al., 1997). A thorough characterization identified distinct meningeal localizations with compacted VAβ deposition that reproducibly occurred in these mice. We compared the effects of a plaque-clearing antibody directed against the N terminus of Aβ (3D6) and a central Aβ epitope-directed antibody with high affinity for soluble Aβ (266) for effects on VAβ levels and the occurrence of microhemorrhage. In contrast to previous findings in significantly older mice treated with an antibodies generated against Aβ28–40 and Aβ33–40 (Wilcock et al., 2004, 2006), we found that nearly complete prevention or clearance of VAβ was achieved by treatment with 3D6, whereas 266 demonstrated no effect. In addition, we found that microhemorrhage was associated with only a subset of vessels with evident amyloid clearance, and that its occurrence was mitigated by reducing antibody exposure. Collectively, our observations suggest that Aβ immunotherapeutic approaches have the potential to favorably impact vascular amyloidogenic pathology in AD at doses that minimize microhemorrhage and maintain other efficacy outcomes.
Materials and Methods
Study design.
The effects of chronic, passive immunization on established VAβ in the PDAPP mouse were examined in two 6-month-long studies. Study A was designed to compare the efficacy of an N-terminal antibody (3D6, Aβ1–5) with a midregion antibody (266, Aβ16–23) at a single dose. Study B was a 3D6 dose–response study. In both studies, 12-month-old, female, heterozygous PDAPP mice were divided into groups of 40; the groups were matched as closely as possible for age and transgenic parent. In a separate assessment, 40 animals from the same colony were examined to determine baseline vascular amyloid levels at 12 months of age. As outlined in Table 1, mice in treatment groups were injected intraperitoneally once weekly with murine monoclonal antibodies 3D6γ2a (at three dose levels), 266γ1, or TY11–15 (as a negative γ2a control). All treated animals received an initial loading dose of 2.5 times the weekly dose to achieve a stable titer at the start of the study. Doses per animal were calculated based on the historical average weight of a PDAPP mouse in this age range, 50 g. After termination of the in-life phase, VAβ and microhemorrhage presence and extent were evaluated.
Table 1.
Study design
| Study | Antibody | n | Load dose | Maintenance |
|---|---|---|---|---|
| A | TY11–15 | 40 | 7.5 mg/kg | 3 mg/kg |
| A | 3D6γ2a | 40 | 7.5 mg/kg | 3 mg/kg |
| A | 266γ1 | 40 | 7.5 mg/kg | 3 mg/kg |
| B | TY11–15 | 40 | 7.5 mg/kg | 3 mg/kg |
| B | 3D6γ2a | 40 | 7.5 mg/kg | 3 mg/kg |
| B | 3D6γ2a | 40 | 0.75 mg/kg | 0.3 mg/kg |
| B | 3D6γ2a | 40 | 0.25 mg/kg | 0.1 mg/kg |
Preparation of antibodies.
Preparation methods for antibodies 3D6 (recognizing Aβ1–5) and 266 (recognizing Aβ16–23) have been described previously (Seubert et al., 1992; Johnson-Wood et al., 1997; Bard et al., 2003). TY11–15 (IgG2a isotype) served as the irrelevant control antibody. It recognizes an unknown human lymphocyte antigen and does not recognize mouse lymphocytes. Antibody 3D6 was labeled with NHS-biotin as described previously (Seubert et al., 1992).
Brain tissue preparation and histochemistry.
Forty micrometer brain sections were prepared, selected, and colabeled for amyloid and hemosiderin as described in the supplemental material (available at www.jneurosci.org). The colabeled tissue was also used to analyze parenchymal amyloid burden as described by Bard et al. (2000).
VAβ quantification.
The meningeal location of VAβ deposits in the PDAPP mice precludes standard percentage area (burden) measurements, which are typically made in the parenchyma and normalized to brain region (e.g., cortical) area. This, coupled with the large sample sizes in this study, prompted us to devise a sensitive counting method of VAβ-containing vessels. We were able to cross-validate the resultant data in a subset of mice using a second measure of VAβ burden, which was normalized to meningeal distance (see below).
3D6-immunoreactive blood vessels were first assessed by counting the number of all amyloid-containing vessels in the sections (n = 4–6/animal) from study A. Vessels were counted if they contained any amount of amyloid, and thus would include both partially cleared and uncleared vessels. Investigators were blinded to the treatment group, and vessels were counted by three investigators in a subset of animals to validate the consistency of the counting method.
The counts were then analyzed using a second method that was statistically determined by using a receiver operating characteristic curve to identify the optimal cutoff that balanced the sensitivity and specificity of the statistical analyses. The two resulting categories reflected the amount of VAβ: “none to little VAβ” (≤3 amyloid-positive vessels per section per animal) or “moderate VAβ” (>3 amyloid-positive vessels in at least one section per animal). Pairwise comparisons using Fisher's exact test (FET) were then performed, and the Hochberg method (Hochberg, 1988) was used to adjust for multiple pairwise comparisons to maintain a statistical significance of 0.05.
Results achieved by this counting method in the dose–response study (study B) were then verified against a percentage area measurement in a subset of animals from the same study. For this, meningeal VAβ area measurements were made using MetaMorph image analysis software to measure the area of vascular Aβ deposits, which was then normalized to the distance along the cortical meninges to allow for comparisons between groups. The 3 mg/kg 3D6 group was not analyzed, because VAβ was nearly zero at this dose.
Hemosiderin quantification.
Each animal was scored on a 0–3 scale for presence, amount, location, and intensity of hemosiderin staining across the sections. A score of “0” indicated very little or no staining, a “1” indicated small punctate or weak staining in a few sections per animal, a “2” was assigned to contiguous accumulations with greater staining intensity in multiple sections, and “3” reflected the darkest observed staining in most of the sections, usually encompassing most of the affected vessel. These ratings were designed to reflect the range of hemosiderin-positive staining observed in the present preclinical animal study and therefore do not represent or translate to ratings of clinical hemorrhagic disorders. Investigators were blinded to the treatment group, and ratings were assigned by three investigators in a subset of animals to validate the rating method. Pairwise comparisons using Fisher's exact test were performed to test for differences between treatment groups. Observations were also made regarding the morphological appearance of the immunolabeled amyloid and its spatial relationship with hemosiderin. Within each study, the Hochberg method was used to adjust for multiple pairwise comparisons.
Results
Vascular Aβ quantitation
VAβ was prominent in discrete foci in the leptomeninges and superficial parenchyma of untreated PDAPP mice as revealed by thioflavin S staining for compact amyloid and by antibody 3D6, which recognizes both compact and diffuse amyloid in the PDAPP mouse and human AD (Fig. 1). Previous analysis of untreated, 12-month-old animals established that Aβ-positive vessels were present in ∼40% of mice, whereas 23% of mice had moderate VAβ deposition. VAβ was largely confined to the leptomeninges and immediately underlying superficial brain layers and was especially predominant in the cortex, where dense deposits were reliably found in midline vessels of the sagittal sinus region (Fig. 1). In the single-dose comparison study of the N-terminal and midregion antibodies (study A), 3D6 at 3 mg/kg completely prevented the formation and/or cleared existing VAβ compared with either 266 or TY11–15 (Fig. 2) (p < 0.0001 for both comparisons). A comparison of the level of moderate VAβ in 3D6-treated mice (0–3%) with the 12 month levels of moderate VAβ (23%) indicates that amyloid was likely removed by treatment. 3D6 treatment also lowered the parenchymal amyloid plaque burden by 98% (p < 0.0001), whereas 266 showed no effect. In the 3D6 dose–response study (study B) (Fig. 3), VAβ was again significantly prevented from developing and/or cleared by 3D6 treatment at the 3.0 mg/kg 3D6 dose level compared with treatment with the TY11–15 control (p < 0.0001). There was also prevention or clearance of VAβ at the intermediate dose level (0.3 mg/kg) versus control (p = 0.016). There was no difference between the 0.1 mg/kg dose group and the TY11–15 control group (p = 0.8037). Similar results were obtained with the quantitation of the percentage area of VAβ (Fig. 3 f). Although the number of vessels was not significantly different, apparent partial clearance of amyloid was observed from vessels in this group at the microscopic level (Fig. 4).
Figure 1.

Meningeal foci of VAβ are found in the PDAPP mouse model and appear similar to human VAβ deposits. a, Thioflavin S staining reveals Aβ in midline vessels of an 18-month-old PDAPP mouse. b, 3D6 antibody detection reveals Aβ in a similar pattern. c, 3D6 immunohistochemistry similarly labels VAβ in a human AD section. d, The leptomeninges and superficial parenchymal vessels are also major sites of VAβ in the PDAPP mouse. Scale bars, 100 μm.
Figure 2.
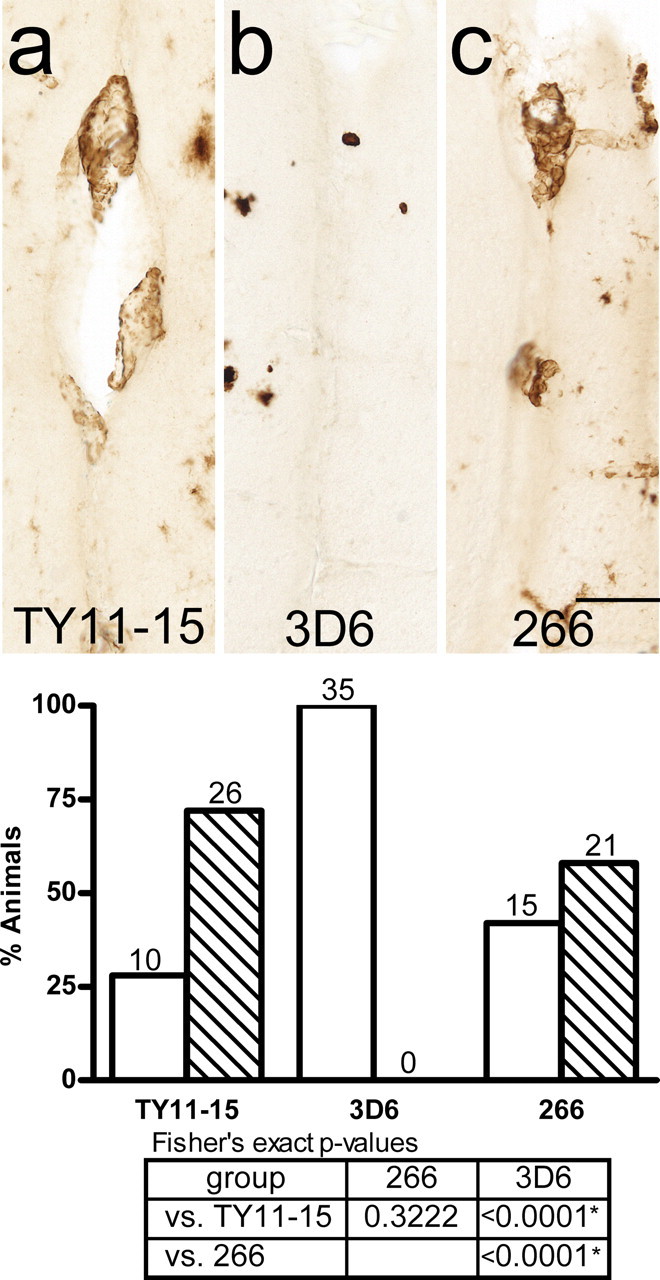
Study A: 3D6 (Aβ1–5), but not 266 (Aβ16–23), clears/prevents VAβ at a dose of 3 mg/kg. Representative observations are shown using the midline vessels as examples. a, VAβ in midline vessels in control-treated animals. b, VAβ in 3 mg/kg 3D6-treated animals is cleared/prevented. c, VAβ in 3 mg/kg 266-treated animals is similar to the control. The graph shows the percentage of animals in each group with none or little VAβ (white bars) and moderate VAβ (hatched bars). Statistical analyses show significant VAβ clearance/prevention by 3D6 compared with the control and 266 groups and no difference in clearance/prevention between the TY11–15 control and 266 groups. FET p values <0.025, indicated by asterisks in the table, are statistically significant using the Hochberg method of multiple comparisons. Scale bar, 50 μm.
Figure 3.

Study B: VAβ is cleared at 3 mg/kg 3D6 and partially cleared at lower doses of 3D6. a, VAβ is abundant in leptomeningeal vessels in control-treated animals. b, VAβ in the 0.1 mg/kg group. c, VAβ in the 0.3 mg/kg group is partially cleared. d, VAβ in the 3 mg/kg group is cleared. e, Percentage of animals in each group with none or little VAβ (white bars) and moderate VAβ (hatched bars). Statistical analyses show significant VAβ clearance/prevention in the 0.3 and 3 mg/kg 3D6-treated groups compared with the other groups. FET p values <0.025, indicated by asterisks in the table, are statistically significant using the Hochberg method of multiple comparisons. f, Quantitation of VAβ by area shows the same result. Brackets and arrows indicate VAβ. Scale bar, 100 μm.
Figure 4.
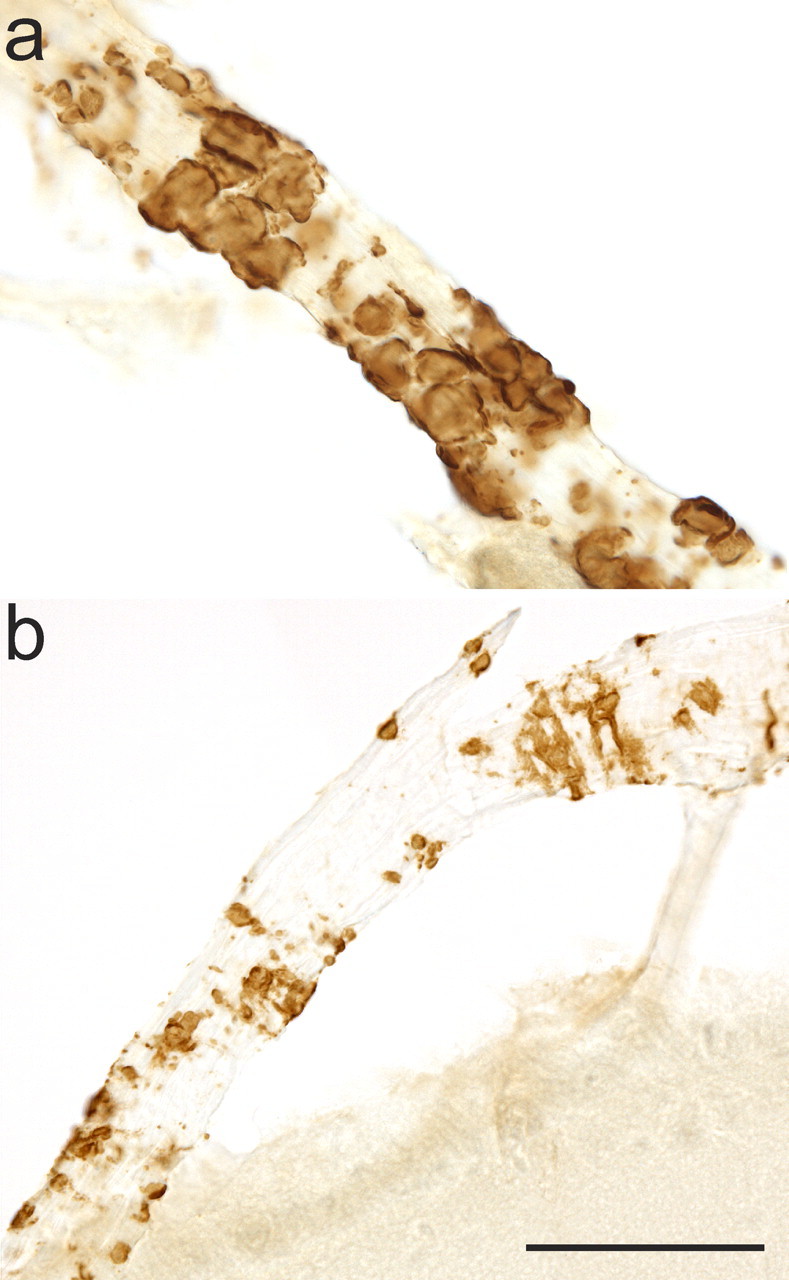
Morphology of VAβ during clearance. a, Rounded masses and bands of intact Aβ encompass an unaffected leptomeningeal vessel. b, VAβ has a patchy, eroded appearance during partial clearance. This morphology is not seen in untreated mice. Both images are from 0.1 mg/kg 3D6-treated animals. Scale bar, 50 μm.
Hemosiderin quantitation
To categorize differences among treatment groups, we developed a hemosiderin rating scale that reflected the range of staining densities found within this study. In 12-month-old mice, hemosiderin, indicative of microhemorrhage, was nearly undetectable. In immunized mice, the hemosiderin staining was limited and confined to the structural boundaries of the vasculature without spread into the surrounding parenchyma. Focal hemosiderin deposits were found in vessels of the leptomeninges of the cortex (Fig. 5) and the hippocampal thalamic interface, vessels of the sagittal sinus at the medial cortex, and a few parenchymal vessels connected to those in the leptomeninges. Hemosiderin was usually concentrated within macrophage-like cells in these areas (Fig. 5 f). The foci of hemosiderin were often associated with altered VAβ morphology. Instead of the characteristic distinct bands and scales of VAβ deposition, the amyloid had an unusual patchy, degraded appearance or was completely absent. These features were particularly notable in the leptomeningeal vessels (Fig. 5) and vessels in the sagittal sinus, which often displayed well developed VAβ morphology in the TY11–15 control groups (Figs. 2, 3).
Figure 5.

VAβ clearance results in microhemorrhage in a subset of vessels. a–c, Partial (a, b) and complete (c) clearance or prevention of VAβ is seen at all doses of 3D6 with no evidence of microhemorrhage in most animals. d–f, In a subset of mice, microhemorrhage was seen at some sites of partial clearance at lower doses (d, e) and at sites of complete clearance at 3 mg/kg 3D6 (f). Arrows indicate phagocytes containing hemosiderin but not amyloid. Scale bar, 100 μm.
Hemosiderin staining was predominantly absent or mild in all treatment groups, with the majority of animals across groups having scores of 0 or 1 (Fig. 6). Scores in the 3D6 3 mg/kg treatment groups of both studies were significantly higher, indicating that these were more likely than the control to have hemosiderin scores >0. In study A, the distributions of hemosiderin scores were similar in the TY11–15 and 266 groups, indicating that treatment with 266 antibody did not increase hemosiderin scores. In the dose–response study, differences between the TY11–15 control group and the 0.1 mg/kg 3D6 and 0.3 mg/kg 3D6 groups were either significantly reduced or substantially minimized, indicating that the low and intermediate doses were not likely to increase hemosiderin ratings beyond baseline levels. These differed from the 3.0 mg/kg group (p = 0.0003 and 0.0043, respectively), which again significantly differed from controls (p < 0.0001). Compared with study A, the higher hemosiderin scores in the 3.0 mg/kg group could be attributable to differences in antibody exposure levels over time and slightly higher baseline levels in the cohort, because scores were slightly elevated in the control group.
Figure 6.
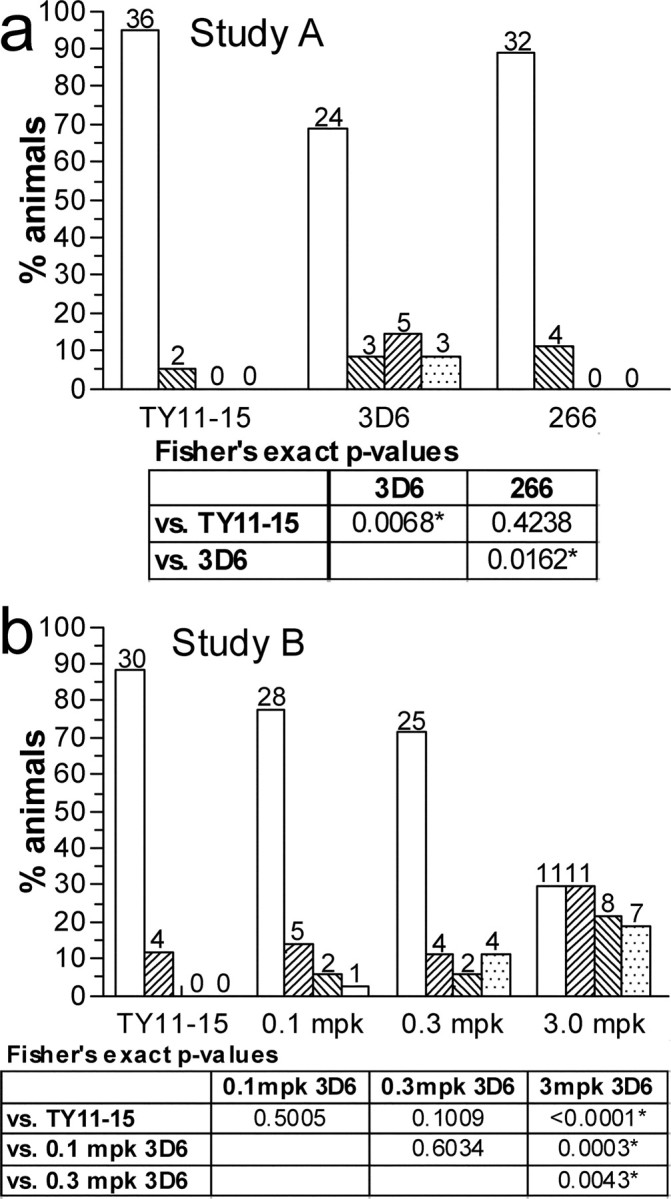
The incidence of microhemorrhage is mitigated by dose. a, Study A: the mean hemosiderin rating of the 3D6 group at 3 mg/kg was significantly higher than the TY11–15 group, and no significant difference was seen between the 266 and TY11–15 groups. b, Study B: the two lower-dose groups did not differ from the control, and the mean hemosiderin rating was significantly higher in the 3D6 3 mg/kg group compared with the TY11–15 and the 0.3 and 0.1 mg/kg 3D6 groups. Hemosiderin ratings 0–3 are represented by the white and hatched bars from left to right in each group. For studies A and B, all FET p values <0.025 and <0.0125, respectively, are statistically significant using the Hochberg method of multiple comparisons.
Association of VAβ clearance and microhemorrhage
The relationship between VAβ clearance and microhemorrhage was established by examining the colocalization of VAβ morphology and hemosiderin staining. Significant reduction of vascular amyloid was observed in the 0.3 and 3.0 mg/kg groups in both studies. The majority of animals in these groups had hemosiderin scores of 0 or 1, indicating that most brains with reduced VAβ had little or no evidence of microhemorrhage. Several examples of hemosiderin-negative vessels with unusually sparse VAβ and an eroded appearance were found in all of the treatment groups (Fig. 4 b). These may be vessels in which amyloid was being cleared in the absence of microhemorrhage. When hemosiderin staining was seen in 0.1 mg/kg 3D6-treated animals (Fig. 5 d), it was typically accompanied by an unusually sparse and uneven perivascular distribution of amyloid, possibly indicative of clearance in progress. These perivascular patches of amyloid occurred at sites of vessel-associated hemosiderin labeling within the cortical meninges, parenchyma, and the sagittal sinus vessels. Hemosiderin staining in the 0.3 mg/kg 3D6-treated animals was also associated with a very scant, perivascular distribution of amyloid (Fig. 5 e). The amyloid morphology was similar to that in the 0.1 mg/kg 3D6-treated animals, but amyloid was less abundant, and some hemosiderin-positive vessels were cleared of amyloid. Both complete and partial amyloid removal were observed at sites of vessel-associated hemosiderin staining, including the cortical meninges and parenchyma. In contrast, hemosiderin-positive vessels in the 3 mg/kg 3D6-treated animals were often completely devoid of amyloid (Fig. 5 f). This hemosiderin-positive, VAβ-negative feature was never observed in untreated mice and likely illustrates a residual hemosiderin “footprint” that occurred in a subset of vessels with complete VAβ removal. Another feature in these areas was the presence of cells that contained hemosiderin inclusions (Fig. 5 f). These macrophage-like cells were not immunoreactive for Aβ and therefore appear to be a separate population from the microglia and macrophages that remove parenchymal amyloid plaques.
Discussion
CAA has been identified as an independent risk factor for cognitive impairment and is associated with significant pathologies such as hemorrhage and ischemic damage (Greenberg et al., 2004). In typical cases, progressive CAA leads to the destruction of smooth muscle cells in the meningeal and parenchymal vasculature, presumably leading to tonal impairment and compromise of both perfusion and perivascular clearance systems (Christie et al., 2001; Preston et al., 2003). Currently Aβ-targeted therapeutic approaches for AD have not been shown to reduce vascular amyloid deposition in chronic in vivo preclinical paradigms. However, there is evidence that the direct application of amyloid antibodies to the brain (Prada et al., 2007) and blocking the apolipoprotein E/Aβ interaction (Sadowski et al., 2006) reduce VAβ.
We show here evidence of the near-complete prevention and/or clearance of VAβ by an N-terminal-specific Aβ antibody (3D6) after chronic treatment with a peripherally administered antibody. The effect was likely dependent on the ability to efficiently bind deposited amyloid, because a midregion Aβ antibody (266), which less avidly binds deposited Aβ in vivo, showed no evidence of clearing or preventing VAβ. Although a growing body of evidence suggests that the formation and composition of vascular amyloid may differ from that of parenchymal plaques (Herzig et al., 2004), antibody 3D6 is competent to prevent and/or clear both forms and thus may have a broad spectrum of amyloid-reducing activity. It would be of interest to determine whether the parenchymal and vascular amyloid-clearing properties of various Aβ antibodies with different epitope configurations are obligately linked.
Previous studies investigating the effects of Aβ immunotherapy and brain vasculature in APP transgenic mice have reported an increased incidence of microhemorrhage. However, a clear cause-and-effect relationship between VAβ and microhemorrhage has not been described. In this report we showed that vascular amyloid was either prevented from developing and/or cleared in a majority of vessels without inducing microhemorrhage. These results augmented earlier observations by demonstrating that the incidence of microhemorrhage was associated with VAβ removal. Moreover, the limited areas with microhemorrhages were focally restricted to the architectural boundaries of the vasculature that did not involve the parenchyma. These were associated with either partial or complete removal of VAβ. Importantly, microhemorrhage could be significantly mitigated by modulating the antibody dose within ranges that still effectively cleared parenchymal amyloid plaques.
Racke et al. (2005) reported the increased occurrence of microhemorrhage in PDAPP mice after a significantly larger dose of 3D6 after a 6 week treatment period using 22/23-month-old animals, significantly older than the mice in the current study. Notably, the extent of the reported microhemorrhage was also larger than any observed incidence in the current study, in keeping with our findings of a positive correlation between antibody dose and microhemorrhage scores. Our observations agree with their findings regarding the inability of 266 to bind deposited amyloid or induce microhemorrhage and extend these findings to show that 266 is also not able to prevent or clear VAβ.
VAβ effects in different APP transgenic mice are likely to vary according to several important features, including the age of animal during the study interval and the nature of the mutated protein. Because the compaction of the deposits over time and the relative degree of Aβ40 versus Aβ42 production likely influence both the aggregation properties of Aβ and the epitope-related binding efficiency of antibodies, these factors need to be considered in cross-comparisons between studies. We used 12-month-old PDAPP mice that do not have widespread, aggressive VAβ pathology, but reliably develop foci of compacted thioflavin S-positive meningeal VAβ that increases over the 6 month treatment interval.
In contrast, APP transgenic mice with very severe VAβ pathology (APP23 mice) develop spontaneous cerebral hemorrhage (Winkler et al., 2001), and, similar to human CAA patients, it is likely attributable to derangement and loss of smooth muscle cells (Winkler et al., 2001) and other destructive consequences of Aβ-related toxicity (Christie et al., 2001). The degree of treatment-related effects such as microhemorrhage will probably vary depending on the severity of baseline VAβ pathology. Passive immunization of 21-month-old APP23 mice using an N-terminal region Aβ antibody exacerbated the incidence and extent of the baseline hemorrhage (Pfeifer et al., 2002). However, subsequent ultrastructural studies did not find structural differences in the vasculature of treated and nonimmunized control mice (Burbach et al., 2007). The conclusion was that immunotherapy did not lead to or exacerbate overt damage to the vascular wall, despite the severity of the baseline VAβ pathology. The present study differs from the previous report by examining PDAPP mice that exhibit little spontaneous microhemorrhage. In addition, Aβ and hemosiderin were measured in the same sections. We documented the colocalization of apparent Aβ removal and microhemorrhage and found that focal microhemorrhage occurred only in a subset of vessels being cleared of amyloid. Because our quantitation method did not distinguish between partially cleared and intact VAβ, the absolute degree of clearance was likely underestimated.
The relationship between clearance of parenchymal and vascular amyloid is not entirely understood. However, recent reports indicate that a comodulatory relationship may exist between the two pathologies, which may be further clarified in the context of plaque removal (Herzig et al., 2004; Wilcock et al., 2004; Nicoll et al., 2006). For example, breeding mutant APP mice with heavy VAβ to those with heavy parenchymal plaque loads actually decreases VAβ, suggesting that plaques can provide a template for Aβ that would otherwise deposit onto the vasculature (Herzig et al., 2004). Conversely, Wilcock et al. (2004) showed an increase in VAβ during the course of parenchymal plaque removal in a passive immunization paradigm in aged mice, suggesting that Aβ displacement from parenchymal to vascular compartments may occur during the course of immunotherapy.
In the current study, we demonstrate that VAβ can be prevented from developing or nearly completely cleared during the course of passive immunization. Our results are in keeping with recently published studies involving the direct application of an N-terminal antibody to the brain in which VAβ was cleared without evidence of microhemorrhage (Prada et al., 2007). The clearance seen in our studies was accompanied by an increased incidence of microhemorrhage that could be controlled by lowering the antibody dosage. Because VAβ was ultimately prevented from developing or cleared by the termination of the study, such microhemorrhages may be transient events in this model and raise the possibility that Aβ immunotherapy may offer prophylactic benefit toward CAA progression. Interestingly, a recent study has shown that immunization with full-length Aβ peptide in TG 2576 mice actually improves the integrity of the blood–brain barrier (i.e., reduced the permeability of Evan's blue), suggesting that other aspects of Aβ immunotherapy might have a positive impact on the vasculature (Dickstein et al., 2006).
We should also note that the mice used in our studies do not develop the equivalent degree of VAβ associated with aggressive CAA and spontaneous hemorrhage. Therefore, extension of these outcomes to all forms of CAA would be premature.
To date, no autopsy information is available from passively immunized patients. However, some information from the autopsy studies of AN1792-immunized patients might be relevant to these findings, because they were found to predominantly produce N-terminal Aβ antibodies. Several reports demonstrate reduction of parenchymal Aβ plaque burden with generally high or even increased CAA (Nicoll et al., 2003; Patton et al., 2006). However, in more recent cases (>5 years after treatment), evidence of reduced CAA (J. Nicoll, personal communication) (Boche et al., 2007) is being documented in treated patients. Thus, the prevention, production, and potential clearance of vascular amyloid in the context of Aβ immunotherapy are likely to be dynamic processes that warrant further outcome measures in preclinical animal models and documentation in treated patients.
Approximately 80% of AD patients are affected by at least mild CAA, in which a subset is more severely affected. These individuals exhibit clinically detrimental consequences of hemorrhage, white matter degeneration, ischemia, and inflammation (Ellis et al., 1996; Greenberg et al., 2004). The findings from our study provide evidence that Aβ immunotherapy can potentially prevent, mitigate, or even reverse the progression of significant vascular pathology in PDAPP mice and raise the possibility for a similar effect in AD patients. Currently, no treatment for CAA is available, and thus these findings extend the potential therapeutic benefits of Aβ immunotherapy.
Footnotes
We thank Drs. Manuel Buttini, Wagner Zago, Jim Callaway, and Michael Grundman for their review and comments on this manuscript. We also thank Sonia Minassian, Dr. Keith Gregg, and their colleagues for development and performance of statistical analyses, and members of the Elan and Wyeth AD immunotherapy teams.
References
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, et al. Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100:2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boche D, DeBeer S, Cox A, Wilkinson D, Holmes C, Neal J, Love S, Esiri M, Bridges L, Weller R, Nicoll J. Evidence of a transient increase in cerebral amyloid angiopathy after Abeta42 immunization in human Alzheimer's disease. Neurodegenerative Dis. 2007;4(Suppl 1):273. [Google Scholar]
- Burbach GJ, Vlachos A, Ghebremedhin E, Del Turco D, Coomaraswamy J, Staufenbiel M, Jucker M, Deller T. Vessel ultrastructure in APP23 transgenic mice after passive anti-Abeta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007;28:202–212. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–433. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa-Jussá F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006;9:133–149. doi: 10.3233/jad-2006-9s316. [DOI] [PubMed] [Google Scholar]
- Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F. Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry. 2004;9:664–683. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Stürchler-Pierrat C, Bürki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castaño EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. 2006:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Prada CM, Garcia-Alloza M, Betensky RA, Zhang-Nunes SX, Greenberg SM, Bacskai BJ, Frosch MP. Antibody-mediated clearance of amyloid-β peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci. 2007;27:1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol. 2003;29:106–117. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-β antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]