

Abstract
A shift of GABAA-mediated responses from hyperpolarizing to depolarizing after neuronal injury leads to GABAA-mediated increase in [Ca2+]i. In addition, central neurons become dependent on BDNF for survival. Whether these two mechanisms are causally interrelated is an open question. Here, we show in lesioned CA3 hippocampal neurons in vitro and in axotomized corticospinal neurons in vivo that posttraumatic downregulation of the neuron-specific K–Cl cotransporter KCC2 leads to intracellular chloride accumulation by the Na–K–2Cl cotransporter NKCC1, resulting in GABA-induced [Ca2+]i transients. This mechanism is required by a population of neurons to survive in a BDNF-dependent manner after injury, because blocking GABAA-depolarization with the NKCC1 inhibitor bumetanide prevents the loss of neurons on BDNF withdrawal. The resurgence of KCC2 expression during recovery coincides with loss of BDNF dependency for survival. This is likely mediated through BDNF itself, because injured neurons reverse their response to this neurotrophin by switching the BDNF-induced downregulation of KCC2 to upregulation.
Keywords: chloride homeostasis, intracellular Ca2+, trophic factors, neuronal survival, KCC2, NKCC1
Introduction
Accumulating evidence is showing that, to regenerate axonal connections and to compensate by collateral sprouting and increased plasticity after injury, adult central neurons reactivate developmental programs (Benowitz and Routtenberg, 1997; Cramer and Chopp, 2000; Raineteau and Schwab, 2001; Harel and Strittmatter, 2006). Whereas an increase in plasticity takes place after trauma, during development the maturation of neuronal circuits is accompanied by a reduction in plasticity, known as a closure of critical periods. One of the key factors in the closure of plasticity is the establishment of functional inhibitory GABAergic transmission (Hensch, 2005). For example, in the visual cortex it has been shown that accelerating the maturation of GABAergic innervation by overexpression of brain-derived neurotrophic factor (BDNF) leads to a premature termination of the plastic state (Huang et al., 1999; Harel and Strittmatter, 2006). Whether developmental-like mechanisms are required for the survival of mature injured CNS neurons is not known.
Immature neurons display strong depolarizing GABAA-mediated responses that become hyperpolarizing in mature neurons (Payne et al., 2003). This developmental change is caused by a decrease in [Cl−]i, which is, in turn, produced by the upregulation of the neuron-specific K–Cl cotransporter-2 (KCC2) (Rivera et al., 1999; Hübner et al., 2001). The depolarizing action of GABAergic transmission during development is thought to provide trophic support for immature neurons via Ca2+ signaling (Franklin and Johnson, 1992; Ikeda et al., 1997; Owens and Kriegstein, 2002). Under pathophysiological conditions such as neuronal trauma or axotomy, mature neurons downregulate the functional expression of KCC2 and, consequently, revert to a depolarizing, immature-like response to GABA (Payne et al., 2003). This depolarizing response is maintained by Cl− uptake mediated by the Na–K–2Cl cotransporter-1 (NKCC1) (Nabekura et al., 2002; Payne et al., 2003).
The regulation of neonatal neuronal survival and plasticity are strongly influenced by neurotrophins (Lewin and Barde, 1996). During early stages of development, neurotrophins predominantly regulate cell fate decisions and neuronal survival (Lewin and Barde, 1996). During maturation, their regulatory repertoire shifts more toward modulation of neuronal plasticity, synaptic transmission, and the maintenance of a differentiated neuronal phenotype (Lewin and Barde, 1996). In particular, the neurotrophic factor BDNF shows a qualitative transition from developmental to adult functions (Davies, 1997). Apart from its effects on GABAergic innervation (Danglot et al., 2006), BDNF can also promote the functional maturation of GABAA-mediated responses by inducing upregulation of KCC2 (Aguado et al., 2003; Carmona et al., 2006). Interestingly, the effect of BDNF on KCC2 expression is the opposite in adult cortical neurons, in which BDNF downregulates KCC2 expression (Rivera et al., 2004). Thus, there are two modes of BDNF-mediated regulation of KCC2 expression: upregulation during development and downregulation in mature central neurons. An interesting finding in this context is that, after peripheral nerve injury, KCC2 expression in lamina 1 of the superficial dorsal horn is reduced, leading to a change in GABAA-mediated responses. This effect has been shown to be mediated by BDNF released from microglia in an ATP-dependent manner (Coull et al., 2003, 2005). Whether similar mechanisms involving a cross talk between BDNF and KCC2 are present in injured cortical neurons is an open question.
Little is known about the detailed mechanisms whereby neurons revert to immature responses after trauma, and whether these mechanisms also affect the survival of lesioned neurons. The present study addresses the question whether the adult-to-immature transition in the survival-promoting action of BDNF requires the reactivation of the ontogenetic shift in GABAA-mediated responses in injured mature central neurons.
We used an in vitro hippocampal model in which CA3 principal neurons are axotomized. Here, we observed that a GABA-induced [Ca2+]i increase is required for neurons to become dependent on BDNF for survival. The in vivo relevance of this finding was examined by investigating the role of BDNF in KCC2 expression and in the survival of retrogradely labeled axotomized corticospinal neurons (CSNs). We show that BDNF acting through TrkB is essential for the survival of lesioned CSNs. After a recovery period, surviving neurons regain KCC2 expression levels and become independent of BDNF for survival. This increase in KCC2 expression is likely mediated by BDNF itself, because injury induces a switch in the effect of BDNF on KCC2 expression from downregulatory to upregulatory.
In summary, we provide evidence that axotomy activates a developmental-like mode of BDNF-mediated effects, and the mature mode of BDNF action is reestablished during recovery.
Materials and Methods
Organotypic culture.
Hippocampal slices from 8- to 9-d-old NMRI mice were prepared as described previously (Ludwig et al., 2003). Briefly, transverse slices were cut from the hippocampi using a tissue chopper. They were immediately placed on sterile Millicell-CM membranes in six-well culture trays with 1 ml of plating medium. Slices were left to recover for 4 d at 37°C under 5% CO2. One day after plating, the medium was changed to an antibiotic-free one, and renewed again 4 d after plating. All of the experiments were performed immediately after the second change of the medium, and slices with dark regions as observed under light microscopy were removed. Slices were cut between CA3 and CA1 regions using a razor blade. BDNF (recombinant human BDNF; Sf 21-derived; R&D Systems) was added to the plating medium of unlesioned and lesioned slices immediately after cutting at a concentration of 10 ng/ml. TrkB-Fc (recombinant human TrkB-Fc chimera; R&D Systems) was used at a concentration of 200 ng/ml. Bumetanide (Tocris) was used at a concentration of 10 μm. After each experiment, the slices were left for 3 d at 37°C under 5% CO2 and then collected for analysis.
Immunohistochemistry.
Slices remained on the Millicell-CM membranes during the staining procedure. They were fixed in 4% paraformaldehyde (PFA) overnight at 4°C. Next day, the slices were washed in PBS and dehydrated through a graded series of methanol in PBS. Thereafter, the slices were incubated in Dent's fixative (80% methanol, 20% DMSO; Sigma-Aldrich), washed in TBSTD (TBS, 0.1% Tween, 5% DMSO), and placed in BSA overnight at 4°C. Afterward, slices were incubated with primary antibody diluted in BSA at 4°C for 48 h. Rabbit anti-KCC2 Ab (Ludwig et al., 2003) was diluted 1:500, and mouse anti-NeuN Ab (Millipore Bioscience Research Reagents) was diluted 1:400. After washing in TBSTD, slices were incubated overnight with Alexa Fluor 488 donkey anti-mouse IgG and Alexa Fluor 568 goat anti-rabbit IgG (1:400). Slices were then washed in TBSTD, incubated in Hoechst (1:1000; Invitrogen), rinsed with TBSTD, and finally mounted with membranes on Superfrost Plus slides in Vectashield mounting medium (Vector Laboratories). Fluorescence images were acquired with Leica TCS SP2 confocal microscope using HCXPL APO 10× air or 20× glycerol objectives. Mean intensity over a given area of the slices was measured and differences were quantified with ImagePro software.
Because the intensity of NeuN staining was used to estimate the number of neurons, it was important to know whether the amount of NeuN protein is influenced by BDNF treatment or axotomy. Optical sections from six slices (three controls and three treated with BDNF or lesioned as described in the experimental procedure), three sections from each slice, were analyzed. From each section, 20 neurons were randomly selected and the density of NeuN was measured. Neither BDNF treatment (83 ± 17% of control; p > 0.1) nor lesion (108 ± 18% of control; p > 0.1) did significantly alter the expression of NeuN protein per neurons.
Real-time PCR.
Slices were collected and stored in the RNAlater reagent (Qiagen). Total RNA was isolated with RNeasy kit (Qiagen) and reverse transcribed with SuperScript II RNase H-Reverse Transcriptase (Invitrogen). cDNA samples were amplified using SYBR Green PCR Master Mix (Applied Biosystems) and detected with the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Primer Express, version 2.0, software (Applied Biosystems) was used for primer design: 5′-TGAAGGGACTGCTCTCTTTGG-3′ (KCC2 reverse), 5′-GCCACCATGCTCAACAACCT-3′ (KCC2 forward), 5′-GCAAAGTGGAGATTGTTGCCAT-3′ [glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward], 5′-CCTTGACTGTGCCGTTGAATTT-3′ (GAPDH reverse). Results were analyzed on 7000 system SDS software (Applied Biosystems).
Western blotting.
Slices were homogenized in homogenization buffer (0.3 m sucrose, 10 mm HEPES, 1 mm EDTA, pH 7.2, containing protease inhibitors, CompleteMini; Roche) and the homogenate incubated with Laemmli loading buffer 45 min at 45°C. The sample was separated in 8% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Blots were probed with rabbit α-wNT NKCC1 (Evans et al., 2000) or rabbit anti-KCC2 (Ludwig et al., 2003) at a concentration of 1:2000 and developed using ECL-plus kit (GE Healthcare). After that, membranes were stripped using DTT/SDS in Tris-HCl, pH 7.0, and probed with rabbit anti-β-tubulin Ab (1:20,000; Sigma-Aldrich). Optical densities of the bands were analyzed with the AIDA imaging software.
Precipitation of TrkB.
Slices were homogenized and incubated overnight with wheat germ lectin Sepharose (GE Healthcare). Next day, Sepharose was washed off with homogenization buffer and incubated in Laemmli loading buffer for 45 min at 45°C. The solution was centrifuged and supernatant was loaded, separated in 8% SDS-PAGE, and electrophoretically transferred to nitrocellulose membranes. Blots were probed with rabbit anti-TrkB polyclonal antibodies that recognize the extracellular portion of both the long and short isoforms of TrkB (Mamounas et al., 2000) and reblotted with mouse anti-human transferrin receptor Ab (Zymed Laboratories) for normalization.
Fluo-4 imaging.
Neurons were loaded with Fluo-4 acetoxyl methyl (10 μm) in extracellular standard solution containing the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 15 d-glucose, 2 CaCl2, 5% CO2/95% O2. Slices were then laid on the glass bottom of a submerged-type chamber, and this was placed on a microscope stage and continuously perfused with standard solution gassed with 95% O2/5% CO2 at a rate of 2–3 ml/min. Fluo-4 fluorescence was excited using a multiwavelength monochromator (Polychron VI; Till Photonics), and the emitted light was filtered using a bandpass filter (480 nm). Fluorescence images were obtained using a 20× objective lens (LCPLanFl; Olympus) and a CCD camera (iXon; Andor Technology) fitted to an upright microscope (IX-71; Olympus). Data were stored for off-line analysis by means of image-processing software (Till Photonics). Variations in [Ca2+]i were observed as changes in intensities at 488 nm. Changes were monitored in the CA3 pyramidal neurons by taking measurements every 30 s. Somata of pyramidal-shaped neurons in the CA3 region were randomly selected for quantification.
Surgical procedures.
Experimental procedures and maintenance of animals were approved by the local animal care committee. Male Sprague Dawley rats weighing 190–330 g were used. The surgical procedures, neurotrophic factor, and antibody application, and coordinates for stereotaxic lesion and intracortical substance delivery have been described previously in detail (Giehl and Tetzlaff, 1996; Bonatz et al., 2000). In brief, to distinguish CSNs from other cortical layer V neurons, they were retrogradely labeled by fluorescence tracer injections to the corticospinal tracts at cervical spinal cord levels C4/5 before axotomy. Fast Blue (FB) (2% in 0.2% DMSO) (rats) and a rhodamine tracer mixture (RDX) (15% rhodamine dextrane 10,000, 10% rhodamine dextrane 3000, and 10% rhodamine-B-isothiocyanate in 0.2% DMSO) (mice) were used as tracers. Mice traced with RDX were killed without lesion 1 week after application of the tracer and processed for the analysis of CSNs numbers (see below). One to 2 weeks after FB injection in rats, CSNs of one side of the brain were axotomized by unilateral internal capsule lesion (ICL). This creates a horizontal, circle-shaped cut through the entire internal capsule through which CSNs send their axons to their spinal cord targets (Bonatz et al., 2000). This lesion results in axotomy of all corticospinal neurons of the sensory motor cortex (CSNs) of the lesion side (Giehl and Tetzlaff, 1996; Bonatz et al., 2000). Surgery was performed under anesthesia with a combination of chloral hydrate (150 mg/kg, i.p.) and sodium pentobarbital (32 mg/kg, i.p.) for rats or an inhalation anesthesia with metofane for mice.
Cortical infusions.
For neurotrophic factor, antibody or vehicle application to intact or axotomized CSNs, a 30 gauge steel cannula connected to an osmotic minipump (Giehl and Tetzlaff, 1996) via a silicone tube was implanted intraparenchymally into the lesion site of the ipsilateral cortex. The following solutions were infused into the cortex at a rate of 1 μg/h for the given periods: 20 mm PBS (vehicle), BDNF (500 μg/ml in PBS), affinity-purified BDNF-neutralizing rabbit polyclonal anti-BDNF antibody (Yan et al., 1997b) (1 mg/ml in PBS), polyclonal TrkB-blocking rabbit anti-TrkB antibody (Kang et al., 1997; Yan et al., 1997a) (serum; 1:7 diluted in PBS). All solutions contained penicillin/streptomycin at a final concentration of 50 U/ml.
Tissue processing.
Seven days after tracer application (intact animals) or after ICL (lesioned animals), the animals were killed by an overdose of sodium pentobarbital and transcardially perfused with PBS followed by 4% PFA solution. The brains were postfixed in 4% PFA (2 h), cryoprotected in 20% sucrose in PBS (12 h), frozen, and cut into 20 μm cryostat serial coronal sections. Only sections from the center of the lesion/treatment area were used.
In situ hybridization.
In situ hybridizations were performed as described previously (Giehl et al., 1998, 2001). Oligonucleotide probes for the individual mRNAs were labeled with 35S-αATP (PerkinElmer Life and Analytical Sciences), using terminal deoxynucleotide transferase (Invitrogen). The probe for KCC2 mRNA (Kanaka et al., 2001) (GenBank accession number U55816) was complementary to bases 2981–3016 of the rat KCC2 mRNA sequence. The controls of the in situ hybridization were performed by competition with an excess of cold probe (data not shown). A 3 week exposure time for KCC2 in Kodak NTB 2 photoemulsion was used in these experiments.
Quantification of mRNA expression.
To quantify KCC2 expression in CSNs, the grain densities over FB-labeled CSNs were measured blindly with the aid of the image analysis system DIGGER (MEDVIS) on unstained sections (Giehl et al., 1998, 2001). For each quantification step, two images were loaded into two separate buffers of the image analysis system. The first buffer contained the fluorescent image of FB-labeled CSNs; the second buffer contained the dark-field image of the respective autoradiography at the same position of the section. The FB-labeled neurons were traced in the first buffer to create a mask that was automatically used for the quantification of grain densities in the second buffer at the corresponding position in the dark-field image. CSNs were traced along their perikaryal borders, excluding the apical dendrites. The analysis of mRNA expression is based on >30,000 CSNs in total.
Analysis of neuronal survival.
The analysis of cell survival of rat and mouse CSNs was performed by a stereologic approach as described previously (Bonatz et al., 2000). In brief, the number of CSNs was assessed by blinded cell counts of every second section collected for cell counts (i.e., every fourth section of the mice and every 10th section of the rat brains were quantified). The criterion for a CSNs was a tracer-filled pyramidal-shaped profile >4 μm (rats) or 3 μm (mice) in diameter (Giehl and Tetzlaff, 1996; Bonatz et al., 2000). The analysis of CSNs survival is based on >1,000,000 cells counted. Similarly, the number of NeuN-positive cells were counted from confocal optical sections (1 μm). Cells were stereologically analyzed from three optical sections in every organotypic slice.
Statistics.
For the in vitro experiments, statistical analysis was performed using the nonpaired t test on Microsoft Excel software, and statistical significance was defined as follows: *p < 0.1, **p < 0.05, and ***p < 0.001. For the in vivo experiments, one-way ANOVA (p = 0.042) followed by a post hoc Newman–Keuls test (NKT) or a Fisher's least significant difference test (FLSD) on SigmaStat software was used to determine the statistical significance of differences between lesion and control sides and the respective experimental groups. Error bars represent SE.
Results
A population of injured CA3 hippocampal neurons requires BDNF for survival
To investigate the mechanisms that underlie the connection between neuronal chloride homeostasis and the survival of axotomized neurons, we used an in vitro model in which organotypic hippocampal slices were cut between the CA3 and CA1 region (Fig. 1A) (see also Materials and Methods). Cutting the Schaffer collateral bundle leaves neurons axotomized in the CA3 region (Fig. 1A, right).
Figure 1.
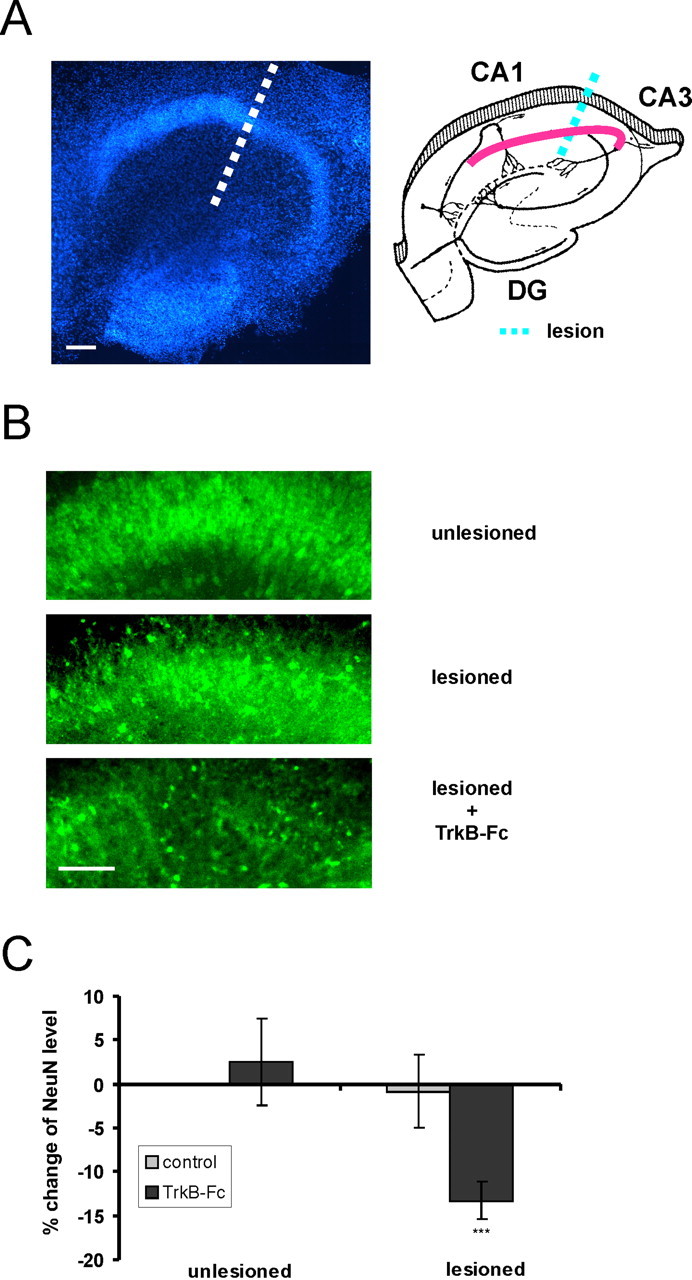
Neurons in organotypic hippocampal slices become dependent on BDNF trophic support after axotomy. A, Left, Hoechst staining of a representative organotypic hippocampal slice. Scale bar, 100 μm. Right, Schematic figure showing the position of Schaffer collaterals and the different regions of the hippocampus CA1, CA3, and dentate gyrus (DG). The site of the lesion is shown as dashed lines. B, NeuN immunostaining of representative unlesioned and lesioned slices, and a lesioned slice treated with the BDNF scavenger TrkB-Fc. Scale bar, 100 μm. C, There is no reduction in NeuN intensity in the CA3 region after administration of TrkB-Fc to unlesioned slices (103 ± 5% of unlesioned control; p > 0.1; n = 14). After axotomy, there is no reduction in NeuN expression of lesioned slices (99 ± 4% of unlesioned control; p > 0.1; n = 23), whereas NeuN immunoreactivity of lesioned slices treated with TrkB-Fc is reduced by 13 ± 2% (lesioned TrkB-Fc-treated vs unlesioned controls; ***p < 0.001; n = 24). n, Number of slices. Error bars indicate SE.
Changes in NeuN immunofluorescence in the CA3 region were used to estimate changes in neuronal number (see Materials and Methods). When we applied the BDNF scavenger TrkB-Fc to injured slices, a significant decrease in the NeuN immunoreactivity occurred compared with injured slices in the absence of TrkB-Fc (Fig. 1) (13 ± 2%; lesioned TrkB-Fc-treated vs unlesioned controls; p < 0.001; n = 24). Thus, a population of neurons of the CA3 region becomes dependent on BDNF after damage. In contrast, in the intact control slices we did not observe a reduction in NeuN immunoreactivity after application of Trkb-Fc (Fig. 1). This fact suggests that the survival of injured, but not intact, CA3 neurons depends on BDNF. This effect is not mediated through changes in TrkB expression levels, because TrkB protein levels did not change after lesion as revealed by Western blot analysis (103 ± 25%; p > 0.1; n = 4). When TrkB-Fc was applied 2 d after lesion combined with BDNF treatment, no reduction in NeuN immunoreactivity occurred compared with lesioned BDNF-treated slices that were not exposed to TrkB-Fc (116 ± 6% of lesioned BDNF-treated slices; p < 0.05; n = 16). This indicates that, after acute injury, BDNF-treated neurons lose their dependency on BDNF.
NKCC1-dependent GABAA-mediated [Ca2+]i increase in lesioned CA3 neurons
KCC2 immunohistochemistry of unlesioned slices showed a distribution of KCC2 expression similar to that in vivo (Rivera et al., 2004), and lesion of the slices induced a pronounced downregulation of KCC2 immunoreactivity by 15 ± 3% (p < 0.05; n = 22) (Fig. 2A). This was also observed in Western blots (downregulation by 22 ± 5%; p < 0.05; n = 6 slices) (Fig. 2B) and at the transcriptional level at which KCC2 mRNA dropped to 18 ± 5% (p < 0.001; n = 16 slices) as determined with real-time RT-PCR. Western blot analysis did not show a significant decrease in the expression of NKCC1 after lesion (p > 0.1; n = 12 slices). KCC2 and NKCC1 are normalized to β-tubulin.
Figure 2.

Lesion induces downregulation of KCC2 protein in organotypic hippocampal slices. A, KCC2 immunostaining of representative unlesioned and lesioned slices. Scale bar, 100 μm. B, Western blot analysis shows that KCC2, but not NKCC1, is downregulated in injured slices.
Next, we addressed the question whether injury in organotypic hippocampal slices produce changes in GABAA-mediated responses. To this end, we monitored GABAA-mediated intracellular Ca2+ ([Ca2+]i) responses. Organotypic hippocampal cultures were loaded with Fluo-4 and the fluorescence intensity changes were measured at 488 nm (Fig. 3A). Somata of pyramidal-shaped neurons in the CA3 region were randomly selected for quantification. In unlesioned slices, application of the GABAA-specific agonist muscimol (100 μm) failed to induce an increase in intracellular Ca2+ (Fig. 3B) (n = 4), as expected for the mature neural tissue. The same procedure induced a robust [Ca2+]i response in lesioned cultures (Fig. 3C) (n = 6). This indicates that GABAA responses are depolarizing in lesioned slices.
Figure 3.

Blocking NKCC1 inhibits the GABAA-mediated increase in intracellular [Ca2+]i in lesioned neurons. A, Fluorescence of Fluo-4 in organotypic hippocampal slices. Scale bar, 100 μm. B, In unlesioned slices, application of glutamate but not muscimol evokes an increase in [Ca2+]i. C, In lesioned slices, 100 μm muscimol evokes an increase in [Ca2+]i. This effect can be blocked by NKCC1 inhibitor bumetanide (10 μm) (D). E, In BDNF-treated unlesioned slices, muscimol evokes an increase in [Ca2+]i, which is blocked by bumetanide. F, In cut slices treated with BDNF, muscimol does not evoke an increase in [Ca2+]i. n = 4. Error bars indicate SE.
Application of bumetanide, a selective NKCC1 inhibitor (Payne, 1997), blocked the GABAA-mediated [Ca2+]i increase (Fig. 3D) (n = 4), indicating that intracellular chloride accumulation and the consequent GABAA depolarization are maintained by NKCC1 in the injured neurons. Expression levels of NKCC1 did not change in the presence of bumetanide as indicated by Western blot analysis (p > 0.1; n = 12 slices).
Lesion-induced BDNF requirement for survival is abolished by blocking GABA-induced depolarization
We next asked whether neurons lose their dependence on BDNF for survival if GABAA-mediated depolarization is blocked. Lesioned TrkB-Fc-treated slices have reduced NeuN immunoreactivity in CA3, as described above. Notably, we found that NeuN intensity in lesioned slices treated both with TrkB-Fc and bumetanide did not differ significantly from the lesion control slices (Fig. 4). We performed Ca2+ measurements to ensure that, on the day of the experiments, the lesioned bumetanide-treated slices did not show a depolarizing response to GABA (Fig. 4). Axotomy in the presence of the BDNF scavenger TrkB-Fc results in a reduction of neuronal number (lesioned TrkB-Fc-treated vs lesioned control, 87 ± 2%; p < 0.001; n = 24). This effect is blocked when slices are treated with bumetanide (lesioned TrkB-Fc plus bumetanide-treated vs lesioned control, 105 ± 4%; p > 0.1; n = 13). The results were corroborated by stereological counting of NeuN staining: lesioned controls, 5137 ± 212 neurons/mm2, n = 9; lesioned plus Trkb-Fc, 4489 ± 389 neurons/mm2, n = 8; lesioned plus Trkb-Fc plus bumetanide, 5216 ± 108 neurons/mm2, n = 9 sections. Thus, GABAA-mediated depolarization is required to establish the need of injured neurons for BDNF trophic support.
Figure 4.

Block of NKCC1 and the consequent GABAA-mediated [Ca2+]i increase inhibits the establishment of BDNF dependence in lesioned slices. The consequences of bumetanide applied right after lesion, preventing the [Ca2+]i increase evoked by muscimol, can be observed 3 d later. Error bars indicate SE.
Damaged neurons require BDNF/TrkB signaling for survival, whereas intact neurons are independent of BDNF trophic supply in vivo
Results using BDNF-neutralizing antibodies and BDNF knock-out mice indicate that BDNF may have a different action after injury (Giehl et al., 1998, 2001). In these studies, inhibition of BDNF signaling did not reduce the number of unlesioned adult CSNs but produced a significant reduction in the number of axotomized neurons. Because not only BDNF but also neurotrophin-4 (NT-4) acts via TrkB (Barbacid, 1995), and both factors are present in the adult cortex (Maisonpierre et al., 1990; Timmusk et al., 1993), it is conceivable that endogenous NT-4 compensates for the reduced BDNF levels in the BDNF knock-outs as well as animals infused with the BDNF-neutralizing antibody. Here, NT-4 levels could sustain a TrkB activity that would be sufficient to support survival of intact, but not lesioned, CSNs.
To resolve this question, we infused the polyclonal TrkB antibody (anti-TrkB) (Kang et al., 1997) into the frontal cortex of intact animals and animals lesioned at the internal capsule level. In lesioned animals, the infusion started immediately after the axotomy and lasted for 7 d. CSNs were identified by retrograde tracing before the experiments (Fig. 5A). TrkB blockade by anti-TrkB significantly reduced the survival of lesioned CSNs, whereas the survival of intact CSNs was not affected (Fig. 5B,C). Similarly, treatment with the BDNF-neutralizing antibody (anti-BDNF) (Yan et al., 1997b; Giehl et al., 2001) reduces the survival of lesioned CSNs, whereas intact CSNs are unaffected after anti-BDNF treatment (Fig. 5D).
Figure 5.

CSNs become dependent on BDNF/TrkB trophic support for survival after axotomy in vivo. A, The corticospinal lesion model and neurotrophic factor treatment scheme for lesioned CSNs. Left, Retrograde tracing of CSNs. The retrograde tracer Fast Blue was microinjected into the corticospinal tracts at the cervical level C4/5 of the spinal cord before lesion to specifically label CSNs. Middle, CSN lesion was made at the internal capsule level with a knife to cause axotomy-induced death of CSNs. Right, Intraparenchymal growth factor delivery via an intracortically implanted applicator connected through silicon tubing to an osmotic minipump. The applicator is located in the frontal cortex at the lesion side. B, Photomicrographs of representative sections of rats treated with control solution (PBS) and receptor-blocking antibodies (anti-TrkB-serum). We used a 1:7 diluted serum for TrkB blockade. Scale bar, 1 mm. C, TrkB signaling is required for the survival of lesioned but not intact rat CSNs [anti-TrkB versus PBS: **p < 0.01 after NKT; CSN survival: PBS (67 ± 2%, n = 11 for lesioned; 105 ± 4%, n = 4 for intact CSNs), anti-TrkB serum (anti-TrkB) (45 ± 4%, n = 5 for lesioned; 100 ± 7%, n = 4 for intact CSNs)]. D, Treating lesioned CSNs with the BDNF-neutralizing antibody resulted in reduced survival of lesioned, but not unlesioned, CSNs [CSN survival: PBS (axotomized), 66 ± 2% (n = 12); anti-BDNF (axotomized), 41 ± 9% (n = 11); PBS (unlesioned), 99 ± 2% (n = 5); and anti-BDNF (unlesioned), 97 ± 5% (n = 3)]. In lesioned animals, anti-BDNF, **p < 0.01 versus PBS after NKT. n, Number of animals. Error bars indicate SE.
These data show that a population of CSNs switch from a BDNF/TrkB-independent to BDNF/TrkB-dependent state after axotomy in vivo. Because also embryonic cortical neurons are supported by endogenous BDNF for survival (Ghosh et al., 1994), this qualitative change in BDNF/TrkB signaling may represent the reactivation of a developmental mechanism after a pathological damage.
Recovery from neuronal injury coincides with recovery of KCC2 expression
Injury-induced death of CSNs does not proceed beyond the first week after the lesion, and more than one-half of the cells survive for at least 42 d (Giehl et al., 1997). This suggests that a significant portion of lesioned CSNs recovers from the injury. To examine whether this recovery is accompanied by a loss of BDNF dependency, we infused the BDNF-neutralizing antibody anti-BDNF to CSNs that had been lesioned 35 d before. This treatment was performed for 7 d and did not result in additional death of CSNs (PBS: 58 ± 4%, n = 6; anti-BDNF: 62 ± 7%, n = 3). Thus, axotomized CSNs lose their injury-induced BDNF dependence after several weeks. KCC2 expression levels in CSNs are significantly decreased (Fig. 6), as was also observed in vitro. Assuming that the neurons return to mature state also regarding their GABAergic responses, the KCC2 expression of lesioned CSNs should return to the levels observed before the axotomy. Indeed, KCC2 mRNA levels were recovered by day 42 after the lesion and even exceeded the KCC2 levels in CSNs of the intact control side (Fig. 6B). Thus, lesioned CSNs appear to reestablish a mature state after an initial postinjury period.
Figure 6.

After the acute injury period, surviving neurons reestablish KCC2 expression levels in vivo. A, Photomicrographs showing KCC2 mRNA expression (yellow grains) as detected by radioactive in situ hybridization and Fast Blue-labeled CSNs in cortical layer V 1 d after the lesion. Scale bar, 100 μm. B, Time course of KCC2 expression showing that KCC2 is rapidly downregulated already 8 h after axotomy, and its expression levels are recovered after 42 d. KCC2 mRNA levels were as follows: 8 h (64%; n = 1), 1 d (56 ± 2%; n = 3), 3 d (51%; n = 1), 5 d (49%; n = 1), 7 d (51 ± 11%; n = 4), 42 d (151 ± 11%; n = 3). Error bars indicate SE.
BDNF-mediated KCC2 downregulation changes to upregulation after axotomy
What is the mechanism for the upregulation of KCC2 during the recovery period? We examined this by infusing BDNF (0.5 μg/μl) in both intact and lesioned frontal cortex. After 7 d of infusion, animals were killed, and KCC2 mRNA levels were quantified in CSNs by in situ hybridization. We found that BDNF had strikingly different effects on KCC2 expression in intact and lesioned CSNs: whereas in intact CSNs it significantly suppressed the KCC2 mRNA levels, in axotomized CSNs it increased KCC2 mRNA (Fig. 7A,B). Without BDNF treatment, KCC2 levels were rapidly and significantly reduced already 8 h after axotomy and the low levels were sustained for at least 7 d (Figs. 6B, 8A,B). The BDNF-induced upregulation of KCC2 in lesioned CSNs was already fully established at day 1 after the injury (Fig. 8A,B). Treating injured neurons with BDNF resulted in higher KCC2 levels than in intact control neurons (Figs. 7, 8), indicating that BDNF increases KCC2 levels rather than prevents downregulation of KCC2 induced by injury.
Figure 7.
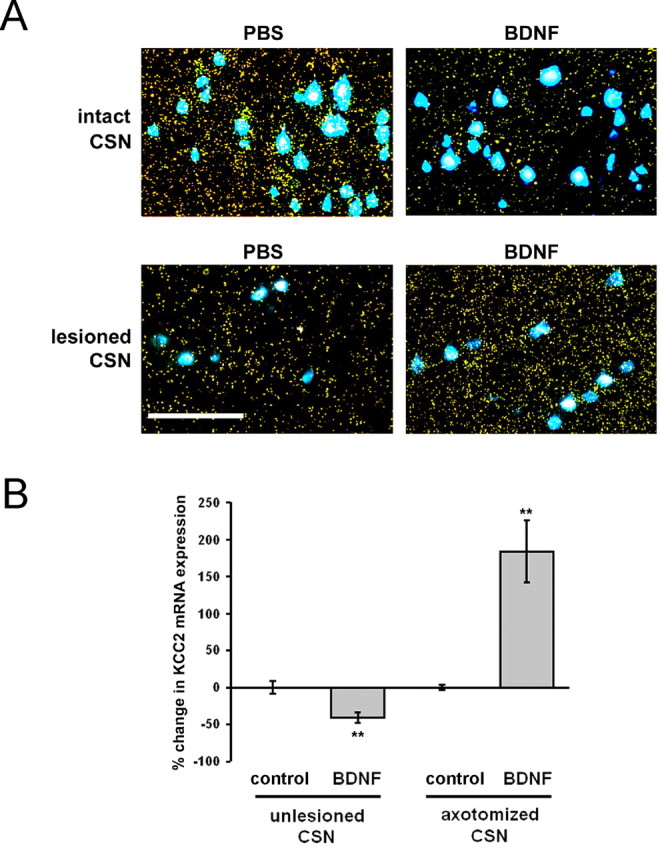
BDNF produced a decrease in KCC2 mRNA expression in intact CSNs and an increase in lesioned CSNs. A, Representative photomicrographs showing KCC2 mRNA expression (yellow grains) as detected by radioactive in situ hybridization and Fast Blue-labeled CSNs in cortical layer V of PBS- and BDNF-treated animals. Scale bar, 100 μm. B, Quantification of the grains over Fast Blue-labeled CSNs. Compared with the average grain density of the respective control group (control, PBS-treated animals), BDNF decreases KCC2 expression by 40% in intact CSNs and increases KCC2 expression by 180% in lesioned CSNs. Changes in KCC2 mRNA levels were as follows: intact CSNs: control, 0 ± 10% (n = 3); BDNF, −41 ± 8% (n = 2); axotomized CSNs: control, 0 ± 5% (n = 3); BDNF, +183 ± 43% (n = 5). n, Number of animals. BDNF versus control, **p < 0.05 after FLSD. Error bars indicate SE.
Figure 8.
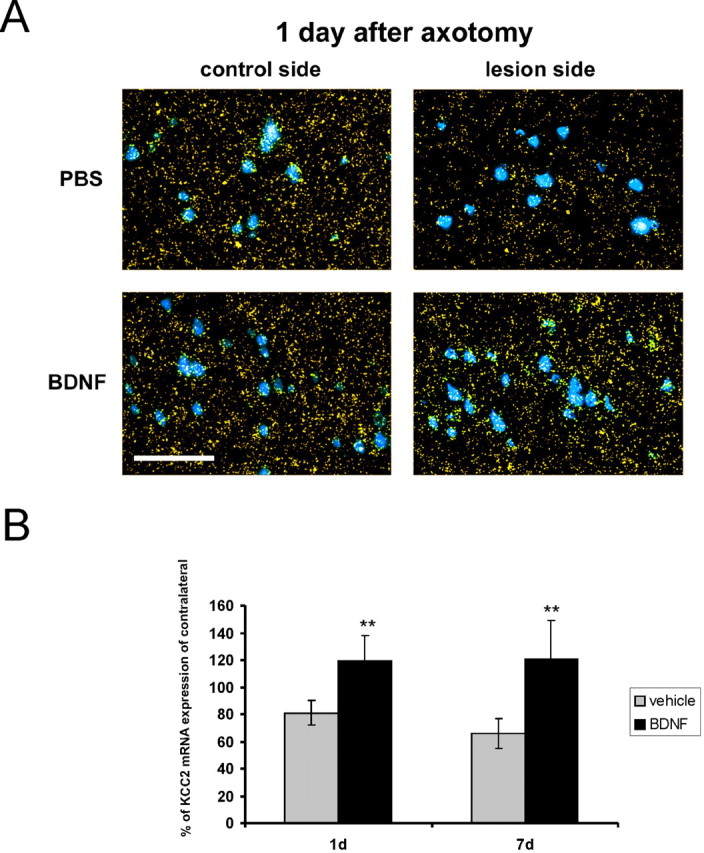
The upregulatory effect of BDNF is present already 1 d after axotomy. A, Photomicrographs showing KCC2 mRNA expression and Fast Blue-labeled CSNs in cortical layer V of PBS- and BDNF-treated animals 1 d after the lesion. Scale bar, 100 μm. B, BDNF treatment increases KCC2 levels compared with the contralateral side (vehicle) 1 and 7 d after the lesion. KCC2 mRNA levels were as follows: vehicle (PBS), 1 d (81 ± 9%; n = 2), 7 d (66 ± 11%; n = 5); BDNF: 1 d (119 ± 19%; n = 2), 7 d (121 ± 28%; n = 5). n, Number of animals. BDNF versus PBS (1 and 7 d): **p < 0.05 after FLSD. Error bars indicate SE.
We previously showed that BDNF can induce downregulation of KCC2 in mature hippocampal organotypic cultures (Rivera et al., 2002). Similar results were obtained in the present study (Fig. 9). Administration of exogenous BDNF (10 ng/ml) leads to downregulation of KCC2 by 12 ± 2% in unlesioned slices (unlesioned plus BDNF-treated vs unlesioned; p < 0.001; n = 19). This observation indicates that our in vitro model mimics the responses of mature neural tissue. During development, the effect of BDNF on KCC2 expression is different and can produce upregulation (Aguado et al., 2003; Carmona et al., 2006). Similar to the results obtained in vivo, application of BDNF on lesioned slices resulted in a significant increase in KCC2 expression compared with control injured levels (Fig. 9) (upregulation of KCC2 by 15 ± 2%; lesioned plus BDNF- treated vs lesioned; p < 0.001; n = 15). Similar results were obtained with real-time RT-PCR, in which BDNF decreased KCC2 levels to 46 ± 13% (p < 0.001; n = 16 slices) in uninjured slices, whereas it upregulated KCC2 to 362 ± 55% (p < 0.05; n = 13 slices) in axotomized control slices. Interestingly, application of TrkB-Fc to injured slices did not significantly affect the expression levels of KCC2 (p > 0.1; n = 11), indicating that the endogenous levels of BDNF were not enough to upregulate KCC2. Application of BDNF 24 h after the axotomy at the time point when KCC2 levels were already downregulated by 12 ± 3% (p < 0.05; n = 9), induced KCC2 upregulation (by 10 ± 2%; p < 0.05; n = 10), verifying that BDNF upregulates and not merely prevents downregulation of KCC2 in injured neurons.
Figure 9.

BDNF induces downregulation of KCC2 in unlesioned hippocampal slices and upregulation of KCC2 in lesioned hippocampal slices. KCC2 immunostaining of representative unlesioned and lesioned slices in the absence and presence of BDNF. Scale bar, 100 μm.
In accordance with the results above, and in line with the downregulatory effect of BDNF on KCC2 expression in intact mature neurons (Rivera et al., 2002), muscimol was able to evoke a [Ca2+]i increase in unlesioned BDNF-treated slices (Fig. 3E) (n = 6). Notably, muscimol failed to evoke [Ca2+]i transients in BDNF-treated lesioned slices (Fig. 3F) (n = 6) indicating that BDNF is able to block the injury-induced change in chloride homeostasis that leads to GABA-induced [Ca2+]i elevation. These results indicate that the effect of BDNF on KCC2 expression is qualitatively reversed after neuronal trauma and is involved in the mechanism for the upregulation of KCC2 during the recovery of injured neurons.
Blocking GABA-induced depolarization abolishes posttraumatic BDNF-mediated KCC2 upregulation
To investigate whether the switch in the effect of BDNF on KCC2 expression is dependent on GABAA-mediated depolarization, we performed experiments in which organotypic slices were lesioned in the presence of 10 μm bumetanide and the effect of BDNF on KCC2 expression was monitored. Strikingly, the upregulatory effect of BDNF on KCC2 was absent in the presence of bumetanide (BDNF without bumetanide: 115 ± 2% of lesioned control, p < 0.001, n = 15; BDNF with bumetanide: 101 ± 3% of lesioned bumetanide control, p > 0.1, n = 18). This result indicates that GABAA-mediated depolarization is required for the posttraumatic action of BDNF on KCC2 expression.
Discussion
In this study, we show that whereas adult intact neurons are not dependent on BDNF, injured neurons become dependent on BDNF trophic support for survival. After axotomy, KCC2 levels decrease, leading to NKCC1-mediated accumulation of intracellular chloride, and this, in turn, leads to a depolarizing response on opening of GABAA channels and elevation of [Ca2+]i (Payne et al., 2003). A striking result was that GABA-induced [Ca2+]i increase is required for the survival-promoting action of BDNF: blocking GABA-induced [Ca2+]i increase by the NKCC1 inhibitor bumetanide prevented neuronal loss caused by the absence of BDNF in injured neurons.
We also observed that, whereas in intact mature neurons BDNF downregulated KCC2 (Rivera et al., 2002, 2004), in injured neurons BDNF upregulated KCC2. Block of GABAA-mediated [Ca2+]i increase with bumetanide also significantly prevented the BDNF-induced KCC2 upregulation, indicating that depolarization is involved in the change of BDNF action after trauma. In addition, upregulation of KCC2 by BDNF in injured neurons prevents GABAA-mediated responses to become depolarizing. The results and conclusions of the present work are summarized in Figure 10.
Figure 10.

Schematic drawing summarizing the results and conclusions obtained in this study. For details, see Discussion.
It is evident from the present results that a population of axotomized central neurons, both CSNs in vivo and hippocampal CA3 neurons in vitro, experiences a pronounced qualitative change in their response to BDNF. Previous results have shown that adult unlesioned CSNs do not require BDNF for survival (Giehl et al., 1998, 2001). These results were obtained in conditions that restricted either the availability or expression of endogenous BDNF. TrkB receptors are also activated by NT-4. Because this neurotrophin is expressed in mature cortex (Timmusk et al., 1993), it is plausible that it could compensate for the absence of BDNF. In the present study, the results obtained with the TrkB neutralizing antibody strongly indicate that unlesioned neurons are not dependent on BDNF/TrkB signaling for survival. Similarly, the residual population of surviving CSNs after axotomy could be supported by NT-4, which would not be disclosed by BDNF neutralizing antibodies. This reasoning is not supported by the present results because no significant change was observed in the population of surviving neurons when using the two different antibodies, anti-TrkB and anti-BDNF.
Interestingly, unlike CSNs in vivo, CA3 hippocampal neurons in vitro survive after axotomy. However, these neurons do have decreased KCC2 levels after axotomy, and, similar to CSNs, they become dependent on BDNF, because their requirement for BDNF is disclosed by scavenging endogenous BDNF. This may indicate that CA3 neurons receive enough autocrine and/or paracrine trophic support, which is enough to promote survival but not to upregulate KCC2. Notably, application of exogenous BDNF to the damaged CA3 neurons in vitro promotes upregulation of KCC2.
Ipsilateral infusion of BDNF to unlesioned CSNs produced a significant decrease in the mRNA expression levels of KCC2. This is in accordance with our previous in vitro results showing that BDNF application to both acute and cultured hippocampal slices induced a dose-dependent downregulation of KCC2, and provides additional evidence that this effect has relevance in vivo. Although in organotypic hippocampal cultures the concentrations of BDNF used were relatively low (10 μg/ml), they resulted in a small but significant decrease in KCC2 expression and GABAA-mediated [Ca2+]i increase. This is also in accordance with our previous results showing that reduced levels of KCC2 expression result in a depolarizing shift in the reversal potential of GABAA-mediated responses (Rivera et al., 2002, 2004). It should be pointed out that BDNF may also affect trafficking and/or net ion transport activity of NKCC1 and KCC2 (Payne et al., 2003).
Embryonic neurons depend on BDNF for survival (Ghosh et al., 1994). During development, KCC2 levels rise and NKCC1 levels decrease, leading to a change in the mode of GABA signaling from depolarization to hyperpolarization (Rivera et al., 1999; Hübner et al., 2001), and mature central neurons do not need BDNF for survival. BDNF downregulates KCC2 in adult mature neurons (Rivera et al., 2002, 2004), but it upregulates KCC2 in both developing neurons (Aguado et al., 2003) and damaged neurons. Thus, traumatized neurons switch to a mode resembling the one that prevails during ontogeny.
Previous results on facial and dorsal motor neurons showed that GABAA-mediated responses become depolarizing after axotomy. These results suggested that the qualitative change in GABAA function affects the survival of damaged neurons (Nabekura et al., 2002; Toyoda et al., 2003). Similar depolarizing responses in immature neurons are caused by a high concentration of intracellular chloride that is maintained by the chloride accumulating activity of the Na–K–Cl cotransporter NKCC1 (Payne et al., 2003). GABA-induced membrane depolarization, in turn, activates voltage-gated calcium channels resulting in [Ca2+]i increase. The depolarizing effect of GABA in immature neurons can be blocked with the NKCC1 specific inhibitor bumetanide (Payne et al., 2003). Similarly, in the present study, the axotomy-induced GABA-mediated [Ca2+]i increase could be blocked by bumetanide, showing that the depolarizing GABA responses are maintained by NKCC1 in the injured neurons.
During development, there are critical periods for the maturation of neuronal circuits (Hensch, 2005). Termination of this time window is correlated with diminished plasticity. It is probable that a posttraumatic return to “developmental state” also takes place for a limited period of time (Cramer and Chopp, 2000), which could be considered to be a “posttraumatic critical period.” Notably, CSNs change their trophic dependence in a qualitative manner twice after injury: immediately after axotomy when CSNs acquire BDNF dependence, and for the second time after the acute postinjury phase when surviving neurons lose their BDNF dependence, presumably reassuming an “intact” state. For example, by day 42, KCC2 levels are recovered and even exceed the KCC2 levels in the intact control side, which indicates that GABAergic transmission has returned to its mature state. We cannot exclude the possibility that KCC2 would be functionally inactive despite the strong expression of KCC2. Considering the published data on KCC2 expression and function (Chudotvorova et al., 2005; Fiumelli et al., 2005; Lee et al., 2005; Farrant and Kaila, 2007), this would be, however, very unlikely.
The relationship between KCC2 expression levels and neuronal survival may not be simple. To prevent GABAA-mediated depolarization, an apparent alternative to bumetanide application would be to overexpress KCC2 in acute injured neurons. From a point of view of energy consumption, the last approach is radically different to the use of bumatanide. Na/K-ATPase consumes ATP to build up and maintain the transmembrane gradients of Na and K that are then used by, for example, NKCC1 and KCC2 to drive chloride transport. Thus, chloride transporters are heavy energy consumers (Buzsáki et al. 2007). Application of bumetanide that blocks chloride uptake through NKCC1 decreases transporter-mediated energy consumption, whereas overexpression of KCC2 results in the opposite. Considering that traumatized neurons are highly sensitive to low ATP levels, overexpression of KCC2 in traumatized neurons may lead to cell death instead of rescue. Perhaps this may be part of the basis behind the gradual upregulation of KCC2 in recovering neurons.
To design appropriate treatment strategies for the damaged CNS, it is crucial to know for what period of time neurons reside in the immature-like state, and when they return to the mature state. The present study suggests that monitoring KCC2 levels and the consequent changes in GABAergic actions can be used for identifying the onset and termination of posttraumatic critical periods in various kinds of neuronal circuits after trauma.
Footnotes
This work was supported by grants from the Academy of Finland, Sigrid Juselius Foundation and Biocentrum Helsinki (C.R., K.K., M.S.), the Deutsche Forschungsgemeinschaft SFB 530 (K.M.G.), a Deutscher Akademischer Austauschdienst studentship (J.T.-C.), and a Helsinki Biomedical Graduate School studentship (A.S.). We thank Dr. Urmas Arumäe for comments on a previous version of this manuscript. We also thank Marjo Heikura, Miika Palviainen, and Britta Leiner for excellent technical support, and Dr. Kimmo Tanhuanpää for imaging assistance. We thank Dr. R. James Turner for kindly providing the α-wNT NKCC1 antibody.
References
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Barbacid M. Neurotrophic factors and their receptors. Curr Opin Cell Biol. 1995;7:148–155. doi: 10.1016/0955-0674(95)80022-0. [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- Bonatz H, Röhrig S, Mestres P, Meyer M, Giehl KM. An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods. 2000;100:105–115. doi: 10.1016/s0165-0270(00)00238-7. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Kaila K, Raichle M. Inhibition and brain work. Neuron. 2007;56:771–783. doi: 10.1016/j.neuron.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona MA, Pozas E, Martínez A, Espinosa-Parrilla JF, Soriano E, Aguado F. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb Cortex. 2006;16:47–63. doi: 10.1093/cercor/bhi083. [DOI] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hubner CA, Pellegrino C, Ben Ari Y, Medina I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol (Lond) 2005;566:671–679. doi: 10.1113/jphysiol.2005.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cramer SC, Chopp M. Recovery recapitulates ontogeny. Trends Neurosci. 2000;23:265–271. doi: 10.1016/s0166-2236(00)01562-9. [DOI] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus. 2006;16:1032–1060. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- Davies AM. Neurotrophin switching: where does it stand? Curr Opin Neurobiol. 1997;7:110–118. doi: 10.1016/s0959-4388(97)80128-6. [DOI] [PubMed] [Google Scholar]
- Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem. 2000;275:26720–26726. doi: 10.1074/jbc.M003753200. [DOI] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Johnson EM., Jr Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Giehl KM, Schacht CM, Yan Q, Mestres P. GDNF is a trophic factor for adult rat corticospinal neurons and promotes their long-term survival after axotomy in vivo. Eur J Neurosci. 1997;9:2479–2488. doi: 10.1111/j.1460-9568.1997.tb01665.x. [DOI] [PubMed] [Google Scholar]
- Giehl KM, Schütte A, Mestres P, Yan Q. The survival-promoting effect of glial cell line-derived neurotrophic factor on axotomized corticospinal neurons in vivo is mediated by an endogenous brain-derived neurotrophic factor mechanism. J Neurosci. 1998;18:7351–7360. doi: 10.1523/JNEUROSCI.18-18-07351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl KM, Röhrig S, Bonatz H, Gutjahr M, Leiner B, Bartke I, Yan Q, Reichardt LF, Backus C, Welcher AA, Dethleffsen K, Mestres P, Meyer M. Endogenous brain-derived neurotrophic factor and neurotrophin-3 antagonistically regulate survival of axotomized corticospinal neurons in vivo. J Neurosci. 2001;21:3492–3502. doi: 10.1523/JNEUROSCI.21-10-03492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel NY, Strittmatter SM. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat Rev Neurosci. 2006;7:603–616. doi: 10.1038/nrn1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Nishiyama N, Saito H, Katsuki H. GABAA receptor stimulation promotes survival of embryonic rat striatal neurons in culture. Brain Res Dev Brain Res. 1997;98:253–258. doi: 10.1016/s0165-3806(96)00183-6. [DOI] [PubMed] [Google Scholar]
- Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience. 2001;104:933–946. doi: 10.1016/s0306-4522(01)00149-x. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Lee H, Chen CX, Liu YJ, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–782. doi: 10.1523/JNEUROSCI.20-02-00771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME. Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci. 2001;2:263–273. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmusk T, Belluardo N, Metsis M, Persson H. Widespread and developmentally regulated expression of neurotrophin-4 mRNA in rat brain and peripheral tissues. Eur J Neurosci. 1993;5:605–613. doi: 10.1111/j.1460-9568.1993.tb00526.x. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Ohno K, Yamada J, Ikeda M, Okabe A, Sato K, Hashimoto K, Fukuda A. Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J Neurophysiol. 2003;89:1353–1362. doi: 10.1152/jn.00721.2002. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997a;378:135–157. [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997b;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]