

Abstract
Brain-derived neurotrophic factor (BDNF) is a major regulator of activity-dependent synapse development and plasticity. Because BDNF is a secreted protein, it has been proposed that BDNF is released from target neurons in an activity-dependent manner. However, direct evidence for postsynaptic release of BDNF triggered by ongoing network activity is still lacking. Here we transfected cultures of dissociated hippocampal neurons with green fluorescent protein (GFP)-tagged BDNF and combined whole-cell recording, time-lapse fluorescent imaging, and immunostaining to monitor activity-dependent dendritic release of BDNF. We found that spontaneous backpropagating action potentials, but not synaptic activity alone, led to a Ca2+-dependent dendritic release of BDNF-GFP. Moreover, we provide evidence that endogenous BDNF released from a single neuron can phosphorylate CREB (cAMP response element-binding protein) in neighboring neurons, an important step of immediate early gene activation. Therefore, together, our results support the hypothesis that BDNF might act as a target-derived messenger of activity-dependent synaptic plasticity and development.
Keywords: BDNF, synaptic plasticity, LTP, development, action potential, neurotrophins
Introduction
Ongoing neuronal activity generated by neuronal networks contributes to the construction of cortical maps and circuits during development, as well to reorganization of preexisting connections in the adult brain. In the last years, evidence has shown that the morphogenic action of neuronal activity is, in part, mediated through the control of neurotrophin synthesis and secretion (Bonhoeffer, 1996; Lu and Figurov, 1997; Poo, 2001; Tyler et al., 2002). In particular, brain-derived neurotrophic factor (BDNF) has been reported to (1) modulate the strength of preexisting synapses and the formation or functional maturation of new synapses (Marty et al., 1997; Lu et al., 2005), (2) reverse the effects of activity deprivation (McAllister et al., 1996; Rutherford et al., 1997; Marty et al., 2000; Seil and Drake-Baumann, 2000), and (3) contribute to the induction of several forms of activity-dependent synaptic plasticity (Schuman, 1999; Huang and Reichardt, 2001; Bramham and Messaoudi, 2005; Lu et al., 2005).
With the observation that BDNF can be targeted into dendritic secretory granules of the regulated pathway of secretion (Lessmann et al., 2003), it was proposed that BDNF is released from the postsynaptic target cells in an activity-dependent manner to modulate synaptic development and plasticity. However, to date, direct evidence for postsynaptic release of BDNF triggered by physiological patterns of electrical activity is still lacking. A calcium-dependent release of BDNF was demonstrated in hippocampal and cortical neuronal cultures (Goodman et al., 1996; Griesbeck et al., 1999; Hartmann et al., 2001; Balkowiec and Katz, 2002; Gärtner and Staiger, 2002; Brigadski et al., 2005) and in acute hippocampal slices (Griesbeck et al., 1999; Aicardi et al., 2004) after the robust and sustained membrane depolarization produced by high potassium concentration, bath-applied glutamate agonist, or global tetanic stimulation. However, during physiological network activity, membrane depolarization occurs mainly as discrete events of short duration, i.e., EPSPs and/or action potentials (APs). Therefore, the current challenge is to determine whether ongoing network activity can lead to dendritic release of BDNF from a single neuron.
In the present study, we transfected cultures of dissociated hippocampal neurons with green fluorescent protein (GFP)-tagged BDNF and combined whole-cell recording time-lapse fluorescent imaging and immunostaining to monitor activity-dependent dendritic release of BDNF. We found that network-driven backpropagating action potentials (b-APs) trigger a calcium-dependent dendritic release of BDNF-GFP. Interestingly b-APs appeared to be necessary and sufficient to trigger BDNF-GFP secretion during ongoing network activity. We further show that APs evoked in single nontransfected neurons trigger endogenous BDNF release that, in turn, induces the phosphorylation of the cAMP response element-binding protein (CREB) in the neighboring neurons. These observations support the view that BDNF might serve as a target-derived regulator of activity-dependent synaptic plasticity.
Materials and Methods
Cell cultures and transfections
All animal experiments have been performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC). Neurons from postnatal day 0 rat hippocampus were dissociated using trypsin and plated on coverslips coated with poly-l-lysine at a density of 70,000 cells/cm2 in minimal essential medium (MEM) supplemented with 10% Nu-Serum (BD Biosciences), 0.8% glucose, 1 mm sodium pyruvate, 2 mm glutamine, and 10 IU/ml penicillin–streptomycin as previously described (Krapivinsky et al., 2003) and incubated at 37°C, 5% CO2. On days 3, 7, and 10 of culture incubation, one-half of the medium was changed to MEM with 2% B27 supplement (Invitrogen).
Eleven days after plating [11 d in vitro (DIV)], neurons were transfected with cDNAs using a Magnetofection kit (OZ Biosciences) and Lipofectamine 2000 (Invitrogen) according to OZ Biosciences protocol (Buerli et al., 2007). After transfection, culture media was replaced with fresh MEM with 2% B27 supplement (Invitrogen), and the cultures were incubated at 30°C, 5% CO2. For most of the experiments, neurons were transfected with green fluorescent protein (pEGFP; BD Biosciences) or BDNF-GFP (Haubensak et al., 1998). The efficiency of transfection as well as the neuronal morphology of the BDNF-GFP-transfected neurons was similar to that of GFP-transfected neurons, suggesting that the overexpression of BDNF-GFP had no detectable effect on neuronal viability and dendritic development. All experiments were performed at 13–14 DIV.
Electrophysiological recordings
Electrophysiological recordings were performed at 13–14 DIV. Neurons were continuously perfused with artificial CSF (ACSF) containing the following (in mm): 140 NaCl, 2.5 KCl, 10 HEPES, 10 d-glucose, and 2.0 CaCl2, and 1 MgCl2, pH 7.4. Recording electrodes (4–6 MΩ) were filled with a solution containing the following for voltage-clamp recording (in mm): 100 cesium gluconate, 20 CsCl, 10 HEPES, 4 Mg-ATP, 0.4 Na-GTP, 10 sodium phosphocreatine, and 1 EGTA, pH 7.2; or the following for current-clamp recording (in mm): 135 potassium gluconate, 10 KCl, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP. Recordings were made using an Axopatch-200A amplifier and pCLAMP acquisition software (Molecular Devices). Series resistance was <20 MΩ. Data were low-pass filtered at 2 kHz and acquired at 10 kHz, and analyzed using Clampfit software (Molecular Devices). All electrophysiology experiments were performed at room temperature.
Time-lapse imaging
Real-time imaging was performed on an inverted epifluorescence microscope (Nikon Diaphot 300) using a 40 × objective. GFP fluorescence was detected with excitation (470–510 nm) and emission (dichroic mirror: 495; 515–545 nm) bandpass filters. Illumination of the probe was controlled with an electronic shutter device (Uni-Blitz). Pictures were captured with a digital CCD camera (Hamamatsu ORCA-ER). Frames for time-lapse imaging were acquired every 10 s using Simple PCI software (Hamamatsu). Fluorescence intensity was measured from dendritic regions containing clusters of BDNF-GFP with ImageJ software (National Institutes of Health; http://rsb.info.nih.gov/ij/) after subtraction of background fluorescence. Clusters in which the fluorescence intensity varied during the 5 min control period by >5% were discarded from the analysis. Fluorescence decreases caused by photobleaching and constitutive release were corrected by subtracting the extrapolation of the fluorescence decrease in the first 5 min over the whole recording time. Values are plotted as the percentage of the fluorescence intensity of the last frame before stimulation. Percentage variation in the text and statistical analysis were calculated by comparing the relative fluorescence of the interval −100 to 0 s (control) with that of 500–600 s (after stimuli). In the high-potassium experiments, 47.5 mm NaCl was substituted by 47.5 mm KCl. In Figure 8, the release of BDNF-GFP was arbitrarily considered significant when the decrease in fluorescence intensity was >3% between 100 and 200 s after the stimuli.
Figure 8.

The probability of BDNF-GFP release increases with the number of APs. The probability to induce a dendritic BDNF-GFP secretion (fluorescence decrease >3% between 100 and 200 s after stimuli) is a function of the number of APs elicited by somatic current injection (at 3.96 ± 0.5 Hz). Inset, Representative trace of APs. In parentheses is the number of experiments.
Immunocytochemistry
Coverslips with transfected neurons were fixed (14 DIV) for 15 min (4% paraformaldehyde) at 4°C and rinsed several times in 0.1 m PBS, pH 7.4. Coverslips were then preincubated in a solution of PBS containing 3% normal goat serum and 0.1% Triton X-100 for 1 h at room temperature before the addition of primary antibody. The coverslips were then incubated overnight with antibodies raised against BDNF (rabbit; 1:1000; N-20; Santa Cruz Biotechnology), or trans-Golgi network 38 (mouse; 1:500; Abcam), or secretogranin II (rabbit; 1:500; Abcam), or MAP2 (mouse; 1:2000; Sigma), or synaptobrevin-2 (rabbit; 1:500; Synaptic Systems). Immunoreactivity was detected with a Cy3-coupled anti-rabbit secondary antibody or Cy3-coupled anti-mouse secondary antibody (both 1:1000; FluoProbes). Control tissues for all immunohistochemical experiments were prepared in an identical manner, but primary or secondary antibodies were omitted from the incubation solutions. Coverslips were examined with a Zeiss confocal microscope using an oil-immersion 63 × 1.4 objective. Confocal micrographs shown here are digital composites of a Z-series scan of 10–15 optical sections through a depth of 4–6 μm. Final images were constructed with ImageJ software.
Phospho-CREB activation and immunocytochemistry of cultured hippocampal neurons
Twenty-four hours before stimulation (13 DIV), one-half of the culture medium was changed to MEM with 2% B27 supplement. We gave special attention to the standardizing of the neuronal densities and maintenance of culturing conditions because any change of any of the parameters affected CREB activation. To reduce the basal level of CREB phosphorylation, cultures were incubated for 4 h in TTX (1 μm); they were then transferred in the recording chamber and perfused with ACSF containing 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 5 μm) and d-APV (40 μm). In the TrkB IgG condition, 5 μg/ml Ig was added both during incubation and recording. Whole-cell recording of one neuron per coverslip was done in current-clamp configuration. For the stimulated group, four bursts (0.2 Hz) of 10 action potentials each (10 Hz) were produced by 10 ms current steps. Five to ten minutes after stimulation, the neurons were fixed, permeabilized, preincubated as described above, and coincubated with rabbit anti-phospho-CREB (pCREB; rabbit; 1:1000; Millipore) and with mouse anti-MAP2 (Sigma). Immunoreactivity for pCREB was detected with a Alexa 488-coupled anti-rabbit secondary antibody (1:500; FluoProbes). Immunoreactivity for MAP2 was detected with a Cy5-coupled anti-mouse secondary antibody (1:500; Jackson ImmunoResearch Laboratories). All procedures were performed in phosphate-free solution containing 140 mm NaCl, 5 mm KCl, and 10 mm HEPES-Na, pH 7.4. CREB immunostaining was performed in parallel cultures treated in the same condition as for pCREB experiment using a rabbit anti-CREB (CREB; rabbit; 1:2000; Cell Signaling Technology)
Surface GFP immunofluorescence staining
Surface-bound BDNF-GFP was detected by immunocytochemical detection against GFP under nonpermeable conditions on neuronal cultures treated with TTX, 4-aminopyridine (4-AP), or glutamatergic receptor antagonists. Treated and control experiments were done in parallel from sister, low-density, neuronal cultures. Moreover, the number of transfected neurons in the different conditions and the average distance between them was not significantly different: (control: 14.9 ± 5.5 transfected neurons, average distance of 396 ± 218 μm; 4-AP: 15.3 ± 6.8 transfected neurons, average distance of 352 ± 236 μm; TTX: 14.5 ± 7.3 transfected neurons, average distance of 378 ± 184 μm; p = 0.53, ANOVA). After washing in ACSF to remove unbounded BDNF-GFP, the still-living cultures were incubated at 4°C for 1 h in the presence of an anti-GFP antibody (10 μg/ml; Invitrogen). Because low temperature is known to inhibit endocytosis, BDNF-binding receptors remain on the surface during this incubation. After the incubation, the neurons were washed with ice-cold 0.1 m PBS, pH 7.4, to remove the unbound antibody, followed by fixation for 15 min with 4% paraformaldehyde/4% sucrose. After fixation, the neurons were exposed to a saturating concentration (10 μg/ml) of anti-rabbit secondary antibody coupled to either Cy3 (FluoProbes) or Cy5 (Jackson ImmunoResearch Laboratories) for 1.5 h under a nonpermeabilized condition. Surface immunocytochemical detection of TrkB receptor was done on cultures subjected to the same experimental manipulation as for surface-bound BDNF-GFP by using the polyclonal rabbit TrkB antibody (1:2000; Abcam).
The coverslips were mounted on slides with an aqueous mounting medium (Gel Mount; Biomeda).
Image acquisition and analysis
Images were acquired with an Olympus Fluoview 500 confocal microscope using a 20× objective and an oil-immersion 40 × 1.0 lens. For this, we first focused on rhodamine-filled neurons. Fluorescent images of rhodamine, GFP, or MAP2 and pCREB were then acquired. Confocal micrographs shown here are digital composites of Z-series scans of 10–15 optical sections through a depth of 4–6 μm. Final images were constructed with ImageJ software.
pCREB analysis.
For analysis of the intensity of pCREB staining in neuronal cells, we first created a binary mask from MAP2-positive cells in the frame containing the rhodamine-filled cell and then analyzed pCREB intensity only in regions overlapping the binary mask. This procedure allowed avoiding detection of pCREB staining on non-neuronal cells. Acquisition parameters were same for every set of experiments.
BDNF-GFP-binding receptor analysis.
Quantification of GFP-Cy3 material colocalized with BDNF-GFP was performed with ImageJ. Coexistence of GFP-Cy3 and BDNF-GFP in transfected neuronal cells is expressed as the ratio between the mean of GFP-Cy3 and BDNF-GFP colocalized area and the mean BDNF-GFP area ± SEM as percentages. The sections were scanned by sequential acquisition of fluorescence, first GFP and then Cy3, to avoid overlap of excitation and emission of fluorescence. Verifications based on the observation of double-labeled details present in the cells close to the coverslip interface showed no visible shifts between GFP and Cy3 fluorescence emissions. The density of GFP-Cy3 immunoreactivity colocalized with BDNF-GFP was indicated by vertical picture of the gray level histograms giving the amounts of immunoreactive material present. The intensity of each pixel, which ranged between 0 and 255 gray levels, was proportional to the number of fluorescent photons emitted by the corresponding point in the section.
Statistical analysis
If not stated otherwise, all population data are expressed as mean ± SEM. Paired Student's t test was used to compare statistical significance between pre- and post-depolarizing pulse (DP) or -AP values, and unpaired Student's t test was used to examine the statistical significance of the differences between groups; ANOVA was used for multiple comparisons. Significantly different values (p < 0.05) are indicated in figures with asterisks.
Results
Activity-dependent release of BDNF-GFP
We first studied the cellular distribution and targeting of BDNF-GFP in primary cultures of hippocampal neurons. As already reported (Haubensak et al., 1998; Hartmann et al., 2001), a patchy distribution of the BDNF-GFP fluorescence was observed in the neuronal processes, whereas in neurons expressing GFP alone, the fluorescence was always distributed uniformly throughout the cytoplasm (Fig. 1a,b). Immunohistochemical labeling of the neurons further showed that BDNF-GFP is targeted to dendritic secretory granules localized to the vicinity of synaptic contacts (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.
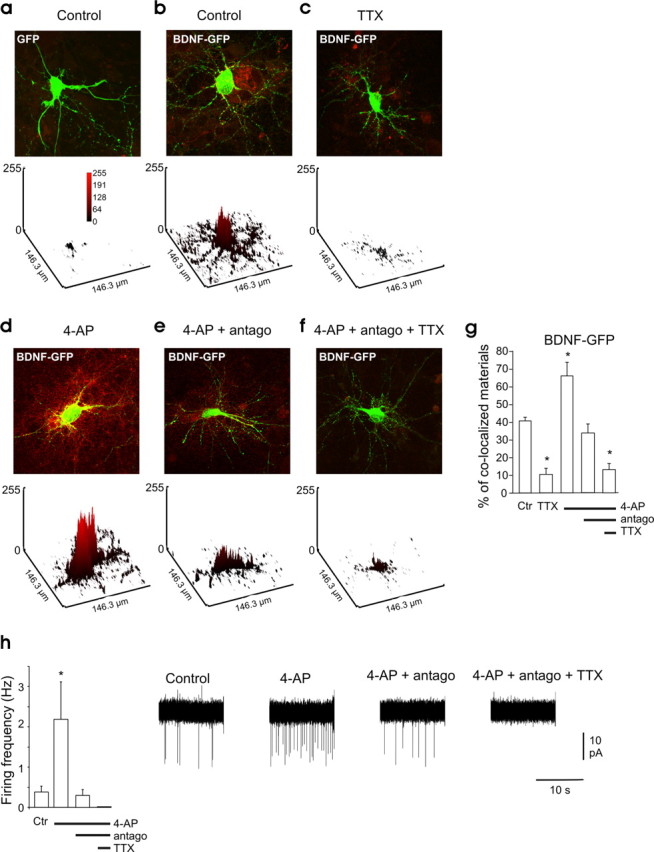
Spontaneous network activity triggers BDNF-GFP release. a–f, Overlapped images showing intracellular GFP fluorescence (green) and secreted BDNF-GFP detected using anti-GFP antibody (red) under nonpermeable conditions. a, Hippocampal neurons transfected with GFP alone. b–f, Neurons transfected with BDNF-GFP. b, Control condition. c, In the presence of TTX (1 μm). d, Stimulated with 4-AP (50 μm). e, Stimulated with 4-AP (50 μm) in the presence of NBQX (5 μm), d-APV (40 μm), and bicuculline (10 μm), indicated in the figure as “antago.” f, Stimulated with 4-AP (50 μm) in the presence of NBQX, d-APV, bicuculline, and TTX (1 μm). Top, Merged picture of both fluorescence channels of neurons transfected with the BDNF-GFP construct (green) and immunocytochemistry staining using an antibody against the GFP (red). Bottom, Quantification of the surface-bound BDNF-GFP on the transfected neuron (yellow signal); the plots are a three-dimensional representation of the mean gray level values. g, Quantitative analysis of surface-bound GFP comparing BDNF-GFP-transfected neurons in different ACSF conditions (n = 4 different cultures, 4 neurons per culture in each condition). h, Average firing rate of the neurons in the different conditions, measured in cell-attached configuration (n = 5 neurons in each condition). Right, Representative electrophysiological traces for each condition. The green signal produced by released BDNF-GFP is not visible in the images presented here because of the low laser intensity used to avoid saturation of the green signal in the transfected neurons. The red signal in the untransfected cell is attributable to the BDNF-GFP secreted by the transfected neurons that bound to membrane TrkB receptors of the neighboring cells. Antago in e, f, and h stands for NBQX (5 μm), APV (40 μm), and bicuculline (10 μm). Ctr, Control. In this and following figures, * indicates p < 0.05 compared with control condition.
We then asked whether ongoing network activity can cause BDNF secretion and how the release is affected by the modification of the activity level of the cultures. For this purpose, released BDNF-GFP was detected by surface immunofluorescence staining (see Materials and Methods). Because the surface-bound BDNF-GFP depends not only on the quantity of the release but also on the density of the cells surrounding the transfected neurons, we compared the different conditions by evaluating exclusively the BDNF-GFP bound on the extracellular membrane of the transfected neurons. For this reason, we use an antibody directed against GFP (red signal) to identify BDNF-GFP that has been released from the transfected cells. In Figure 1, released BDNF-GFP that bound to the extracellular membrane of BDNF-GFP-expressing and nontransfected neurons appears respectively as yellow and red signal. Yellow staining surrounding BDNF-GFP-expressing neurons was observed in control conditions (41 ± 2% surface colocalized material) (Fig. 1b). In contrast, surface GFP immunofluorescence was not detectable on GFP-only-expressing neurons (3.5 ± 1.5% surface colocalized material) (Fig. 1a). Adding TTX (1 μm) to the culture medium for 3 h resulted in a marked decrease in the surface-bound BDNF-GFP, compared with control nontreated cultures (Fig. 1c,h).
Ten minutes of 4-AP (50 μm) application led to an increase in postsynaptic firing activity (Fig. 1i) and dramatically increased the density of surface-bound BDNF-GFP (Fig. 1d,h). In the presence of 4-AP, the application of the ionotropic glutamatergic and GABAergic receptor antagonists NBQX, d-APV, and bicuculline decreases both neuronal firing (Fig. 1i) and the density of surface-bound BDNF-GFP (Fig. 1e,h) Finally, a further application of TTX abolished neuronal firing (Fig. 1i) and largely reduced density of surface-bound BDNF-GFP (Fig. 1f,h). Immunolabeling of the extracellular domain of the TrkB receptors under nonpermeabilized conditions shows that membrane TrkB receptor expression was not modified by the different treatments (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), further supporting that the modifications in surface-bound BDNF-GFP are consequences of BDNF-GFP release changes.
Altogether, these observations show that, in hippocampal cultures, spontaneous neuronal firing is required to trigger BDNF-GFP release.
Repeated postsynaptic depolarization triggers a dendritic Ca2+-dependent release of BDNF-GFP
Next we combined whole-cell recording and time-lapse fluorescence imaging of transfected hippocampal neurons to directly determine whether BDNF can be secreted from the dendrites after short direct postsynaptic depolarization activating voltage-dependent Ca2+ channels (VDCCs). To prevent possible effects of excitatory synaptic activity, voltage-clamp whole-cell recordings were performed in the presence of the glutamatergic receptor antagonists NBQX (5 μm) and d-APV (40 μm). After a stable control period, 20 DPs (50–60 mV amplitude, 500 ms duration at 0.1 Hz) were applied to the cell through the recording pipette. Each DP triggered an inward current (Fig. 2a). DPs applied to BDNF-GFP-expressing neurons led to a significant decrease of fluorescence intensity (−7.8 ± 1.6% change 10 min after the DPs; p = 0.0005 compared with pre-DPs; n = 11) (Fig. 2a,b). When applied on GFP-only-expressing neurons, DPs only led to a transient nonsignificant decrease in fluorescence (1 ± 1% change 10 min after DPs; p = 0.42 compared with pre-DPs; p = 0.0001 compared with post-DPs in BDNF-GFP release experiments; n = 9) (Fig. 2c,d), thus ruling out possible direct effect of DPs on GFP fluorescence properties.
Figure 2.

Depolarizing steps trigger dendritic secretion of BDNF-GFP. a, Decrease of fluorescence intensity from BDNF-GFP granules localized in the dendrites was produced by 20 depolarizing steps of 50–60 mV depolarization, 500 ms long, given at 0.01 Hz. Note that fluorescence decreased within the white circle and did not increase in the surrounding area, indicating that the decrease in fluorescence intensity was not attributable to lateral movements in the x–y-axes. Left, Representative traces of a DP; note the inward Ca2+ current produced by the depolarization. Right, Example of dendritic BDNF-GFP granules (indicated by circles) before and after the DPs. b, Average time course of dendritic fluorescence change evoked by the DPs (n = 11). c, d, DP failed to produce significant variation of fluorescence in GFP-only-transfected neurons (n = 9). e, The effect of DPs on dendritic fluorescence was abolished by bath-applied Cd2+ (200 μm; n = 8); note the absence of Ca2+ current in response to DP (superimposed control and Cd2+ traces). f, Postsynaptic loading of GDP β-S (0.6 mm), a G-protein inhibitor that blocks granular secretion, prevented the DP-induced decrease of fluorescence in the dendrites of BDNF-GFP-transfected cells (n = 8); note that the Ca2+ current in response to DP was not affected (superimposed control and Cd2+ traces). All the experiments were performed in the presence of NBQX and APV. Rel., Relative.
Because an intracellular Ca2+ influx through VDCCs is necessary for dendritic BDNF release (Hartmann et al., 2001), we investigated the effect of Cd2+, a broad spectrum blocker of VDCCs. Bath-applied CdCl2 (100 μm) blocked the DP-induced inward currents (Fig. 2e, inset) and prevented the decrease of fluorescence produced by the DPs (−2 ± 1.4% change; p = 0.33 compared with pre-DPs; p = 0.008 compared with post-DPs in control group; n = 8) (Fig. 2e). The Ca2+ dependency of the DP-induced decrease in fluorescence and the neuritic accumulation of BDNF-GFP in secretory granule (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) suggested that BDNF is released by membrane fusion and exocytosis. To test this hypothesis, we investigated the effect of GDPβS, a potent inhibitor of exocytosis (Zilberter et al., 1999). GDPβS (0.6 mm) was dissolved in the pipette recording solution. In GDPβS-loaded cells, the DPs had no effect on the BDNF-GFP fluorescence intensity (0 ± 1.5%; p = 0.48 compared with pre DP; p = 0.001 compared with control; n = 8) (Fig. 2f). GDPβS had no effect on inward Ca2+ currents produced by the DPs (Fig. 2f).
The release of BDNF-GFP by DPs was confirmed by GFP surface immunostaining (Fig. 3). Before recording, the culture medium was washed three times, followed by 3 h in TTX, to remove BDNF-GFP released by spontaneous activity. The culture was then transferred in the recording chamber in the presence of NBQX (5 μm) and d-APV (40 μm). The recorded cell was loaded with the fluorescent dye rhodamine for post hoc identification. BDNF-GFP-transfected cells were stimulated either with the standard suprathreshold DPs or with a subthreshold DPs (steps of 5 mV) that did not produced inward Ca2+ currents, and the culture was immediately processed for immunohistochemistry (Fig. 3). Suprathreshold DPs induced GFP staining surrounding the BDNF-GFP-expressing cells that spread to neighboring nontransfected cells. With subthreshold DPs, surface-bound GFP was restricted to proximal BDNF-GFP expressers, possibly because of constitutive release (Brigadski et al., 2005) (Fig. 3a). Quantification of surface-bound BDNF-GFP on transfected neurons show that suprathreshold DPs significantly increased BDNF-GFP released (subthreshold DPs, 9 ± 2% of colocalization; suprathreshold DPs, 56 ± 4% of colocalization; p = 0.0001).
Figure 3.

Surface staining confirmed the BDNF-GFP secretion produced by the depolarizing steps. BDNF-GFP-expressing neurons received either 20 suprathreshold or 20 subthreshold DPs applied through the recording pipette. The intracellular solution contained rhodamine for post hoc identification. Surface staining of released BDNF-GFP was detected by immunocytochemical staining against GFP under nonpermeabilized conditions (blue staining).
Altogether, these results show that repeated, short postsynaptic membrane depolarizations trigger a decrease in dendritic fluorescence in BDNF-GFP-expressing neurons that reflects a Ca2+-dependent release of BDNF.
Spontaneous backpropagating action potentials trigger dendritic release of BDNF
As illustrated in Figure 4, both spontaneous and evoked somatic action potentials travel back through the dendritic tree as b-APs (n = 4; average distance, 100 μm). We therefore investigated whether spontaneous b-APs can lead to dendritic BDNF secretion. Therefore, we performed time-lapse-imaging on BDNF-GFP-expressing neurons recorded in current-clamp mode. During the control period, the cells were kept hyperpolarized at around −80 mV by steady current injection, to prevent action potential firing. After the control period, neurons were depolarized to −52 ± 1 mV (n = 14; range, −49 to −60 mV). In five neurons, this procedure led to the generation of synaptically driven firing activity; in these cases, we observed a decrease in the dendritic BDNF-GFP fluorescence (6 ± 1% decrease in fluorescence intensity 10 min after the first AP; n = 5; p = 0.002 compared with prespike period) (Fig. 5a). On average, the cells fired 65 ± 22 APs at 0.5 ± 0.12 Hz (5.7 ± 1.4 Hz of maximum instantaneous frequency, quantified during the first 2 min after the first AP). On the other hand, in the neurons in which synaptic activity did not reach the threshold for AP generation (n = 5), or in which APs were prevented by the Na+ channel blocker 5-N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX314; 5 mm) in the pipette solution (n = 4), no variation in BDNF-GFP fluorescence was observed (0.2 ± 0.9% increase; n = 9; p = 0.7 compared with control; p = 0.003 compared with firing group) (Fig. 5b).
Figure 4.

Dendritic backpropagation of APs in cultured neurons. a, Example of paired somatic (s) and dendritic (d) whole-cell recording of a neuron in culture. b, APs elicited by somatic current injection backpropagate into the dendrite. c, Spontaneous AP generated in the soma backpropagates into the dendrite. Note the delay between somatic and dendritic AP.
Figure 5.

Synaptically driven b-APs trigger dendritic BDNF secretion through membrane depolarization. a, Synaptically induced b-APs, occurring when neurons were held at −52 ± 1 mV, produced a dendritic BDNF-GFP secretion (n = 5); the arrow indicates the arrival of the first AP. Left, Representative trace. b, BDNF-GFP secretion did not occur when APs were prevented by QX314 in the recording pipette (n = 9). Left, Representative trace. c, Membrane depolarization to −40 mV, with QX314 in the pipette, produced a BDNF-GFP secretion even in the absence of APs (n = 6). Left, Representative trace. Rel., Relative.
A recent report has provided evidence for BDNF secretion from a single neuron produced by steady depolarization to −40 mV in voltage-clamp recording (Magby et al., 2006). To exclude possible direct interaction of QX314 with the process of BDNF secretion, transfected neurons were depolarized from −80 to −40 mV in current-clamp conditions, while the firing activity was prevented by intracellular application of QX314. This protocol produced dendritic release of BDNF-GFP not statistically different from that induced by firing activity (4 ± 1% decrease in fluorescence intensity; n = 6; p = 0.02 compared with control period; p = 0.24 compared with firing induced release) (Fig. 5c).
These observations show that backpropagating action potentials are the principal trigger of dendritic BDNF-GFP release induced by spontaneous network activity.
Spike-induced BDNF release required Ca2+ from VDCC but not from intracellular Ca2+ stores
We next examined the minimal requirements for AP-induced BDNF-GP secretion. First we investigated the contribution of VDCCs for the spike-induced BDNF secretion. Because VDCCs blockers can affect presynaptic transmitter release and thus, indirectly, postsynaptic APs generated by ongoing synaptic activity, intrinsic b-APs were directly induced by short (200 ms to 1 s) current injections through the recording electrode. In control conditions, the b-APs led to a decrease in dendritic BDNF-GFP fluorescence (4.8 ± 1.2% decrease in fluorescence intensity; n = 17; p = 0.01) (Fig. 6a). This decrease was abolished by bath application of Cd2+ 200 μm (0.7 ± 0.8% decrease; n = 9; p = 0.42 compared with prespike period; p = 0.03 compared with control) (Fig. 6b) or in zero extracellular calcium and 3 mm EGTA (0.2 ± 1% decrease; n = 4; p = 0.79 compared with prespike period; data not shown) (see Hartmann et al., 2001).
Figure 6.

VDCC activation, but not intracellular Ca2+ stores, is required for b-AP-induced BDNF secretion. a, BDNF-GFP secretion was induced by firing activity produced by somatic current injection. Left, Representative trace of step-induced firing activity (n = 17). b, BDNF-GFP release was prevented by bath application of the VDCC blocker CdCl (200 μm; n = 9). c, Thapsigargin (10 μm) did not prevent BDNF-GFP release elicited by b-APs (n = 8). Left, Representative traces of step-induced firing activity. Arrows indicate the arrival of the first action potential. In a–c, Vh = −70 mV. Rel., Relative.
Several studies have stressed the important role of internal Ca2+ stores in amplifying the initial Ca2+ influx for the release of BDNF by high extracellular K+ or tetanic stimulation (Griesbeck et al., 1999; Balkowiec and Katz, 2002; Kolarow et al., 2007). We therefore investigated the possible contribution of intracellular Ca2+ stores in BDNF release induced by b-APs. Twenty minutes of preincubation of the neuronal cultures with 10 μm thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+-ATPase, had no effect on dendritic BDNF-GFP release induced by b-APs (5 ± 1% decrease in fluorescence intensity; n = 8; p = 0.002) (Fig. 6c). Similarly, thapsigargin had no effect on BDNF secretion occurring during spontaneous synaptic activity (Fig. 7). Thapsigargin was, however, able to (1) produce a detectable rise in resting intracellular Ca2+ concentration (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), (2) reduce the amplitude of glutamate-induced Ca2+ rise (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and (3) as already reported (Kolarow et al., 2007), prevent BDNF-GFP released induced by bath application of 50 mm potassium (KCl-induced decrease of fluorescence in control = 7.8 ± 1.3%; n = 11; p = 0.0002 compared with pre-KCl period; KCl-induced decrease of fluorescence in the presence of thapsigargin = 2 ± 2%; n = 8; p = 0.3 compared with pre-KCl period; p = 0.02 compared with control) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We concluded that, unlike in experiments with bath application of KCl, the release of BDNF induced by b-APs does not require activation of other than VDCC calcium sources.
Figure 7.

Intracellular Ca2+ stores are not required for BDNF secretion produced by ongoing activity. a–c, Surface immunofluorescence staining on neuronal cultures in control condition (a) and incubated in TTX (b) or thapsigargin (10 μm; c) for 3 h. d, Quantitative analysis of surface-bound GFP (yellow signal/green signal) in the different conditions (n = 3 cultures, 4 neurons per cultures in each condition). Thapsi, Thapsigargin.
We next investigated the minimum number of APs necessary to produce a significant BDNF-GFP fluorescence loss. To control the exact number of APs produced for each neuron, APs were induced by short (10 ms) somatic current injections applied at a frequency of 4 Hz in the presence of NBQX and APV. As shown in Figure 8, increasing the number of APs increases the probability of BDNF secretion reaching a plateau at eight APs.
Altogether, these observations show that backpropagating action potentials trigger a dendritic, Ca2+-dependent release of BDNF-GFP through the activation of VDCCs.
b-APs trigger endogenous BDNF release
To date, experiments on activity-dependent release of BDNF in cultured neurons have been performed on transfected cells that overexpress BDNF (Goodman et al., 1996; Griesbeck et al., 1999; Hartmann et al., 2001; Balkowiec and Katz, 2002; Gärtner and Staiger, 2002; Brigadski et al., 2005). However, such overexpression could have modified the release properties of BDNF. We therefore asked whether b-APs are able to trigger secretion of native BDNF in nontransfected cells. To this end, we used pCREB as a sensor of BDNF release. Once released, BDNF interacts with high-affinity TrkB receptors to activate downstream signaling pathways. One of the most common is the ERK (extracellular signal-regulated protein kinase) pathway, which leads to the phosphorylation of CREB (Ghosh et al., 1994). We reasoned that BDNF released from one single stimulated neuron will bind to TrkB receptors in the neighboring cells and induce the phosphorylation of CREB. Nontransfected cultures were incubated with TTX for 4 h to reduce the basal levels of pCREB and then transferred in the recording chamber in the presence of CNQX and d-APV to prevent postsynaptic firing generated by spontaneous synaptic activity. Four bursts of 10 APs (10 Hz; 5 s between bursts) were produced by 10 ms current step in one single neuron per culture, a protocol that induces BDNF-GFP release from transfected neurons (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). The neuron was loaded with rhodamine for post hoc identification. Five minutes after stimulation, the cultures were fixed and processed for immunocytochemistry. APs induced an increase in pCREB level in MAP2-positive cells (Fig. 9d–f,j). pCREB level was significantly (p = 0.001) lower in cultures in which the cells were patched but not stimulated (Fig. 9a–c,j). Finally, the increase in pCREB induced by APs was prevented in the presence of the BDNF scavenger TrkB-IgG (5 μg/ml) in the culture medium, showing that CREB phosphorylation results from the release of BDNF (Fig. 9g–i,j). In all experiments, pCREB level was not modified in the MAP2-negative cells (i.e., glial cells) (p = 0.973). No differences in the total amount of CREB was observed for the different conditions (mean gray values: control, 236 ± 18; n = 3 cultures, 20 cells per culture; TrkB, 230 ± 20; n = 3 cultures, 20 cells per culture; stimulated, 232 ± 17; n = 3 cultures, 20 cells per culture; p = 0.32, ANOVA; data not shown).
Figure 9.
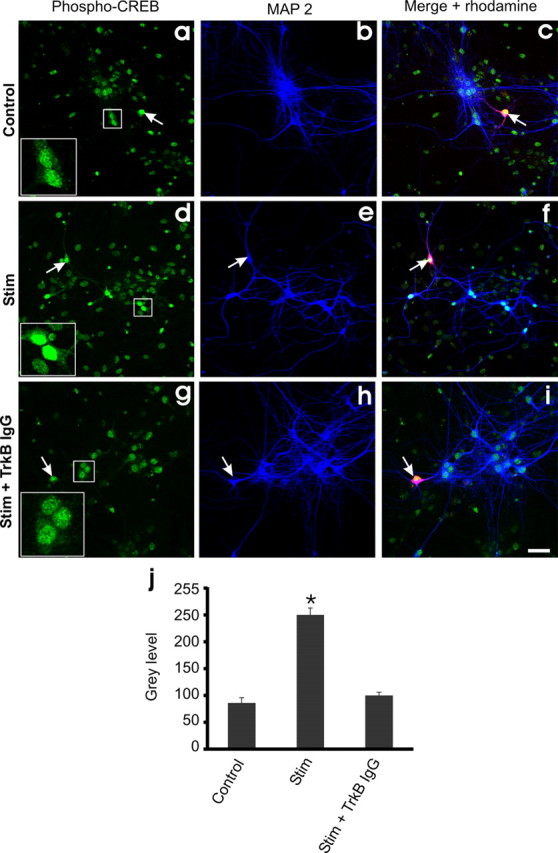
Endogenous BDNF is released by firing activity. Shown is activation of pCREB in nontransfected neuronal cells after electrophysiological stimulation. Left and middle, Images of pCREB (left) and MAP2 (middle), in neurons neighboring the patched rhodamine-filled cell (arrows). Right, Merged pictures of the rhodamine-filled cell immunofluorescence (red) in the presence of MAP2 and pCREB immunostaining (blue) for the same field of view. Scale bar, 45 μm. a–c, Control condition; cell patched but not stimulated. d–f, Cell patched and stimulated to fire (40 spike at 10 Hz). g–i, Cell patched and stimulated to fire (40 spikes at 10 Hz) in the presence of the BDNF scavenger TrkB IgG. j, Histogram showing the pCREB level activation by measurement of fluorescence intensity (mean gray level values) (n = 3 cultures in each condition. The amount of pCREB was calculated through a distance of 250 μm from the recorded neuron. A total of 47, 56, and 49 neurons, respectively, were used for analysis of the intensity of pCREB in control, stimulated, and TrkB-IgG conditions). Electrophysiology was performed in ACSF supplemented with NBQX and APV. Stim, Stimulated.
As illustrated in Figure 10, pCREB-positive somata are not only restricted to the vicinity of the stimulated cell but can be observed at long distance (up to 2 ± 0.5 mm; n = 3) (Fig. 10a). One possible explanation for such broad distribution of pCREB-positive neurons relies on the fact that both dendrites and axons can release BDNF when stimulated (Lessmann et al., 2003) and that BDNF can bind to TrkB receptors located on both axons and dendrites of the “recipient” cells (Ginty and Segal, 2002; Nagappan and Lu, 2005). Given the broad and complex dendritic and axonal distribution of the cells in cultures (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), pCREB-positive somata can be observed at a distance from the stimulated cell. To restrict the release of BDNF to proximal neurites, in another series of experiments, depolarizing pulses (20 DPs of 50 mV, 500 ms duration at 0.1 Hz) were applied through the recording pipette, in the presence of TTX 1 μm, to directly activate proximal VDCCs while reducing the spread of the depolarization into distal neurites. In these conditions, DPs induced an increase in pCREB level restricted to the neighboring MAP2-positive cells (Fig. 10b) (radius from the soma of the stimulated cell = 230 ± 50 μm; n = 3).
Figure 10.
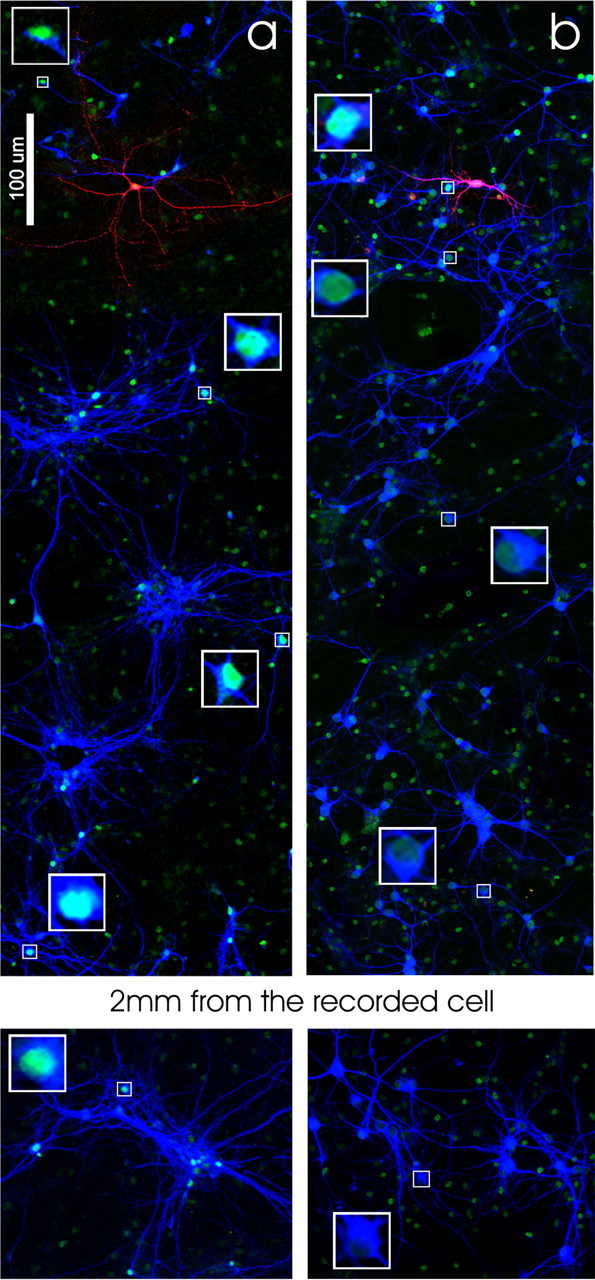
Distribution of pCREB-positive neurons after single-cell stimulation. a, CREB phosphorylation produced by neuronal stimulation (40 spikes at 10 Hz) in current clamp is distributed in the culture and can be observed at a distance from the stimulated cell. b, CREB phosphorylation produced by neuronal stimulation (20 DP of 50 mV at 0.1 Hz) in voltage clamp in the presence of TTX is limited to the neurons surrounding the stimulated cell. Squares show nuclei at higher magnification.
These experiments therefore show that spike firing in one “donor” single cell can induce the release of endogenous BDNF that activates TrkB receptors on “recipient” neurons
Discussion
Several studies have shown that impairing network activity produces structural and functional alterations that are reverted by exogenous application of BDNF (McAllister et al., 1996; Rutherford et al., 1997; Marty et al., 2000; Seil and Drake-Baumann, 2000), suggesting that activity-dependent secretion of BDNF can act as a regulatory mechanism of network development and plasticity. However, direct evidence for dendritic BDNF release induced by ongoing network activity was still lacking. Our study is the first direct demonstration of cell autonomous, Ca2+-dependent, dendritic release of BDNF in response to spontaneous activity. Moreover, we provide evidence that endogenous BDNF released from a single neuron can phosphorylate CREB in neighboring neurons, an important step of immediate early gene activation (Finkbeiner et al., 1997). Therefore, together, our results support the general hypothesis that BDNF can serve as a target-derived messenger for activity-dependent development and plasticity in response to single-neuron activation (Tanaka et al., 2008).
b-APs are necessary and sufficient to trigger activity-dependent dendritic release of BDNF
Our results show that b-APs are necessary and sufficient to trigger activity-dependent dendritic release of BDNF. Thus, we found that (1) ionotropic glutamatergic receptor antagonists were not able to abolish activity-dependent release of BDNF (Fig. 1), (2) in the presence of QX314, to block APs in the recorded cell, synaptically induced membrane depolarization was not able to produce any detectable release of BDNF-GFP (Fig. 5b), and (3) evoked b-APs triggered in single cells induced dendritic release of BDNF-GFP (Fig. 6a). Using phosphorylation of CREB in neighboring neurons as a sensor for BDNF release, to avoid the possible artifactual consequences of overexpressing tagged proteins, we also show that APs induced in nontransfected neurons can lead to the release of endogenous BDNF in the presence of ionotropic glutamatergic receptor antagonists (Fig. 9).
Our results therefore suggest that depolarization mediated by the activation of ionotropic glutamatergic transmission during ongoing synaptic activity does not directly lead to BDNF secretion. We, however, cannot exclude a contribution of receptor-mediated depolarization on BDNF secretion in a more intact system, for instance, during physiological or pathological network oscillations. Thus, a dendritic release of BDNF-GFP evoked by presynaptic tetanic stimulation of glutamatergic terminals in neuronal cultures was blocked by AMPA and NMDA receptor antagonists (Hartmann et al., 2001). This suggests that a receptor-mediated depolarization of postsynaptic neurons, caused by summation of glutamatergic postsynaptic potentials and possibly the generation of APs, can trigger the postsynaptic release of BDNF during strong presynaptic tetanic stimulation (Hartmann et al., 2001). Therefore, although dendritic BDNF release in the absence of neuronal firing can be triggered by experimentally induced membrane depolarization of sufficient amplitude (Fig. 5c) [but see also Magby et al. (2006) and Kolarow et al., (2007)], during ongoing spontaneous network activity b-APs generation is a required and sufficient step to trigger dendritic BDNF release. In the present study, we show that as few as eight b-APs at a frequency of 4 Hz are sufficient to produce a secretion of BDNF. In a previous study, Gärtner and Staiger (2002) reported that a minimum of 100 electrical shocks induced at a frequency of 50 Hz by field stimulation were required to detect BDNF secretion. This difference could be explained by a lower sensitivity of the ELISA technique used in this former study to measure the extracellular BDNF. Interestingly, hippocampal pyramidal neurons fire spontaneous bursts of three to seven APs in vivo (Ranck, 1973). Thus, our data describe that a physiological pattern of neuronal firing can trigger dendritic secretion of BDNF. This provides a new perspective on studies of the role of ongoing neuronal activity in the maintenance of endogenous BDNF levels in neuronal cultures and brain.
Several studies have stressed the important role of internal Ca2+ stores in amplifying the initial Ca2+ influx for the release of BDNF after electrical field stimulation (Balkowiec and Katz, 2002) or high K+ concentration (Griesbeck et al., 1999; Kolarow et al., 2007). Here, we show that the inhibitor of the endoplasmic reticulum Ca2+-ATPase thapsigargin, which efficiently prevents BDNF secretion triggered by high-potassium application (Kolarow et al., 2007; present study), does not prevent BDNF secretion triggered by b-APs or ongoing synaptic activity. This result suggests that the mechanism underlying Ca2+-dependent secretion of BDNF could differ depending on the stimulating protocol. The reason for this dissimilarity is presently unknown. Difference in kinetic and/or amplitude between KCl- or AP-induced Ca2+ rises could explain why amplification of the Ca2+ signal by intracellular stores is required in one case but not the other. Internal stores might be more important for potassium-induced release because of the inactivation of the VDCC after such prolonged depolarization, whereas this inactivation is absent after repetitive (i.e., phasic) stimulation. It should be pointed out that thapsigargin has been reported to increase the decay time course but not the amplitude of the transient Ca2+ signal evoked by b-APs, suggesting that internal Ca2+ stores participated in dendritic clearance of Ca2+, but not in the Ca2+ rise (Markram et al., 1995) (but see Sandler and Barbara, 1999).
Functional significance
Backpropagating APs in hippocampal and cortical pyramidal cells evoke a transient dendritic increase in intracellular Ca2+ concentration, providing an associative signal for long-term synaptic plasticity (Magee and Johnston, 1997; Markram et al., 1997). The b-APs can also trigger a Ca2+-dependent dendritic release of different messengers, leading to short-term control of synaptic transmission efficacy on both pyramidal cells and interneurons (Zilberter et al., 2005). The observation that b-APs can trigger a dendritic Ca2+-dependent secretion of BDNF, and that BDNF secretion from one single neuron can activate the immediate early genes upstream regulator CREB in neighboring neurons, provides another mechanism by which target neurons can affect the development or efficacy of impinging synaptic connections. Accordingly, several forms of synaptic plasticity evoked by postsynaptic firing (Gubellini et al., 2005) or correlated presynaptic and postsynaptic activity (Kang et al., 1997; Mu and Poo, 2006; Walz et al., 2006; Mohajerani et al., 2007) require the presence of endogenous BDNF. Moreover, BDNF released from target neurons locally promotes synapse formation (Ohba et al., 2005; Kohara et al., 2007; Tanaka et al., 2008). Although extrapolations of data from neuronal cultures to a more intact system are limited, our finding that b-APs or short postsynaptic depolarization triggers dendritic release of BDNF supports the notion that BDNF can act as a target-derived messenger for synaptic plasticity and development.
Footnotes
This work was supported by Inserm, Centre National de la Recherche Scientifique, Agence Nationale pour la Recherche (ANR), the Deutsche Forschungsgemeinschaft (SFB 553), Stiftung Rheinland-Pfalz, and the Schram-Stiftung. N.K. was the recipient of a Fondation pour la Recherche Médicale Fellowship and an ANR grant. We thank Drs. M. Colonnese and Y. Ben-Ari for critical reading of this manuscript.
References
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, Canossa M. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 2004;101:15788–15792. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Brigadski T, Hartmann M, Lessmann V. Differential vesicular targeting and time course of synaptic secretion of the mammalian neurotrophins. J Neurosci. 2005;25:7601–7614. doi: 10.1523/JNEUROSCI.1776-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerli T, Pellegrino C, Baer K, Lardi-Studler B, Chudotvorova I, Fritschy J, Medina I, Fuhrer C. Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat Protoc. 2007;2:3090–3101. doi: 10.1038/nprot.2007.445. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Gärtner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Canossa M, Campana G, Gärtner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H. Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech. 1999;45:262–275. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<262::AID-JEMT10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Ben-Ari Y, Gaïarsa JL. Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J Neurosci. 2005;25:5796–5802. doi: 10.1523/JNEUROSCI.0824-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci. 1998;111:1483–1493. doi: 10.1242/jcs.111.11.1483. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69:341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Magby JP, Bi C, Chen ZY, Lee FS, Plummer MR. Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J Neurosci. 2006;26:13531–13536. doi: 10.1523/JNEUROSCI.4576-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons [see comments] Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol. 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Marty S, Berzaghi MdaP, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrlé R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophic regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E. Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc Natl Acad Sci U S A. 2007;104:13176–13181. doi: 10.1073/pnas.0704533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Poo MM. Spike timing-dependent LTP/LTD mediates visual experience-dependent plasticity in a developing retinotectal system. Neuron. 2006;50:115–125. doi: 10.1016/j.neuron.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Ohba S, Ikeda T, Ikegaya Y, Nishiyama N, Matsuki N, Yamada MK. BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition. Cereb Cortex. 2005;15:291–298. doi: 10.1093/cercor/bhh130. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Ranck JB., Jr Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp Neurol. 1973;41:461–531. doi: 10.1016/0014-4886(73)90290-2. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Barbara JG. Calcium-induced calcium release contributes to action potential-evoked calcium transients in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:4325–4336. doi: 10.1523/JNEUROSCI.19-11-04325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–109. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J Neurosci. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002;9:224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz C, Jüngling K, Lessmann V, Gottmann K. Presynaptic plasticity in an immature neocortical network requires NMDA receptor activation and BDNF release. J Neurophysiol. 2006;96:3512–3516. doi: 10.1152/jn.00018.2006. [DOI] [PubMed] [Google Scholar]
- Zilberter Y, Kaiser KM, Sakmann B. Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron. 1999;24:979–988. doi: 10.1016/s0896-6273(00)81044-2. [DOI] [PubMed] [Google Scholar]
- Zilberter Y, Harkany T, Holmgren CD. Dendritic release of retrograde messengers controls synaptic transmission in local neocortical networks. Neuroscientist. 2005;11:334–344. doi: 10.1177/1073858405275827. [DOI] [PubMed] [Google Scholar]