

Abstract
Spinal muscular atrophy (SMA) is an inborn neuromuscular disorder caused by low levels of survival motor neuron protein, and for which no efficient therapy exists. Here, we show that the slower rate of postnatal motor-unit maturation observed in type 2 SMA-like mice is correlated with the motor neuron death. Physical exercise delays motor neuron death and leads to an increase in the postnatal maturation rate of the motor-units. Furthermore, exercise is capable of specifically enhancing the expression of the gene encoding the major activating subunit of the NMDA receptor in motor neurons, namely the NR2A subunit, which is dramatically downregulated in the spinal cord of type 2 SMA-like mice. Accordingly, inhibiting NMDA-receptor activity abolishes the exercise-induced effects on muscle development, motor neuron protection and life span gain. Thus, restoring NMDA-receptor function could be a promising therapeutic approach to SMA treatment.
Keywords: spinal muscular atrophy, NMDA receptor, neuroprotection, motor-unit development, exercise, MK-801
Introduction
Spinal muscular atrophy (SMA) is a neurodegenerative disease characterized by the loss of spinal cord motor neurons. No efficient therapy is presently available for SMA, which represents a common genetic cause of death in childhood. SMA can be classified into three types on the basis of the age of onset, time course and range of motor function loss. Deletion or mutation of the telomeric copy of the survival-of-motor-neuron (SMN) gene (Smn1) causes SMA (Hahnen et al., 1995; Lefebvre et al., 1995; Rodrigues et al., 1995). The expression of SMN protein encoded by a centromeric copy of the Smn gene (Smn2) only partially compensates for the lack of Smn1 function. Indeed, the predominant SMN protein isoform encoded by Smn2 lacks its C terminus because of the alternative splicing of exon 7 (Lorson et al., 1999; Monani et al., 1999), which results in a protein that cannot oligomerize efficiently (Lorson et al., 1998) and becomes degraded (Lorson and Androphy, 2000). Knocking out the single copy Mouse Smn gene causes developmental arrest and embryonic death (Schrank et al., 1997). Introduction of human Smn2 transgenes on either a Smn null background (Monani et al., 2000) or on a Smn-delta7 background (Hsieh-Li et al., 2000; Le et al., 2005) rescues the embryonic lethal phenotype and results in mice with severe SMA.
One well documented role of the SMN protein is linked to the assembly of small nuclear ribonucleoproteins (snRNPs), the essential components of the pre-mRNA splicing machinery (Meister et al., 2001; Yong et al., 2004; Wan et al., 2005), suggesting that SMA might originate from an alteration of the transcriptome. To this respect, the SMN deficiency induced alteration of a mechanism that is essential for the motor neuron development has recently emerged as an hypothesis to explain some cellular aspects of SMA, notably during the postnatal period (Fan and Simard, 2002; Gabanella et al., 2005). First, SMN is found in motor axons of normal and SMA-like mice in vitro (Fan and Simard, 2002; Sharma et al., 2005; Zhang et al., 2006). Second, the knockdown of SMN in zebrafish led to cell-autonomous axonal defects (McWhorter et al., 2003). Yet, the activity-dependent maturation of motor neurons is dependent on the activation of the NMDA receptor by l-glutamate, the major excitatory neurotransmitter in the mammalian CNS (Kalb and Hockfield, 1992; Kalb, 1994). Interestingly, the activation of the NMDA receptor induces a significant increase in SMN levels and gem number in neurons (Andreassi et al., 2002).
We have previously evaluated the running-based training efficiency on a type 2 SMA-like mouse model (Grondard et al., 2005). The results have shown a gain in motor function and life span for type 2 SMA-like mice. Exercise leads to an increase in exon7-containing SMN transcripts. In the present study, we performed a complete analysis of all parts of the motor units in trained versus untrained type 2 SMA-like mice and we provide experimental data that strongly substantiate the hypothesis of a direct relationship between the maturation state of the motor units and the resistance to neuronal cell death. Most importantly, we report here that the exercise-induced neuroprotection in SMA-like mice results from the trigger of NMDA signaling pathway.
Materials and Methods
Mice.
The knock-out transgenic SMA-like mice (SmnΔ7+/+ Smn2) were obtained from the Institute of Molecular Biology (Academia Sinica, Taipei, Taiwan). These mice derive from the Smn2 transgenic line 2 from Hsieh-Li et al. (2000), which carry two Smn2 transgene copies as originally determined by Southern blots (Hsieh-Li et al., 2000; Tsai et al., 2006). These mice were subsequently purified in an FVB/NRj (Janvier, Le Genest-St-Isle, France) genetic background (10 generations). Heterozygous Smn knock-out (SmnΔ7+/−) mice were bred with heterozygous transgenic-knock-out (SmnΔ7+/− Smn2) mice. The offspring were genotyped by PCR assay of DNA obtained from tail tissue, as described previously (Hsieh-Li et al., 2000), and SMA-like mice were classified as type 1, 2, or 3 at 9 d of age based on their phenotype, particularly on their age of survival (Hsieh-Li et al., 2000). Breeder mice that gave only type 2 SMA-like mice in their progeny were selected for this study. To standardize the type 2 phenotype, mutant mice with a weight ranging from 3 to 4 g at 9 d of age were used for this study (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Other mutant mice were used to check that the life span did not exceed 3 weeks. To further confirm the phenotype of the type 2 SMA-like mice, the amount of full-length SMN transcript in the spinal cord was systematically evaluated at the end of each experiment (9 or 12 d of age), as reported previously (Grondard et al., 2005). An untrained group (n = 47) and a trained group (n = 68) of type 2 SMA-like mice were randomly constituted in a blind systematic manner to minimize bias. The control mice were heterozygous knock-out for Smn with the human Smn2 transgene (Smn+/−Smn2; n = 66). The care and treatment of animals followed the national authority (Ministère de la Recherche et de la Technologie, Paris, France) guidelines for the detention, use and the ethical treatment of laboratory animals.
To evaluate the role of NMDA receptor in exercise-induced neuroprotection, P9 neonatal mice were injected subcutaneously with 0.25 mg/kg (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine hydrogen maleate (MK-801) in 0.9% NaCl (Sigma, Saint Quentin Fallavier, France).
Training protocol.
Exercise was performed in an elastomer-covered wheel with a controlled speed essentially as described previously (Grondard et al., 2005). The protocol was started at 9 d of age. The mice were progressively accustomed to wheel training for a period of 2 d (first day, four periods of 5 min running separated by rest periods of 30 min; second day, two periods of 10 min running separated by a rest period of 30 min). Then, the mice were continuously trained, individually for 20 min/d. The speed was fixed at 2 m min−1 for the first 2 d and then progressively increased by 1 m min−1 per training day. The MK-801-treated mice did not show evidence of any motor behavior worsening and were thus trained exactly as described above.
Histological and immunohistochemical analysis.
Dissected spinal cord were directly frozen in isopentane cooled in liquid nitrogen (−140°C) and then the lumbar region (between L1 and L5) was selectively cut on a cryostat microtome. Two hundred serial 14-μm-thick cross sections of the lumbar spinal cords were performed (2800 μm total length), among which one of every five sections (40 sections examined) was processed for immunohistochemical analysis. Tissue sections were fixed with 2% paraformaldehyde/0.1 m sodium phosphate buffer, pH 7.2, for 20 min, rinsed, and incubated for 1 h at room temperature in a blocking solution [7% normal donkey serum with 0.3% Triton X-100 in Tris buffer solution (TBS)]. Incubations in the primary antiserums, namely anti-NR2A (1:300; Upstate Biotechnology, Euromedex, Mundosheim, France), anti-NR1 (1:100; Millipore, Temecula, CA), anti-5-HT2AR (1:200; PharMingen, Le-Pont-de-Claix, France), and anti-L-type calcium channel (α1c subunit) (1:200; Sigma, St. Louis, MO) were performed during 3 d at 4°C. To stain motor neurons, immunodetection of choline acetyltransferase (ChAT) (polyclonal goat anti-ChAT; 1:200; Millipore) was performed in single- or double-labeling analysis. Sections were washed between each subsequent step with 0.3% Triton X-100 in TBS. Sections were subsequently incubated with an Alexa Fluor 568 donkey anti-goat Ig or an Alexa Fluor 488 donkey anti-rabbit IgG (1:400; Invitrogen, Carlsbad, CA) for 1 h at room temperature. The sections were washed three times for 10 min in TBS and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). The staining specificity was checked in control incubations performed in the absence of the primary antibody.
For the apoptosis evaluation in the spinal cord, the apoptotic nuclei were observed after terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining, as described previously (Grondard et al., 2005).
For motor end-plate labeling, whole-mount preparations of muscle fibers were stained using Alexa Fluor 568-conjugated α-bungarotoxin (4 mg/ml in PBS with 4% bovine serum albumin). Presynaptic motor nerve terminals were stained with monoclonal antibody directed against the 145 kDa isoform of neurofilament protein (NF-M; 1:200; NN18, Millipore). The whole-mount preparations were subsequently incubated with an Alexa Fluor 488 goat anti-rabbit IgG (1:400; Invitrogen) for 1 h at room temperature. To compare neuromuscular junction (NMJ) maturation between fast and slow muscle fibers, α-bungarotoxin-labeled whole-mount preparations of muscle were analyzed for succinate dehydrogenase activity, as described previously (Pool et al., 1979).
For muscle typology, cryostat sections were incubated with mouse monoclonal antibodies against myosin heavy chains (MyHCs) reacting either with (1) slow type I (NCL-MHCS; Novocastra, Newcastle, UK), (2) all fast epitopes (NCL-MHCF), (3) fast type IIA (SC71), (4) fast type IIB (BF-F3), and (5) embryonic (F1.652; from Helen M. Blau, Stanford University, Stanford, CA) and (6) neonatal MHC isoforms (N3.36; from Helen M. Blau) at a dilution of 1:20 for 1 h at room temperature, washed three times in PBS containing 0.1% Tween 20 (PBST). Sections were then incubated at room temperature with a rabbit anti-mouse Ig conjugated with fluorescein isothiocyanate (1:20; Sigma) for 1 h. After washing with PBST, sections were mounted in Vectashield mounting medium (Vector Laboratories). The proportion of fiber IIX was determined as the difference between the total number of type II fibers and the number of fibers IIA plus IIB.
For muscle-fiber cross-sectional analysis, frozen soleus, plantaris, and tibialis muscles from mice at 9 and 12 d of age were collected and sectioned into 10-μm-thick cryostat sections. Muscle sections were stained with hematoxylin and eosin, dehydrated through an ethanol gradient (70, 90, and 100%), and mounted in Eukitt (VWR International, Strasbourg, France).
Retrograde labeling of motor neurons projecting in soleus, plantaris, and tibialis muscles.
Nine-day-old mice were anesthetized with halothane (Belamont; Mundipharma SAS, Boulogne Billancourt, France). A total volume of 50 nl of fluorogold (Fluorochrome, Denver, CO) in PBS was injected in three different parts of each muscle (median, proximal, and distal) using an oil-based microinjector (Nanoject; Drummond Scientific, Broomall, PA) as described previously (Grondard et al., 2005). At 12 d of age, mice were perfused and processed for histological analysis.
In situ hybridization.
To detect mRNAs for NMDA-receptor subunits, antisense oligonucleotide probes were synthesized as reported previously (Oshima et al., 2002; Fukaya et al., 2005): oligoNR3b-1, oligoNR3b-3, and oligoNR3b-4 for NR3B cDNA, oligo-ζ1Ex11 for NR1 cDNA, oligo-ε1L for NR2A cDNA, oligo-ε2L for NR2B cDNA, oligo-ε3L for NR2C cDNA, and oligo-ε4L for NR2D cDNA. Under deep chloral hydrate anesthesia, the spinal cord was obtained from 12-d-old SMA-like mice. Frozen sections (14 μm thickness) were mounted on glass slides coated with 3-aminopropyltriethoxysilane. Probe labeling and hybridization were performed exactly as described previously (Vassias et al., 2005). Sections were exposed to Nuclear Track emulsion (NTB-2; Kodak, Rochester, NY) for 8 weeks. Emulsion-dipped sections were stained with toluidine blue.
Real-time reverse transcription-PCR analysis.
RNA isolation and reverse transcription (RT) were performed as described previously (Grondard et al., 2005). The real-time RT-PCR were performed using an ABI Prism 7700 (Applied Biosystems, Courtaboeuf, France) and the following forward and reverse primers: NR2A, forward, 5′-AGCCCCCTTCGTCATCGTAGA-3′, reverse, 5′-ACCCCTTGCAGCACTTCTTCAC-3′; GAPDH, forward, 5′-TCCTGCACCACCAACTGCTTAGCC-3′, reverse, 5′-TAGCCCAAGATGCCCTTCAGTGGG-3′). PCR conditions and sample value comparisons using the relative Ct (cycle threshold) method were described previously (Grondard et al., 2005).
Microscopy.
All immunofluorescence and ISH images, except L-type calcium-channel-labeled sections and double-labeled NMJ images, were collected with a CCD camera (Retiga 2000R Fast, Cooled Mono 12 bit; QImaging Burnaby, British Columbia, Canada) mounted on an Olympus (Tokyo, Japan) microscope (BX51) using the Image Pro-Plus version 6.0 software (MediaCybernetics, Bethesda, MD) with 40× (4× Olympus objective UPlan FL N 0.13), 100× (10× Olympus objective UPlan FL N 0.3), 200× (20× Olympus objective FL N 0.5), 400× (40× Olympus objective UPlan FL N 0.75), 600× (60× Olympus objective UPlanS Apo 1.35 oil), and 1000× (100× Olympus objective UPlanS Apo 1.4 oil) magnifications. The L-type calcium-channel-labeled sections were examined with a Zeiss (Oberkochen, Germany) LSM 510 Meta confocal microscope at 100× Zeiss objective (UPlanS Apo 1.4 oil). Double-labeled NMJ images were collected with a Nikon (Tokyo, Japan) 3D TE2000E microscope equipped with a CoolSnap HQ2 CCD camera (Roper Instruments, Tucson, AZ) with a 100× Nikon objective (PlanApo numerical aperture, 1.4 oil).
Images measurements.
For spinal cord immunofluorescence images, all cells were counted within the ventral horn of the lumbar spinal cord (L1–L5) below an arbitrary horizontal line drawn from the central canal. L-type calcium channel confocal images were analyzed with Zeiss LSM 5 Image Examiner software. TUNEL-positive cells were counted in 30 sections of the lumbar spinal cord of each mouse. These counts were then compared with the total number of nuclei determined after DAPI staining. Approximately 150 neuromuscular junctions were counted for each mouse in any given muscle. Images of double-labeled NMJs were processed by three-dimensional deconvolution using the AutoDeblur and Visualize version X1.4 software (MediaCybernetics). Muscle immunofluorescence images were assembled using Photoshop version 7.0 (Adobe Systems, San Jose, CA). The highest number of myofibers per muscle section was retained for statistical analysis.
All counts were performed using NIH ImageJ software version 1.37. Color images were tinted using Image Pro-Plus software, where identical brightness, contrast, and color balance adjustments were applied to all groups.
Statistical analysis.
All values are presented as means and SD within each group (Systat version 8.0; SPSS, Chicago, IL). A Kolmogorov–Smirnov normal distribution analysis was performed on all data followed by either a Student's t test for normally distributed data or a nonparametric Kruskal–Wallis test, to verify significant differences between groups. Survival analysis was performed using Kaplan–Meier analysis. For statistical evaluation of motor neuron number, identified by ChAT immunoreactivity, the number of cells present in each ventral horn of the L1–L5 spinal cord was counted and corrected according to the method of Abercrombie (1946), which compensates for double counting in adjacent sections.
Results
Comparative analysis of the skeletal-muscle development in trained and sedentary SMA-like mice
Type 2 SMA-like mice (n = 26) were submitted to a regular exercise protocol, which consisted of a forced run in a wheel. Based on the ratio between embryonic and neonatal fibers, and the relative persistence of type I fibers in fast-twitch muscles (Fig. 1f, supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we showed a dramatic change in fiber-type distribution in the untrained type 2 SMA-like mice compared with controls (Fig. 1g,h). Furthermore, the muscular maturation between 9 and 12 d of age of the two extensor muscles (i.e., the soleus and the plantaris) proved to be abolished in SMA as demonstrated by the absence of neonatal fiber gain (31.5% in the soleus and 70% in the plantaris at 9 and 12 d of age, respectively). Comparatively, in controls, the proportion of neonatal fibers increased from 41 to 46.4% in the soleus and from 82.2 to 88% in the plantaris during the same period. The flexor tibialis was less affected by SMA compared with the two extensors. Despite the proportion of neonatal fibers being lower compared with the corresponding control muscles [64.6% in SMA vs 75.9% in control and 84.9% in SMA vs 93.8% in controls, at 9 (p < 0.01) and 12 (p < 0.05) d, respectively], a significant increase in the percentage of neonatal fibers was evidenced in the tibialis of SMA-like mice between 9 and 12 d (p < 0.01). Thus, the range of the SMA-induced effect was muscle-specific. Interestingly, in controls, the maturation in the tibialis proved to be more advanced than in the two extensor muscles, soleus and plantaris.
Figure 1.

Muscle typology analysis of the extensor slow-twitch soleus, the extensor fast-twitch plantaris, and the flexor fast-twitch tibialis from type 2 SMA-like mice. a–e, Immunodetection of MyHC isoforms revealed an increase in embryonic muscle fiber proportion in the soleus from type 2 SMA-like mice (b) compared with control mice (a) at 9 d of age and an increase in type I muscle fiber proportion in the plantaris from untrained SMA-like mice (d) compared with trained SMA-like mice (e) and control mice (c) at 12 d of age. Scale bars, 100 μm. f, Schematic presentation of the myofiber transitions during postnatal development of the motor unit including the role of motor neuron innervations on adult phenotype acquisition. MN, Motor neuron; Emb, embryonic; Neo, neonatal. g, h, The quantitative analysis of the muscle typology variation during the postnatal development of soleus, plantaris, and tibialis muscles from type 2 SMA-like mice compared with control at 9 d of age (g) and from untrained and trained type 2 SMA-like mice compared with controls at 12 d of age (h) shows a muscle-function-specific delay in skeletal-muscle maturation in SMA that could be counteracted by the exercise protocol (n = 6; *p < 0.05; **p < 0.01; ***p < 0.001).
The exercise program partially corrects the typology of the three muscles in type 2 SMA-like mice, as witnessed by the decrease in the number of embryonic fibers in trained muscles. In addition to this effect, physical exercise also alters the adult muscle typology in the SMA models. For the fast-twitch muscles plantaris and tibialis, the maintenance of the percentage of type I fiber percentage over control values contrasts with a return to normal values for type IIA, IIX, and IIB fiber-relative numbers (Fig. 1h). These last results demonstrate the training-induced acceleration of the adult phenotype acquisition. For the slow-twitch soleus, training increases the ratio of type I to type II fibers. This transition to a slower phenotype is generally observed in healthy adult muscles, submitted to endurance training (Pette and Staron, 1997). Surprisingly, neither an exercise-induced acceleration of the muscle maturation nor an exercise-induced change in the adult MyHC isoform expressing myofiber proportion was observed in control muscles.
Comparative analysis of the neuromuscular junction structure in trained and sedentary SMA-like mice
To ensure an accurate description of the motor unit developmental pattern in trained versus untrained type 2 SMA-like mice, we have analyzed the structure of the neuromuscular junctions (NMJ) during the postnatal period (Fig. 2). The changes in the synapse shape were determined for innervated NMJ using α-bungarotoxin staining of the three muscles. Up to 63% of the NMJ were elliptical, made of uniform plaques of acetylcholine receptors (AChRs) in the two extensors muscles of the sedentary type 2 SMA-like mice at 12 d of age. Only 28% of immature NMJ were found in control muscles (p < 0.001) (Fig. 2e), regardless of slow or fast type muscle fibers (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, in the flexor tibialis at the same age, uniform plaques of AChRs were found in only 30% of the NMJ. Moreover, at 9 d of age, only a small difference in the NMJ structure was found in the tibialis, when compared with control muscles (p < 0.05) (Fig. 2d). All these data revealed that the NMJ maturation defect observed in untrained type 2 SMA-like versus control mice was muscle-function dependent. Accordingly, in controls, the maturation of the tibialis NMJs proved to be more advanced than those of the extensor muscles, soleus and plantaris. The exercise paradigm improved the NMJ morphology in the three muscles in type 2 SMA-like mice, as demonstrated by the exercise-induced perforation of the NMJ on the muscle fibers (Fig. 2e). The exercise-induced effect on the NMJ maturation in SMA-like mice was further demonstrated by the analysis of the synaptic gutter through the use of a neurofilament staining (Fig. 2f–h). A significant reduction (∼16%; p < 0.001) of immature large terminal axons was revealed in plantaris and soleus of trained type 2 SMA-like mice, compared with untrained mutant mice.
Figure 2.
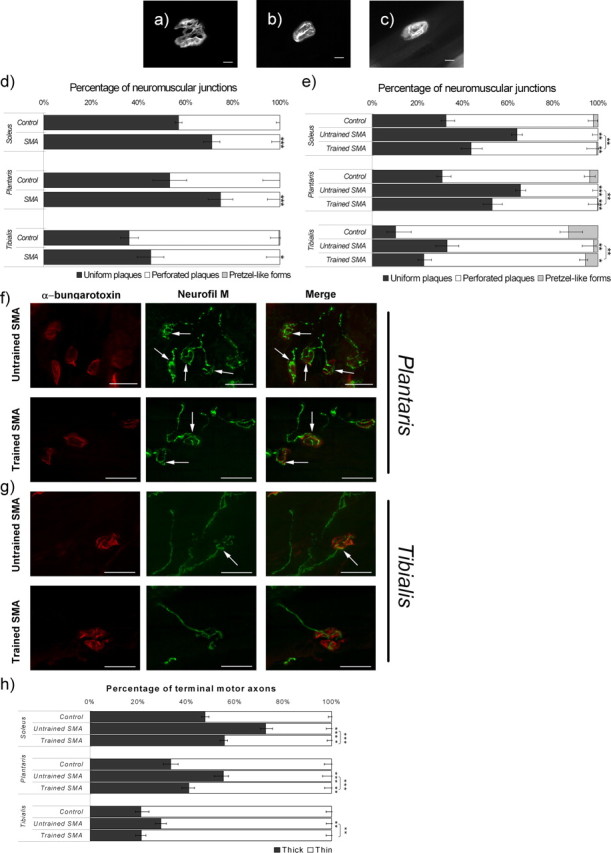
Morphological analysis of the NMJs in the soleus, plantaris, and tibialis from type 2 SMA-like mice. a–c, Uniform immature plaque of NMJ are frequently observed in tibialis from untrained SMA-like mice (b) compared with the more perforated plaque, which is preferentially observed in trained SMA-like mice (c) and the pretzel-like form observed in control mice at 12 d of age (a). Scale bars, 10 μm. d, e, Changes in the NMJ morphology computed after a whole-mount α-bungarotoxin labeling of soleus, plantaris, and tibialis muscles from untrained and trained type 2 SMA-like mice compared with control mice at 9 d (d) and 12 d of age (e) reveal an NMJ maturation delay in SMA that could be counteracted by the exercise protocol. f, AChR (red) and neurofilament (Neurofil M) (green) labeling in the plantaris muscle reveal the presence of thick terminal axons (arrows) at the NMJ of untrained (top) type 2 SMA-like mice, whereas thin terminal axons are more frequently observed in trained type 2 SMA-like mice (bottom) at 12 d of age. g, These morphological differences are less evident in the tibialis muscle. Scale bars, 25 μm. h, Quantification of thick terminal axons filled with neurofilament in the soleus, plantaris, and tibialis muscles from untrained and trained type 2 SMA-like mice and from control mice (n = 6; *p < 0.05; **p < 0.01; ***p < 0.001).
Comparative analysis of the motor neuron postnatal maturation in trained and sedentary SMA-like mice
To determine whether the exercise paradigm also induced an acceleration of motor neuron maturation, we have analyzed the expression patterns of the L-type calcium channels and of the serotonin receptor 2A (5-HT2AR) in the lumbar spinal cord of trained versus untrained type 2 SMA-like mice and controls. The L-type calcium channels were shown to progressively migrate from perinuclear to membrane locations during the early postnatal development of mouse motor-neurons (Jiang et al., 1999). At 12 d of age, the motor neurons of the sedentary mutant mice displayed a predominantly perinuclear localization of the L-type calcium channels (76% in SMA vs 65% in controls; p < 0.01) and only a faint labeling could be detected within or in the vicinity of the neuron plasma membrane (Fig. 3a,b). In the trained SMA-like mice, a lower number of motor neurons displaying perinuclear L-type calcium channel labeling was recorded compared with untrained SMA-like mice (65 vs 76%, respectively; p < 0.01). This result, indicating an exercise-induced acceleration of the motor neuron maturation, was further substantiated by the analysis of the time course of the 5-HT2A-receptor gene expression. The postnatal appearance of the 5-HT2AR was recently shown to occur between postnatal days 7 (P7) and P14 in the rat (Vult von Steyern and Lomo, 2005), which approximately corresponds to days 5 and 12 in the mouse. In the sedentary type 2 SMA-like mice at 12 d of age, only a small number of motor neurons displayed an immunoreactivity for 5-HT2AR, when compared with controls at the same age (55 vs 84%, respectively; p < 0.001) (Fig. 3c,d). In contrast, in the lumbar spinal cord of trained SMA-like mice, a clear 5-HT2AR labeling, albeit lower than in the control spinal cord, was shown in the cell body and in the dendrites of a large number of motor neurons (69 vs 84%, in trained SMA-like and control mice, respectively; p < 0.01).
Figure 3.
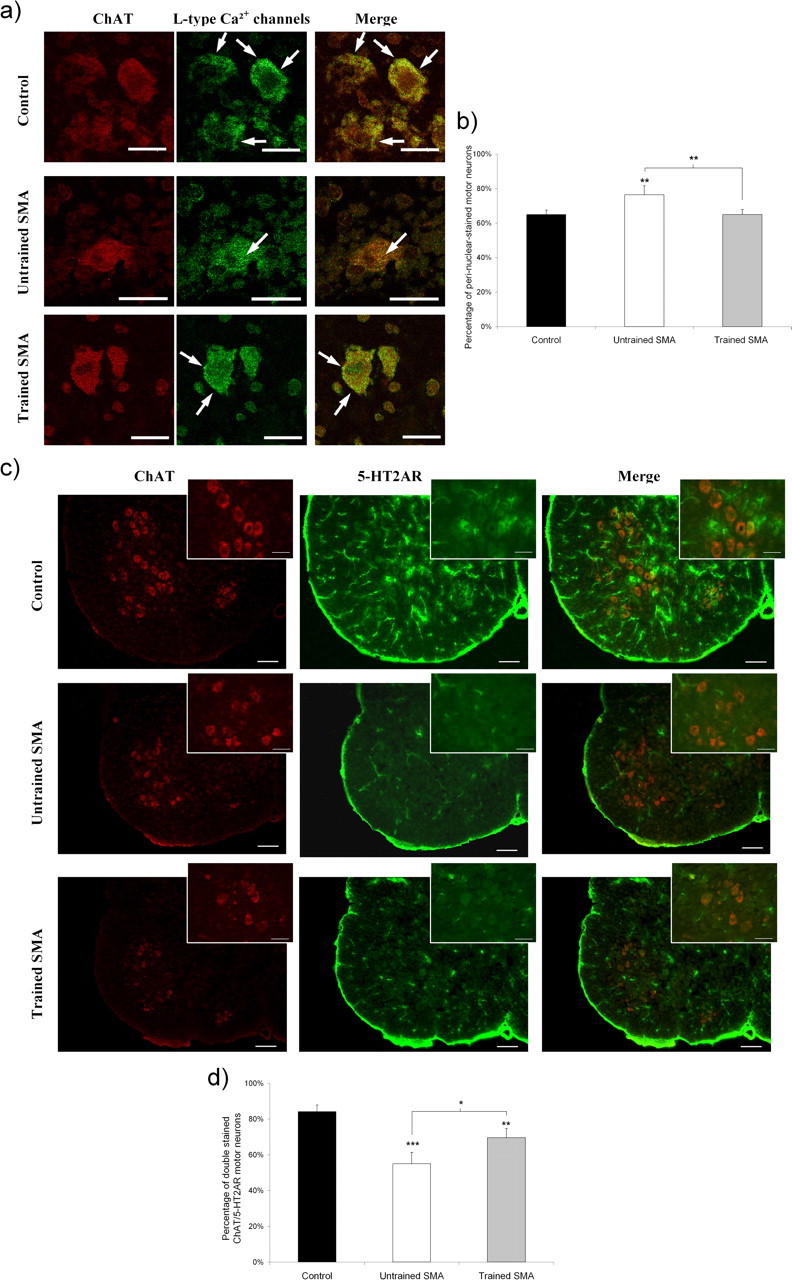
Analysis of motor neuron postnatal maturation in untrained and trained type 2 SMA-like mice. a, The subcellular location of the L-type calcium channels as determined by confocal immunofluorescence imaging is preferentially a perinuclear one (arrows), in the motor neurons of the lumbar spinal cord from untrained type 2 SMA-like mice (middle) compared with membrane-labeled motor neurons (arrows) from trained SMA-like mice (bottom) and control mice (top) at 12 d of age. Scale bars, 10 μm. b, The proportion of perinuclear-stained motor neurons recorded in the spinal cord of untrained SMA-like mice is significantly decreased in the spinal cord of trained SMA-like and control mice at 12 d of age (n = 6, **p < 0.01). c, The motor neuron maturation rate was further evaluated by the immunodetection level of the 5-HT2AR over the lumbar spinal cord of the untrained (middle) versus trained (bottom) SMA-like mice and control mice (top). Scale bars: low-magnification views, 100 μm; high-magnification views, 25 μm. d, The quantification of 5-HT2AR-expressing motor neurons highlights a delay in 5-HT2AR appearance in the lumbar spinal cord from untrained type 2 SMA-like mice when compared with their trained counterparts (n = 4; *p < 0.05; **p < 0.01; ***p < 0.001).
Analysis of muscle-specific motor neuron survival in trained and sedentary SMA-like mice
To test a causal relationship between exercise-induced acceleration of the motor-unit maturation and exercise-induced motor-neuron protection, we have analyzed the motor-neuron connectivity through the counts of Fluorogold retrogradely labeled neurons projecting into the soleus, plantaris, and the tibialis muscles in trained versus untrained type 2 SMA-like mice (Fig. 4). In untrained type 2 SMA-like mice at 12 d of age, a 40.4 and 33.1% decrease in the cell counts was noted for the motor neurons innervating the soleus and plantaris muscles respectively (p < 0.001), compared with controls, contrasting with <17% in the tibialis (p < 0.001) (Fig. 4d). The faster maturation of the motor units in the tibialis compared with the two extensor muscles further substantiate the concept that the maturation of the motor unit decreased the sensitivity of motor neurons to SMA. With a decrease of only 15.6, 12.4, and <6.3% of the motor neurons innervating the soleus, plantaris, and tibialis muscles, respectively, compared with controls (p < 0.01), the exercise regimen efficiently delayed motor neuron death, regardless of the motor-unit maturation level.
Figure 4.

Analysis of the muscle-specific motor-neuron populations in untrained and trained type 2 SMA-like mice. a–c, A loss of the soleus-innervating motor neurons, specifically labeled using a fluorogold retrolabeling technique, is observed in the spinal cord from untrained type 2 SMA-like mice (b) compared with trained type 2 SMA-like (c) and control mice (a) at 12 d of age. Scale bars, 20 μm. d, The quantification of surviving motor neurons innervating each muscle clearly indicates that the extent of motor neuron loss in type 2 SMA-like mice at 12 d of age is muscle-specific and that, in each muscle, the exercise protocol exerts an efficient neuroprotective effect in SMA (n = 6; *p < 0.05; **p < 0.01; ***p < 0.001).
Expression analysis of the NMDA-receptor subunit genes in trained and sedentary SMA-like mice
Accumulating experimental data that strongly suggest a major involvement of the NMDA receptor in motor neuron postnatal maturation prompted us to investigate the expression patterns of the main NMDA-receptor subunit genes in the lumbar spinal cord i.e., NR1, NR2A and NR3B (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), of trained versus untrained type 2 SMA-like mice, and controls (Fig. 5). Surprisingly, NR2A mRNA and protein displayed a specific decrease in gene expression in lumbar motor neurons of the untrained type 2 SMA-like mice (Fig. 5a,d,e), contrasting with a conserved gene expression levels for NR1 (Fig. 5c,f) and NR3B subunits (Fig. 5b) compared with the corresponding controls. Physical exercise enhanced the expression of the NR2A subunit gene expression in the motor neurons of trained type 2 SMA-like and control mice without any detectable effect on the gene expression levels of NR1 and NR3B subunits.
Figure 5.
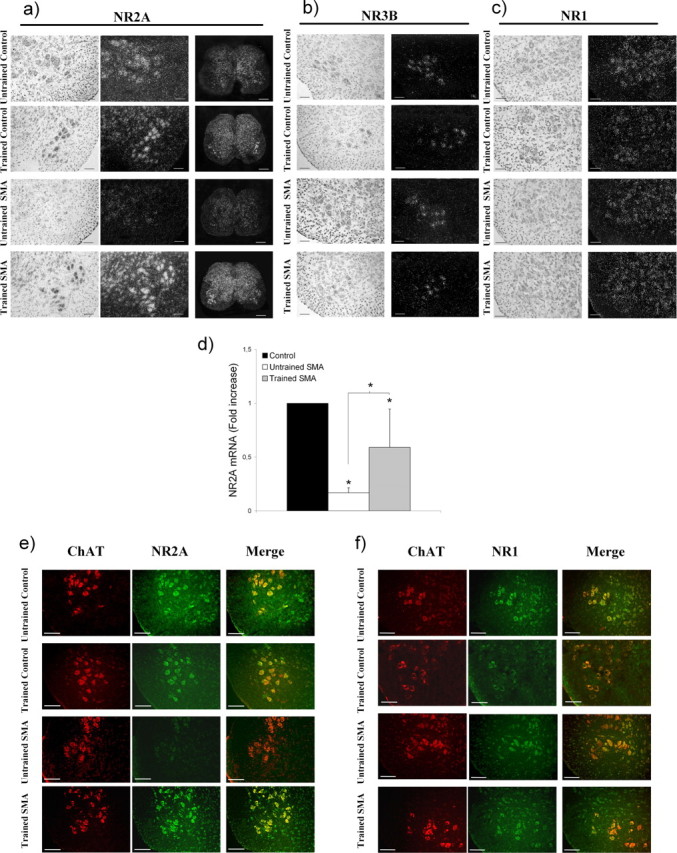
Expression profiles of NMDA-receptor isoforms in the lumbar spinal cord from trained and untrained type 2 SMA-like mice. a–c, An important decrease in the NR2A subunit expression level is observed in the lumbar spinal cord of the type 2 SMA-like mice at 12 d of age at the level of the transcripts compared with controls (a) whereas the expression of either the NR3B (b) or the NR1 (c) subunit genes remains unchanged. The exercise regimen proves to specifically enhance the NR2A subunit gene expression in SMA-like as well as in control mice. Scale bars: 50 μm for all images, except 250 μm for the large views of the spinal cord. d, The quantification of NR2A transcripts in the spinal cord at 12 d of age by real-time RT-PCR further substantiates the assumption of an exercise-induced reactivation of the NR2A gene expression in trained SMA-like mice (n = 3; *p < 0.05). e, The immunodetection level of NR2A in motor neurons, identified for their immunoreactivity for ChAT, is in agreement with the in situ hybridization data. The low NR2A labeling in motor neurons from untrained type 2 SMA-like mice is to be compared with the high detection of this protein in the motor neurons of trained SMA-like mice, at 12 d of age. Scale bars, 50 μm. f, In contrast, no difference in the NR1 distribution in the spinal cord of SMA-like and control mice could be evidenced (n = 4).
We then used real-time RT-PCR to evaluate more accurately the modulation of the NR2A gene expression in the spinal cord of trained and untrained type 2 SMA-like mice. We found that the relative amount of the NR2A mRNA was markedly decreased in the spinal cord of untrained SMA-like mice by ∼6-fold compared with control mice. The exercise regimen enhanced the NR2A gene expression by ∼3.5-fold in SMA-like spinal cords limiting the NR2A mRNA decrease to half of the control (Fig. 5d).
NMDA-receptor involvement in the exercise-induced neuroprotection of SMA-like mice
To test whether the beneficial effects of exercise were dependent on the NMDA-receptor activity, we evaluated the effects of MK-801, a specific noncompetitive NMDA-receptor antagonist on the exercise-induced promotion of the motor unit development and protection in trained type 2 SMA-like mice (Fig. 6). The NMDA-receptor blockade resulted in a drastic reduction of the exercise-induced increase of survival (Fig. 6a) and weight (Fig. 6b). Importantly, the MK-801 treatment led to a decrease of the motor neuron number in the whole lumbar spinal cord compared with vehicle-treated trained SMA-like mice (6.2 vs 7.7 motor neurons/anterior horn/section, respectively; p < 0.01) (Fig. 6c–f). Because this number of motor neurons is comparable with the number found in untrained type 2 SMA-like mice at 13 d of age (Grondard et al., 2005), it could be assumed that blocking the NMDA receptor results in the abolishment of the major part of the exercise-induced protection of the spinal motor neurons.
Figure 6.

NMDA-receptor involvement in the exercise-induced neuroprotection in type 2 SMA-like mice. a–e, SMA-like mice that were submitted to a MK-801 treatment (MK801; n = 21) before training have lost the benefits of the exercise regimen compared with the vehicle-treated trained mice (Vehicle; n = 9) in terms of life span (12.8 and 22.3 d, respectively), as revealed by the Kaplan-Meier curve (a); growth gain, as revealed by the growth curve (b); and motor neuron protection, as revealed by ChAT-immunodetection (c–e). Scale bars, 50 μm. f, The quantification of the motor neurons was performed in the anterior horn of the lumbar spinal cord (L1–L5) from the MK-801- or vehicle-treated trained SMA-like mice (n = 7 and n = 6, respectively) and compared with MK-801- or vehicle-treated trained control mice (n = 6) (*p < 0.05; **p < 0.01; ***p < 0.001). g, The MK-801 treatment does not promote motor neuron death at 12 d of age in the spinal cord of sedentary type 2 SMA-like mice, treated by MK-801 or vehicle, as revealed by the count of Chat-positive cells (n = 3; nonsignificant). h, j, Immunodetection of MyHC isoforms revealed an increase in embryonic myofiber proportion in the soleus from MK-801-treated (i) versus vehicle-treated (j) trained type 2 SMA-like mice and control mice (h) at 12 d of age. Scale bars, 100 μm. k, The comparison of the muscle typology of the soleus, plantaris, and tibialis muscles in MK-801- versus vehicle-treated trained type 2 SMA-like and control mice at 12 d of age (n = 8, 6, 6, and 6, respectively) indicate that the NMDA-receptor blockade results in the abolishment of the exercise-induced acceleration of muscle maturation (*p < 0.05; **p < 0.01; ***p < 0.001).
Therefore, the NMDA-receptor blockade resulted in a loss of the exercise-induced acceleration of the soleus, plantaris, and tibialis muscle maturation, as shown by the observed delay in the embryonic to neonatal myofiber switch (Fig. 6k). The ratios of embryonic fibers is comparable in MK-801-treated SMA-like mice and in untrained SMA-like mice (78 vs 90%, p < 0.05; 25 vs 32%, and 17 vs 18%, nonsignificant; in the soleus, plantaris, and tibialis muscles, respectively). With only 60, 18 and 13% of embryonic fibers in the soleus, plantaris, and tibialis muscles, respectively, the data obtained in vehicle-treated SMA-like mice are in close agreement with the ratio observed in trained type 2 SMA-like mice (compare Figs. 1h, 6k).
Because a proapoptotic effect of MK-801 has been reported previously during development (Ikonomidou et al., 1999; Lladó et al., 1999) that could artificially worsen the phenotype of type 2 SMA-like mice through an additional and nonspecific increase in neuronal death, we analyzed the effects of the MK-801 treatment on the motor neuron populations from sedentary type 2 SMA-like mice. No significant MK-801-induced increase of motor neuron death was observed in the lumbar spinal cord of sedentary type 2 SMA-like mice (Fig. 6g). We then counted the lumbar apoptotic cells in the spinal cord of MK-801-treated control mice. No apoptotic cells could be detected through TUNEL assays (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Together, these data reinforce the role of the NMDA-receptor activation in the exercise-induced neuroprotection of the motor neurons of trained type 2 SMA-like mice.
Discussion
We have shown, in the present study, that the deficient expression of the SMN protein induces a structural alteration in each part of the motor unit i.e., the motor neuron, the NMJ and the muscle fibers in type 2 SMA-like mice, which can be partially corrected by exercise. The range of these alterations was correlated with the extent of motor neuron death. This is clearly demonstrated by the observed range of neuronal loss in the motor neuron populations that innervate the extensors soleus and plantaris compared with the flexor tibialis. Indeed, the neuronal loss for the extensors is twice the neuronal loss recorded for the tibialis. The analysis of the muscle typology unambiguously revealed that the postnatal maturation is faster in the tibialis than in the soleus and plantaris of control and SMA-like mice. This result is consistent with previous electrophysiological data that highlight specific time courses of electrical property acquisition in extensor versus flexor motor neurons during their postnatal maturation in the rat (Vinay et al., 2000). Determining whether the defects in the SMA-like motor-units were a consequence of the spinal denervation and/or a lack of motor activity or alternatively the result of the specific development disruption requires further investigation.
Moreover, our data clearly show that physical exercise leads to a delay in the motor-neuron death regardless of the rate of postnatal motor-unit maturation. This exercise-induced effect could originate from the restoration of a basal level of activity needed for the proper motor-unit postnatal maturation (Navarrette and Vrbová, 1993). Basically, exercise could delay motor-neuron death directly through a specific neuroprotective effect, thus potentially promoting reinnervation of the NMJ and consequently the remodeling of the motor unit structure. Nevertheless, it remains that the exercise-induced protection of the motor neuron populations is concomitant with an acceleration of their postnatal maturation which is evident when comparing the phenotype of each part of the motor unit in trained versus untrained SMA-like mice. To this respect, the lack of effect of exercise on healthy muscles, at the same ages is puzzling. The greatest plasticity of SMA muscles compared with controls could arise from a greatest plasticity of the motor neurons, accounting for a maturation delay in SMA motor neurons. However, it might be assumed that the exercise workload, although unable to induce motor-unit adaptation in wild mice, was efficient in restoring a normal muscle phenotype in the weak SMA-like mice.
The greater sensitivity of immature motor neurons to SMA is still unexplained. During the development of the nervous system, the apparently spontaneous cell death of spinal motor neurons is a commonly occurring phenomenon that is, in all likelihood, determined by the presence and the size of their peripheral targets (Hamburger, 1958; Tanaka and Landmesser, 1986). Although this natural event in the spinal cord is no longer observed after birth in rat and mouse (Oppenheim, 1991), both motor neurons and sensory neurons undergo apoptosis after experiments of target deprivation or peripheral nerve axotomy in the neonates (Kashihara et al., 1987). The dying predisposition of embryonic and neonatal motor neurons during development suggests a greater sensitivity to cell death unlike adult motor neurons, which are capable of resisting an apoptotic stimulus. Therefore, the delay in motor unit maturation observed at P12 in SMA-like mice could increase the sensitivity of the motor neuron to death and the exercise-induced acceleration of motor unit maturation would result in the motor neuron protection against cell death.
One molecular explanation of the exercise-induced effects in SMA-like mice could originate from the modulation of NMDA-receptor activity in motor neurons. Indeed, the specific inhibition of the NMDA-receptor activity leads to the suppression of the exercise-induced benefits in the trained type 2 SMA-like mice, in terms of motor neuron protection, acceleration of muscle maturation, and lifespan gain. Furthermore, the activation of the NMDA receptor was shown to induce a significant increase in SMN levels in neurons (Andreassi et al., 2002). Our data suggest that the exercise-induced triggering of the NMDA signaling may result in an enhanced Smn gene expression in motor neurons, leading to an increase in the level of exon-7 containing SMN transcripts and consequently to an increase in SMN protein level as previously described in trained type 2 SMA-like mice (Grondard et al., 2005). However, these data substantiate the role of exercise in accelerating motor-unit postnatal development in SMA-like mice. Indeed, the role of the NMDA-receptor signaling pathway in the activity-dependent maturation of motor neurons during the postnatal developmental period is well established (Kalb and Hockfield, 1992; Kalb, 1994). Thus, if exercise triggers NMDA signaling in motor neurons, it is tempting to speculate that it may accelerate the postnatal development of the motor units leading to their protection.
Surprisingly, we have observed the defective expression of the major activating factor of the NMDA receptor, namely the NR2A subunit, in the spinal cord motor neurons of neonate type 2 SMA-like mice. Functional NMDA receptors are heterodimeric complexes containing NR1, the ion channel, and the regulatory subunits NR2 (NR2A-NR2D) and (or) NR3 (NR3A-NR3B), which respectively activates or potentially inhibits NR1 (Seeburg, 1993; Andersson et al., 2001; Nishi et al., 2001). The major regulatory subunit of NMDA receptor switches from NR2A to NR3B in motor-neurons during the early postnatal period (Fukaya et al., 2005) leading to potential changes in channel properties such as current kinetics and channel conductance (Monyer et al., 1994; Nishi et al., 2001; Matsuda et al., 2002). The NR2A expression defect in SMA-like spinal cord could originate from spinal denervation or alternatively might be a direct and specific consequence of SMA. Nevertheless, the modulation of the NMDA-receptor activity, because of the likely absence of NR2A, might also be involved in the motor unit defects seen in sedentary SMA-like mice. Unquestionably, this assumption needs to be experimentally confirmed, e.g., through the overexpression of NR2A in SMA-like mice using transgenic approaches and through the analysis of the NR2A expression patterns during embryonic stages in SMA-like mice and in the spinal cord of NR2A−/− mice. Indeed, in these mutant mice, if no developmental defect has been detected in various brain regions (Sakimura et al., 1995) in which NR2A mRNA is only expressed after birth (Watanabe et al., 1992), the motor unit development, during which the NR2A expression peaks at embryonic day 15 (Fukaya et al., 2005), should be thoroughly studied.
In addition, we report that the exercise program enhanced the NR2A gene expression in motor neurons of trained SMA-like mice, in agreement with previous findings in the hippocampus of wild mice using a microarray approach (Molteni et al., 2002). However, the exercise program did not modify the NR2B expression pattern in the SMA-like mice. Importantly, Liu et al. (2007) reported previously that the activation of NR2A-containing NMDA receptors promotes neuronal survival in mature cortical cultures but activating NR2B-containing NMDA receptors results in excitotoxicity and neuronal apoptosis. Accordingly, the exercise-induced expression of the NR2A subunit gene in the motor neurons of SMA-like mice would result in an increased number of NR2A-containing NMDA receptors, leading to neuroprotection. Moreover, because the NR2A subunit is the major activating subunit of the NMDA receptor in the ventral spinal cord of neonatal mice (Fukaya et al., 2005), the exercise-induced novel expression of the NR2A subunit in lumbar motor neurons in type 2 SMA-like mice would contribute to the NMDA-receptor activation.
Together, the present results would indicate that the activation of the NMDA-receptor activity is beneficial in promoting motor neuron survival in SMA-like spinal cords. This finding is provocative given the extensive amount of literature advocating the use of NMDA-receptor antagonists to protect immature motor neurons from programmed cell death (Mentis et al., 1993; Greensmith et al., 1994; Dick et al., 1995). The opposite effects of the NMDA signaling in neonatal motor neurons i.e., the promotion of apoptosis in injured motor neurons and the neuroprotection of SMA-like motor neurons might be related to the level of the NMDA-receptor activity. Indeed, the well described proapoptotic potential effect of NMDA signaling was shown to occur in a context of nerve injury in neonates, leading to an important release of glutamate in the medium and consequently to a hyper-activation of the NMDA receptor (Mentis et al., 1993; Greensmith et al., 1994; Dick et al., 1995). This proapoptotic side effect of the NMDA signaling in motor neurons was not activated when glutamate receptors are downregulated (Lladó et al., 1999). In this case, a MK-801 treatment induced massive neuron death and a NMDA chronic treatment promoted motor neuron survival (Lladó et al., 1999). In SMA, where the alteration of the NMDA-receptor subunit composition is likely to induce a low level of NMDA signaling, triggering the NMDA receptor unexpectedly induced glutamatergic excitotoxicity but promoted neuron survival. In addition, the MK-801 treatment had no effect in the sedentary type 2 SMA-like, presumably because of the very low activity of the NMDA receptor when deprived of its NR2A subunit.
In the future, it will be useful to examine whether the NMDA signaling is altered in other SMA mouse models. In particular, studying the severe SMA mouse model developed by Le et al. (2005), which is close to the model used in the present study in terms of genetic background and phenotype, represents a good opportunity to evaluate how specific is the influence of the NMDA signaling in SMA features. In the hypothesis of a SMA-induced specific disruption of the NMDA signaling, restoring the NR2A expression in embryo rather than in the postnatal period could represent a new therapeutic approach to treat the disease. The use of histone deacetylase inhibitor such as valproic acid (VPA) that was shown to increase SMN protein levels in cells from SMA patients (Brichta et al., 2003; Sumner et al., 2003) and in SMA-like mouse models (Tsai et al., 2006) seems promising. Indeed, the VPA-induced expression of SMN might result from the change in NMDA subunit gene expression, such as the NR2A gene, through acetylation of nucleosomal DNA. Thus, evaluating the NMDA-receptor activity in VPA-treated SMA-like mice would have important implications for developing therapeutic approaches for SMA.
Footnotes
This work was supported by the Association Française contre les Myopathies. O.B. and C.G. are the recipients of a fellowship from the Ministere de la Recherche et de la Technologie. We thank C.-L. Gallien and P.-P. Vidal for their comments and advice, P. Ascher for fruitful discussion about the NMDA-receptor study, J. Gilleron and D. Carette for deconvolution imaging, and V. Mouly for the muscle antibodies.
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Andersson O, Stenqvist A, Attersand A, von Euler G. Nucleotide sequence, genomic organization, and chromosomal localization of genes encoding the human NMDA receptor subunits NR3A and NR3B. Genomics. 2001;78:178–184. doi: 10.1006/geno.2001.6666. [DOI] [PubMed] [Google Scholar]
- Andreassi C, Patrizi AL, Monani UR, Burghes AH, Brahe C, Eboli ML. Expression of the survival of motor neuron (SMN) gene in primary neurons and increase in SMN levels by activation of the N-methyl-d-aspartate glutamate receptor. Neurogenetics. 2002;4:29–36. doi: 10.1007/s10048-001-0128-y. [DOI] [PubMed] [Google Scholar]
- Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- Dick J, Greensmith L, Vrbova G. Blocking of NMDA receptors during a critical stage of development reduces the effects of nerve injury at birth on muscles and motoneurones. Neuromuscul Disord. 1995;5:371–382. doi: 10.1016/0960-8966(94)00088-q. [DOI] [PubMed] [Google Scholar]
- Fan L, Simard LR. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Hayashi Y, Watanabe M. NR2 to NR3B subunit switchover of NMDA receptors in early postnatal motoneurons. Eur J Neurosci. 2005;21:1432–1436. doi: 10.1111/j.1460-9568.2005.03957.x. [DOI] [PubMed] [Google Scholar]
- Gabanella F, Carissimi C, Usiello A, Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet. 2005;14:3629–3642. doi: 10.1093/hmg/ddi390. [DOI] [PubMed] [Google Scholar]
- Greensmith L, Mentis GZ, Vrbová G. Blockade of N-methyl-d-aspartate receptors by MK-801 (dizocilpine maleate) rescues motoneurones in developing rats. Brain Res Dev Brain Res. 1994;81:162–170. doi: 10.1016/0165-3806(94)90302-6. [DOI] [PubMed] [Google Scholar]
- Grondard C, Biondi O, Armand AS, Lécolle S, Della Gaspera B, Pariset C, Li H, Gallien CL, Vidal PP, Chanoine C, Charbonnier F. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2005;25:7615–7622. doi: 10.1523/JNEUROSCI.1245-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahnen E, Forkert R, Marke C, Rudnik-Schoneborn S, Schonling J, Zerres K, Wirth B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet. 1995;4:1927–1933. doi: 10.1093/hmg/4.10.1927. [DOI] [PubMed] [Google Scholar]
- Hamburger V. Regression versus peripheral control of differentiation in motor hypoplasia. Am J Anat. 1958;102:365–409. doi: 10.1002/aja.1001020303. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Rempel J, Li J, Sawchuk MA, Carlin KP, Brownstone RM. Development of L-type calcium channels and a nifedipine-sensitive motor activity in the postnatal mouse spinal cord. Eur J Neurosci. 1999;11:3481–3487. doi: 10.1046/j.1460-9568.1999.00765.x. [DOI] [PubMed] [Google Scholar]
- Kalb RG. Regulation of motor neuron dendrite growth by NMDA receptor activation. Development. 1994;120:3063–3071. doi: 10.1242/dev.120.11.3063. [DOI] [PubMed] [Google Scholar]
- Kalb RG, Hockfield S. Activity-dependent development of spinal cord motor neurons. Brain Res Brain Res Rev. 1992;17:283–289. doi: 10.1016/0165-0173(92)90020-m. [DOI] [PubMed] [Google Scholar]
- Kashihara Y, Kuno M, Miyata Y. Cell death of axotomized motoneurones in neonatal rats, and its prevention by peripheral reinnervation. J Physiol (Lond) 1987;386:135–148. doi: 10.1113/jphysiol.1987.sp016526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lladó J, Calderó J, Ribera J, Tarabal O, Oppenheim RW, Esquerda JE. Opposing effects of excitatory amino acids on chick embryo spinal cord motoneurons: excitotoxic degeneration or prevention of programmed cell death. J Neurosci. 1999;19:10803–10812. doi: 10.1523/JNEUROSCI.19-24-10803.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Kamiya Y, Matsuda S, Yuzaki M. Cloning and characterization of a novel NMDA receptor subunit NR3B: a dominant subunit that reduces calcium permeability. Brain Res Mol Brain Res. 2002;100:43–52. doi: 10.1016/s0169-328x(02)00173-0. [DOI] [PubMed] [Google Scholar]
- McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Hannus S, Plöttner O, Baars T, Hartmann E, Fakan S, Laggerbauer B, Fischer U. SMNrp is an essential pre-mRNA splicing factor required for the formation of the mature spliceosome. EMBO J. 2001;20:2304–2314. doi: 10.1093/emboj/20.9.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Greensmith L, Vrbová G. Motoneurons destined to die are rescued by blocking N-methyl-d-aspartate receptors by MK-801. Neuroscience. 1993;54:283–285. doi: 10.1016/0306-4522(93)90253-c. [DOI] [PubMed] [Google Scholar]
- Molteni R, Ying Z, Gomez-Pinilla F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci. 2002;16:1107–1116. doi: 10.1046/j.1460-9568.2002.02158.x. [DOI] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Navarrette R, Vrbová G. Activity-dependent interactions between motoneurones and muscles: their role in the development of the motor unit. Prog Neurobiol. 1993;41:93–124. doi: 10.1016/0301-0082(93)90041-p. [DOI] [PubMed] [Google Scholar]
- Nishi M, Hinds H, Lu HP, Kawata M, Hayashi Y. Motoneuron-specific expression of NR3B, a novel NMDA-type glutamate receptor subunit that works in a dominant-negative manner. J Neurosci. 2001;21:RC185. doi: 10.1523/JNEUROSCI.21-23-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Oshima S, Fukaya M, Masabumi N, Shirakawa T, Oguchi H, Watanabe M. Early onset of NMDA receptor GluR epsilon 1 (NR2A) expression and its abundant postsynaptic localization in developing motoneurons of the mouse hypoglossal nucleus. Neurosci Res. 2002;43:239–250. doi: 10.1016/s0168-0102(02)00035-4. [DOI] [PubMed] [Google Scholar]
- Pette D, Staron RS. Mammalian skeletal muscle fiber type transitions. Int Rev Cytol. 1997;170:143–223. doi: 10.1016/s0074-7696(08)61622-8. [DOI] [PubMed] [Google Scholar]
- Pool CW, Diegenbach PC, Scholten G. Quantitative succinate-dehydrogenase histochemistry. III. Variations in histochemical succinate dehydrogenase activity in different cross-sections of the same muscle fibre. Histochemistry. 1979;64:251–262. doi: 10.1007/BF00495027. [DOI] [PubMed] [Google Scholar]
- Rodrigues NR, Owen N, Talbot K, Ignatius J, Dubowitz V, Davies KE. Deletions in the survival motor neuron gene on 5q13 in autosomal recessive spinal muscular atrophy. Hum Mol Genet. 1995;4:631–634. doi: 10.1093/hmg/4.4.631. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Landmesser LT. Cell death of lumbosacral motoneurons in chick, quail, and chick-quail chimera embryos: a test of the quantitative matching hypothesis of neuronal cell death. J Neurosci. 1986;6:2889–2899. doi: 10.1523/JNEUROSCI.06-10-02889.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LK, Tsai MS, Lin TB, Hwu WL, Li H. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol Dis. 2006;24:286–295. doi: 10.1016/j.nbd.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Vassias I, Lecolle S, Vidal PP, de Waele C. Modulation of GABA receptor subunits in rat facial motoneurons after axotomy. Brain Res Mol Brain Res. 2005;135:260–275. doi: 10.1016/j.molbrainres.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Vinay L, Brocard F, Clarac F. Differential maturation of motoneurons innervating ankle flexor and extensor muscles in the neonatal rat. Eur J Neurosci. 2000;12:4562–4566. doi: 10.1046/j.0953-816x.2000.01321.x. [DOI] [PubMed] [Google Scholar]
- Vult von Steyern F, Lomo T. Postnatal appearance of 5-HT2A receptors on fast flexor and slow extensor rat motor neurons. Neuroscience. 2005;136:87–93. doi: 10.1016/j.neuroscience.2005.07.059. [DOI] [PubMed] [Google Scholar]
- Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G. The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol. 2005;25:5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. NeuroReport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- Yong J, Wan L, Dreyfuss G. Why do cells need an assembly machine for RNA-protein complexes? Trends Cell Biol. 2004;14:226–232. doi: 10.1016/j.tcb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]