

Abstract
Abnormal accumulation of soluble oligomers of amyloid β (Aβ) is believed to cause malfunctioning of neurons in Alzheimer's disease. It has been shown that Aβ oligomers impair synaptic plasticity, thereby altering the ability of the neuron to store information. We examined the underlying cellular mechanism of Aβ oligomer-induced synaptic modifications by using a recently described stable oligomeric Aβ preparation called “Aβ1–42 globulomer.” Synthetically prepared Aβ1–42 globulomer has been shown to localize to neurons and impairs long-term potentiation (Barghorn et al., 2005). Here, we demonstrate that Aβ1–42 globulomer does not affect intrinsic neuronal properties, as assessed by measuring input resistance and discharge characteristics, excluding an unspecific alteration of membrane properties. We provide evidence that Aβ1–42 globulomer, at concentrations as low as 8 nm, specifically suppresses spontaneous synaptic activity resulting from a reduction of vesicular release at terminals of both GABAergic and glutamatergic synapses. EPSCs and IPSCs were primarily unaffected. A detailed search for the precise molecular target of Aβ1–42 globulomer revealed a specific inhibition of presynaptic P/Q calcium currents, whereas other voltage-activated calcium currents remained unaltered. Because intact P/Q calcium currents are needed for synaptic plasticity, the disruption of such currents by Aβ1–42 globulomer may cause deficits in cellular mechanisms of information storage in brains of Alzheimer's disease patients. The inhibitory effect of Aβ1–42 globulomer on synaptic vesicle release could be reversed by roscovitine, a specific enhancer of P/Q currents. Selective enhancement of the P/Q calcium current may provide a promising strategy in the treatment of Alzheimer's disease.
Keywords: Aβ globulomer, Aβ oligomers, P/Q-type calcium channel, amyloid, Alzheimer's disease, hippocampal neurons
Introduction
Alzheimer's disease (AD) is a neurodegenerative ailment accompanied by a progressive loss of memory and by a variety of histopathological changes. Among those are extracellular deposits of amyloid β (Aβ) peptide, so called amyloid plaques that occur predominantly in parts of the brain relevant for memory-related processes, such as the hippocampus and the basal forebrain. Multiple lines of evidence suggest that Aβ crucially contributes to the pathology of the disease (Scheuner et al., 1996; Bales et al., 1999). However, soluble Aβ forms, rather than amyloid plaques, correlate with the severity of cognitive impairment in humans (Lue et al., 1999; McLean et al., 1999). Thus, soluble oligomeric β-amyloid forms may significantly contribute to the cellular pathology of the disease. For example, artificially generated soluble Aβ oligomers bind to synapses and are detrimental to neurons (Lacor et al., 2004, 2007). Acute application of Aβ oligomers suppresses synaptic plasticity in vitro (Lambert et al., 1998; Wang et al., 2004) and in vivo (Walsh et al., 2002), thereby decreasing the ability of neurons to store information. Cleary et al. (2005) could show that oligomers directly impair learning and memory after intraventricular injection into rat brain. For these reasons, it has now become widely accepted that soluble oligomeric forms of Aβ account at least in part for the cognitive deficits in AD patients (Selkoe, 2002) and that reduction of soluble Aβ might be a promising strategy in the therapeutic intervention of AD (McLean et al., 1999).
We described recently a method for the preparation of a highly stable oligomeric Aβ complex called “Aβ1–42 globulomer”(Barghorn et al., 2005), which mimics binding properties and pathology of other described oligomeric Aβ forms. For example, Aβ1–42 globulomer binds to primary hippocampal neurons and blocks long-term potentiation (LTP) in hippocampal slices. The pathological relevance of Aβ1–42 globulomer is supported by the observation that specific antibodies detect globulomer epitopes in brains of AD patients and Aβ-overproducing transgenic mice (Barghorn et al., 2005). These data suggest that a naturally occurring form of Aβ1–42 globulomer causes a defective information storage of hippocampal synapses and thus contributes to the cognitive deficits in brains of AD patients.
Although the detrimental effect of different oligomeric species on synaptic plasticity is well described, less is known about the molecular mechanism that leads to malfunctioning of synapses. It has been suggested recently that oligomeric Aβ may disturb synaptic function via NMDA-related cascades (De Felice et al., 2007; Shankar et al., 2007). However, a direct molecular target of Aβ oligomers has not yet been described. Therefore, we examined the effect of Aβ1–42 globulomer on intrinsic neuronal properties and on synaptic transmission and found evidence that the pathological molecular mechanism of Aβ oligomers is based on a specific suppression of presynaptic P/Q calcium current.
Materials and Methods
Cell culture
Primary hippocampal cell cultures were prepared from Wistar rat embryos at embryonic day 19 (E19) in accordance with the protocol described previously by Banker and Cowan (1977). Briefly, pregnant rats were deeply anesthetized by ether narcosis and decapitated. Embryos were rapidly removed, and brains were dissected under constant cooling with chilled (∼4°C) PBS. Then, both hippocampi were taken out and washed twice with ice-cold PBS followed by a wash with PBS at room temperature. Hippocampi were triturated using three siliconized pipettes with decreasing tip diameters. Cells were then plated on coverslips (density 60000 cells/coverslip, coated with 0.01% poly-l-lysine solution) and stored at 37°C in an incubator gassed with 5% CO2 in normal air. The culture medium contained 0.25% penicillin/streptomycin, 2% B27, 0.25% l-glutamine (Invitrogen, Karlsruhe, Germany). Cells were cultured for 10–21 d until used for experiments.
Throughout culturing, we added 0.5 μm ω-conotoxin MVIIA to the culture medium to block N-type calcium channels and to stabilize the expression of P/Q-type currents. Previous experiments on pharmacologically naive cells revealed similar results, although with high variability between cells and between different culture preparations. Addition of ω-conotoxin MVIIA yielded more stable results without altering the qualitative results. For example, Aβ1–42 globulomer reduced the frequency of spontaneous postsynaptic currents (sPSCs) to 47 ± 10% (n = 11) without ω-conotoxin preincubation compared with 38 ± 5% in cells that were raised in the blocker. This effect of ω-conotoxin is well consistent with the highly variable relative contribution of N- and P/Q-type channels to synaptic transmission in developing neuronal networks (Scholz and Miller, 1995; Iwasaki et al., 2000; Reid et al., 2003).
Current recording
Currents were measured under whole-cell voltage-clamp conditions at room temperature using borosilicate pipettes of 2–4 MΩ resistance. Electrode solution contained the following (in mm): 10 NaCl, 100 KCl, 0.25 CaCl2, 5 EGTA, 10 HEPES, 40 glucose, pH set at 7.3, when used for recordings of synaptic events. A low-chloride solution was used for experiments in which GABA-induced currents were elicited, which consists of the following (in mm): 130 Cs-gluconate, 10 CsCl, 0.5 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES, 2 Mg-ATP, pH 7.3. Using this solution, the calculated equilibrium potential for chloride-ions was −54 mV. During calcium current measurements, the solution contained the following (in mm): 110 CsCl, 10 EGTA, 25 HEPES, 10 tris-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, and 20 U/ml creatine-phosphokinase, pH 7.3. Osmolarity was adjusted to 295 mOsm/L, when necessary, by adding glucose. Bath solutions contained the following (in mm): 156 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 16.5 Glucose, 10 HEPES, pH set to 7.3, for recordings of synaptic events; and 140 tetraethylammonium (TEA)-Cl, 10 BaCl2, 10 HEPES, and 20 Glucose, pH 7.3 for calcium currents, respectively. The bath perfusion was stopped for 10 min before the application of the Aβ1–42 globulomer, and baseline activity was recorded. Subsequently, Aβ1–42 globulomer (164 nm with respect to the 12mer complex) was added to the bath by means of a micro pump, yielding a final concentration of 82 nm. In some experiments, we added a 10-fold lower concentration of Aβ1–42 globulomer yielding a final amount of 8.2 nm. TTX, ω-agatoxin IVA, ω-conotoxin MVIIA, roscovitine (Alomone Labs, Jerusalem, Israel), and nifedipine (Sigma, Deisenhofen, Germany) were added directly to the bath solution at the concentrations indicated.
Currents were measured with an Axopatch 200B (Molecular Devices, Union City, CA) or an EPC-7 amplifier (HEKA, Lambrecht, Germany), digitized by a CED 1401 micro-analog/digital converter (Cambridge Electronics Design, Cambridge, UK) and stored on a personal computer (sample frequency, 20 kHz). All recorded currents were low-pass filtered with a cutoff frequency of 3 kHz. Capacitive transients and series resistances were compensated on-line (∼50–60% compensation) during the calcium current measurements. No compensation was performed during recordings of synaptic events. Data were evaluated off-line using Spike5 and Signal3 software (Cambridge Electronics Design, Cambridge, UK). All calcium current traces were corrected for aspecific linear leak (reversal potential, 0 mV) determined at holding potential using ±5 mV potential steps.
Current analysis
Calcium currents.
All cells were voltage clamped at a holding potential of −80 mV, and calcium ions were substituted by Barium ions to increase the amplitude of the current flow through the calcium channels. Peak amplitudes of the currents (I) evoked with the activation protocol were plotted as a function of membrane potential (V). The resulting I–V relationships were fitted with a combination of a first-order Boltzmann activation function and the Goldman-Hodgkin-Katz (GHK) current–voltage relationship (Kortekaas and Wadman, 1997) as follows:
 |
with α = F/RT and g max = α FP 0 [Ba+]out, where g max is the maximal membrane conductance (which is proportional to the maximal permeability and the extracellular concentration of barium), Vh is the potential of half-maximal activation, and Vc is proportional to the slope of the curve at Vh. F represents the Faraday constant, R represents the gas constant, P 0 is the maximal permeability, and T the absolute temperature. The intracellular concentration of Ba2+ was assumed to be 0.01 μm. Assuming higher values of up to 0.1 mm did not significantly change the resulting values of the parameters.
The voltage dependence of steady-state inactivation of the barium current was estimated from the relationship of peak current amplitude versus the prepotential. This relationship was well described by a Boltzmann function, which normalized the current as follows:
 |
where N(V) is the level of steady-state inactivation determined from the current amplitude I(V) normalized to I max, V is the prepulse potential, Vh is the potential of half-maximal inactivation, and Vc is a factor proportional to the slope of the curve at Vh.
Synaptic events
For these recordings, all cells were voltage clamped at a holding potential of −70 mV. Synaptic events triggered by the release of GABA were inwardly directed (E Cl is approximately −10 mV) because of the use of high-chloride concentrations in the pipette and the bath. Routinely, 10 min of baseline activity was acquired, serving as control data, before any drug application was started. Synaptic events were then analyzed off-line for frequency and amplitude, using a custom-made, template-based algorithm.
Evoked IPSCs
Synaptic responses were elicited by extracellular stimulation using a conventional patch pipette (2–4 MΩ resistance, filled with bath solution), positioned close to the soma of a neuron in the vicinity of the recorded cell. Solution of the recording pipette contained 5 mm N-ethyl bromide quaternary salt in these experiments. After optimizing electrode position and stimulation strength, IPSCs could be reliably induced (stimulus intensity, 70–100 V; duration, 50–200 μs; isolated stimulator, NPI Electronics, Tamm, Germany). Evoked IPSCs were recorded with 10 s intervals, and 31 events per condition were averaged for analysis. In these experiments, the bath solution contained CNQX (20 μm), APV (30 μm), and (2S)-3-([(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid (CGP 55845) (2 μm).
Preparation of Aβ1–42 globulomer and monomers
Aβ1–42 globulomer was prepared as described previously (Barghorn et al., 2005). In brief, lyophilized Aβ1–42 synthetic peptide was disaggregated by using 100% 1,1,1,3,3,3 hexafluoro-2-propanol. After evaporation, Aβ1–42 was resuspended at a concentration of 5 mm in dimethylsulfoxide, diluted to a final concentration of 400 μm in PBS containing 0.2% SDS. After 6 h incubation at 37°C, the sample was diluted with three volumes of H2O and incubated for another 18 h at 37°C. The sample was concentrated by ultrafiltration (30 kDa cutoff), dialyzed against 5 mm NaH2PO4 35 mm NaCl, pH 7.4, centrifuged at 10,000 × g for 10 min, and the supernatant containing the 48 kDa Aβ1–42-globulomer withdrawn. Aβ1–42-globulomer was diluted in extracellular solution at the concentration indicated immediately before experiments. Currents were measured before and immediately after addition of Aβ1–42 globulomer to the bath solution.
For control experiments, synthetic monomeric Aβ1–42 peptide (H-1368; Bachem, Bubendorf, Switzerland) was dissolved in 0.1% NaOH, yielding a 1 mm stock solution, which was frozen at −80°C. Immediately before the experiment, this solution was dissolved at 1:500 in bath solution, which was added to the bath by means of a micro-pump, resulting in a final concentration of 1 μm.
Statistics
Values are presented as the mean ± SEM. Statistical comparisons were made with Student's t test after testing for normality. A p value <0.05 was used to indicate significant differences.
Results
Aβ1–42 globulomer reduces spontaneous synaptic activity in hippocampal cell cultures
We measured spontaneous synaptic activity in cultured hippocampal neurons using whole-cell voltage-clamp techniques (V hold, −70 mV). Under our ionic conditions, all synaptic events appeared as inward currents (sPSCs) with a mean frequency of 216 ± 71/min (n = 11). Bath application of 82 nm Aβ1–42 globulomer (globulomer molarities calculated with respect to the 12mer complex) rapidly reduced the frequency of sPSCs to 36 ± 5% of control (p < 0.05; n = 11) (Figs. 1 A, 2 A). This effect was partially reversible after washout in two of three cells tested (61 ± 16%). The median amplitude of events was 77 ± 18 pA and was reduced to 88 ± 12% under Aβ1–42 globulomer (p < 0.05; n = 11) (Fig. 2 B). Similar, but slightly weaker, effects were seen after application of 8.2 nm Aβ1–42 globulomer (frequency reduced to 63 ± 9%; p < 0.05; median amplitude, 94 ± 5% of control, n = 8, n.s.). Thus, the suppression of spontaneous synaptic activity by Aβ1–42 globulomer is dose dependent and starts at low nanomolar concentrations. Input resistance was not affected by Aβ1–42 globulomer (control, 120.9 ± 13.6 MΩ; Aβ1–42, 131.6 ± 13.7 MΩ).
Figure 1.

A, Synaptic activity is reversibly suppressed by Aβ1–42 globulomer. Original recording of sPSCs (A) and mPSCs (B) in cultured hippocampal neurons before (control), during (glob), and after (wash) application of Aβ1–42 globulomer. C, Cumulative probability plot of interevent intervals of mPSCs from the cell shown in B. Note the rightward shift of the curve induced by the globulomer, whereas the amplitude remained stable (D).
Figure 2.
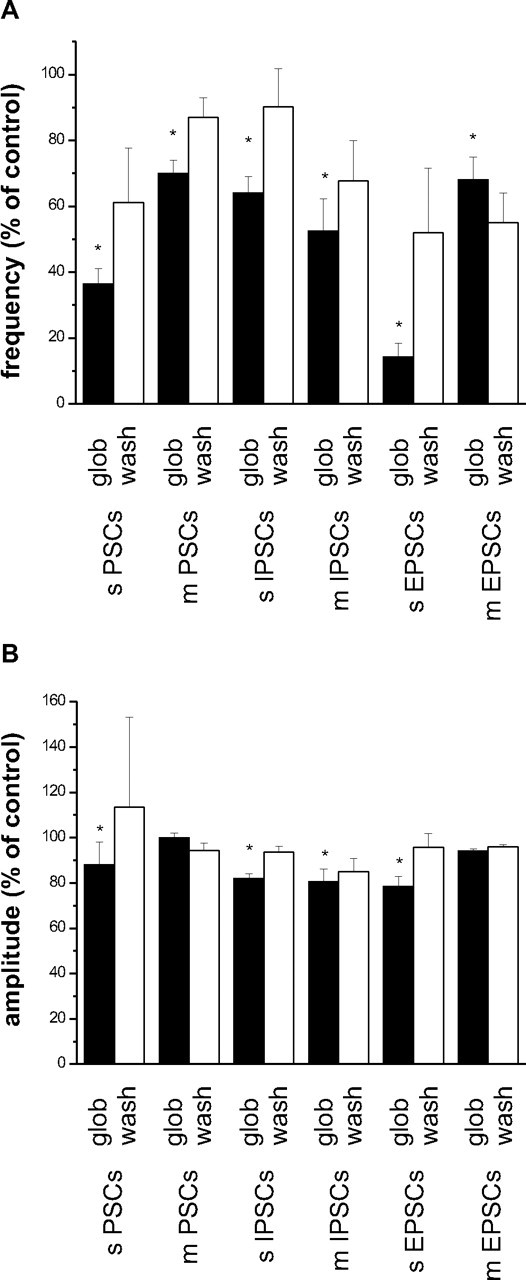
Effects of Aβ1–42 globulomer on different types of synaptic currents in cultured hippocampal neurons. Black bars, Effect of Aβ1–42 globulomer (glob); white bars, washout for at least 10 min (wash). A, Reduction of event frequency as percentage of previously recorded control currents (100%). B, Effects of Aβ1–42 globulomer on median amplitude of the respective currents. sPSCs, Spontaneously occurring pharmacologically naive PSCs; mPSCs, pharmacologically naive mPSCs recorded in the presence of TTX.
Suppression of synaptic currents by an agent may be caused by changes in neuronal activity or, alternatively, by specific synaptic interactions. We therefore tested for effects of Aβ1–42 globulomer on active discharge properties by recording action potentials (APs) in current-clamp mode. Action potentials elicited by current injection showed no difference in amplitude, shape, or kinetics when compared before and after Aβ1–42 globulomer application (Fig. 3 A). In detail, the threshold for firing was −22.5 ± 8.2 versus −24.2 ± 9.8 mV, and the amplitude of the AP (baseline to peak) amounted to 119.9 ± 11.2 versus 110.9 ± 16.7 mV. Likewise, kinetic parameters did not differ: values for the half-width time were 3.5 ± 1.6 versus 4.0 ± 2.9 ms, maximal rate of rise 100.5 ± 46.4 versus 84.2 ± 50.0 V/s, and maximal rate of repolarization 46.0 ± 18.6 versus 47.4 ± 19.3 V/s (n = 16 action potentials from four cells before and after Aβ1–42 globulomer, respectively) (Fig. 3 B). It thus appears that the alteration of synaptic activity by Aβ1–42 globulomer may be caused by a direct interaction with presynaptic or postsynaptic proteins, rather than by an unspecific alteration of cellular excitability.
Figure 3.
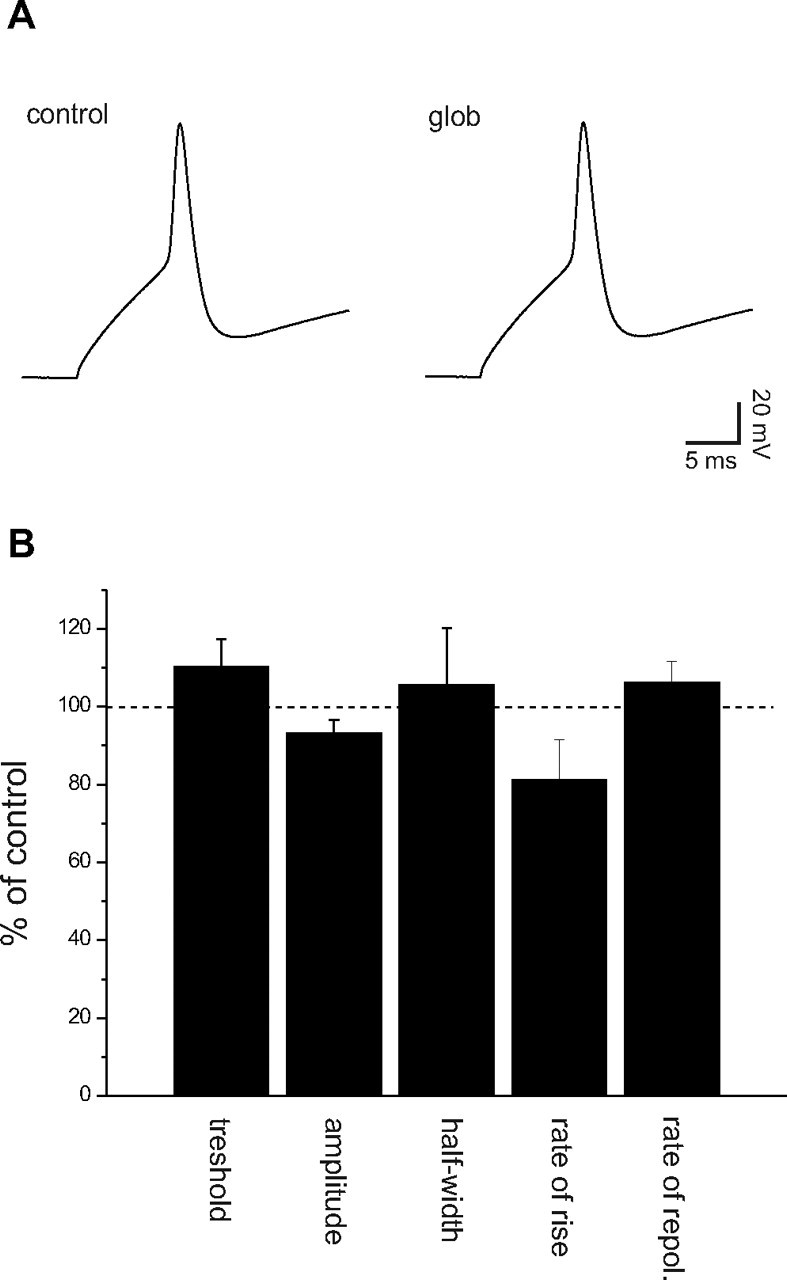
Aβ1–42 globulomer does not change intrinsic discharge properties of hippocampal neurons. A, Current-clamp recordings of action potentials revealed no difference between the evaluated parameters in control solutions and during the application of the Aβ1–42 globulomer (glob). B, Normalized values for action potential threshold (threshold); mean amplitude of action potentials (amplitude); half-width of action potentials (half-width); and rates of rise and repolarization of the action potentials.
This hypothesis was corroborated by recordings of spontaneously occurring miniature PSCs (mPSCs) in the presence of TTX. Similar to spontaneous “macroscopic” PSCs, these currents were reduced in frequency by 82 nm Aβ1–42 globulomer (yielding 70 ± 4% of control; n = 9; p < 0.05) (Figs. 1 B, 2 A). However, the amplitude of mPSCs was unaltered (median amplitude, 29.7 ± 2.5 pA under control conditions vs 29.6 ± 2.4 pA in the presence of Aβ1–42 globulomer; n = 9) (Fig. 2 B). After washout for 10 min, the effect on event frequency recovered partially to 87 ± 6% of control (n = 4; wash, 4 of 9). Together, these data suggest that Aβ1–42 globulomer interferes with the presynaptic machinery of transmitter release.
Effects on spontaneous and miniature IPSCs
Pharmacologically naive synaptic currents reflect a mixture of glutamatergic (excitatory) and GABAergic (inhibitory) events. To differentiate between these components, we isolated IPSCs by adding CNQX (20 μm) and dl-APV (30 μm) to the bath solution. The frequency of spontaneously occurring IPSCs was suppressed by Aβ1–42 globulomer (yielding 64 ± 5% of control; p < 0.05; n = 12), and the median amplitude was reduced to 82 ± 2% of control (p < 0.05) (Fig. 2 A,B). These reductions could be reversed to some degree after withdrawal of the globulomer (frequency, 90 ± 12%; amplitude, 94 ± 2%).
Miniature IPSCs (mIPSCs) (recorded in 0.5 μm TTX) did also show a similar reduction in frequency after application of Aβ1–42 globulomer (52 ± 10% of control; p < 0.05; n = 6). This effect was partially reversible after washout, yielding 68 ± 12% of control (Fig. 2 A). In addition, we observed a reduction of mIPSC amplitude (81 ± 6% of control; p < 0.05; no washout in 3 of 3 cells, 85 ± 6%) (Fig. 2 B).
Effects on postsynaptic GABAA receptors
To test for potential effects of Aβ1–42 globulomer on postsynaptic GABAA receptors, we applied a high (100 μm) concentration of GABA by brief pressure pulses directly onto the cell. Repetitive application of GABA to cultured cells elicited large (>1 nA) inward currents, which showed only minor rundown with time. This behavior was unaltered after application of Aβ1–42 globulomer for 5 min, indicating that GABAA receptors are not affected by the agent (Fig. 4 A,B).
Figure 4.
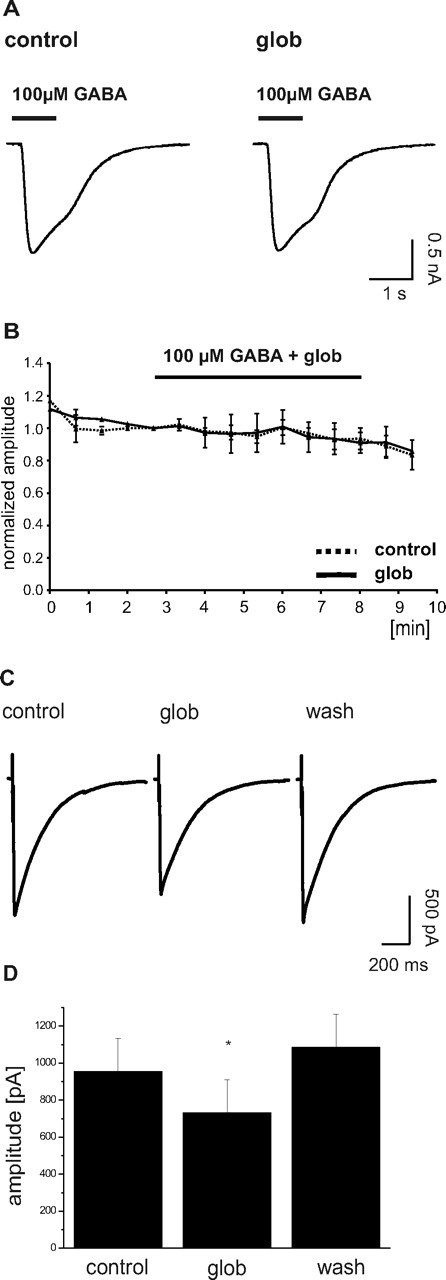
Stability of GABAA receptor-mediated currents toward Aβ1–42 globulomer. A, Repetitive application of 100 μm GABA to a cultured hippocampal neuron yields stable inward currents in the absence and presence of the oligomer. B, Time course of GABA-induced currents from five cells recorded in control solution (dashed line) and from three neurons where Aβ1–42 globulomer (glob) was applied (continuous line, time of application indicated by bar). Amplitudes normalized to the last GABA-induced current before application of Aβ1–42 globulomer. Note the stability of response in the absence and presence of Aβ1–42 globulomer. C, Averaged traces (5 events each; from one cell) of stimulus-evoked IPSCs before, during, and after the application of Aβ1–42 globulomer. Stimulation artifacts are truncated. The amplitude of these synaptic currents was reduced to 78 ± 3% of control by the action of the globulomer. D, Bar graph showing the mean reduction in amplitude of eIPSCs induced by the globulomer.
Evoked IPSCs
To test for the effect of Aβ1–42 globulomer on action potential-dependent transmitter release, we recorded evoked IPSCs (eIPSCs) in the presence of CNQX, APV, and CGP 55845. Stimulation of synaptic inputs at 0.1 Hz over a period of 5 min in control conditions elicited eIPSCs with a median amplitude of 955 ± 252 pA (n = 6) (Fig. 4 C). Application of 82 nm Aβ1–42 globulomer reduced the amplitude of eIPSCs to 78 ± 3% (i.e., 730 ± 179 pA) of control within 7 min of exposure (n = 6; p < 0.05) (Fig. 4 D). After washout for 10 min, the effect on eIPSC amplitude recovered to 109 ± 14% of control (n = 4; wash, 4 of 6). Application of the bath solution without Aβ1–42 globulomer did not change the amplitude of eIPSCs (105 ± 11% of control; n = 3).
Effects on spontaneous and miniature EPSCs
Finally, we isolated EPSCs in the presence of 5 μm gabazine (a GABAA receptor antagonist). Basal frequency of these events was 386 ± 124/min. Their frequency was reduced by Aβ1–42 globulomer to 14 ± 4% of control (p < 0.05; n = 6) (Fig. 2 A). Likewise, the amplitude was reduced to 79 ± 4% of control (n = 6; p < 0.05) (Fig. 2 B). The effect was partially reversible during washout (frequency increasing to 52 ± 19% of control, amplitude to 96 ± 6%; n = 6). The frequency of miniature EPSCs (mEPSCs) was likewise suppressed to 68 ± 7% of control (n = 6; p < 0.05), whereas the amplitude of mEPSCs remained stable (94 ± 1% of control; wash out, 96 ± 1%). The frequency suppression did not recover after wash out (55 ± 9%; n = 6) (Fig. 2 A,B).
Together, these data indicate that Aβ1–42 globulomer depresses vesicular release at GABAergic and glutamatergic synapses, most likely by decreasing the probability of vesicle exocytosis from presynaptic terminals.
Lack of effect of monomeric Aβ1–42 peptide on mPSCs
To test for the specificity of the Aβ1–42 globulomer effect, a preparation of synthetic monomeric Aβ1–42 peptide was applied while recording mPSCs in the presence of TTX. A temporarily stable monomer solution was prepared by dissolving synthetic Aβ1–42 in 0.1% NaOH (see Materials and Methods). A Coomassie-stained SDS-PAGE confirmed the presence of Aβ1–42 monomer and the Aβ1–42 globulomer at the expected molecular weights in the respective preparations (Fig. 5 A).
Figure 5.
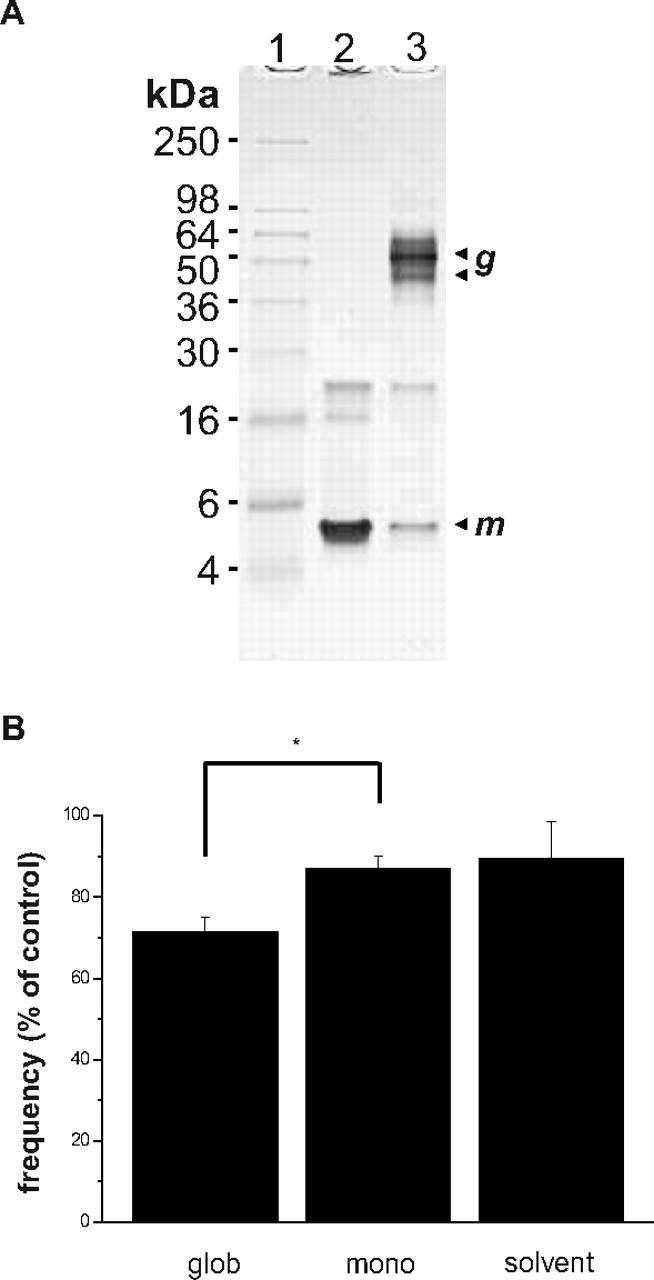
A, Coomassie-stained SDS-PAGE showing the Aβ1–42 globulomer (glob) and Aβ1–42 monomer (mono) as used in the electrophysiological experiments. In each case, 4.5 μg was loaded onto a 18% Tris glycine gel (Invitrogen), which was subsequently coomassie stained using standard protocols. Aβ1–42 globulomers (g; column 3) are visualized at an apparent molecular mass of 38/48 kDa, whereas the majority of Aβ1–42 monomer (m; column 2) runs at ∼5 kDa. B, Bar diagram showing no effect of the monomer on mPSC frequency compared with the significant reduction in frequency induced by the globulomer. The right bar shows that the solvent alone (0.0001% NaOH) does not affect the frequency.
The monomeric preparation was bath-applied at an initial concentration of 1 μm Aβ1–42 monomer, which equals the amount of monomer contained in the 82 nm globulomer preparation. Frequency of mPSCs was 87 ± 3% of control in the presence of monomeric Aβ1–42 (n = 7; n.s.) (Fig. 5 B), which was similar to the frequency observed by application of the solvent alone (0.1% NaOH diluted 1:1000 in bath solution; 89 ± 9% of control; n = 7) (Fig. 5 B). The amplitude of mPSCs was unaltered after application of the monomer preparation (median amplitude, 34.2 ± 3.0 pA under control conditions vs 33.7 ± 3.0 pA in the presence of Aβ1–42 monomer) or its respective solvent (median amplitude, 32.4 ± 1.5 pA under control conditions vs 32.3 ± 1.1 pA in the presence of the solvent).
Note that, in general, Aβ1–42 peptide can hardly be maintained in its monomeric state in physiological buffers, because it aggregates within minutes to protofibrils and fibrils. We used 0.1% NaOH as the initial solubilization buffer for the synthetic Aβ1–42 peptide, which is the most suitable buffer for solubilizing and maintaining Aβ1–42 peptide in a monomeric state under our experimental conditions. Although great care was taken to minimize Aβ1–42 peptide aggregation, we observed aggregation at the final dilution of 0.0001% NaOH in the bath solution when samples were retrieved after the actual experiments. Therefore, the applied monomeric Aβ1–42 peptide is likely a mixture of Aβ1–42 aggregation states (i.e., Aβ1–42 monomer, Aβ1–42 protofibrils, and Aβ1–42 fibrils). Furthermore, aggregated Aβ1–42 peptide within the monomeric Aβ1–42 preparation can also be seen in the SDS-PAGE gel loading pocket (Fig. 5 A). Preparations of Aβ1–42 tend to adhere to surfaces and therefore may reach lower final effective concentrations at the target cells. Therefore, we representatively determined the Aβ1–42 content after the experiment and found that in both Aβ1–42 monomer and globulomer preparations, >50% of the initial Aβ1–42 peptide were present during the electrophysiological recordings.
Effects on voltage-activated calcium currents
Presynaptic vesicle release is triggered by an influx of calcium ions into the presynaptic terminal (Zucker, 1993). We therefore speculated that Aβ1–42 globulomer might act on presynaptic calcium signaling. A common pathway for release of both glutamatergic and GABAergic vesicles is presynaptic calcium influx via N-type or P/Q-type calcium channels. We therefore analyzed the effects of Aβ1–42 on whole-cell calcium currents in cultured hippocampal neurons. Typical P/Q channel-mediated currents could be reliably elicited in somatic whole-cell recordings under our culture conditions. In these experiments, 10 mm Ba2+ was used as charge carrier in the extracellular solution (see Materials and Methods). Measurements were performed in the presence of 10 μm nifedipine (an L-type calcium channel blocker), ω-conotoxin MVIIA (an N-type calcium channel blocker), and blockers of other voltage-gated ion channels (0.5 μm TTX, 140 mm TEA, Cs+-based intracellular solution). Rundown of these currents was avoided by adding 20 U/ml phosphocreatine kinase and 10 mm tris-phosphocreatine to the pipette solution. Under these conditions, P/Q-type currents were evoked by a depolarizing voltage step to −10 mV (Fig. 6 B) (mean amplitude, 1015 ± 145 pA). Aβ1–42 globulomer reduced the amplitude of these currents to 62 ± 7% of control (n = 10). This effect was partially reversible in three of three cells (Fig. 6 A,B).
Figure 6.
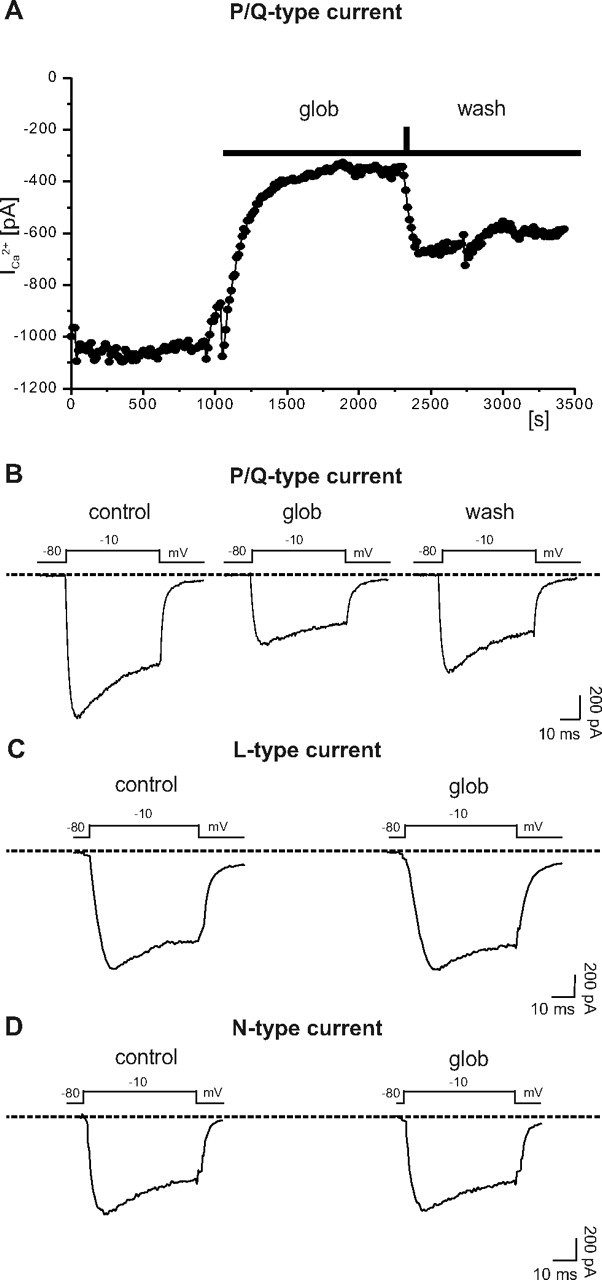
Selective suppression of P/Q-type calcium currents by Aβ1–42 globulomer. A, Time course of P/Q current amplitudes after application of globulomer (glob). Currents were elicited by voltage steps to −10 mV. B, Example traces of P/Q-type currents before, during, and after application of the globulomer, showing a prominent reduction of the P/Q current amplitude induced by the globulomer. Other types of calcium currents (N- and L-type) were not affected by the globulomer. C, L-type current recorded in the presence of 0.5 μm ω-conotoxin and ω-agatoxin (0.5 μm). D, N-type current recorded in the presence of 10 μm nifedipine and ω-agatoxin (0.5 μm).
In addition, we analyzed the effects of Aβ1–42 globulomer on N-, and L-type calcium currents. Besides blockers for Na+ and K+ channels (see above), we added 0.5 μm ω-agatoxin IVA to block P/Q channels. L-type calcium currents were isolated by addition of 0.5 μm ω-conotoxin MVIIA. Voltage pulses from −80 to −10 mV elicited inward currents of 597.7 ± 230.9 pA amplitude, which remained stable after addition of Aβ1–42 globulomer (573.0 ± 225.6 pA; n = 3) (Fig. 6 C). When N-type currents were isolated by adding nifedipine (10 μm) instead of ω-conotoxin, the same voltage-clamp protocol elicited inward currents, which were, again, insensitive to Aβ1–42 globulomer (amplitude in control solution, 1368.9 ± 332.8; amplitude in Aβ1–42 globulomer, 1399.8 ± 376.4 pA; n = 3) (Fig. 6 D). When all blockers were added together, the remaining calcium current (possibly R-type) was too small for a detailed analysis (<100 pA), indicating that this component was only marginally expressed in the cultured hippocampal neurons.
We further characterized the effect of Aβ1–42 globulomer on P/Q-type calcium currents by steady-state activation and inactivation protocols (Fig. 7 A–C, inset). The reduction of the current amplitude by the globulomer was not limited to the test potential of −10 mV but was present over a wide range of voltages (−20… +60 mV) (Fig. 7 A). A graphical fit of the resulting I–V relationship by the GHK equation (see Materials and Methods) gives a maximal conductance (g max) of 61.7 ± 2.4 nS (control) versus 27.2 ± 3.2 nS (Aβ1–42 globulomer; p < 0.05; n = 6) (Fig. 7 D). In contrast to this marked reduction in conductance (and current amplitude), other kinetic parameters were not affected by Aβ1–42 globulomer. Steady-state activation was characterized by Vh of −15.4 ± 1.1 mV, which was not changed after application of Aβ1–42 globulomer (Vh, −17.3 ± 1.3 mV; n = 6) (Fig. 7 B,D). The slope of the fitted first-order Boltzmann equation Vc was −7.8 ± 0.3 mV in control solution and −10.8 ± 0.5 mV in Aβ1–42 globulomer (not different; n = 6). Likewise, steady-state inactivation was not affected by Aβ1–42 globulomer, as indicated by stable values for the voltage at half-maximal inactivation (−29.2 ± 0.6 mV in control; −32.4 ± 1.2 mV in Aβ1–42 globulomer; n = 4) and for the slope Vc (−11.0 ± 0.9 mV vs −12.6 ± 1.1 mV) (Fig. 7 C,D). Thus, Aβ1–42 globulomer reduces the current amplitude by reducing the conductance of the P/Q channels, without affecting the voltage dependence of activation or inactivation.
Figure 7.
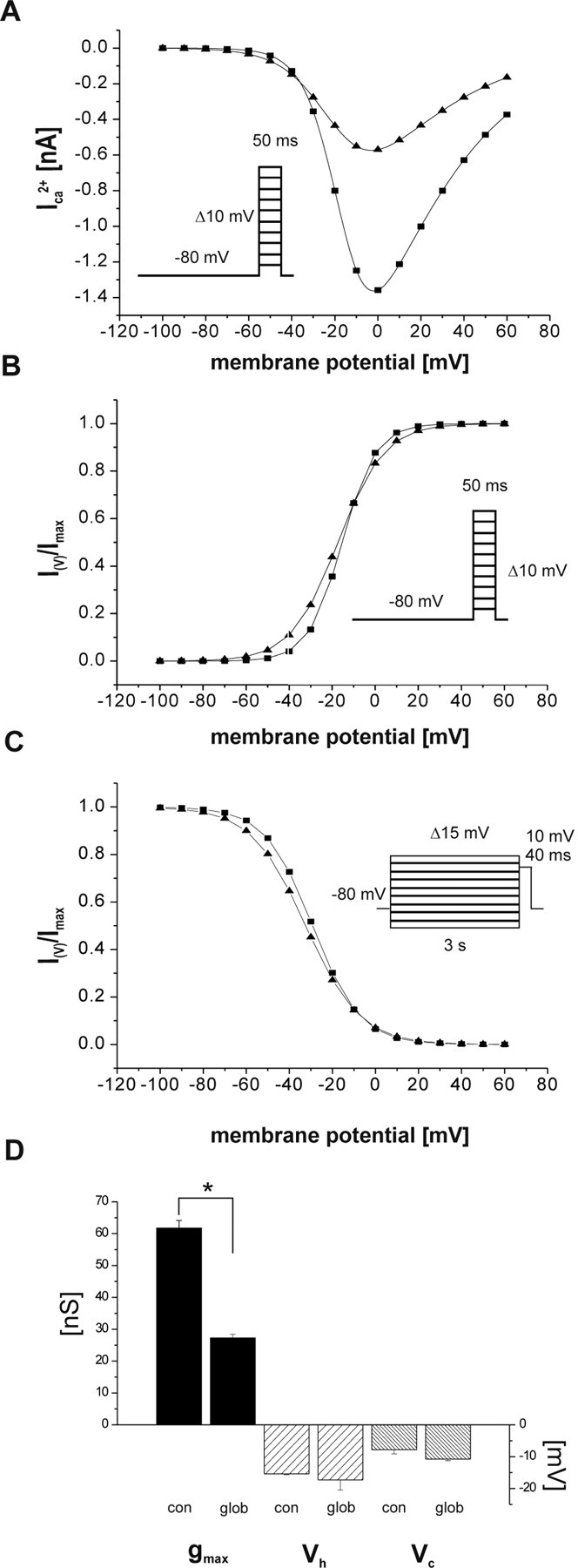
Steady-state activation and inactivation parameters of P/Q currents. Curves are reconstructed from the averaged parameters Vh, Vc, and g max, respectively (see Materials and Methods). A, Current–voltage relationship before globulomer (squares) and during Aβ1–42 (triangles). A reduction of the current amplitudes over the entire voltage range, where the current could be activated, was observed after application of the globulomer. B, C, No difference in steady-state activation (B) and inactivation (C) curves for P/Q channel-mediated barium currents in the absence and presence of Aβ1–42 globulomer. D, A significant decrease in maximal conductance (g max) of the P/Q channels was induced by Aβ1–42 globulomer (glob), whereas other activation parameters (Vh, Vc) remained unchanged. Insets represent the command voltage protocols.
If the effect of Aβ1–42 globulomer on synaptic currents is mediated by a block of P/Q-type calcium channels, it should be mimicked and occluded by the selective P/Q-type calcium channel blocker ω-agatoxin IVA. Indeed, this toxin (0.5 μm) reduced the frequency of mPSCs to 27 ± 7% (n = 3; amplitude, 90 ± 7%), similar to the effect of Aβ1–42 globulomer (Fig. 8 A,B). After preincubation with ω-agatoxin IVA, Aβ1–42 globulomer had no additional effect on the synaptic currents (n = 6; frequency, 108 ± 15%; amplitude, 102 ± 7% of currents after ω-agatoxin IVA control) (Fig. 8 A,B). These data suggest that ω-agatoxin IVA and Aβ1–42 globulomer share the same molecular mechanism.
Figure 8.
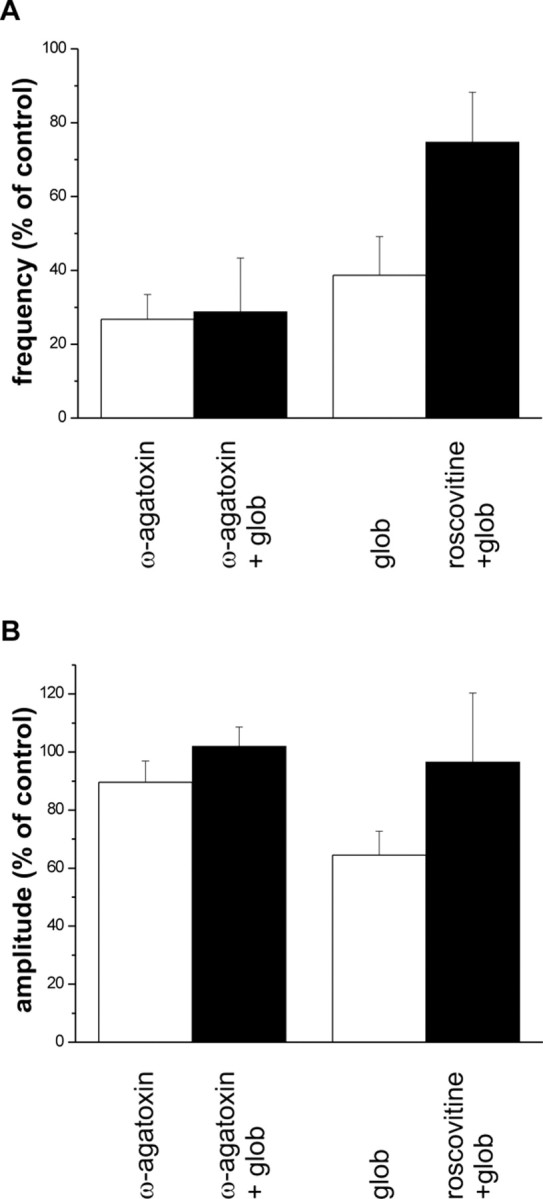
Pharmacological modulation of the effect of Aβ1–42 globulomer by agents interacting with P/Q-type calcium channels. A, Left bars, ω-Agatoxin reduces the frequency of spontaneous synaptic currents (white bar). Additional application of the globulomer (glob) does not exert an additional effect (black bar). Right bars, The reduction in frequency by Aβ1–42 globulomer can partially be recovered by application of the P/Q current enhancer roscovitine. B, Effects on median amplitude. Values are given relative to data in control solution.
Rescue by roscovitine
Recent evidence indicates that currents through P/Q-type calcium channels can be selectively enhanced by roscovitine (Yan et al., 2002). Application of roscovitine in the presence of Aβ1–42 globulomer did indeed partially recover the frequency of synaptic currents. Although in these experiments, Aβ1–42 globulomer reduced the frequency of sPSCs to 38 ± 10% of control, application of roscovitine (20 μm) restored this parameter to 75 ± 13% (n = 5) (Fig. 8 A).
Together, these data indicate that Aβ1–42 globulomer reduces the frequency of spontaneous and miniature synaptic currents by suppression of presynaptic calcium influx via P/Q-type calcium channels.
Discussion
This study characterizes the cellular mechanisms by which a defined oligomeric Aβ1–42 species carrying an epitope detected in AD patients, the Aβ1–42 globulomer, exhibits its pathophysiological effects on hippocampal neurons. For the first time, we are able to define a molecular target of oligomeric Aβ by providing evidence for a direct suppression of a presynaptic calcium current that is essentially involved in neurotransmission and synaptic plasticity in the brain. This effect is specific for Aβ oligomers, because the same effect was not observed when using a conventional preparation of Aβ1–42 monomers containing also protofibrils and fibrils.
We have shown recently that Aβ1–42 globulomer inhibits LTP in rat hippocampal slices (Barghorn et al., 2005). Other Aβ oligomer preparations affected synaptic plasticity in a similar manner (Lambert et al., 1998; Walsh et al., 2002). Various studies indicated that oligomeric Aβ specifically binds to synapses of hippocampal neurons (Lacor et al., 2004; Barghorn et al., 2005). We therefore assumed that the mode of action of oligomeric Aβ species may lie in the synapse itself. This inevitably leads to the question: which molecular target, possibly at synaptic entities, is modulated by soluble Aβ oligomers?
Although the described findings hint toward a synaptic mechanism, we also tested whether intrinsic neuronal properties were affected by globulomer. Previous publications using different preparations of Aβ1–42 or Aβ1–40 peptides at high concentrations pointed toward pore-forming capabilities of amyloid peptides (Lashuel et al., 2002; Kayed et al., 2004). Although these preparations did not contain stable oligomeric forms of Aβ, we found it indispensable to exclude globulomer-induced changes of intrinsic properties.
Aβ1–42 globulomer did not affect passive membrane properties, such as input resistance, when used at 80 nm concentration. Thus, it is unlikely that this Aβ species integrates into the membrane, thereby affecting intrinsic neuronal properties. There was also no alteration of the action potential and its underlying currents. Instead, we observed an instantaneous change in the presynaptic release probability when cultured neurons were incubated with globulomer. We examined the affected synaptic mechanism in detail and provide evidence that Aβ1–42 globulomer strongly impairs presynaptic P/Q-type calcium currents at both glutamatergic and GABAergic synapses.
Our data do not unambiguously prove that Aβ1–42 globulomer directly interacts with P/Q-channel subunits. It is also possible that binding occurs at other synaptic proteins, which then causes a modification of the P/Q current, perhaps by interacting with the auxiliary subunits of the channel. However, the immediate effect of globulomer on the isolated P/Q current suggests a direct modulation of the channel.
The present study was restricted to cultured hippocampal neurons and does not allow us to predict the precise effects of Aβ1–42 globulomer in native networks in the brain. Presynaptic transmitter release is mediated by three main types of calcium channels (P/Q, N, R) with variable contribution at different synapses and with different functional regulation (for review, see Reid at al., 2003; Evans and Zamponi, 2006). In our previous study (Barghorn et al., 2005), we found unchanged field EPSPs after application of Aβ1–42 globulomer to the CA1 region of rat hippocampal slices while LTP was selectively impaired. In our present study on cultured hippocampal neurons, synaptic transmission was reduced at the level of miniature, spontaneous, and evoked activity. This apparent discrepancy may be explained by various differences in the preparations and experimental approaches. First, in our present experiments, neurons were cultured in a monolayer preparation and were thus directly exposed to Aβ1–42 globulomer. In contrast, previous experiments on LTP were performed in hippocampal slices, which only allow limited and slow access of large molecules to neuronal target structures. Therefore, it can be inferred that presynaptic calcium channels were exposed to much lower effective concentrations of Aβ1–42 globulomer than in our present experiments. Second, the net effect of reduced calcium influx through P/Q-type calcium channels depends on their fractional expression at Schaffer collateral synapses (Wu and Saggau, 1994) as well as on their presence on inhibitory terminals, which might partially cancel effects of reduced excitatory transmission. In our culture preparation, P/Q-mediated synaptic transmission had been stabilized by addition of ω-conotoxin MVIIA, ensuring a constant and significant contribution of this presynaptic calcium channel subtype to synaptic transmission (see Materials and Methods). After sustained and strong synaptic activation, however, Aβ1–42 globulomer does reduce LTP at Schaffer collateral-CA1 synapses (Barghorn et al., 2005), in accordance with previous studies using oligomer preparations (Lambert et al., 1998; Walsh et al., 2002).
The detrimental effect of altered P/Q currents may not be restricted to LTP and associated synaptic information storage. Certain pathways that are critically involved in the dementia symptoms of AD depend on intact P/Q channel-mediated calcium signaling, and a general decrease in the transmitter release in such pathways may be detrimental to cognitive function. For example, Momiyama and Fukazawa (2007) could show that P/Q-type calcium channels facilitate glutamate release onto cholinergic basal forebrain neurons, thereby altering cholinergic transmission. A suppression of P/Q-type calcium currents may therefore result in modified cortical acetylcholine levels. Cholineric neurons originating in the basal forebrain substantially contribute to learning and memory (Everitt and Robbins, 1997), and deficits in cholinergic transmission of basal forebrain neurons is one key feature of AD (Kasa et al., 1997).
One may speculate that an enhancement of P/Q currents would offset the oligomer-induced deficits in affected neuronal pathways, thereby improving impaired cognitive function in AD. Roscovitine has been shown to enhance P/Q-type calcium currents by slowing the deactivation kinetics of the channel (Yan et al., 2002). We were able to demonstrate that roscovitine reversed the globulomer-induced deficits on vesicle release in hippocampal neurons, most probably by enhancing the P/Q-type calcium current. This suggests that P/Q current-enhancing compounds could at least in part offset synaptic deficits in AD. It is feasible that an improvement of Aβ oligomer-induced deficits in neurotransmission could translate into enhanced cognitive function in patients. Additional studies need to confirm that agonism of the P/Q channel can reverse cognitive deficits in animal models of AD.
According to our data, Aβ1–42 globulomer acutely impairs spontaneous synaptic transmission. It has been described in numerous studies that long-term reduction of synaptic activity leads to a degeneration of synapses (for review, see Segal and Andersen, 2000). Chronic reduction of P/Q calcium signaling may thus result in a retraction of synaptic processes. AD is accompanied by a loss of synapses, which correlates with the severity of the symptoms (Coleman and Yao, 2003). It will be interesting to see whether restoring P/Q-channel activity may reverse such deficits in synapse morphology.
Our study advances the understanding of the pathophysiology of soluble oligomeric Aβ1–42 in AD. By using the Aβ1–42 globulomer, an oligomeric Aβ conformation detected in AD patients and APP-overexpressing mice (Barghorn et al., 2005), we identified a molecular target for its pathological effect on neurons. Although a conventional preparation of Aβ1–42 monomer was not able to interfere with the P/Q channel, it will be interesting to determine whether other oligomeric forms of Aβ also affect P/Q calcium channels, which would result in a unified pathological mechanism of Aβ oligomers. This is probable, because Aβ1–42 globulomer mimics other oligomers in terms of the effect on synaptic plasticity and binding properties, which suggests that all Aβ oligomers target the same synaptic mechanism. However, it has been shown recently that certain oligomer preparations disturb NMDA receptor-related postsynaptic cascades (De Felice et al., 2007; Shankar et al., 2007). Although we do not have indications from our study that the NMDA receptor is directly affected by Aβ1–42 globulomer, the tight interplay of presynaptic and postsynaptic mechanisms suggests that more than one functional pathway is involved in the detrimental cascade of events initiated by suppressed P/Q currents. In case of LTP, for example, it is known that both vesicle release and postsynaptic NMDA currents are closely linked and necessary for its induction.
In summary, we provided compelling evidence for an Aβ oligomer-mediated mechanism of synaptic failure in AD, which could initiate a pathological cascade of synaptic loss and cognitive deficits. Future studies need to address whether other Aβ oligomer-induced impairments, like NMDA receptor-associated deficits, are downstream of the P/Q-channel blockade or, alternatively, are independent effects of Aβ oligomer.
Footnotes
We acknowledge Sean Turner for critically reading this manuscript.
References
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. Globular amyloid beta-peptide oligomer–a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Evans RM, Zamponi GW. Presynaptic Ca2+ channels–integration centers for neuronal signalling pathways. Trends Neurosci. 2006;29:617–624. doi: 10.1016/j.tins.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer's disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Kortekaas P, Wadman WJ. Development of HVA and LVA calcium currents in pyramidal CA1 neurons in the hippocampus of the rat. Brain Res Dev Brain Res. 1997;101:139–147. doi: 10.1016/s0165-3806(97)00059-x. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Momiyama T, Fukazawa Y. D1-like dopamine receptors selectively block P/Q-type calcium channels to reduce glutamate release onto cholinergic basal forebrain neurones of immature rats. J Physiol (Lond) 2007;580:103–117. doi: 10.1113/jphysiol.2006.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 2003;26:683–687. doi: 10.1016/j.tins.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. J Neurosci. 1995;15:4612–4617. doi: 10.1523/JNEUROSCI.15-06-04612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Andersen P. Dendritic spines shaped by synaptic activity. Curr Opin Neurobiol. 2000;10:582–586. doi: 10.1016/s0959-4388(00)00123-9. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Pharmacological identification of two types of presynaptic voltage-dependent calcium channels at CA3-CA1 synapses of the hippocampus. J Neurosci. 1994;14:5613–5622. doi: 10.1523/JNEUROSCI.14-09-05613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Chi P, Bibb JA, Ryan TA, Greengard P. Roscovitine: a novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J Physiol (Lond) 2002;540:761–770. doi: 10.1113/jphysiol.2001.013376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Calcium and transmitter release. J Physiol (Paris) 1993;87:25–36. doi: 10.1016/0928-4257(93)90021-k. [DOI] [PubMed] [Google Scholar]