

Abstract
In slice preparations, electrical stimulation of the superficial gray layer (SGS) of the superior colliculus (SC) induces EPSC bursts in neurons in the intermediate gray layer (SGI) when GABAA receptor (GABAAR)-mediated inhibition is reduced. This preparation has been used as a model system to study signal processing involved in execution of short-latency orienting responses to visual stimuli such as saccadic eye movements. In the present study, we investigated the role of GABAB receptors (GABABRs) in modulating signal transmission in the above pathway with whole-cell patch-clamp recordings in SC slices obtained from GAD67-GFP knock-in mice. Perfusion of the slice with the GABABR antagonist CGP52432 (CGP) greatly prolonged the duration of the EPSC bursts. Local application of CGP to the SGS but not to the SGI produced similar effects. Because SGS stimulation elicited bursts in GABAergic neurons in the SGS when GABAARs were blocked, these results suggest that GABA released after bursts activates GABABRs in the SGS, leading to reduced burst duration. We found both postsynaptic and presynaptic actions of GABABRs in the SGS; activation of postsynaptic GABABRs induced outward currents in narrow-field vertical cells, whereas it caused shunting inhibition in distal dendrites in wide-field vertical cells. On the other hand, activation of presynaptic GABABRs suppressed excitatory synaptic transmissions to non-GABAergic neurons in the SGS. These results indicate that synaptically released GABA can activate both presynaptic and postsynaptic GABABRs in the SGS and limit the duration of burst responses in the SC local circuit.
Keywords: superior colliculus, GABAB receptors, bursts, GAD67-GFP knock-in mouse, saccadic eye movement, patch clamp
Introduction
The mammalian superior colliculus (SC) is a pivotal structure for visuomotor transformation to execute orienting behaviors, including saccadic eye movements. Its superficial layers [stratum griseum superficiale (SGS) and stratum opticum (SO)] receive visual information from the retina and primary visual cortex, and its deeper layers [stratum griseum intermediale (SGI) and stratum griseum profundum (SGP)] send descending motor commands to the brainstem reticular formation and spinal cord. Previous studies showed that stimulation of the superficial layers induces excitatory synaptic responses in the deeper layers in slice preparations (Lee et al., 1997, 2001; Isa et al., 1998; Helms et al., 2004). In particular, when the slices were treated with GABAA receptor (GABAAR) antagonists such as bicuculline or gabazine, stimulation with single current pulses of the superficial layers elicits bursting responses in deeper layer neurons including crossed tecto-reticular neurons, output neurons projecting to the brainstem gaze center (Saito and Isa, 2003; Sooksawate et al., 2005). This direct activation of deeper layer neurons by the superficial layers has been supposed to underlie the execution of extremely short-latency visuomotor responses such as express saccades (Lee et al., 1997; Isa et al., 1998; Isa, 2002), which are generated by introducing a temporal “gap” between the fixation offset and the target onset (Fischer and Boch, 1983; Fischer and Weber, 1993).
This preparation has been used as an experimental model system to study visuomotor signal processing in the SC local circuits (Isa and Saito, 2001; Isa, 2002; Isa and Sparks, 2006). Our group has shown that an intrinsic self-excitation mechanism via a local excitatory network and NMDA receptor (NMDAR)-dependent synaptic transmission could regulate burst activity (Isa et al., 1998; Saito and Isa, 2003). In addition, it is also possible that excitatory afferent inputs to the SC may regulate the burst activity. For example, the brainstem saccade generator is suggested to send excitatory feedback inputs to the SC via the central mesencephalic reticular formation (Soetedjo et al., 2002). On the other hand, the role of inhibitory mechanisms, particularly that of GABABRs, in regulating burst activity within the SC local circuits remains unclear.
Postsynaptic and presynaptic GABABR activation leads to long-latency, long-duration hyperpolarization and reduction of transmitter release, respectively [for review, see Misgeld et al. (1995) and Ulrich and Bettler (2007)]. These GABABR mechanisms have accounted for response habituation observed in the SC (Binns and Salt, 1997). In addition to this functional role, the electrophysiological characteristics together with dense expression of GABABRs in the SC (Bowery et al., 1987; Chu et al., 1990) suggest that such long-lasting, slow responses may contribute to the regulation of burst activity, particularly to the termination of bursts. Here we show that GABABR-mediated responses, which are induced by synaptically released GABA during bursts of GABAergic neurons, limit the duration of burst activity in SC neurons. Blockade of both presynaptically and postsynaptically expressed GABABRs in the SGS prolongs burst duration in SGS/SO cells in an NMDAR-dependent manner, which in turn results in the generation of long-lasting bursts in SGI projection neurons.
Materials and Methods
This study was approved by the Animal Research Committee of the Okazaki National Research Institutes. All efforts were made to minimize the suffering and number of animals used in this study.
Animals.
The procedures for generating and genotyping the two strains of GAD67-GFP knock-in mice are described previously (Tamamaki et al., 2003). GAD67-GFP knock-in mice retain a loxP-flanked neomycin-resistance cassette (PGK-Neo), whereas GAD67-GFP (Δneo) knock-in mice lack the PGK-Neo cassette. We used both strains and refer to them both as GAD67-GFP knock-in mice for simplicity. We saw no differences between the two strains. Mice heterozygous for the GAD67-GFP allele were mated with C57BL/6 wild-type mice to obtain the heterozygous mice. Fifteen- to 22-d-old heterozygous mice were used in the experiments.
Slice preparations.
The mice were killed with ether and decapitated. The brains were quickly removed and submerged for 2–3 min in ice-cold modified Ringer's solution containing (in mm) 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 11 glucose, and bubbled with 95% O2/5% CO2, pH 7.4. Parasagittal slices (300–350 μm thick) of the SC were cut with a Microslicer (DTK-2000; Dosaka EM, Kyoto, Japan) and incubated at room temperature for >1 h before recording in standard Ringer's solution containing (in mm) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 25 glucose, and bubbled with 95% O2/5% CO2, pH 7.4.
Electrophysiological recordings.
Slices were mounted in a recording chamber on an upright microscope (DM LFS; Leica, Wetzlar, Germany) and continuously superfused with the standard Ringer's solution at a flow rate of 2–2.5 ml/min. Whole-cell patch-clamp recordings were obtained from the neurons in the SGS, SO, and SGI by visual control of patch pipettes. GFP-positive and -negative neurons were selected using fluorescent optics, and then a whole-cell configuration was obtained using bright-field optics. Patch pipettes were prepared from borosilicate glass capillaries and were filled with an internal solution containing (in mm) 145 K-gluconate, 2 MgCl2, 4 Na2ATP, 0.3 Na3GTP, 0.2 EGTA, 10 HEPES, and 0.1 spermine, pH 7.3. To stain the recorded neurons, biocytin (5 mg/ml; Sigma, St. Louis, MO) was dissolved in the solution. In some experiments, we added QX-314 (5 mm) and GDPβS (0.6 mm) instead of Na3GTP to the intracellular solution. Both QX-314 and GDPβS are known to block postsynaptic GABABR-mediated responses (Nathan et al., 1990; Andrade, 1991; McLean et al., 1996). The resistance of the electrodes was 4–8 MΩ in the Ringer's solutions. The actual membrane potentials were corrected by the liquid junction potential of −10 mV. In voltage-clamp recordings, we held membrane potentials between −60 and −65 mV, aiming to detect both EPSCs and GABABR-mediated outward currents that are primarily mediated by G-protein-coupled inwardly rectifying K+ channels. E K+ was −107 mV under present experimental condition. All recordings were performed at 32–34°C. Data were acquired with an EPC9 patch-clamp amplifier and PULSE8.5 software (Heka, Lambrecht, Germany) or a Multiclamp 700B amplifier and pClamp10 (Molecular Devices, Sunnyvale, CA).
Electrical stimulation of the SGS was applied as a cathodal square-wave pulse of 200 μs duration with an intensity of up to 150 μA using a glass electrode filled with normal Ringer's solution. In our previous studies, SGS stimulation evoked burst activities in SGS, SO, and SGI neurons during the blockade of GABAARs (Isa et al., 1998, Saito and Isa, 2003). In this study, we used gabazine (10 μm; Sigma) as a selective GABAAR antagonist, which produced the same effects as bicuculline (Saito and Isa, 2003). To examine the involvement of GABABR-mediated responses in burst activity, a GABABR antagonist, CGP52432 (CGP; 3 μm; Tocris, Ellisville, MO), was bath applied. Depending on the requirements for each experiment, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm; Tocris) and dl-2-amino-5-phosphonovaleric acid (APV; 50 μm; Sigma) were also bath perfused. In some experiments, CGP52432 (1 mm), APV (5 mm), glutamate (Glu; 1 mm; Sigma) and/or baclofen (Bac; 1 mm; Tocris) were puff applied with air pressure pulses (5–10 psi, 50–500 ms duration) through a micropipette with a tip diameter of 2–3 μm.
Data were analyzed with the Igor Pro program (WaveMetrics, Lake Oswego, OR) or Clampfit (Molecular Devices). To obtain decay time constants of the EPSC bursts, traces were fitted to single exponential curves using Clampfit. Values are given as the means ± SE. Statistical significance was examined with a two-tailed Student's paired t test and the difference was considered significant if p < 0.05.
Visualization of recorded neurons.
After recording, the slices were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, for more than 1 d at 4°C. After fixation, biocytin-filled neurons were visualized by the ABC method (Vectastain; Vector Laboratories, Burlingame, CA). Details have been described previously (Isa et al., 1998).
Immunohistochemistry.
Three male adult GAD67-GFP knock-in mice were deeply anesthetized (sodium pentobarbital, 80 mg/kg, i.p.), perfused transcardially with 0.05 m PBS, pH 7.4, followed by fixative containing 4% paraformaldehyde, 15% saturated picric acid, and 0.05% glutaraldehyde in 0.1 m PB, pH 7.4. The brains were cryoprotected in 30% sucrose in 0.1 m PB, and 40-μm-thick parasagittal and coronal sections were cut on a freezing microtome. After washes in PBS, sections were incubated in PBS containing 10% normal goat serum (NGS) and 0.1% Triton X-100 at room temperature for 1 h, and then with anti-GABABR1a/b (1.0 μg/ml) in PBS containing 3% NGS and 0.1% Triton X-100 overnight at 4°C. The antibody used was a kind gift from Dr. Shigemoto (National Institute for Physiological Sciences, Okazaki, Japan), and its characteristics and specificity have been described previously (Kulik et al., 2002, 2003). After washes in PBS, the sections were incubated with biotinylated goat anti-rabbit IgG antibody at room temperature for 2 h, washed, and reacted with avidin–biotin peroxidase complex (ABC elite kit; Vector Laboratories) at room temperature for 2 h. Subsequently, the sections were incubated in 50 mm Tris-HCl, pH 7.3, containing 0.025% 3–3-diaminobenzidine tetrahydrochloride (DAB; Wako, Osaka, Japan) and 0.003% hydrogen peroxide. Specificity of the immunolabeling was demonstrated by the absence of labeling for GABABR1a/b when the primary antibody was omitted. Furthermore, sections of cerebellum were included in each incubation plate to act as a positive control for the GABABR1a/b immunolabeling.
Results
GABABRs regulate burst duration in SGI non-GABAergic neurons
Previous studies in rat slice preparations have shown that when GABAAR-mediated inhibition was reduced, stimulation with single current pulses delivered to the SGS and/or SO evoked bursts in SGI neurons (Isa et al., 1998; Saito and Isa, 2003). Using this system, we first explored the role of GABABRs in regulating burst activity in SGI neurons. In slice preparations from GAD67-GFP knock-in mice (Tamamaki et al., 2003), we recorded from GFP-negative, non-GABAergic neurons in the SGI and examined the effects of GABABR antagonists on burst activity. Under control conditions, SGS stimulation evoked EPSPs (n = 3) (Fig. 1 A1), single action potentials (n = 1), or no response (n = 1), whereas the same stimulation induced bursts in these same neurons after bath application of the GABAAR antagonist gabazine (10 μm) (Fig. 1 A2), as has been reported previously (Isa et al., 1998; Saito and Isa, 2003). Additional application of the GABABR selective antagonist CGP (3 μm) significantly increased the number of spikes within a burst and the duration of bursts compared with gabazine application alone (spike number, 26.5 ± 8.2 in gabazine, 70.7 ± 17.3 in gabazine + CGP, p < 0.05, n = 5; burst duration, 865.4 ± 396.3 ms in gabazine, 1965.2 ± 27.09 ms in gabazine + CGP, p < 0.05, n = 5) (Fig. 1 A3). Voltage-clamp recordings revealed that SGS stimulation elicited a population of EPSCs, hereafter referred to as EPSC bursts, when gabazine was bath applied (Fig. 1 B1,B2). The EPSC bursts demonstrate that this neuron receives a number of inputs arising from a population of excitatory neurons located in the SGI and/or superficial layers (Isa et al., 1998; Özen et al., 2000; Saito and Isa, 2003). Addition of CGP greatly prolonged the duration of EPSC bursts (Fig. 1 B3) without changing basal holding currents. As shown in Figure 1, B4 and C, the decay time constant but neither the peak amplitude nor rise time of EPSC bursts were significantly increased by CGP application compared with gabazine application alone. Another selective GABABR antagonist CGP55845 (1 μm) produced similar effects (n = 6, data not shown). These results indicate that synaptically released GABA activated GABABRs and that blockade of GABABRs greatly prolonged the duration of bursts in non-GABAergic neurons in the SGI.
Figure 1.
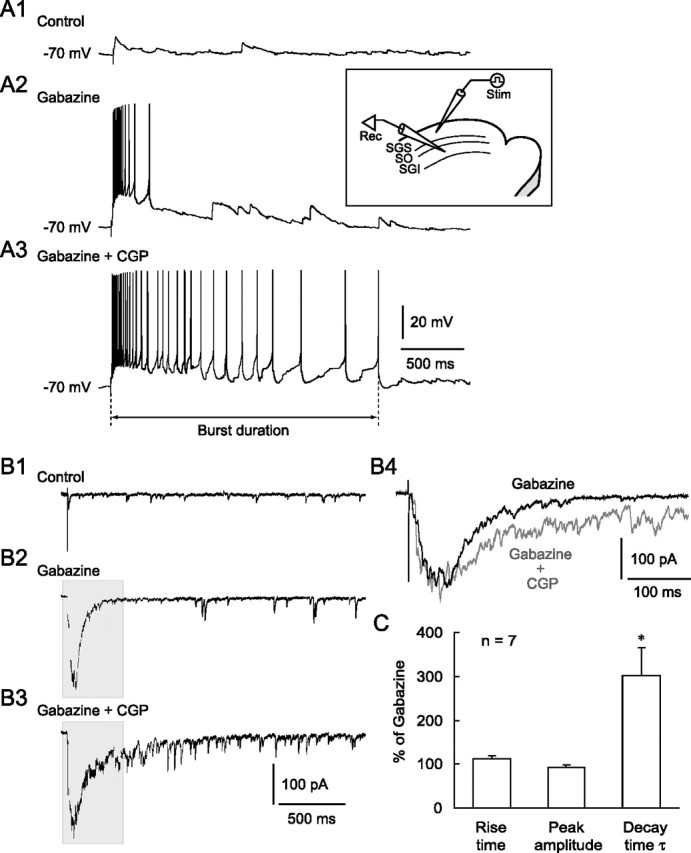
Blockade of GABABRs prolonged the duration of burst activity. A1, SGS stimulation evoked EPSPs under control conditions in an SGI non-GABAergic neuron. Inset, Schematic showing the electrode arrangement. A stimulating (Stim) electrode was placed in the SGS. Whole-cell recordings (Rec) were obtained from SGI non-GABAergic neurons. A2, The SGS stimulation evoked bursts in this neuron after bath application of the GABAAR antagonist gabazine (10 μm). A3, Additional application of the GABABR antagonist CGP (3 μm) greatly prolonged burst duration, defined as the period between stimulation onset and the peak of the last spike. B1, Voltage-clamp recordings from the same neuron shown in A revealed that SGS stimulation evoked EPSCs in the control. Membrane potential was held at −60 mV. B2, Bath application of gabazine induced a burst of EPSCs in response to SGS stimulation. B3, Additional application of CGP increased the duration, but not the amplitude, of the EPSC bursts. B4, Expanded traces from shaded areas in B2 and B3 showed a similar peak amplitude but a prolonged decay time of the EPSC bursts when CGP was added. C, Summary graph showing that the rise time and peak amplitude of the EPSC bursts were not significantly changed, whereas the decay time constant (τ) was significantly prolonged by CGP. *p < 0.05, compared with gabazine alone (paired t test).
These results raised the question of whether blockade of GABABRs could induce long-lasting burst activity. To address this issue, we first bath applied CGP alone and then added gabazine during the recording of five SGI non-GABAergic neurons. As shown in Figure 2 A–C, CGP application alone did not elicit any significant effects on SGS stimulation-evoked, presumably monosynaptic, EPSCs in three of five neurons. However, addition of gabazine induced long-lasting EPSC bursts in response to the same SGS stimulation (Fig. 2 D). In the remaining two neurons, SGS stimulation induced no response in the control condition (data not shown). CGP application also had no effect on these neurons. However, additional application of gabazine induced long-lasting EPSC bursts similar to those seen in the other three neurons (data not shown). These results indicate that blockade of GABABRs alone was not sufficient to evoke long-lasting bursts, an effect that was only accomplished when both GABAARs and GABABRs were blocked.
Figure 2.

SGS stimulation did not evoke EPSC bursts in the presence of CGP alone in SGI non-GABAergic neurons. A, Under control conditions, presumably monosynaptic EPSCs were evoked by SGS stimulation. B, Bath application of CGP (3 μm) had no effect on the EPSCs. C, Expanded traces from the control and CGP conditions showed nearly the same EPSCs. D, When gabazine was added to the bath, SGS stimulation evoked EPSC bursts. Membrane potential was held at −65 mV.
GABAergic neurons generate burst activity when GABAARs are blocked
The findings thus far have led us to hypothesize that bursts in GABAergic neurons, which might be evoked by SGS stimulation in the presence of gabazine, may be necessary for release of a large amount of GABA that can activate GABABRs expressed in the SC. To examine this possibility, we recorded from five GFP-positive, GABAergic neurons in the SGI. Figure 3 A shows an example. This neuron, and two others, exhibited almost no response with single SGS stimulation under control conditions. In the remaining two neurons, SGS stimulation evoked small EPSPs (data not shown). After bath application of gabazine, the same SGS stimulation evoked bursts in all five GABAergic neurons (Fig. 3 A). Four of five neurons were successfully stained with biocytin and appeared to be multipolar cells (Fig. 3 C).
Figure 3.

Bursts of GABAergic neurons in the SGI and SGS. A, B, Under control conditions no clear responses were evoked in either SGI (A) or SGS (B) GABAergic neurons by SGS stimulation (top traces), whereas in the presence of gabazine (10 μm), the stimulation evoked bursts in both types of neurons (bottom traces). Arrows indicate stimulus timing. C, D, Somatodendritic morphology of the biocytin-filled SGI (C) and SGS (D) GABAergic neurons shown in A and B, respectively. Scale bar in C applies to D.
We also recorded from GFP-positive, GABAergic neurons in the SGS. These neurons also generated bursts in response to single SGS stimulation when GABAARs were blocked by gabazine (Fig. 3 B) (n = 6). Four of six neurons were morphologically identified. They consisted of two horizontal cells, one of which is depicted in Figure 3 D, and two piriform cells. In our slice preparations, GFP-positive cells did not include stellate cells (Endo et al., 2003), although they are considered to be GABAergic neurons (Mize, 1992). Instead, three GFP-negative neurons were determined to be stellate cells with biocytin staining. These neurons also generated bursts with SGS stimulation in the presence of gabazine (data not shown). Thus, when GABAARs are blocked, GABAergic neurons in both the SGI and SGS generate bursts in response to SGS stimulation, as do the non-GABAergic neurons. These results suggest that bursts in GABAergic neurons might contribute to a large release of GABA that leads to activation of GABABRs in the SC.
Primary site of action of GABABR antagonist is in the SGS
We next investigated the possible site of action of the GABABR antagonist in the SC. Previous autoradiographic (Bowery et al., 1987; Chu et al., 1990) and immunohistochemical studies (Fritschy et al., 1999) in rats suggested that GABABRs are expressed in both the SGS and SGI with an intense expression in the SGS. To reveal the site of CGP action in the above-mentioned experiments, we applied CGP locally either to the SGS or SGI with air pressure while recording from non-GABAergic neurons in the SGI (Fig. 4 A). When we puff applied CGP (1 mm) locally to the SGS close to the stimulation site, the duration of EPSC bursts evoked by SGS stimulation in the presence of gabazine was significantly prolonged; neither the rise time nor the amplitude were affected (Fig. 4 B1,C1). This effect was reversible (Fig. 4 B2). We then moved the same puff pipette to the SGI and locally applied CGP (Fig. 4 A2). Although the distance between the recording and the puff pipettes in the SGI (155.5 ± 31.7 μm) was shorter (p < 0.01, n = 4) than that in the SGS (281.2 ± 38.3 μm), CGP failed to affect EPSC bursts in SGI (Fig. 4 B2,C2). These results indicate that the primary site of action of CGP is in the SGS.
Figure 4.

The primary site of action of the GABABR antagonist is in the SGS. A1, A2, Schematic showing the arrangement of recording (Rec) and stimulating (Stim) electrodes and a puff pipette containing 1 mm CGP (CGP puff) on the SC slices. The puff pipette was placed either in the SGS (A1) or SGI (A2). B1, SGS stimulation evoked EPSC bursts in an SGI non-GABAergic neuron in the presence of gabazine (10 μm) (black trace). When CGP was puff applied locally into the SGS, the decay time of the EPSC bursts was greatly prolonged without changing peak amplitude (gray trace). B2, After recovery from these effects (black trace), puff application of CGP into the SGI induced almost no effect (gray trace). Membrane potential was held at −65 mV. C1, C2, Summary graphs showing the effects of local puff application of CGP into SGS (C1) or SGI (C2). *p < 0.05.
To further confirm the possible site of CGP action, we examined expression of GABABRs in the SC of GAD67-GFP knock-in mice with immunohistochemistry using an antibody against GABABR1a/b. The characteristics and specificity of the antibody have been well documented previously (Kulik et al., 2002, 2003). As shown in Figure 5, we found dense GABABR1a/b immunoreactivity in the SGS, particularly in its most dorsal region. The level of GABABR1a/b immunoreactivity in the SGI is much less pronounced than in the SGS (Fig. 5). These data support the idea that the effect of CGP was primarily exerted by blockade of GABABRs in the SGS.
Figure 5.

Dense expression of GABABRs in the SGS. A, B, Distribution of immunoreactivity for GABABR1a/b in parasagittal (A) and coronal (B) sections. Strong immunoreactivity for GABABR1a/b was detected in the SGS. Scale bar in B applies to A. C, D, Higher magnifications of SGS (C) and SGI (D) obtained from the framed areas in B, respectively. Scale bar in D applies to C.
The results above prompted us to investigate how SGS and SO neurons behave in response to SGS stimulation during application of GABAAR and GABABR antagonists. We recorded from six and four non-GABAergic neurons in the SGS and SO, respectively. Biocytin staining allowed us to identify the morphology of five of the six SGS neurons. These included two narrow-field vertical (NFV) cells, two wide-field vertical (WFV) cells, and one multipolar cell. In the SO, we focused on WFV cells, somata of which are mainly located in the SO. Previous studies showed that this cell type is glutamatergic and may play important roles in signal transmission from the superficial to deeper layers and in burst generation (Mize, 1992; Isa et al., 1998, Saito and Isa, 2004, 2005). We confirmed morphologically that all of the four recorded cells were actually WFV cells. As shown in Figure 6, A and B, both SGS and SO non-GABAergic neurons generated bursts in response to SGS stimulation during the blockade of GABAARs with gabazine. Addition of CGP significantly increased the number of spikes within a burst and prolonged the duration of the bursts in these neurons without affecting resting membrane potentials (SGS neuron, spike number, 7.3 ± 1.7 in gabazine, 22.5 ± 3.9 in gabazine + CGP, p < 0.01, n = 6; burst duration, 108.8 ± 21.7 ms in gabazine, 769.2 ± 191.4 ms in gabazine ± CGP, p < 0.05, n = 6; SO neuron, spike number, 13.5 ± 3.4 in gabazine, 65.7 ± 15.7 in gabazine + CGP, p < 0.05, n = 4; burst duration, 96.0 ± 16.9 ms in gabazine, 1029.5 ± 247.4 ms in gabazine + CGP, p < 0.05, n = 4) (Fig. 6 A,B). These results, together with the data of local CGP puff application, suggest that the prolonged bursts observed in the SGI are attributable to the prolonged bursts in the non-GABAergic neurons with somata in the SGS and SO.
Figure 6.

Bursts evoked in non-GABAergic neurons in the SGS and SO. A1, B1, Under control conditions, SGS stimulation evoked single action potentials in both SGS (A1) and SO (B1) non-GABAergic neurons. Insets, Voltage responses of these neurons to various current pulses. The bottom traces indicate the injected currents. A2, B2, In the presence of gabazine (10 μm), SGS stimulation evoked bursts in both neurons. A3, B3, Addition of CGP (3 μm) greatly increased the duration of bursts.
GABA released from SGS GABAergic neurons activates GABABRs
We next investigated the source of GABA that activates GABABRs in the SGS. One possible source is GABAergic neurons in the SGS and the other is those in the SGI because a recent study has shown that SGI GABAergic neurons project to the SGS (Lee et al., 2007). To distinguish these two possible sources, we prepared “isolated SGS” slices by knife cuts between the SGI and SGS and recorded from non-GABAergic neurons in the SGS piece (Fig. 7 A). In five of six slices, SGS stimulation evoked short bursts (duration, 66.8 ± 12.7 ms, n = 5) after bath application of gabazine (Fig. 7 B,C). Addition of CGP considerably prolonged the duration of bursts to 502.5 ± 108.4 ms (Fig. 7 D) (p < 0.05, compared with gabazine alone), as has been observed in normal slice preparations, although the duration of the burst seemed slightly shorter than that observed in normal slices, probably attributable to damage caused by the knife cut. These results suggest that the main source of GABA that activates GABABRs in the SGS may be GABAergic neurons in the SGS.
Figure 7.
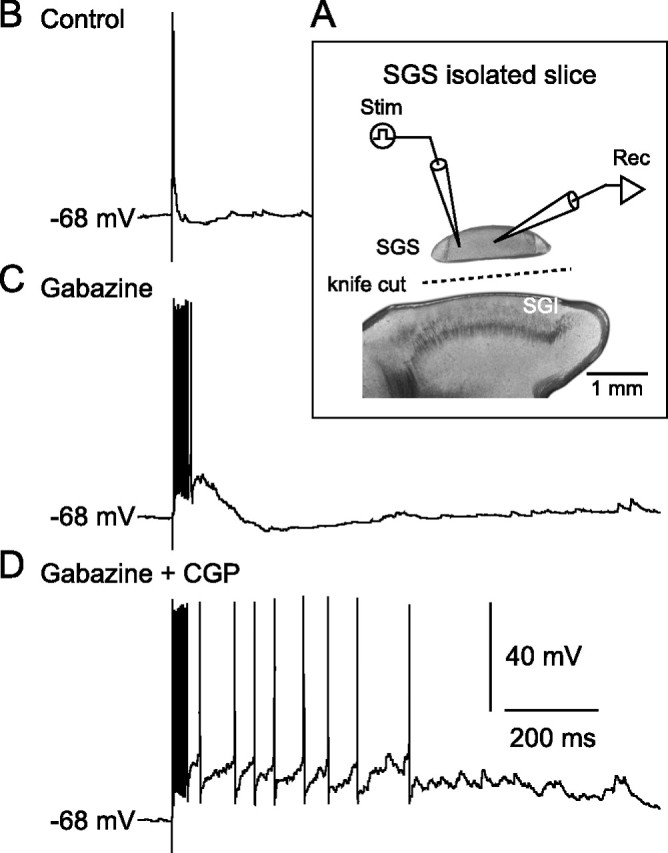
GABA released from SGS GABAergic neurons activates GABABRs. A, Schematic showing the arrangement of stimulating and recording electrodes on the SGS-isolated slice. B, In control, SGS stimulation evoked a single action potential in an SGS non-GABAergic neuron. C, After gabazine application, the stimulation induced bursts. D, Addition of CGP increased spike number and burst duration.
Postsynaptic GABAB mechanisms
The results above indicated that GABA released during bursts of GABAergic neurons in the SGS may activate GABABRs in the SGS, resulting in the limitation of burst duration in the SC local circuit. To determine whether postsynaptic and/or presynaptic GABABRs are involved in the limitation of burst activity, we delivered repetitive stimulations to the SGS to mimic the bursts of SGS GABAergic neurons and tested whether synaptically released GABA could actually activate GABABRs. We reasoned that this GABABR activation would then induce GABABR-mediated responses that might explain the shortening of the burst duration. In the first series of experiments, voltage-clamp recordings were obtained from non-GABAergic neurons in the SGS and SO in the presence of the NMDAR antagonist APV (50 μm), the AMPA/kainate receptor antagonist CNQX (10 μm), and gabazine (10 μm). In this series of experiments, we were unable to use a Cs+-based intracellular pipette solution with QX-314 because this solution abolishes postsynaptic GABAB responses (Nathan et al., 1990; Andrade, 1991; McLean et al., 1996). Thus, we used a K+-based electrolyte and applied small-intensity stimuli (≤60 μA) to avoid generating action potentials with direct somatodendritic stimulation (even in voltage-clamp configuration, strong stimulation evoked a large current corresponding to an action potential, probably as a result of direct activation of dendrites).
Repetitive stimulation with 10 pulses at 100 Hz delivered to the SGS evoked clear outward currents in four of five non-GABAergic neurons in the SGS (Fig. 8 A1,C). Bath application of CGP abolished the outward currents and unmasked unidentifiable inward currents, which were blocked by TTX (1 μm, data not shown). This indicates that postsynaptic GABABRs mediated the outward currents. In the fifth neuron, repetitive stimulation did not induce clear outward currents (Fig. 8 C). However, even in this neuron, CGP application unmasked inward currents like those observed in the other four neurons (Fig. 8 C), indicating the existence of small outward currents through the activation of GABABRs. After visualization with biocytin, we confirmed that the neurons that showed CGP-sensitive outward currents were NFV cells (Fig. 8 B). On the other hand, WFV cells showed almost no or very small, nonsignificant outward currents after repetitive SGS stimulation (Fig. 8 A2,C).
Figure 8.

Local repetitive stimulation elicited CGP-sensitive currents in NFV but not WFV cells. A1, Voltage-clamp recordings obtained from an NFV cell show that repetitive local stimulation in the SGS evoked outward currents (black trace, Control), which were abolished by CGP application (gray trace, CGP). Recordings were performed in the presence of CNQX (10 μm), APV (50 μm), and gabazine (10 μm) at a holding membrane potential of −60 mV. Note that ionotropic glutamate receptor antagonist-insensitive inward currents were unmasked by CGP application. The effect of CGP was recovered after washout (black trace, Wash). A2, SGS repetitive stimulation did not evoke a clear CGP-sensitive current in a WFV cell. Membrane potential was held at −60 mV. B, Morphology of the biocytin-filled NFV cell, from which the responses shown in A1 were obtained. Soma and dendrites are drawn as thick lines and axons as thin lines. C, A summary graph showing the effect of CGP on the repetitive stimulation-induced currents. Open circles represent individual neurons. Bar graphs indicate average current amplitude of each condition. *p < 0.05. NS, Not significant.
Because our previous study demonstrated that the GABABR-selective agonist baclofen induced a hyperpolarization in many types of superficial layer neurons, including WFV cells (Endo and Isa, 2002), we presumed that synaptically released GABA might elicit small outward currents at distal dendrites of WFV cells. Thus, the currents might be difficult to detect with somatic recordings using the K+-based intracellular electrolyte because of difficulties in space clamping WFV cells. We therefore tried to investigate the effects of activating postsynaptic GABABRs on excitatory inputs in WFV cells by examining the effects of local applications of baclofen and glutamate under current-clamp recordings. Puff application of glutamate (1 mm) locally into the dorsal portion of the SGS, in which distal dendrites of WFV cells are located, induced a depolarization in WFV cells in the presence of TTX (1 μm) and gabazine (10 μm) (Fig. 9 A,B1). On the other hand, local application of 1 mm baclofen in the vicinity of the glutamate-containing puff pipette (∼20 μm) evoked a slight hyperpolarization (Fig. 9 B2). Baclofen applied 100 ms before glutamate application significantly reduced the amplitude of the glutamate-evoked depolarization (Fig. 9 B3,C). Bath application of CGP reversed this reduction (Fig. 9 B4,C). A digital summation of the glutamate-evoked depolarization and baclofen-evoked hyperpolarization exhibited a much larger amplitude than the amplitude of responses induced by combined application (Fig. 9 B5), indicating that the effect of baclofen was not merely the summation of the depolarization and hyperpolarization. Rather, this demonstrated that the GABABR-mediated increase in membrane conductance might reduce the amplitude of the depolarization by a shunting mechanism.
Figure 9.

Increase in GABABR-mediated conductance at distal dendrites reduced glutamate-evoked excitation via shunting inhibition in WFV cells. A, Reconstruction of a biocytin-filled WFV cell showing the locations of recoding electrode (Rec) and puff pipettes for application of glutamate (Glu puff) and baclofen (Bac puff). DS, Dorsal surface of the slice. B, Local glutamate application (Glu, 1 mm, 10 psi, 30 ms) into the SGS, dendritic fields of the WFV cells, evoked depolarization (B1, Glu) in the presence of TTX (1 μm) and gabazine (10 μm). Baclofen (1 mm, 10 psi, 60 ms) applied 20 μm away from the glutamate application site evoked a slight hyperpolarization (B2, Bac). Glutamate application preceded by baclofen application (100 ms) elicited a smaller depolarization than glutamate application alone (B3, Bac Glu). This baclofen effect was blocked by CGP bath application (B4, Bac Glu in CGP). Note that the amplitude of depolarization evoked by combined application of glutamate and baclofen (B3) is smaller than the amplitude of the digital sum of the traces obtained from puff application of glutamate and that of baclofen (B5). The recordings were obtained from the cell shown in A. C, A summary graph showing that the amplitude of depolarization elicited by glutamate puff application was reduced by the preceding baclofen puff application. *p < 0.05.
Presynaptic GABAB mechanism
We next examined whether synaptically released GABA could activate GABABRs localized in glutamatergic presynaptic terminals and then suppress glutamate release, which may contribute to the limitation of burst duration. To investigate this hypothesis, we used the protocol shown in Figure 10 A. First, we applied conditioning stimuli consisting of 10 pulses at 100 Hz in the SGS, whereby a large release of GABA is expected, as seen in the previous section. Subsequently, at an interval of 100–1200 ms, test stimuli of paired pulses at 50 ms interval were delivered with the same stimulating electrode. By comparing the amplitudes of EPSCs evoked by conditioning and test stimuli, we monitored whether GABA release accompanying the conditioning stimuli affect glutamatergic transmission elicited by the test stimuli. To isolate AMPA/kainate receptor-mediated EPSCs and to avoid generation of EPSC bursts, we bath applied APV (50 μm) and gabazine (10 μm). Voltage-clamp recordings at a holding potential of −65 mV were obtained from non-GABAergic neurons in the SGS. As shown in Figure 10 B, the amplitude of the EPSC evoked by the first pulse of the test stimuli delivered 100 ms after the conditioning stimulation was significantly smaller than that of the conditioning stimulus (234.3 ± 60.8 pA for first conditioning stimulus, 158.1 ± 59.9 pA for first test stimulus, p < 0.05, n = 5). The first test stimulus-evoked EPSC amplitudes increased gradually as the interval from the conditioning stimulation increased, and there were no differences between them at intervals of 800 and 1200 ms and the first conditioning stimulus-evoked EPSC amplitude (Fig. 10 B,C). Bath application of CGP significantly reversed the reduced amplitude of EPSC evoked by the first test stimuli delivered at intervals of 100–400 ms. Because in this series of experiments we used a pipette solution containing QX-314 (5 mm) and GDPβS (0.6 mm) to block postsynaptic GABAB responses (Nathan et al., 1990; Andrade, 1991; McLean et al., 1996), these results indicate that the reduced test stimuli EPSCs were caused by activation of presynaptic GABABRs localized on glutamatergic terminals by GABA synaptically released during the conditioning stimulations.
Figure 10.

Synaptically released GABA activates presynaptic GABABRs and reduces glutamatergic transmission. A, Left, Schematic showing the stimulation protocol. Conditioning stimulations consisting of 10 pulses at 100 Hz were delivered to the SGS. Subsequently, after an interval of 100–1200 ms (Δt), test stimuli (2 pulses with an interval of 50 ms) were delivered to the SGS via the same stimulating electrode. Right, Schematic showing the electrode arrangement. A stimulating electrode was placed in the SGS, and whole-cell recordings were obtained from SGS non-GABAergic neurons. B, Conditioning stimulations greatly reduced the amplitude of the first test stimulation-evoked EPSCs at intervals of 100–400 ms, but not at 800 and 1200 ms, compared with that evoked by the first conditioning stimulus (black traces, Control). Application of CGP greatly increased the amplitude of the test stimulus-evoked EPSCs at intervals of 100–400 ms, but not at 800 and 1200 ms (gray traces, CGP). Voltage-clamp recordings were performed in the presence of gabazine (10 μm) and APV (50 μm) at a holding potential of −65 mV. Pipette solution contained 5 mm QX-314 and 0.6 mm GDPβS to block postsynaptic GABAB responses. C, A summary showing the normalized amplitude ratios plotted against intervals of test stimuli. EPSCs amplitudes were normalized with the amplitude of EPSCs evoked by the first conditioning stimulus under the control condition. The amplitude of EPSCs evoked by the first test stimulus at intervals of 100–400 ms, but not at 800 and 1200 ms, was significantly smaller than that evoked by the first conditioning stimulus. † p < 0.05. At intervals of 100–400 ms, the amplitude ratios were significantly increased by CGP application (closed circles, *p < 0.05, compared with control), whereas they were not significantly affected at intervals of 800 and 1200 ms. D, A summary showing paired pulse ratios (amplitude of the second EPSC relative to the first) plotted against intervals between conditioning and test stimuli. In the control, paired pulse ratios gradually decreased as the intervals increased (open circles). The paired pulse ratio after an interval of 1200 ms was significantly different from those at 100–200 ms in control. † p < 0.05. CGP application (closed circles) significantly reduced the paired pulse ratio after intervals of 100–400 ms. *p < 0.05, compared with control.
To further confirm this hypothesis, we calculated the paired pulse ratios of EPSCs evoked by the paired test stimuli, changes in which should implicate a presynaptic mechanism. The paired pulse ratio of responses to test stimuli delivered after an interval of 100 ms in the control condition was relatively high, indicating a low release probability (Fig. 10 B,D). The ratio gradually decreased as the interval increased, but was nearly constant between the intervals of 800 and 1200 ms (Fig. 10 B,D). When CGP was bath applied, the ratios at intervals of 100–400 ms, but not of 800 and 1200 ms, were less than those in the controls. Also, there was no significant difference among the ratios at intervals of 100–1200 ms during CGP treatment (p = 0.67) (Fig. 10 D). These results strongly suggest that there was a reduction in release probability of glutamate after the conditioning stimulation. Together, it would appear that the reduction in test stimuli-evoked EPSCs was mediated by activation of presynaptic GABABRs under the present conditions.
NMDAR-dependent bursts in SGS are necessary for generation of bursts in SGI
Because our results demonstrated that CGP is effective in the SGS and that non-GABAergic neurons in the SGS generate long-lasting bursts when both GABAARs and GABABRs are blocked, we hypothesized that the long-lasting bursts elicited in the SGS might be responsible for the long-lasting bursts in the SGI. To address this issue, we investigated the effects of local suppression of long-lasting bursts in the SGS on the expression of such bursts in the SGI. Because previous studies indicated that NMDARs play an important role in generating bursts in both the SGS and SGI (Isa et al., 1998; Saito and Isa, 2003), we first examined effects of local application of APV on bursts in the SGS. When APV (5 mm) was puff applied locally into the SGS 0.5 s before SGS stimulation, long-lasting bursts evoked by the stimulation in the presence of gabazine and CGP were significantly suppressed (n = 4) (Fig. 11). This result indicates that activation of NMDARs in the SGS contributes to the induction of long-lasting bursts.
Figure 11.
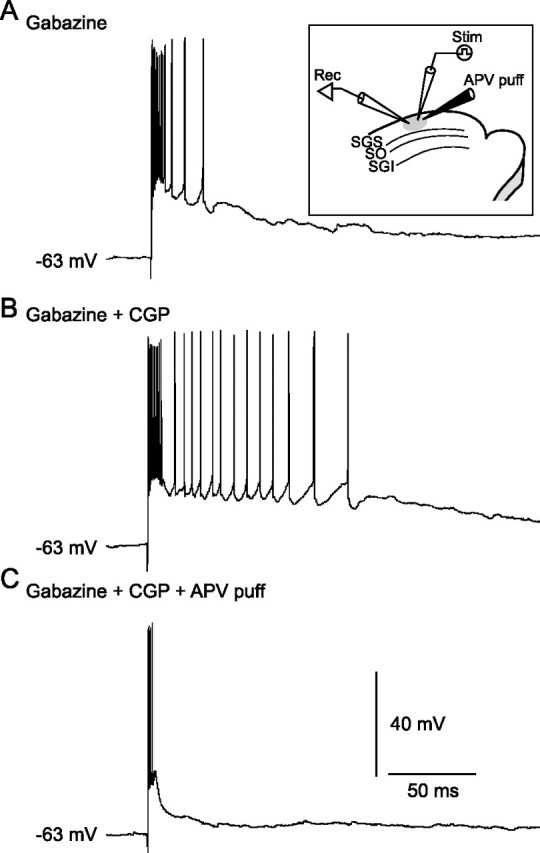
NMDARs are a prerequisite for evoking long-lasting bursts in SGS non-GABAergic neurons. Inset, Schematic showing the arrangement of recording and stimulating electrodes and puff pipette for application of APV (5 mm). A, In the presence of gabazine, SGS stimulation evoked bursts in an SGS non-GABAergic neuron. B, Addition of CGP greatly prolonged burst duration. C, Local puff application of APV (5 psi, 300 ms) profoundly inhibited the bursts.
We next recorded from non-GABAergic neurons in the SGI and examined the effects of local application of APV into the SGS. In the presence of gabazine and CGP, SGS stimulation evoked long-lasting bursts (Fig. 12 A1). Local puff application of APV into the SGS 0.5 s before SGS stimulation significantly decreased both the spike number within a burst and the burst duration (Fig. 12 A2,B,C). To confirm that the APV effect was caused by the blockade of NMDA receptors expressed in the SGS and not in the SGI, we then moved the puff pipette to the vicinity of the recorded neuron (i.e., SGI). In contrast to the local application into the SGS, local application of APV into the SGI 0.5 s before the SGS stimulation only slightly reduced the number of action potentials within a burst and the burst duration (Fig. 12 A4,B,C). Thirty seconds after the application of APV, however, both the spike number and burst duration were reduced (Fig. 12 A5,B,C). However, local application of APV into the SGI was less effective than that into the SGS, indicating that the effect of local application of APV into the SGS is attributable to the blockade of NMDARs expressed in the SGS. These results indicate that the long-lasting bursts in the SGS are dependent on NMDAR activation and are a prerequisite for the generation of long-lasting bursts in the SGI.
Figure 12.

NMDAR-dependent bursts in the SGS are necessary for long-lasting burst generation in the SGI. A1, In the presence of gabazine and CGP, SGS stimulation evoked long-lasting bursts in an SGI non-GABAergic neuron. A2, The stimulation induced only two action potentials and slow depolarization 0.5 s after puff application of APV locally to the SGS. Inset, Schematic showing the arrangement of recording and stimulating electrodes and puff pipette for application of APV (5 mm) into the SGS. A3, The effect of APV was reversible upon washout. A4, In the same SGI neuron, puff application of APV into the SGI 0.5 s before stimulation slightly inhibited the bursts. Inset, Schematic showing the arrangement of recording and stimulating electrodes and puff pipette for application of APV (5 mm) into the SGI. A5, The burst was further inhibited 30 s after APV application. B, A summary graph showing the effects of local APV application into the SGS or SGI on the number of spikes within a burst. The puff application of APV into the SGS significantly reduced the spike number. C, A summary graph showing the effect of local APV application into the SGS or SGI on the burst duration. Again, the puff application of APV into the SGS significantly decreased the burst duration. *p < 0.05, compared with control.
Discussion
Our main findings, summarized in Figure 13, were as follows: (1) synaptically released GABA after bursts of SGS GABAergic neurons can activate GABABRs in SGS, (2) GABABR activation limits the burst duration in SGS and SGI, (3) both postsynaptic and presynaptic GABABR mechanisms may contribute to the burst regulation, (4) the GABABR blockade-induced burst prolongation in the SGS depends on NMDARs, and (5) the prolonged SGS bursts are necessary for the generation of long-lasting bursts in SGI. These results indicate that the SC local circuit has an intrinsic mechanism to regulate burst duration by GABABRs in the SGS.
Figure 13.

Schematic of local circuit underlying GABABR-mediated regulation of bursts in the SC. Postsynaptic GABABRs expressed both in NFV and WFV cells and presynaptic GABABRs located on glutamatergic synaptic terminals in the SGS are activated by synaptically released GABA during bursts of SGS GABAergic neurons. Hyperpolarization in NFV cells, shunting inhibition in WFV cells, and reduction of glutamate release may contribute to the limitation of burst duration. When GABABRs are blocked, burst duration in the SGS may be prolonged in an NMDAR-dependent manner, and then, the prolonged burst may spread to the SGI. Thus the burst duration of the SGI may also be prolonged. Note that the localization of the receptors and synaptic connections essential to explain the present results are depicted, and other components, such as AMPA/kainate receptors and GABAARs, are not drawn for clarity.
Synaptically released GABA activates GABABRs in the SC
In SGI non-GABAergic neurons, CGP application alone did not affect the EPSCs evoked by SGS stimulation with single pulses, suggesting that the amount of GABA released by this stimulation was insufficient to activate GABABRs. In contrast, when CGP was coapplied with gabazine, the same stimulation induced EPSC bursts, the duration of which were significantly longer than those evoked in the presence of gabazine alone. Because all GABAergic neurons tested generated bursts in the presence of gabazine, a large amount of GABA release accompanying the bursts of GABAergic neurons should be necessary to activate GABABRs. This is in accordance with previous findings that GABABRs are localized in perisynaptic (Kulik et al., 2002) and extrasynaptic (Kulik et al., 2003) sites, although it remains unclear whether this is also true of the SC, and that affinity of GABA for GABABRs is lower than that for GABAARs (Curtis et al., 1970; Chu et al., 1990). We also found that repetitive stimulation was required to activate GABABRs in the SC, as has been observed in various brain regions (Johnson et al., 1992; Isaacson et al., 1993; Mitchell and Silver, 2000; Saitoh et al., 2004; Kaneda and Kita, 2005).
CGP did not affect the rise time or peak amplitude, but prolonged the decay time of EPSC bursts in SGI neurons, suggesting that GABABR-mediated responses have slow kinetics (Misgeld et al., 1995; Ulrich and Bettler, 2007), and competed against late components of EPSC bursts. The slow inhibition by GABABRs might function as a negative feedback to excessive excitation of the SC local circuit.
Although we found that SGS GABAergic neurons generated bursts in response to SGS stimulation during blockade of GABAARs, there is no in vivo study that addressed activity of SGS GABAergic neurons during or in preparation for saccades. Thus, future in vivo studies are necessary to investigate actual activity of GABAergic neurons in relation to saccades.
Previous studies suggested that GABACRs expressed predominantly in SGS GABAergic neurons have a role in disinhibitory circuitry (Lee et al., 2001; Schmidt et al., 2001). We investigated the effect of muscimol, which activates GABACRs and inhibits specifically GABAergic neurons, on the bursts evoked under our experimental condition. We obtained inconsistent results: burst activity was reduced, enhanced, or unchanged in non-GABAergic neurons (our unpublished observations). To conclude the role of GABACRs in the burst activity, further studies would be required.
GABABRs in the SGS are responsible for regulation of burst duration
Local CGP application into the SGS but not SGI effectively prolonged the duration of EPSC bursts in SGI neurons. Also, our immunohistochemical analysis as well as previous receptor binding studies (Bowery et al., 1987; Chu et al., 1990) demonstrated dense expression of GABABRs in the SGS. These findings strongly suggest that the primary site of CGP action is in the SGS. In our preliminary experiments, local repetitive stimulation was unable to induce any CGP-sensitive currents in SGI non-GABAergic neurons via activation of either postsynaptic or presynaptic GABABRs (our unpublished observations). Thus, GABABRs sparsely expressed in the SGI may not be essential for the burst regulation.
In the isolated SGS slices, where somata of SGI GABAergic neurons are eliminated, stimulation with single pulses still evoked long-lasting bursts in non-GABAergic neurons in the presence of CGP and gabazine. Because bursts of GABAergic neurons are necessary for GABABR activation, GABA released from SGS but not SGI GABAergic neurons most likely activated GABABRs in the SGS.
Postsynaptic mechanisms
Two postsynaptic mechanisms may contribute to the burst regulation. In NFV cells, repetitive stimulation evoked CGP-sensitive outward currents, suggesting that released GABA activates postsynaptic GABABRs, presumably leading to the activation of potassium channels. Thus, blockade of GABABR-mediated hyperpolarization could prolong the burst duration in NFV cells. Because NFV cells have dense axon collaterals in the SGS and also project to the SGI (Hall and Lee, 1993; Lee and Hall, 1995; Isa et al., 1998; Özen et al., 2000), the prolonged bursts in NFV cells may activate other cell types in the superficial layers as well as SGI neurons.
In contrast, repetitive stimulation of the SGS did not induce significant outward currents in WFV cells. We assumed that GABABR-mediated currents elicited in distal dendrites under present experimental conditions might be too small to detect with somatic recordings because of attenuation via passive membrane properties (Gulledge et al., 2005). Our results obtained by combined application of baclofen and glutamate suggest that GABABR activation did not show marked hyperpolarizations but rather shunted glutamate-evoked excitatory responses by increasing membrane conductance. Given that excitatory inputs can evoke dendritic action potentials with the cooperation of sodium channels and I h channels in distal dendrites of WFV cells (Endo et al., 2004), this shunting inhibition may reduce firings of WFV cells. Similar to NFV cells, axon collaterals of WFV cells arborize densely in both the SGS and SGI (Hall and Lee, 1993; Lee and Hall, 1995; Isa et al., 1998). Thus, long-lasting bursts of WFV cells may have a significant impact on neuronal activities not only in the SGS but also in the SGI. Together, these data strongly suggest that the activation of GABABRs expressed in the two major excitatory neurons in the SGS/SO may limit burst duration in the SC local circuit.
Presynaptic mechanisms
The actual expression sites of presynaptic GABABRs (e.g., glutamatergic terminals from the retina, cortex, or SGS neurons) are not clear at this point. However, we have shown that synaptically released GABA can activate presynaptic GABABRs, resulting in reduction of glutamatergic transmission to SGS neurons. Consistent with this, baclofen reduced EPSCs in the SGS (Endo and Isa, 2002). Such heterosynaptic modulation of glutamate release by GABA seemed to persist for at least 400 ms. Cooperation between this long-lasting reduction of glutamate release and the slow postsynaptic GABAB responses might shorten the burst duration effectively.
GABABR blockade-induced burst prolongation depends on NMDARs
Local application of APV into the SGS but not SGI significantly suppressed prolonged bursts in both SGI and SGS neurons. This difference might arise from different lateral interactions within each layer. The lateral interaction in the SGS is narrower than that in the SGI (Saito and Isa, 2004). Moreover, field potential recordings with a 64 channel electrode system revealed that the propagation area of burst activity in the SGS is more restricted than that in the SGI (Phongphanphanee et al., 2006). Therefore, the area covered by APV puff application may have been wide enough to block NMDARs in the SGS, but insufficient for the SGI. Thirty seconds after the puff application of APV into the SGI, the bursts in SGI neurons were diminished. One explanation for this might be that a large extent of the SGI circuit has to be blocked by APV for significant suppression of SGI bursts. Alternatively, APV might have diffused to the SGS and then blocked the SGS bursts. In either case, the present results support the notion that the inhibitory effect of APV local application into the SGS on SGI bursts is caused by the inhibition of burst activity in the SGS. Therefore, the prolonged bursts in SGS may lead to burst prolongation in the SGI.
We proposed previously that NMDAR-mediated synaptic transmission in the SGI is central to burst generation in the SC (Isa and Saito, 2001; Isa, 2002; Isa and Sparks, 2006). However, the present results suggest that we should not underestimate the contribution of NMDARs in the SGS to the burst generation. Stimulation with single pulses evoked APV-sensitive long-lasting bursts in the SGS when GABAARs and GABABRs are blocked. This suggests that recurrent interactions among non-GABAergic neurons such as NFV and WFV cells in the superficial layers are crucial for the maintenance of burst activity, and that glutamatergic local axon collaterals of these neurons may activate NMDARs.
Functional implications
Direct signal transmission from the SGS to SGI has been suggested to generate short-latency orienting responses, such as express saccades (Lee et al., 1997; Isa et al., 1998; Isa, 2002). In this context, the frequency and duration of bursts in the SGS should substantially affect the activity of SGI neurons, which may determine the accuracy of saccades (Stanford et al., 1996). If SGS bursts persist excessively, then the duration of SGI bursts may also be prolonged abnormally. We showed that GABABR-mediated inhibition in the SGS regulates the burst duration not only in the SGS but also in the SGI. Thus, this inhibition might be one of the mechanisms underlying accurate saccadic eye movements.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research 18021039 and 19700361 (K.K.), 18300102 and 18019007 (Y.Y.), and 18200027 (T.I.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, as well as by a grant from the Human Frontier Science Program Organization.
References
- Andrade R. Blockade of neurotransmitter-activated K+ conductance by QX-314 in the rat hippocampus. Eur J Pharmacol. 1991;199:259–262. doi: 10.1016/0014-2999(91)90467-5. [DOI] [PubMed] [Google Scholar]
- Binns KE, Salt TE. Different roles for GABAA and GABAB receptors in visual processing in the rat superior colliculus. J Physiol (Lond) 1997;504:629–639. doi: 10.1111/j.1469-7793.1997.629bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG, Hudson AL, Price GW. GABAA and GABAB receptor site distribution in the rat central nervous system. Neuroscience. 1987;20:365–383. doi: 10.1016/0306-4522(87)90098-4. [DOI] [PubMed] [Google Scholar]
- Chu DC, Albin RL, Young AB, Penney JB. Distribution and kinetics of GABAB binding sites in rat central nervous system: a quantitative autoradiographic study. Neuroscience. 1990;34:341–357. doi: 10.1016/0306-4522(90)90144-s. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Duggan AW, Felix D, Johnston GAR. GABA, bicuculline and central inhibition. Nature. 1970;226:1222–1224. doi: 10.1038/2261222a0. [DOI] [PubMed] [Google Scholar]
- Endo T, Isa T. Postsynaptic and presynaptic GABAB receptor-mediated inhibition in rat superficial superior colliculus neurons. Neurosci Lett. 2002;322:126–130. doi: 10.1016/s0304-3940(02)00100-3. [DOI] [PubMed] [Google Scholar]
- Endo T, Yanagawa Y, Obata K, Isa T. Characteristics of GABAergic neurons in the superficial superior colliculus in mice. Neurosci Lett. 2003;346:81–84. doi: 10.1016/s0304-3940(03)00570-6. [DOI] [PubMed] [Google Scholar]
- Endo T, Notomi T, Tarusawa E, Shigemoto R, Isa T. Hyperpolarization-activated cation current and its modification of dendritic spike initiation in wide field vertical cells of the rat superficial superior colliculus. Soc Neurosci Abstr. 2004;30:647–3. [Google Scholar]
- Fischer B, Boch R. Saccadic eye movements after extremely short reaction times in the monkey. Brain Res. 1983;260:21–26. doi: 10.1016/0006-8993(83)90760-6. [DOI] [PubMed] [Google Scholar]
- Fischer B, Weber H. The time of secondary saccades to primary targets. Exp Brain Res. 1993;97:356–360. doi: 10.1007/BF00228706. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Kiener T, Bouilleret V, Loup F. GABAergic neurons and GABAA-receptors in temporal lobe epilepsy. Neurochem Int. 1999;34:435–445. doi: 10.1016/s0197-0186(99)00040-6. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Kampa BM, Stuart GJ. Synaptic integration in dendritic trees. J Neurobiol. 2005;64:75–90. doi: 10.1002/neu.20144. [DOI] [PubMed] [Google Scholar]
- Hall WC, Lee P. Interlaminar connections of the superior colliculus in the tree shrew. I. The superficial gray layer. J Comp Neurol. 1993;332:213–223. doi: 10.1002/cne.903320206. [DOI] [PubMed] [Google Scholar]
- Helms MC, Özen G, Hall WC. Organization of the intermediate gray layer of the superior colliculus. I. Intrinsic vertical connections. J Neurophysiol. 2004;91:1706–1715. doi: 10.1152/jn.00705.2003. [DOI] [PubMed] [Google Scholar]
- Isa T. Intrinsic processing in the mammalian superior colliculus. Curr Opin Neurobiol. 2002;12:668–677. doi: 10.1016/s0959-4388(02)00387-2. [DOI] [PubMed] [Google Scholar]
- Isa T, Saito Y. The direct visuo-motor pathway in mammalian superior colliculus; novel perspective on the interlaminar connection. Neurosci Res. 2001;41:107–113. doi: 10.1016/s0168-0102(01)00278-4. [DOI] [PubMed] [Google Scholar]
- Isa T, Sparks D. Microcircuit of the superior colliculus: a neuronal machine that determine timing and endpoint of saccadic eye movements. In: Grillenr S, Graybiel AM, editors. Microcircuits; the interface between neurons and global brain function. Cambridge, MA: MIT; 2006. pp. 1–34. [Google Scholar]
- Isa T, Endo T, Saito Y. The visuo-motor pathway in the local circuit of the rat superior colliculus. J Neurosci. 1998;18:8496–8504. doi: 10.1523/JNEUROSCI.18-20-08496.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Johnson SW, Mercuri NB, North RA. 5-hydroxytryptamine1B receptors block the GABAB synaptic potential in rat dopamine neurons. J Neurosci. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda K, Kita H. Synaptically released GABA activates both pre- and postsynaptic GABAB receptors in the rat globus pallidus. J Neurophysiol. 2005;94:1104–1114. doi: 10.1152/jn.00255.2005. [DOI] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Nyíri G, Notomi T, Malitschek B, Bettler B, Shigemoto R. Distinct localization of GABAB receptors relative to synaptic sites in the rat cerebellum and ventrobasal thalamus. Eur J Neurosci. 2002;15:291–307. doi: 10.1046/j.0953-816x.2001.01855.x. [DOI] [PubMed] [Google Scholar]
- Kulik A, Vida I, Luján R, Haas CA, López-Bendito G, Shigemoto R, Frotscher M. Subcellular localization of metabotropic GABAB receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus. J Neurosci. 2003;23:11026–11035. doi: 10.1523/JNEUROSCI.23-35-11026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Hall WC. Interlaminar connections of the superior colliculus in the tree shrew. II: Projections from the superficial gray to the optic layer. Vis Neurosci. 1995;12:573–588. doi: 10.1017/s0952523800008464. [DOI] [PubMed] [Google Scholar]
- Lee PH, Helms MC, Augstine GJ, Hall WC. Role of intrinsic synaptic circuitry in collicular sensorimotor integration. Proc Natl Acad Sci USA. 1997;94:13299–13304. doi: 10.1073/pnas.94.24.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Schmidt M, Hall WC. Excitatory and inhibitory circuitry in the superficial gray layer of the superior colliculus. J Neurosci. 2001;21:8145–8153. doi: 10.1523/JNEUROSCI.21-20-08145.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Sooksawate T, Yanagawa Y, Isa K, Isa T, Hall WC. Identity of a pathway for saccadic suppression. Proc Natl Acad Sci USA. 2007;104:6824–6827. doi: 10.1073/pnas.0701934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean HA, Caillard O, Ben-Ari Y, Gaiarsa JL. Bidirectional plasticity expressed by GABAergic synapses in the neonatal rat hippocampus. J Physiol (Lond) 1996;496:471–477. doi: 10.1113/jphysiol.1996.sp021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol. 1995;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. GABA spillover from single inhibitory axons suppresses low-frequency excitatory transmission at the cerebellar glomerulus. J Neurosci. 2000;20:8651–8658. doi: 10.1523/JNEUROSCI.20-23-08651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mize RR. The organization of GABAergic neurons in the mammalian superior colliculus. Prog Brain Res. 1992;90:219–248. doi: 10.1016/s0079-6123(08)63616-x. [DOI] [PubMed] [Google Scholar]
- Nathan T, Jensen MS, Lambert JD. GABAB receptors play a major role in paired-pulse facilitation in area CA1 of the rat hippocampus. Brain Res. 1990;531:55–65. doi: 10.1016/0006-8993(90)90757-3. [DOI] [PubMed] [Google Scholar]
- Özen G, Augustine GJ, Hall WC. Contribution of superficial layer neurons to premotor bursts in the superior colliculus. J Neurophysiol. 2000;84:460–471. doi: 10.1152/jn.2000.84.1.460. [DOI] [PubMed] [Google Scholar]
- Phongphanphanee P, Kaneda K, Isa T. Analysis of spread of activity in the local circuit of superior colliculus. Soc Neurosci Abstr. 2006;32:345.1. [Google Scholar]
- Saito Y, Isa T. Local excitatory network and NMDA receptor activation generate a synchronous and burst command from the superior colliculus. J Neurosci. 2003;23:5854–5864. doi: 10.1523/JNEUROSCI.23-13-05854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Isa T. Laminar specific distribution of lateral excitatory connections in the rat superior colliculus. J Neurophysiol. 2004;92:3500–3510. doi: 10.1152/jn.00033.2004. [DOI] [PubMed] [Google Scholar]
- Saito Y, Isa T. Organization of interlaminar interactions in the rat superior colliculus. J Neurophysiol. 2005;93:2898–2907. doi: 10.1152/jn.01051.2004. [DOI] [PubMed] [Google Scholar]
- Saitoh K, Isa T, Takakusaki K. Nigral GABAergic inhibition upon mesencephalic dopaminergic cell groups in rats. Eur J Neurosci. 2004;19:2399–2409. doi: 10.1111/j.0953-816X.2004.03337.x. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Boller M, Özen G, Hall WC. Disinhibition in rat superior colliculus mediated by GABAC receptors. J Neurosci. 2001;21:691–699. doi: 10.1523/JNEUROSCI.21-02-00691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetedjo R, Kaneko CR, Fuchs AF. Evidence that the superior colliculus participates in the feedback control of saccadic eye movements. J Neurophysiol. 2002;87:679–695. doi: 10.1152/jn.00886.2000. [DOI] [PubMed] [Google Scholar]
- Sooksawate T, Saito Y, Isa T. Electrophysiological and morphological properties of identified crossed tecto-reticular neurons in the rat superior colliculus. Neurosci Res. 2005;52:174–184. doi: 10.1016/j.neures.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Stanford TR, Freedman EG, Sparks DL. Site and parameters of microstimulation: evidence for independent effects on the properties of saccades evoked from the primate superior colliculus. J Neurophysiol. 1996;76:3360–3381. doi: 10.1152/jn.1996.76.5.3360. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa T, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Bettler B. GABAB receptors: synaptic functions and mechanisms of diversity. Curr Opin Neurobiol. 2007;17:298–303. doi: 10.1016/j.conb.2007.04.001. [DOI] [PubMed] [Google Scholar]