

Abstract
Microglial activation is a hallmark of all neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). Here, a detailed characterization of the microglial cell population within the spinal cord of a mouse model of familial ALS was performed. Using flow cytometry, we detected three distinct microglial populations within the spinal cord of mice overexpressing mutant superoxide dismutase (SOD1): mature microglial cells (CD11b+, CD45low), myeloid precursor cells (CD11b+, CD45int), and macrophages (CD11b+, CD45high). Characterization of cell proliferation within the CNS of SOD1G93A mice revealed that the expansion in microglial cell population is mainly attributable to the proliferation of myeloid precursor cells. To assess the contribution of proliferating microglia in motor neuron degeneration, we generated CD11b-TKmut-30; SOD1G93A doubly transgenic mice that allow the elimination of proliferating microglia on administration of ganciclovir. Surprisingly, a 50% reduction in reactive microglia specifically in the lumbar spinal cord of CD11b-TKmut-30; SOD1G93A doubly transgenic mice had no effect on motor neuron degeneration. This suggests that proliferating microglia-expressing mutant SOD1 are not central contributors of the neurodegenerative process in ALS caused by mutant SOD1.
Keywords: ALS, microglia, motor neuron, degeneration, ablation, transgenic mice
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, adult-onset neurodegenerative disorder that affects motor neurons in the brain and spinal cord. Degeneration of these neurons leads to atrophy of skeletal muscle and, ultimately, to paralysis and death within 1–5 years. ALS occurs in both sporadic and familial forms, which are clinically and pathologically similar. Missense mutations in the gene encoding the free radical-scavenging metalloenzyme, copper, zinc superoxide dismutase (SOD1) are responsible for 20% of familial ALS cases (Rosen et al., 1993). To date, >140 mutations have been found in the SOD1 gene, and transgenic mice overexpressing various SOD1 mutants develop an ALS-like phenotype through a gain of unknown toxic properties (Gurney et al., 1994). Several mechanisms have been proposed to explain motor neuron death in ALS, including glutamate-induced excitotoxicity (Van Den Bosch et al., 2006), cytoskeletal abnormalities (Julien et al., 2005), protein aggregation (Julien, 2001), oxidative stress (Cleveland and Rothstein, 2001), angiogenic factors (Lambrechts et al., 2007) and, more recently, toxicity via extracellular SOD1 (Urushitani et al., 2006).
Although ALS is characterized by the degeneration of motor neurons, the role of nonneuronal cells in disease pathology is now formally recognized (Pramatarova et al., 2001; Clement et al., 2003). Indeed, wild-type nonneuronal cells can extend the survival of mutant SOD1 transgenic mice (Clement et al., 2003). Also, the expression of mutant SOD1 in microglia and astrocytes contributes to the progression motor neuron degeneration (Beers et al., 2006; Boillée et al., 2006; Van Damme et al., 2007; Yamanaka et al., 2008). However, despite the findings of the toxicity of SOD1 mutants through action in glial cells, the exact role of glia, themselves, and how they influence the neurodegenerative process remains to be further defined.
Microglia are the resident myeloid cells of the CNS and account for 5–12% of the total glial population (Ling et al., 1973). One of the hallmarks of virtually all neurodegenerative conditions, including ALS, is the notable microgliosis. Here, we have characterized the CD11b+ myeloid component in the SOD1G93A and SOD1WT transgenic mouse spinal cord. We found that the CD11b+ population constitutes three different microglial cell groups. Aside from mature microglia, which adopt phenotypical features of dendritic cells during disease progression, and macrophages, we found the presence of Gr1+ myeloid precursor cells. Interestingly, the significant increase in CD11b+ cells in the spinal cord of SOD1G93A mice at late phase of disease is mostly attributable to an increase in the proliferation of these myeloid precursor cells. Moreover, to study the role of microglia in mice expressing mutant SOD1, we have used a novel approach based on the reduction of microglia numbers.
To decrease microglial cell numbers, we generated CD11b-TKmut-30; SOD1G93A doubly transgenic mice that allow the elimination of proliferating microglia on administration of the nucleoside analog ganciclovir. The CD11b-TKmut-30 model has previously been used to determine the role of proliferating microglia in several mouse models of CNS disease, including cerebral ischemia, gliomas, Alzheimer's disease, CNS viral infection, and axonal regeneration (Carmen et al., 2006; Simard et al., 2006; Galarneau et al., 2007; Lalancette-Hébert et al., 2007; Barette et al., 2008). In all of these paradigms, the selective ablation of proliferating microglia was deleterious. Surprisingly, here we report that a 50% reduction in activated microglia specifically in the lumbar spinal cord of CD11b-TKmut-30; SOD1G93A transgenic mice had no effect on motor neuron degeneration. Our interpretation of these results is that elimination of mutant SOD1-expressing microglial cells, which are functionally abnormal, did not alter the overall balance of neuroprotective and neurotoxic factors in the system.
Materials and Methods
Animals.
SOD1G93A [B6SJL-TgN(SOD1-G93A)1Gur/J;stock number 002726] and SOD1WT overexpressors [B6SJL-Tg(SOD1)2Gur/J;stock number 002297] were acquired from The Jackson Laboratory. CD11b-TKmt-30 mice were generated and genotyped in the laboratory of Dr. Jean-Pierre Julien as described previously (Gowing et al., 2006a). CD11b-TKmt-30 transgenic mice expressing human mutant SOD1G93A were obtained by breeding heterozygous (Tg/+) CD11b-TKmt-30 mice with heterozygous (Tg/+) SOD1G93A. Hence, all analyses were performed with littermate controls. SOD1G93A were genotyped in accordance with Jackson Laboratory protocols. The use and maintenance of the mice described in this article were performed in accordance to the Guide of Care and Use of Experimental Animals of the Canadian Council on Animal Care or the ethical committee of the Katholieke Universiteit Leuven, Leuven, Belgium.
Surgical procedures.
For intraspinal delivery of ganciclovir (10 mg/ml, Cytovene; Roche) or saline, mice at 85 d of age were anesthetized with isoflurane. Skin and muscle was then cut to expose the lumbar vertebra. A laminectomy was then performed to expose the lumbar spinal cord at the level of L4–L5 segment. A steel canula (0.8 mm in length, 28 ga) connected to an Alzet osmotic minipump (model 2004; Durect) was then cemented to the dorsal vertebra surrounding the laminectomy. Mice were injected intraperitoneally with ganciclovir at a dose of 100 mg/kg/d on the day of surgery as well as 1 d before and after the surgical procedure. At 115 d of age, after 30 d of intraspinal infusion with ganciclovir, mice were killed for analysis.
Analysis of disease progression.
Measurements of body weight and hindlimb reflex was used to score the clinical effects because the rotarod test presented a risk of fall-down resulting in pump damage in the case of CD11b-TKmt-30 transgenic mice. The extensibility and postural reflex of the hindlimbs when mice were pulled up with their tails were scored as described previously (Urushitani et al., 2006). Scoring was performed in a blind manner by animal technicians who had no information about the treatment but have experience in grading SOD1 mice paralysis.
Flow cytometric phenotypical studies.
Transgenic mice were anesthetized using Nembutal (10 mg/kg) and transcardially perfused with ice-cold PBS to remove all blood from the nervous tissue. Spinal cords were surgically removed. Tissue was then mechanically dissociated and filtered through a 70 μm cell strainer to obtain a single cell suspension (BD Biosciences). To isolate mononuclear cell fraction, the single cell suspensions were loaded on a 40–80% Percoll gradient (GE Healthcare) and centrifuged for 20 min at 1500 rpm. After centrifugation, the mononuclear cells from the interface were recovered and were stained with fluorescein isothiocyanate, phycoerythrin (PE), peridinin chlorophyll protein, PE-cyanine 5, PE-Cy7, or allophycocyanin-conjugated antibodies against CD45, CD11b, CD11c, Gr1, CD40, CD86, bromodeoxyuridine (BrdU) (antibodies were purchased from BD Biosciences), and F4/80 (eBiosciences). Samples were then analyzed on a flow cytometer FACStar Plus or fluorescence-activated cell sorting (FACS) Canto Cytometer (BD Biosciences). Cells were gated using Annexin-5 (BD Biosciences) and 7-AAD (BD Biosciences) to eliminate apoptotic/necrotic cells and select for viable cells. To validate the flow cytometric identification of the macrophage population in the mononuclear fraction as CD11b+, F4/80+ cells, we performed a peritoneal lavage of the SOD1G93A mice and obtained peritoneal lavage fluid known to be enriched in macrophages (Takeyama et al., 1999), which was then used as a positive control. For proliferation analysis with FACS, SOD1G93A and SOD1WT mice were intraperitoneally injected with BrdU (10 mg/kg) for 5 consecutive days and killed 2 d after the last injection. Mononuclear cells were isolated as described above and stained for the markers CD11b, CD45, and BrdU according to BrdU flow kit protocol (BD Biosciences). Non-BrdU-treated animals were used as negative controls.
Fluorescence activated cell sorting.
Twenty SOD1WT mice were intracardially perfused and spinal cords isolated. Mononuclear cells were collected and loaded on a flow cytometer, FACSAria (BD Biosciences), as described above. Viable cells were analyzed for expression of the markers CD45 and CD11b. The CD11b+CD45int population was gated, sorted, and collected. The sorted cells were loaded on coated slides by cytospin (800 rpm, 10 min) then dried overnight and stained using the May-Grünwald-Giemsa staining protocol. Pictures were taken with a bright light microscope (Leica DM LB2 microscope; Leica Microsystems) on a 10 × 50 magnification.
Tissue collection for immunohistochemical analyses.
Mice were anesthetized and transcardially perfused with NaCl 0.9% and fixed with 4% paraformaldehyde. Dissected spinal cord and muscle tissue were postfixed for 24 h in 4% paraformaldehyde and equilibrated in a solution of PBS-sucrose (20%) for 48 h. Spinal cord tissue were then cut in 20 μm horizontal or transverse sections with a Leica frozen microtome and kept in a cryoprotective solution at −20°C. Gastrocnemius muscle was cut in 40 μm cryostat sections. Dissected dorsal root ganglia (DRG) was then postfixed in a solution of 3% glutaraldehyde for a period of 48 h, washed in PBS, treated with 1% osmium tetroxide for 2 h, and dehydrated through graded alcohol solutions. Before Epon plastic embedding, DRG were further dissected to ensure that all ventral root axons would be sampled at a distance of 3 mm from the DRG cell body. Semithin cross-sections (1 μm) were stained with toluidine blue, rinsed, and coverslipped.
Immunohistochemistry.
Spinal cord sections were stained with the following antibodies: anti-CD11b (Serotec), anti-NG2 (Millipore), anti-GFAP (Dako or Millipore), anti-Iba1 (Wako), anti-BrdU (Immunosource), and anti-CD3 (BD Pharmingen) according to standard techniques. Of note, we found perfect colocalization of all CD11b+ and Iba1+ cells by double immunofluorescence in the spinal cord of SOD1G93A transgenic mice. For light microscopy, sections were developed with Vectastain ABC kit, reacted with nickel-diaminobenzidine (Vector Laboratories). For immunofluorescence, sections were stained with the fluorophore-coupled secondary antibody Alexa-555, Alexa-488, or Alexa 594 (Invitrogen). For BrdU experiment, mice were intraperitoneally injected with BrdU (10 mg/kg) for 5 consecutive days and killed 2 d after the last injection. Immunohistochemistry for BrdU was performed according to standard protocols.
Quantitative analyses.
For quantification of CD11b, GFAP, NG2, and BrdU reactivity, a minimum of four 20 μm spinal cord transversal sections were analyzed per age group on an upright fluorescence microscope (DMIRB; Leica). CD11b+ cells double positive for 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) and CD11b+, GFAP+ and NG2+ cells double positive for BrdU in the gray matter of the lumbal spinal cord were counted using NIS Elements software. In short, in overlay pictures of NG2/GFAP/CD11b with DAPI and/or BrdU, double-positive cells were counted. For microglial cell ablation experiment, every sixth sample section of horizontal spinal cord sections were immunostained for Iba1 and Mac-2 for microglial cell counts or Nissl stained to identify motor neurons in the lumbar spinal cord. The L3–L5 spinal cord sections were individually traced with a 4× objective and sampled using a 40× objective. Lumbar segments were identified during dissection and stereological analysis. Briefly, L3–L5 DRG were dissected and cut immediately above the L2 level of the spinal cord (as identified by L2 ventral and dorsal exit point) and immediately below L5 spinal cord level. Measurement of the distance from between L3–L4 and L4–L5 were measured on many mice both male and female. An average distance for each segment was determined and was applied during stereological analyses. The density of labeled cells was estimated by the optical fractionator method using Stereo Investigator software (MBF Bioscience). For Iba1-, Mac-2-, GFAP-, CD3-, or NG2-positive cells to be counted, a distinct cell body had to be within the optical dissector height. The counting parameters were the distance between counting frames (600 μm), the counting frame size (150 × 150 μm), the dissector height (8 μm), and the guard zone thickness (1 μm). Motor neurons were identified on the basis of their correct anatomical location (ventral horn/laminas 9), required a distinct nucleolus within the plane of the optical dissector, and had a cross-sectional area ≥250 μm2. Counting parameters were identical to Iba1 cells counts except for the distance between counting frames (300 μm). Bilateral L5 ventral root axons were counted at a magnification of 60× using Stereo Investigator software; interior of axons were marked in each frame until the entire ventral root section had been sampled. Counts represent the mean axonal count for the left and right ventral roots. Bilateral gastrocnemius muscles were sampled and stained by immunofluorescence with rhodamine bungarotoxin (Invitrogen) anti-neurofilament M (MAB5254; Millipore) and SV2 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) antibodies. End plates were scored as “innervated” if there was complete overlap with axon terminal or as “denervated” if the end plate was not associated with an axon. Partial overlap or association with preterminal axon only was scored as intermediate innervation. Every fourth section was systematically sampled to evaluate all neuromuscular junctions present. Statistical analyses were performed using StatsDirect statistical software (StatsDirect) or GraphPad Prism software. Data are presented as mean ± SEM.
Results
Characterization of the CD11b+ cell population in SOD1G93A spinal cord with disease progression
The “microglial reaction,” referring to the activation of CD11b myeloid cells within the CNS, is a well known process occurring in several neurodegenerative diseases, such as ALS. To get a better understanding of the activation pattern of myeloid cells in ALS, CD11b immunoreactivity was quantified in the SOD1G93A and SOD1WT transgenic mice at several stages of the disease. In the SOD1G93A mouse, CD11b+ cells were counted at the age of 60 and 80 d, when mice are clinically asymptomatic, at 100 d, when motor neurons degenerate with a clear loss of motor performance, and at 120 d, when mice develop severe hind limb paralysis attributable to complete loss of motor function. The SOD1WT mouse, aged 130 d, which does not develop overt motor neuron degeneration, was used as a control. At the age of 60 and 80 d, no difference could be found in the number of CD11b+ cells in the spinal cord of SOD1G93A compared with SOD1WT control mice (Fig. 1A). However, at later stages of the disease, a significant increase in the number of CD11b+ cells was observed in SOD1G93A when compared with control mice. Indeed, a 1.7-fold increase in the number of CD11b+ microglia occurred between the asymptomatic (60 d) and the late symptomatic phase (120 d) of the disease. Interestingly, the most notable increase in CD11b+ cells occurred between 60 and 100 d of age with no significant increase occurring in the SOD1G93A between 100 and 120 d of age (Fig. 1A). Furthermore, a notable change in the morphology of the CD11b+ cells could be observed (data not shown). At the age of 60 d, almost all CD11b+ cells had a small cell body from which highly ramified processes were extending, reflecting the nonactivated state of these cells (Cuadros and Navascues, 1998). However, at the age of 120 d, almost all CD11b+ cells had upregulated their CD11b expression and had fewer, thicker processes, reflecting their “activated” state (Cuadros and Navascues, 1998; Streit et al., 1999). These results are in line with a previous study by Alexianu et al. (2001) where microglial activation in SOD1 transgenic mice had been qualitatively assessed.
Figure 1.

Expansion of the CD11b+ cell population, which consists of three different subpopulations, occurs early in the disease. A, Spinal cord (S.C.) sections of SOD1G93A and SOD1WT were stained for the myeloid marker CD11b. Gray matter immunopositive cells were counted at the age of 60, 80, 100, and 120 d for SOD1G93A and 130 d SOD1WT. *p < 0.05, ***p < 0.001, Significant differences versus SOD1WT control; †p < 0.05, significant versus SOD1G93A at 60 d; †††p < 0.001, significant versus SOD1G93A 60 d; and ##p < 0.01, significant compared with SOD1G93A at 80 d. All values are mean ± SEM. B, Isolated spinal cord mononuclear cells were analyzed using 5-color flow cytometry. Cells were gated (R1, B1) using Annexin-5 and 7-AAD to eliminate apoptotic and necrotic cells (data not shown) and further analyzed for viability using 7-AAD (B2). Viable cells were analyzed for CD11b and CD45 expression (B3). Three CD11b+ populations were found: macrophages (orange, R4), myeloid precursor cells (purple, R3), and mature microglia (blue, R2). Because there are only few macrophages present in the mononuclear cell isolate, only myeloid precursor cells and mature microglia were further analyzed. C, Expression of the markers Gr1 and F4/80 in the different cellular subsets. Mature microglia were negative for both Gr1 and F4/80 (C1). Myeloid precursor cells were positive for Gr1 (C3) and negative for F4/80 (C3). Isotype stains are shown in C2 and C4, respectively. D, Contribution of macrophages (black), myeloid precursors (dark gray), and mature microglia (light gray) to the expansion of the CD11b+ cell population at different ages. At the age of 60 d, mature microglia are twice as numerous as the myeloid precursor cells (p < 0.05). At the age of 120 d, the proportion of myeloid precursor cells had greatly increased compared with the age of 130 d SODWT (p < 0.01). At 120 d, there were almost twice as many myeloid precursor cells than mature microglial cells present in the CD11b cell population (p < 0.0001). All values are mean ± SEM; n = 3–9 for all experiments.
Because a myeloid CD11b+ cell population can encompass different cell types, the mononuclear cell population was isolated from total spinal cord, and single cell suspensions of both SOD1G93A and SOD1WT mice at different stages of the disease were analyzed by flow cytometry. Within the CD11b+ cell group, at all time points three distinct populations could be identified. These subpopulations of CD11b+ cells differed in expression level of the pan-leukocyte marker CD45. The first group of cells were CD11b+, CD45low, corresponding to mature microglial cells (Sedgwick et al., 1991; Carson et al., 1998) (Fig. 1B3, blue and R2). These cells neither express the macrophage marker F4/80, nor the granulocyte marker Gr1 (Fig. 1C1). The second group identified was CD11b+ and intermediate for CD45 (CD45int) (Fig. 1B3, purple and R3). These were large cells with a high degree of granularity on forward scatter/side scatter (data not shown) and were identified as myeloid precursor cells: they expressed high levels of Gr1, were negative for F4/80 (Fig. 1C3), and after FACSorting they showed the characteristic doughnut-like nucleus, typical for myeloid precursor cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These cells have previously been described in mouse models of myeloid precursor expansion (Almand et al., 2001; Billiau et al., 2003). The third and smallest group were CD11b+, CD45high, identified as CNS macrophages (Fig. 1B3, orange and R4) as reported previously (Cruse et al., 2004; Stirling and Yong, 2008). Only very few of these cells could be found in the spinal cord of SOD1G93A transgenic mice (Fig. 1D). The flow cytometric identification of this population in the SOD1G93A spinal cord (Fig. 1B3) was validated using peritoneal lavage fluid, known to be enriched in macrophages (Takeyama et al., 1999), from SOD1G93A mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Macrophages were clearly F4/80 positive and Gr1 negative, as described previously (Austyn and Gordon, 1981; Stirling and Yong, 2008).
To establish to what extent these three separate CD11b+ populations contribute to CD11b+ immunoreactivity in the spinal cord of SOD1G93A mice, the relative contribution of the different cell types in the CD11b+ population at different ages was quantified by flow cytometry (Fig. 1D). Interestingly, the different populations of CD11b+ cells in the spinal cord of SOD1G93A mice changed significantly during disease progression. At the age of 60 d, the CD11b+ population consists of mostly mature microglial cells which are twice as numerous as the myeloid precursor cells. At the age of 120 d, the proportion of myeloid precursor cells in SOD1G93A had significantly increased, compared with both earlier time points (60 and 80 d), and was significantly higher than in control SOD1WT mice. Furthermore, at 120 d of age, there was a 1.8-fold higher number of myeloid precursor cells compared with mature microglia present in the CD11b+ cell population in SOD1G93A mice. In the SOD1WT mouse, no significant changes in myeloid precursor cell contributions were detected over time (data not shown). This data therefore suggest that in an early phase of the disease, the CD11b+ population consists mostly of mature microglial cells, whereas with disease progression the myeloid precursor cell population expands significantly. Furthermore, macrophages did not significantly contribute to the myeloid population: only few of these cells were detected within the CD11b+ immunoreactive population compared with the amount of mature microglia and myeloid precursor cells (Fig. 1B3, R4). Thus, within the myeloid population of the spinal cord, there is, in addition to mature microglia and macrophages, the presence of myeloid precursor cells, which contribute to a large extent to the CD11b+ cell population in the course of ALS.
Mature microglia acquire a dendritic cell phenotype
To gain further insight into the functional properties of microglia during disease progression, the phenotype of the mature and myeloid precursor populations was characterized by flow cytometry. Because the activation of microglia leads to the acquirement of antigen presenting and/or phagocytic properties, the expression of surface molecules characteristic for antigen presenting cells was analyzed in the SOD1G93A and SOD1WT transgenic mice at different time points. The following markers were used: CD11c, a dendritic cell marker, CD40 and CD86, two costimulatory molecules for signaling during antigen presentation, and F4/80, a macrophage marker (Fig. 2).
Figure 2.
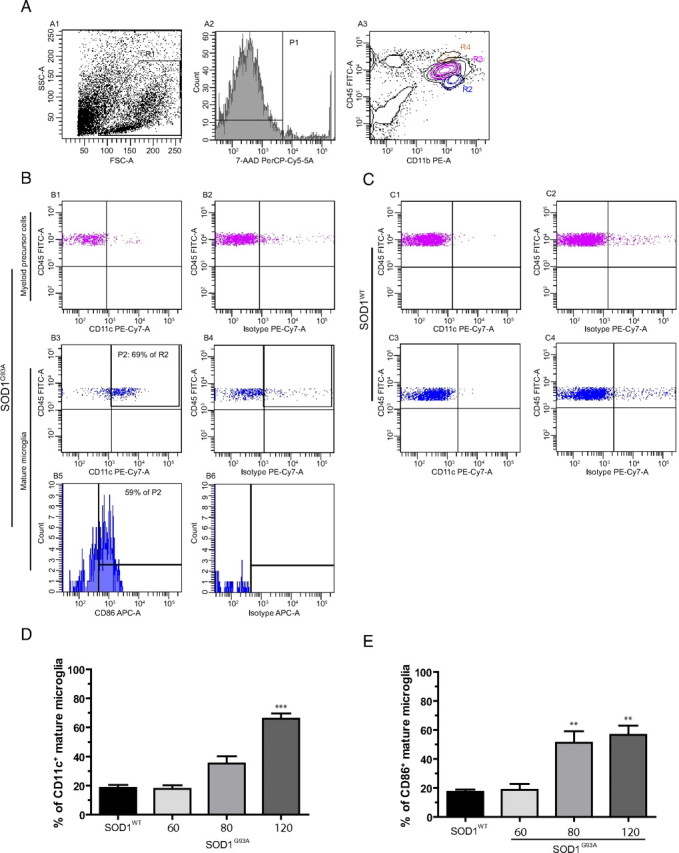
Mature microglia obtain a dendritic cell phenotype as disease progresses. Five-color flow cytometric analysis of microglial cells using different markers of microglial activation. A, Isolated mononuclear spinal cord cells were gated (R1; A1) and viable cells (R1) selected using 7-AAD (P1; A2). A3, Cells were analyzed for CD11b/CD45 expression. Because there were only few macrophages present (R4, orange), we only analyzed myeloid precursor cells (purple, R3) and mature microglia (blue, R2). B, Myeloid precursor cells from SOD1G93A end-stage mice were CD11c negative (B1), whereas mature microglia (B3) were clearly positive for CD11c. B2, B4, Isotype controls for B1 and B3, respectively. B5, Fifty-nine percent of gated CD11c+ mature microglial cells coexpressed CD86. B6, Isotype control for B5. C, However, in SOD1WT mouse, both myeloid precursor cells (C1, compare with C2 isotype control) and mature microglia (C3, compare with C4 isotype control) did not express CD11c. D, E, Quantification of CD11c (D) and CD86 (E) expression of microglial cells at different ages both in SOD1G93A and SOD1WT mice. D, ***p < 0.0001 compared with SOD1WT at 130 d and SOD1G93A at 60 d and p < 0001 compared with SOD1G93A 80 d. E, **p < 0.01 compared with SOD1WT 130 d of age and SOD1G93A 60 d of age; all values are mean ± SEM. For CD11c expression, n = 3–13; for CD86 expression, n = 5–7.
For the SOD1G93A mature microglial (CD11b+, CD45low) cell population, there was a statistically significant increase in CD11c expression at the age of 120 d compared with the age of 60 and 80 d (Fig. 2D). Within the CD11c-positive cell population at 120 d in the SOD1G93A, 59% also stained positive for CD86 (Fig. 2B5). Indeed, the percentage of mature microglia positive for CD86 was almost threefold higher at the age of 120 d compared with the age of 60 d (Fig. 2E). The expression of both CD11c and CD86 tended to increase starting at the age of 80 d, before apparent motor neuron degeneration. Changes in CD11c (Fig. 2C3) or CD86 (data not shown) expression were not found in the mature microglia of the SOD1WT spinal cord at the different ages examined. A significant difference in the expression of CD11c (Fig. 2B1,C1) or CD86 (data not shown) was not found among the myeloid precursor cell population (CD11b+, CD45int) between SOD1G93A and SOD1WT. At all time points, F4/80 or CD40 immunoreactivity was absent in both the myeloid precursor cell and mature microglia population (data not shown). This data indicate that in the SOD1G93A mouse mature microglial cells acquire the phenotype of dendritic cells, whereas evolution toward mature macrophages is not seen.
Characterization of cell proliferation in the spinal cord of SOD1G93A transgenic mice
As CD11b+ cells increase in number with disease progression, the origin of these cells was analyzed in detail. Proliferation was analyzed in the gray matter of SOD1G93A and SOD1WT spinal cords at different stages of disease (Fig. 3). BrdU labeling was used as a proliferation marker. At the age of 80 d, when mice were presymptomatic, a 2.8-fold increase in BrdU-positive nuclei were found in SOD1G93A mice compared with SOD1WT control (Fig. 3B). This further increased to a 4.8-fold and an 11.2-fold at the ages of 100 and 120 d, respectively. Hence, a steady increase in cell proliferation takes place during disease progression with a peak occurring between 100 and 120 d of age.
Figure 3.
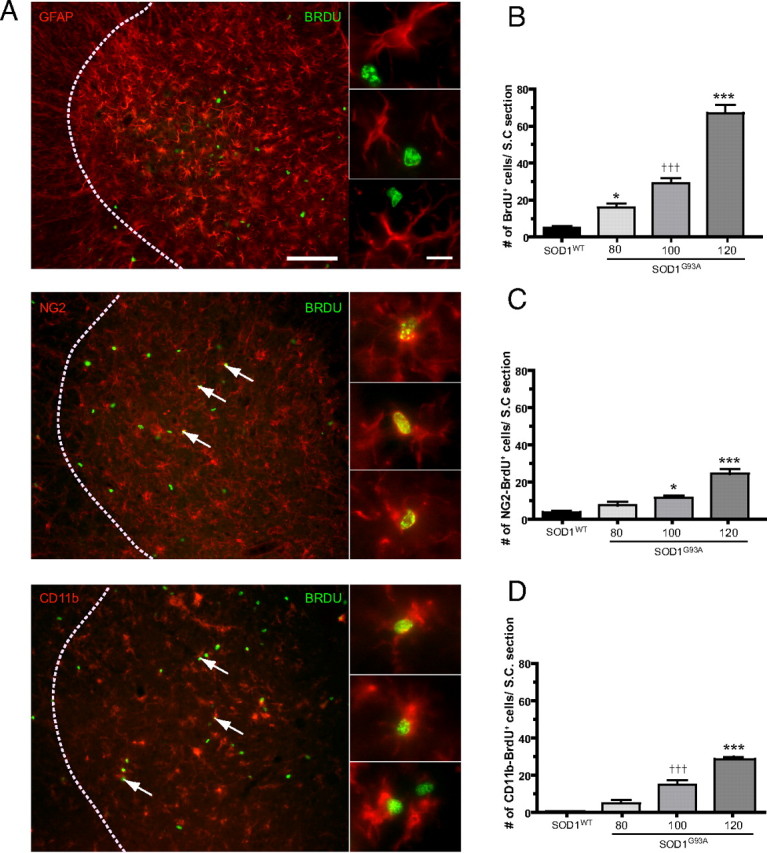
Proliferation in the SOD1G93A lumbar gray matter spinal cord. A, Double immunofluorescence for GFAP, NG2, or CD11b and BrdU in the lumbar spinal cord of SOD1G93A transgenic mice (arrows indicate double-labeled cells; dotted lines indicate the border between white matter and gray matter). Scale bars: 100 μm (insets); 10 μm. B–D, Quantification of cell proliferation at different ages in the SOD1G93A spinal cord compared with SOD1WT spinal cord. B, BrdU+ cells were counted at the ages of 80, 100, and 120 d in the SOD1G93A mouse and at 130 d in the SOD1WT mouse. ***p < 0.0001 compared with SOD1WT 130 d and SOD1G93A at 80 and 100 d; †††p < 0.0001 with SOD1WT 130 d; *p < 0.05 with SOD1G93A at 100 d and SOD1WT at 130 d. C–D, Number of BrdU cells double+ for NG2 (C) and CD11b (D) at the different ages examined. C, ***p < 0.0001 with SOD1WT 130 d and SOD1G93A at 80 d, and p < 0.001 with SOD1G93A 100 d. *p < 0.05 with SOD1WT. D, ***p < 0.0001 with SOD1WT 130 d and SOD1G93A at 80 d, and p < 0001 with SOD1G93A at 100 d. †††p < 0.001 with SOD1WT 130 d and p < 0.05 with SOD1G93A 80 d. All values are mean ± SEM; n = 4–9 for all experiments.
To investigate which cell types contributed to this proliferation, double immunofluorescence for the markers GFAP, NG2, and CD11b with BrdU were performed and quantified (Fig. 3). These markers stain astrocytes, glial precursors, and microglia, respectively. At the age of 80 d, a twofold increase in the number of BrdU-NG2+ doubly labeled cells was observed in SOD1G93A compared with control SOD1WT mice (Fig. 3C). This rose to a 3-fold and a 7.3-fold increase at the ages of 100 and 120 d, respectively, compared with control mice (Fig. 3C). For double-labeled CD11b-BrdU+ cells, a fivefold increase in proliferation of microglia could already be observed at 80 d of age in SOD1G93A compared with SOD1WT controls, but this was not statistically significant (Fig. 3D). The number of CD11b+–BrdU+ cells was further increased by 15- and 29-fold at 100 and 120 d of age, respectively (Fig. 3D). GFAP-labeled cells were not found to proliferate to a significant extent (data not shown) (Fig. 3A). As almost all BrdU+ cells are NG2+ or CD11b+, this suggests that proliferation in the SOD1G93A spinal cord gray matter is responsible for the expansion of both the NG2 and the CD11b population, whereas astrocytes do not proliferate. SOD1G93A and SOD1WT animals treated with BrdU were analyzed using FACS for BrdU incorporation as a measure of proliferation. Both mature microglia and myeloid precursor cells contributed to the CD11b proliferation as both cell types had incorporated BrdU. There was no significant difference in percentage proliferating myeloid precursor cells (SOD1WT, 24.10 ± 5.13; SOD1G93A, 19.93 ± 4.77; p > 0.05) or mature microglia (SOD1WT, 9.15 ± 1.73; SOD1G93A, 16.55 ± 3.81; p > 0.05) between SOD1G93A and SOD1WT.
Ablation of proliferating microglia in the lumbar spinal cord of SOD1G93A transgenic mice
Here, we take advantage of the substantial microglial cell proliferation in the spinal cord of SOD1G93A transgenic mice to directly assess the role of these cells in ALS caused by SOD1 mutations. To accomplish this, CD11b-TKmut-30 transgenic mice were bred with SOD1G93A to generate mice heterozygous for both the TKmut-30 and SOD1G93A transgenes. In this way, administration of the nucleoside analog ganciclovir will cause the ablation of proliferating mature microglia and myeloid precursor cells in the spinal cord of doubly transgenic TKmut-30; SOD1G93A (Tg/+; Tg/+) animals compared with control WT; SOD1G93A (+/+; Tg/+) littermates. As reported previously, chronic and systemic injection of the nucleoside analog ganciclovir is lethal for CD11b-TKmut-30 transgenic mice, and ganciclovir is not highly diffusible within CNS tissue (Gowing et al., 2006a) (G. Gowing and J.-P. Julien, unpublished data). To overcome this constraint, we proceeded with direct delivery of ganciclovir to the spinal cord via an osmotic pump connected to a canula located, in the intrathecal space, at the level of L4–L5 segments from day 85 to day 115 (Fig. 4A,B). Delivery of ganciclovir during this symptomatic stage of the disease resulted in a 34% decrease of the Iba1+ microglial cell population in the lumbar spinal cord of TKmt-30; SOD1G93A transgenic mice compared with WT; SOD1G93A littermates (TKmt-30; SOD1G93A, 29,790 ± 2017 cells/mm3; WT; SOD1G93A, 33,640 ± 2140 cells/mm3; p < 0.001). Elimination of microglia was more pronounced in the L5 segment of the spinal cord with a 40% decrease in Iba1+ cells (Fig. 4C). Mac-2, a marker of activated microglia, has been reported to be preferentially expressed by proliferating and neurotrophic microglia in a mouse model of cerebral ischemia (Lalancette-Hébert et al., 2007). We therefore wanted to investigate whether the number of Mac-2+ microglia were significantly altered in our model. Interestingly, a 51% decrease in Mac-2-positive cells was found in the lumbar spinal cord of TKmt-30; SOD1G93A compared with WT; SOD1G93A (TKmt-30; SOD1G93A, 6572 ± 1410 cells/mm3; WT; SOD1G93A, 12 850 ± 1288 cells/mm3; p < 0.05). A higher degree of ablation of Mac-2-positive cells was also obtained in L4 and L5 segments of the spinal cord compared with distal L3 segment (Fig. 4D).
Figure 4.

Ablation of proliferating microglia in TKmut-30; SOD1G93A transgenic mice. A, Schematic representation of mice receiving ganciclovir or saline intrathecally via an osmotic pump at L4–L5 spinal level. B, Immunohistochemistry for Iba1 and Mac-2 at the L5 level in TKmut-30; SOD1G93A or WT; SOD1G93A transgenic animals receiving ganciclovir. Magnification 20×. Scale bar, 100 μm. C–D, Quantifications of microglial cell ablation stained with Iba1 (C) or Mac-2 (D). *p < 0.05; ***p < 0.001. All values are mean ± SEM; n = 8–12 mice per analysis.
Effect of microglial cell ablation on motor neuron degeneration
To determine whether microglial cell ablation in lumbar spinal cord influenced neurodegeneration in SOD1G93A transgenic mice, motor neurons in the vicinity of ganciclovir delivering canula (L3–L5) were quantified. No significant difference in the counts of Nissl-stained motor neuron cell bodies was observed in any region between TKmut-30; SOD1G93A doubly transgenic and WT; SOD1G93A control mice treated with ganciclovir (TKmut-30; SOD1G93A, 1588 ± 129.3 cells/mm3; WT; SOD1G93A, 1483 ± 94.76 cells/mm3; p > 0.05). Because degeneration begins at the distal axon in ALS and follows a “dying back” pattern (Fischer et al., 2004), we also proceeded to count the L5 ventral root axons and assessed the innervation at the neuromuscular junction of the gastrocnemius muscle. No significant difference was observed in the number of ventral root axons (TKmut-30; SOD1G93A, 502.1 ± 17.6 axons; WT; SOD1G93A, 452.5 ± 19.20 axons; p > 0.05) or in the innervation level of gastrocnemius muscle (TKmut-30; SOD1G93A: innervated, 9.23 ± 3.83%; intermediate innervations, 13.83 ± 2.95%; denervated, 76.87 ± 6.75%; WT; SOD1G93A: innervated, 4 ± 2.27%; intermediate innervations, 11.07 ± 3.08%; denervated, 84.33 ± 5.3%; p > 0.05) between TKmut-30; SOD1G93A doubly transgenic mice and control WT; SOD1G93A mice treated with ganciclovir. Hence, elimination of one-third of the proliferating microglial cell population did not influence motor neuron degeneration. Furthermore, reflex scores and body weight analyses were not significantly different between WT; SOD1G93A and TKmut-30; SOD1G93A transgenic mice treated with ganciclovir (data not shown).
Influence of decreased microglial cell numbers on astrocytes, glial progenitors, and T cells
Activated microglia can secrete numerous factors such as inflammatory cytokines, nitric oxide, and excitotoxins that can induce astrocytosis, influence fate of glial progenitors, and T cell infiltration into the CNS (Giulian et al., 1994a,b; Ridet et al., 1997; Holmøy, 2008). Hence, we investigated whether the changes in the microglial cell population influenced these processes in the spinal cord of TKmut-30; SOD1G93A compared with WT; SOD1G93A controls. Interestingly, quantification by stereology revealed a decrease in the number of GFAP-positive astrocytes in the lumbar spinal cord of TKmut-30; SOD1G93A compared with WT; SOD1G93A controls (TKmut-30; SOD1G93A, 15 770 ± 828.5; WT; SOD1G93A, 20 480 ± 1497; p < 0.05) (Fig. 5). However, no change in the number of glial progenitors (NG2+ cells) was observed between TKmut-30; SOD1G93A and WT; SOD1G93A controls (TKmut-30; SOD1G93A, 21 150 ± 1212; WT; SOD1G93A, 20 480 ± 1497; p > 0.05) (Fig. 5). At this phase of disease, T cells are not evenly distributed in the spinal cord of SOD1G93A transgenic mice. Nevertheless, a tendency for an increased number of CD3+ T cells could be observed in TKmut-30; SOD1G93A compared with WT; SOD1G93A controls but it was not statistically significant (TKmut-30; SOD1G93A, 1685 ± 158.4; WT; SOD1G93A, 945 ± 305.6; p > 0.05) (Fig. 5).
Figure 5.
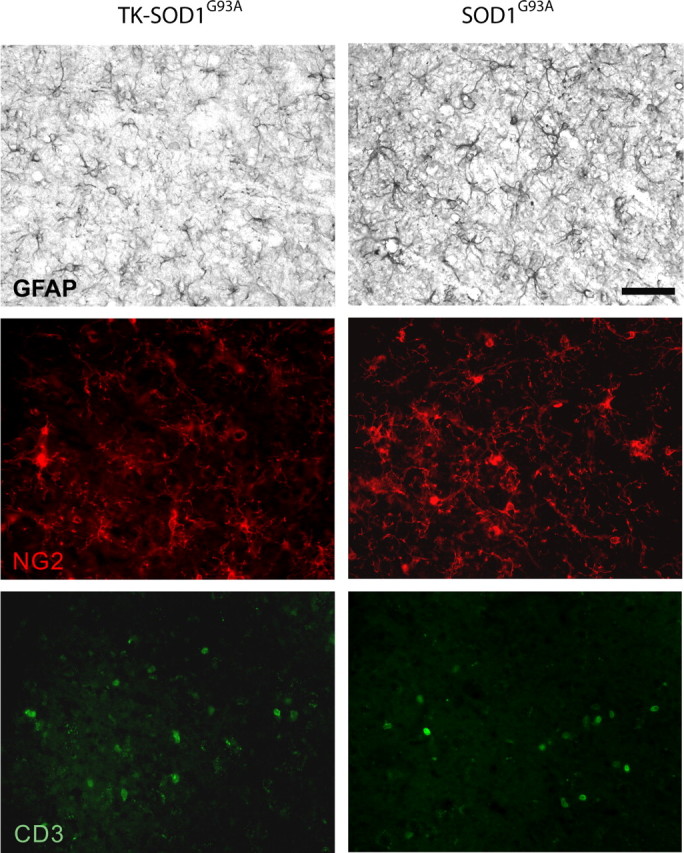
Influence of decreased microgliosis on astrocytes, glial progenitors, and T cell numbers. A, Immunohistochemistry for GFAP, NG2, and CD3 in the lumbar spinal cord of TKmut-30; SOD1G93A and WT; SOD1G93A. Magnification, 40×. Scale bar, 50 μm.
Discussion
Microgliosis can be observed in virtually all CNS pathologies including ALS. However, the role of the microglial response in many disorders of the CNS remains ambiguous as these cells can display both neuroprotective and neurotoxic potential (Wyss-Coray and Mucke, 2002). CD11b-TKmut-30 transgenic mouse model has been used with success to selectively deplete proliferating microglia/macrophages in several paradigms of CNS disorders including cerebral ischemia, gliomas, Alzheimer's disease, CNS viral infection, and axonal regeneration (Carmen et al., 2006; Simard et al., 2006; Galarneau et al., 2007; Lalancette-Hébert et al., 2007; Barette et al., 2008). In all these models, ablation of proliferating microglia was deleterious indicating that microglia are essentially neuroprotective. Here, by using this novel approach, we directly addressed the role of proliferating microglial cells and their influence on motor neuron degeneration in an ALS mouse model. Surprisingly, we found that eliminating 34% of Iba1+ microglia (51% of Mac-2+ microglia) by ganciclovir infusion, in the lumbar spinal cord of CD11b-TKmut-30; SOD1G93A transgenic mice for a period of 30 d after disease onset, had no effect on the progression of motor neuron degeneration. This was an unexpected result as it has been recently proposed that microglia contribute to disease progression in ALS (Boillée et al., 2006). Accordingly, elimination of proliferating microglia, in our model, should have increased motor neuron survival. Conversely, if microglia in mutant SOD1 mice models of ALS were neuroprotective, as recently proposed by Kang et al. (2007), elimination of proliferating microglia should have increased neurodegeneration. However, we observed neither exacerbation of motor neuron degeneration nor neuroprotection after a substantial reduction in the number of activated microglia in the lumbar spinal cord of SOD1G93A transgenic mice. Thus, our results suggest that the population of proliferating microglia in SOD1G93A mice are not key contributors of motor neuron degeneration. Nonetheless, it remains possible that eliminating a larger number of SOD1-positive microglia in our model may have influenced the motor neuron degeneration.
A recent study by Boillée et al. (2006) suggested a detrimental role for microglia in ALS. However, this study did not directly address the role of microglia themselves in the context of disease process but rather revealed the existence of a toxic action of mutant SOD1 in microglia. Moreover, it is likely that by lowering mutant SOD1 expression within microglia, Boillée et al. (2006) rendered these cells neuroprotective as they become phenotypically similar to wild-type microglia. This is supported by a study in which wild-type microglia were found to extend the survival of PU.1 knock-out mice with familial ALS (Beers et al., 2006). Of particular interest to our study, the investigation of the properties of microglia carrying the SOD1G93A mutant gene have found these cells to have reduced neurotrophic and greater neurotoxic potential compared with wild-type microglia (Xiao et al., 2007). Therefore, it would seem that microglia are fundamentally neuroprotective cells but that expression of mutant SOD1 renders them neurotoxic. Because SOD1G93A-expressing microglial cells are less likely to be neuroprotective compared with wild-type microglia, this could explain why partial elimination of these cells did not exacerbate disease.
Although ablation of proliferating microglia did not alter neurodegenerative processes, the reduction in microglial cells numbers significantly decreased the number of GFAP+ astrocytes in the spinal cord of doubly transgenic mice treated with ganciclovir compared with controls. It has been recently proposed that mutant SOD1 expressing astrocytes can induce and amplify the inflammatory response in microglia leading to damage and accelerated disease progression (Yamanaka et al., 2008). Indeed, in the study by Yamanaka et al. (2008), reduced expression of mutant SOD1 in astrocytes significantly decreased the number of Mac-2+ microglia in the spinal cord of SOD1G37R transgenic mice and extended the survival of SOD1G37R transgenic mice. Yet, it is also likely that microglia, as pathological sensors of the CNS, may respond to an initial signal or signals from degenerating/distressed motor neurons or from other cells including astrocytes. Activation of microglia and secretion of various cytokines such as TNF-α and INF-γ could then promote NG2+ glial progenitors differentiation into reactive astrocytes (Magnus et al., 2008). Supporting this view, here, a 51% decrease in activated Mac-2 positive microglia caused a substantial reduction in the number of reactive astrocytes in the spinal cord of TKmut-30; SOD1G93A compared with WT; SOD1G93A controls. However, in contrast to the study by Yamanaka et al. (2008), this reduction in reactive astrocytes and microglia was not sufficient to affect the motor neuron degeneration in SOD1 transgenic mice.
Activated microglia can present diverse phenotypes indicative of their functional diversity (Hanisch and Kettenmann, 2007). Microglia in the healthy or inflamed CNS can be divided into three distinct populations as assessed by the differential expression of the pan-leukocyte marker CD45 (Sedgwick et al., 1991; Cruse et al., 2004). In our study, we found that at late symptomatic stage of the disease there was a significant increase in the contribution of immature myeloid precursor cells to the microglial cell population in SOD1G93A mice. Both morphologically and phenotypically, the myeloid precursor cells displayed an immature undifferentiated phenotype without clear expression of mature cell markers (F4/80−, CD11c−, CD40−, and CD86−). The relevance of this myeloid precursor cell expansion is unsure, but as these cells are abundantly present and therefore most vulnerable to ganciclovir treatment, we suggest these cells do not contribute to disease progression, as inhibiting their proliferation had no effect on neurodegeneration. Moreover, among the population of mature microglia, an increase in the expression of the dendritic cell marker CD11c and the dendritic cell maturation marker CD86 occurs with disease progression. This is in line with previously published reports where an increase in transcripts encoding dendritic cell markers was observed in ALS patients and in late symptomatic or end-stage mice (Henkel et al., 2004, 2005). The acquirement of a dendritic cell phenotype may indicate a role for adaptive immunity in ALS, as these cells are antigen presenting cells required for efficacious activation of naive and memory B and T cell (Banchereau and Steinman, 1998). Interestingly, the presence of microglia-expressing molecules associated with antigen presentation have been associated with neuroprotective effects in various models of CNS injury (Byram et al., 2004; Shaked et al., 2004; Ziv et al., 2007). However, the importance of this microglial cell phenotype in ALS remains to be investigated. Here, we also found that macrophages did not contribute significantly to the myeloid cell population in SOD1G93A transgenic mice spinal cord. The expansion in CD11b+ cells is most likely attributable to proliferation of resident myeloid cells rather than infiltration of the CNS by bone marrow derived cells as suggested by chimeric mouse studies obtained by parabiosis (Ajami et al., 2007). Nevertheless, this expansion in microglial cells does not seem to contribute directly to motor neuron degeneration, as inhibiting this process with administration of ganciclovir in the spinal cord of TKmut-30; SOD1G93A did not influence degeneration of motor neurons.
The interest into the role of microglia in CNS pathology is an old and still active matter of debate. In cases of injury to the CNS, evidence indicates that activation of microglia can have beneficial or detrimental effects (Nguyen et al., 2002; Block and Hong, 2005; Hanisch and Kettenmann, 2007). To date, most studies have investigated the role of microglia in ALS via pharmacological inhibition of microglial activation (Sargsyan et al., 2005; Dewil et al., 2007). It has also been shown that stimulation of innate immunity can exacerbate motor neuron degeneration and reduces lifespan in the SOD1G37R mouse model (Nguyen et al., 2004). Some compounds that attenuate neuroinflammation were found to be neuroprotective in ALS mouse models (Kriz et al., 2002; Van Den Bosch et al., 2002; Zhu et al., 2002; West et al., 2004; Kiaei et al., 2006). However, it must be taken into consideration that many compounds have numerous actions and do not selectively and exclusively target inflammatory processes (Wyss-Coray and Mucke, 2002). Furthermore, to our knowledge, no anti-inflammatory drug has yet proven to be efficacious in ALS clinical trials. The specific targeting of individual and particularly potent proinflammatory targets such as TNF-α and IL-1β by gene knock-out strategies has proven inefficacious in halting disease progression in ALS mice (Nguyen et al., 2001; Gowing et al., 2006b). These and other studies therefore suggest that no single endogenous cytokine can be held responsible for the exacerbation of motor neuron degeneration in ALS but that it is rather the combination of these factors enhanced by the lack of neurotrophic support that can exacerbate motor neuron disease (Kim et al., 2006).
An important pathological hallmark of ALS is the marked expansion of the microglial cell population. To date, the functional role of microgliosis in mutant SOD1-mediated motor neuron degeneration has been a subject of debate. Recent publications have promoted microglia as active contributors in motor neuron degeneration, whereas others maintain an essentially protective role for these cells in ALS (Boillée et al., 2006; Kang and Rivest, 2007). However, the results presented here suggest that proliferating microglia-expressing mutant SOD1 are not key contributors of the neurodegenerative process in ALS caused by SOD1 mutations and therefore might not constitute an appropriate therapeutic target.
Footnotes
This work was supported by the Robert Packard Center for ALS Research at John Hopkins University, the Canadian Institutes of Health Research (CIHR), the Stem Cell Institute Leuven, grants from Katholieke Universiteit Leuven, and the Funds for Scientific Research Flanders (FWO-F). J.-P.J. holds a Canada Research Chair in Neurodegeneration. W.R. is supported through the E von Behring Chair for Neuromuscular and Neurodegenerative Disorders and by the Interuniversity Attraction Poles program P6/43 of the Belgian Federal Science Policy Office. G.G. is the recipient of a CIHR Doctoral Research Award. T.P. is supported by the Institute for the Promotion of Innovation through Science and Technology in Flanders. B.V.W. is a doctoral and A.D.B. a postdoctoral fellow of the FWO-F. We thank Steve Lacroix, Nadia Fortin, Renée Paradis, Genevieve Soucy, Roxanne Larivière, Melanie Simard, Marie-Claude Richer, Ben Sprangers, and Lien De Somer for their assistance. The SV2 antibody developed by Dr. Buckley was provided by the Developmental Studies Hybridoma Bank under the National Institute of Child Health and Human Development and maintained by the University of Iowa, Iowa City, Iowa. The HSV-1 TKmut 30 vector was provided by Dr. M. E. Black (Washington State University, Pullman, Washington,).
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barette B, Hébert MA, Filali M, Lafortune K, Vallières N, Gowing G, Julien JP, Lacroix S. Requirement of myeloid cells for axon regeneration. J Neurosci. 2008;28:9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU. 1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau AD, Fevery S, Rutgeerts O, Landuyt W, Waer M. Transient expansion of Mac1+Ly6-G+Ly6-C+ early myeloid cells with suppressor activity in spleens of murine radiation marrow chimeras: possible implications for the graft-versus-host and graft-versus-leukemia reactivity of donor lymphocyte infusions. Blood. 2003;102:740–748. doi: 10.1182/blood-2002-06-1833. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen J, Gowing G, Julien JP, Kerr D. Altered immune response to CNS viral infection in mice with a conditional knock-down of macrophage-lineage cells. Glia. 2006;54:71–80. doi: 10.1002/glia.20359. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Cruse J, Lewis R, Wang H. Immunology guide book. Amsterdam: Elsevier; 2004. [Google Scholar]
- Cuadros MA, Navascués J. The origin and differentiation of microglial cells during development. Prog Neurobiol. 1998;56:173–189. doi: 10.1016/s0301-0082(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Dewil M, Van Den Bosch L, Robberecht W. Microglia in amyotrophic lateral sclerosis. Acta Neurol Belg. 2007;107:63–70. [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Galarneau H, Villeneuve J, Gowing G, Julien JP, Vallières L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007;67:8874–8881. doi: 10.1158/0008-5472.CAN-07-0177. [DOI] [PubMed] [Google Scholar]
- Giulian D, Li J, Leara B, Keenen C. Phagocytic microglia release cytokines and cytotoxins that regulate the survival of astrocytes and neurons in culture. Neurochem Int. 1994a;25:227–233. doi: 10.1016/0197-0186(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Giulian D, Li J, Li X, George J, Rutecki PA. The impact of microglia-derived cytokines upon gliosis in the CNS. Dev Neurosci. 1994b;16:128–136. doi: 10.1159/000112099. [DOI] [PubMed] [Google Scholar]
- Gowing G, Vallières L, Julien JP. Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia. 2006a;53:331–337. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006b;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Siklos L, Appel SH. The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol Cell Neurosci. 2005;31:427–437. doi: 10.1016/j.mcn.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Holmøy T. T cells in amyotrophic lateral sclerosis. Eur J Neurol. 2008;15:360–366. doi: 10.1111/j.1468-1331.2008.02065.x. Holmøy T. [DOI] [PubMed] [Google Scholar]
- Julien JP. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Julien JP, Millecamps S, Kriz J. Cytoskeletal defects in amyotrophic lateral sclerosis (motor neuron disease) Novartis Found Symp. 2005;264:183–192. discussion 192- 196, 227–230. [PubMed] [Google Scholar]
- Kang J, Rivest S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J Cell Biol. 2007;179:1219–1230. doi: 10.1083/jcb.200705046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Martinez T, Deshpande DM, Drummond J, Provost-Javier K, Williams A, McGurk J, Maragakis N, Song H, Ming GL, Kerr DA. Correction of humoral derangements from mutant superoxide dismutase 1 spinal cord. Ann Neurol. 2006;60:716–728. doi: 10.1002/ana.21034. [DOI] [PubMed] [Google Scholar]
- Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Robberecht W, Carmeliet P. Heterogeneity in motoneuron disease. Trends Neurosci. 2007;30:536–544. doi: 10.1016/j.tins.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Ling EA, Paterson JA, Privat A, Mori S, Leblond CP. Investigation of glial cells in semithin sections. I. Identification of glial cells in the brain of young rats. J Comp Neurol. 1973;149:43–71. doi: 10.1002/cne.901490104. [DOI] [PubMed] [Google Scholar]
- Magnus T, Carmen J, Deleon J, Xue H, Pardo AC, Lepore AC, Mattson MP, Rao MS, Maragakis NJ. Adult glial precursor proliferation in mutant SOD1G93A mice. Glia. 2008;56:200–208. doi: 10.1002/glia.20604. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, D'Aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2004;24:1340–1349. doi: 10.1523/JNEUROSCI.4786-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sargsyan SA, Monk PN, Shaw PJ. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. 2005;51:241–253. doi: 10.1002/glia.20210. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I, Porat Z, Gersner R, Kipnis J, Schwartz M. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J Neuroimmunol. 2004;146:84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. 2008;86:1944–1958. doi: 10.1002/jnr.21659. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Takeyama Y, Nishikawa J, Ueda T, Hori Y, Yamamoto M, Kuroda Y. Involvement of peritoneal macrophage in the induction of cytotoxicity due to apoptosis in ascitic fluid associated with severe acute pancreatitis. J Surg Res. 1999;82:163–171. doi: 10.1006/jsre.1998.5535. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007;104:14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–1070. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1068–1082. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- West M, Mhatre M, Ceballos A, Floyd RA, Grammas P, Gabbita SP, Hamdheydari L, Mai T, Mou S, Pye QN, Stewart C, West S, Williamson KS, Zemlan F, Hensley K. The arachidonic acid 5-lipoxygenase inhibitor nordihydroguaiaretic acid inhibits tumor necrosis factor alpha activation of microglia and extends survival of G93A-SOD1 transgenic mice. J Neurochem. 2004;91:133–143. doi: 10.1111/j.1471-4159.2004.02700.x. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Zhao W, Beers DR, Yen AA, Xie W, Henkel JS, Appel SH. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007;102:2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Finkelstein A, Geffen Y, Kipnis J, Smirnov I, Shpilman S, Vertkin I, Kimron M, Lange A, Hecht T, Reyman KG, Marder JB, Schwartz M, Yoles E. A novel immune-based therapy for stroke induces neuroprotection and supports neurogenesis. Stroke. 2007;38:774–782. doi: 10.1161/01.STR.0000255784.27298.23. [DOI] [PubMed] [Google Scholar]