

Abstract
Aged canines (dogs) accumulate human-type β-amyloid (Aβ) in diffuse plaques in the brain with parallel declines in cognitive function. We hypothesized that reducing Aβ in a therapeutic treatment study of aged dogs with preexisting Aβ pathology and cognitive deficits would lead to cognitive improvements. To test this hypothesis, we immunized aged beagles (8.4–12.4 years) with fibrillar Aβ1–42 formulated with aluminum salt (Alum) for 2.4 years (25 vaccinations). Cognitive testing during this time revealed no improvement in measures of learning, spatial attention, or spatial memory. After extended treatment (22 vaccinations), we observed maintenance of prefrontal-dependent reversal learning ability. In the brain, levels of soluble and insoluble Aβ1–40 and Aβ1–42 and the extent of diffuse plaque accumulation was significantly decreased in several cortical regions, with preferential reductions in the prefrontal cortex, which is associated with a maintenance of cognition. However, the amount of soluble oligomers remained unchanged. The extent of prefrontal Aβ was correlated with frontal function and serum anti-Aβ antibody titers. Thus, reducing total Aβ may be of limited therapeutic benefit to recovery of cognitive decline in a higher mammalian model of human brain aging and disease. Immunizing animals before extensive Aβ deposition and cognitive decline to prevent oligomeric or fibrillar Aβ formation may have a greater impact on cognition and also more directly evaluate the role of Aβ on cognition in canines. Alternatively, clearing preexisting Aβ from the brain in a treatment study may be more efficacious for cognition if combined with a second intervention that restores neuron health.
Keywords: beagle, executive function, memory, oligomers, reversal learning, spatial attention, visual discrimination learning
Introduction
Alzheimer's disease (AD) is associated with progressive cognitive decline and the accumulation of senile plaques and neurofibrillary tangles (Mirra et al., 1991). Senile plaques contain β-amyloid (Aβ), which is a peptide thought to play a causative role in the disease (Hardy and Higgins, 1992; Selkoe, 1996). Thus, a number of therapeutics are currently being developed that may reduce the production and deposition or enhance clearance of Aβ in the brains of patients with AD (Selkoe and Schenk, 2003). In transgenic mouse models of AD, deposition of Aβ may be prevented or significantly reduced after immunization with fibrillar Aβ1–42 (Schenk et al., 1999; Janus et al., 2000; Morgan et al., 2000; Das et al., 2003). Furthermore, learning and memory is improved by either active (Janus et al., 2000; Morgan et al., 2000; Sigurdsson et al., 2004) or passive (Dodart et al., 2002; Kotilinek et al., 2002; Morley et al., 2002; Wilcock et al., 2004b) immunization. These results strongly suggest a link between Aβ and behavioral dysfunction in transgenic mice.
We extended the anti-Aβ immunotherapeutic approach to a canine (beagle) model of human brain aging and β-amyloidogenesis that naturally produces human-type Aβ (Selkoe et al., 1987; Johnstone et al., 1991). Aged canines show a decline in memory and learning (Milgram et al., 1994, 2002b; Tapp et al., 2003a,b) and a corresponding increase in Aβ pathology (Cummings et al., 1996a,b; Head et al., 1998, 2000). Furthermore, the extent of diffuse Aβ plaques in canine brain is correlated with the severity of cognitive decline (Cummings et al., 1996b; Head et al., 1998). The recent publication of the canine genome suggests that the sequence of the β-amyloid precursor protein is ∼98% similar to human (http://www.ensembl.org/Canis_familiaris/), suggesting similar processing events leading to Aβ production. We hypothesized that Aβ immunization would reduce Aβ in the brains of aged canines and would be associated with a corresponding improvement in cognitive function.
Using previously established immunization procedures for canines (Head et al., 2006), we began a longitudinal study in 20 aged beagles (8.4–12.4 years) using a series of sophisticated cognitive tasks that can be administered for extended periods of time to examine effects on maintenance of cognitive function. To avoid possible adverse immune T helper 1 (Th1) responses, we used fibrillar Aβ1–42 formulated with aluminum salt (Alum), which is a Th2 type adjuvant that is approved for use in humans (Cribbs et al., 2003; Lindblad, 2004). We selected cognitive tasks that have been established previously to be sensitive to age, extent of Aβ, and treatment in dogs (Head et al., 1995, 1998; Cotman et al., 2002; Milgram et al., 2002b, 2005). Animals were immunized for over 2 years, and cognition was monitored until the end of the study when brain Aβ was measured.
Materials and Methods
Animals
As shown in Table 1, the longitudinal study included 20 beagles with 17 animals from Harlan (Riglan Farms, Mount Horeb, WI) and three from the Lovelace Respiratory Research Institute (LRRI) (Albuquerque, NM). We found from experience with several sources of dogs that the contribution of two different sources of animals to treatment outcomes is minimal (Milgram et al., 2005). All of the Harlan beagle dogs and the single female LRRI dog were reproductively intact. At the start of baseline cognitive testing, the ages of the animals ranged from 8.4 to 12.4 years (mean ± SD, 9.35 ± 0.96 years). Because of availability, there was a range in age of animals at the start of the study. However, all beagles should have had significant prefrontal Aβ pathology (Head et al., 2000), and, based on our previous work, we predicted that animals would have significant pathology at the end of the study. Furthermore, age at the start of the study was counterbalanced between treatment groups. Dogs were housed singly in kennel buildings with indoor/outdoor runs measuring 91 × 600 cm and were fed Wayne Mini Lab Dog Diet 8759 dog food once daily (Teklad Pioneer Lab Diets, Madison, WI). Water was available at all times. All animals were thoroughly examined before inclusion in the study and were determined to be in good health. Examinations included physical examination, neurological examination, and analysis of blood biochemistry values. All procedures were conducted in accordance with LRRI-approved animal protocols and the National Institutes of Health Policy on Humane Care and Use of Laboratory Animals.
Table 1.
Animals used in the study
| Dog number | Animal | Sex | Age at start of study (years) | Group | Dose (mg) | Age at end of the study (years) | Total time on treatment (years) | Number of injections | Antibody titer at death |
|---|---|---|---|---|---|---|---|---|---|
| 1609Aa | 1 | Male | 8.4 | Control | 0 | 11.6 | 2.4 | 25 | 1:100 |
| D296 | 2 | Female | 8.5 | Control | 0 | 11.8 | 2.4 | 25 | 1:100 |
| D317 | 3 | Female | 8.7 | Control | 0 | 12 | 2.4 | 25 | 1:100 |
| 1603Ba | 4 | Male | 8.8 | Control | 0 | 11.2 | 1.5 | 25 | 1:100 |
| D299 | 5 | Female | 8.8 | Control | 0 | 12.1 | 2.4 | 16 | 1:100 |
| D292 | 6 | Female | 8.9 | Control | 0 | 12.2 | 2.4 | 25 | 1:100 |
| D290 | 7 | Female | 8.9 | Control | 0 | 12.1 | 2.4 | 25 | 1:100 |
| D308 | 8 | Female | 9.8 | Control | 0 | 13.1 | 2.4 | 25 | 1:100 |
| D305 | 9 | Female | 10.1 | Control | 0 | 13.4 | 2.4 | 25 | 1:100 |
| D311 | 10 | Female | 10.2 | Control | 0 | 11.5 | 0.3 | 4 | 1:100 |
| D315 | 11 | Female | 10.7 | Control | 0 | 14 | 2.4 | 25 | 1:100 |
| Means | 9.3 | 12.3 | 2.1 | ||||||
| 1606Ta | 12 | Female | 8.7 | Fibrillar | 0.5 | 11.8 | 2.4 | 25 | 1:100,000 |
| D293 | 13 | Female | 8.7 | Fibrillar | 0.5 | 12 | 2.4 | 25 | 1:25,000 |
| D316 | 14 | Female | 8.7 | Fibrillar | 0.5 | 12 | 2.4 | 25 | 1:6400 |
| D310 | 15 | Female | 8.8 | Fibrillar | 0.5 | 12 | 2.4 | 25 | 1:400,000 |
| D313 | 16 | Female | 9.2 | Fibrillar | 0.5 | 12.5 | 2.4 | 25 | 1:6400 |
| D301 | 17 | Female | 9.3 | Fibrillar | 0.5 | 12.6 | 2.4 | 25 | 1:25,000 |
| D302 | 18 | Female | 9.7 | Fibrillar | 0.5 | 13 | 2.3 | 25 | 1:25,000 |
| D295 | 19 | Female | 9.8 | Fibrillar | 0.5 | 13 | 2.4 | 25 | 1:6400 |
| D291 | 20 | Female | 12.4 | Fibrillar | 0.5 | 15.7 | 2.4 | 25 | 1:100,000 |
| Means | 9.5 | 12.7 | 2.4 |
aAnimals from Lovelace Respiratory Research Institute; all others are from are Harlan (Riglan Farms).
For each cognitive task and Aβ outcome measure, we estimated that, overall, a minimum sample size of eight to nine animals per group would provide 80% power to detect a 25% improvement in cognition or decrease in pathology. To arrive at these sample sizes, we estimated effect sizes based on reports in transgenic mice (Janus et al., 2000; Morgan et al., 2000). In many of the treatment studies (i.e., immunotherapy in aged animals), behavioral improvements ranged from 23 to 80%, and sample sizes ranged from 6 to 12 animals per group. Second, we used our own previous work in canines to estimate individual variability in each of our cognitive tasks, and we also estimated sample size based on treatment effects established in aged canines treated with either or both an antioxidant enriched diet or behavioral enrichment (Cotman et al., 2002; Milgram et al., 2002b, 2005). In the canine studies, we detected significant improvements in cognition based on sample sizes ranging from 6 to 12 animals. As one example, the oddity discrimination learning task used in the current study, which was sensitive to an antioxidant diet in aged dogs (Cotman et al., 2002), has an 80% power to detect a 20% reduction in error scores with a sample size of six animals per group.
Cognitive testing methods
Procedure
All dogs were given a series of baseline tests and ranked on the basis of error scores. Subsequently, two groups were formed matching for total error scores such that each group contained both good and poor performers. After this time, the immunization protocol was started, and cognitive testing was conducted for a 2 year period of time while treatment was ongoing. Table 2 shows the study design for the cognitive testing phases of the study.
Table 2.
Cognitive tasks used during the study
| Cognitive task | Time on treatment (months) | Number of injections |
|---|---|---|
| Baseline | 0.0 | 0 |
| Landmark discrimination, acquisition | 0.9 | 2 |
| Landmark discrimination, variable distance | 4.3 | 5 |
| Spatial two-choice task, maximal memory | 5.5 | 6 |
| Spatial two-choice task, working memory | 8.3 | 8 |
| Spatial three-choice task, maximal memory | 9.6 | 9 |
| Spatial three-choice task, working memory | 12.2 | 11 |
| Oddity discrimination | 13.3 | 12 |
| Spatial two-choice task, maximal memory | 15.4 | 14 |
| Spatial two-choice task, working memory | 17.9 | 17 |
| Spatial three-choice task, maximal memory | 19.4 | 18 |
| Spatial three-choice task, working memory | 21.9 | 20 |
| Size discrimination and reversal | 23.4 | 22 |
| Landmark retention | 26.4 | 24 |
| Death | 28.3 | 25 |
Testing apparatus
As described previously (Milgram et al., 1994), the test apparatus was a 0.609 × 1.15 × 1.08 m wooden box constructed from press board coated with melamine. The box was equipped with a sliding black Plexiglas tray containing three food wells. Vertical stainless steel bars, which could be adjusted to provide openings appropriate for individual dog sizes, served as the front of the box. The experimenter sat behind a barrier and observed the dog through a one-way mirror. The bottom of the barrier was hinged to allow a sliding tray to be pushed either toward or out-of-view of the dog. A 60 W light was placed above the presentation tray to light the objects. Data acquisition was controlled using a customized program developed in the ASYST (ASYST Software Technologies, Rochester, NY) programming language. This program controlled all randomization procedures and timing, indicated the location of the reward, and stored all of the data in data files. Each trial was started when the experimenter pressed a key and simultaneously presented the tray to the dog. The dogs' responses were recorded by pressing either the left or right mouse button, which also indicated the end of the trial and began the intertrial interval. One teaspoon (∼4 ml) of wet dog food was formed into a ball and served as the food reward. Each dog was given either 10 or 12 trials a day (depending on the task), with trials separated by a 30 s intertrial interval. Dogs were tested 5 d a week.
Baseline cognitive testing
All animals in the study were given a battery of cognitive tests to establish baseline level of cognitive function. Dogs were given a reward and object approach learning task and then a simple object discrimination and reversal learning task. After discrimination learning, dogs were given a spatial non-matching-to-position memory task.
Reward and object approach learning.
All dogs received a standard four-phase pretraining protocol (Milgram et al., 1994). This procedure included a phase to expose the dogs to the testing apparatus, a phase to teach dogs that a reward was always present in one of the food wells (reward approach learning), a phase that manually shaped dogs to manipulate the objects, and a phase to teach dogs to visually locate the object and approach it (object approach learning). Dogs completed all of these four phases of pretraining before testing for object discrimination learning.
Object discrimination learning.
The first test session was used to establish object preferences. On each trial, the hinged door of the barrier separating the tester and the dog was raised and the presentation tray was pushed forward. The left and right food wells were covered by the two objects. The food reward was placed inside both the left and right food wells beneath each stimulus. Dogs were required to displace an object to obtain the hidden reward. Ten trials were given with two objects presented simultaneously (yellow plastic coffee jar lid and blue plastic Lego block) and with both objects baited. The objects appeared randomly five times each on the left or right side. The preferred object was whichever object was chosen more frequently (i.e., six or more times). Subsequently, the preferred object was used as the positive stimulus. After establishing preferences, each subject was given 10 daily trials with the food reward located beneath the designated positive object. To prevent the dogs from using olfactory cues, the food odors was smeared over both wells, and a piece of food was pressed inside the negative stimulus such that the dog could smell it but not see it or eat it. A correct response was recorded when dogs approached and displaced the positive stimulus. An error was committed if dogs chose the negative object. One correction per test session was allowed, and subsequent errors resulted in an immediate withdrawal of the tray leaving the dog unrewarded. Dogs were trained until one of two criterion levels was met: 9 of 10 correct on 1 d or 8 of 10 correct on 2 consecutive days. An additional 3 d of testing was provided to ensure that animals maintained an average of 70% or better correct. A maximum of 400 trials were given if a dog could not reach criterion.
Reversal learning.
After dogs had reached criterion in the object discrimination problem, the reward contingencies of the positive and negative stimuli were reversed. Testing was continued on this task until the same criterion as for the object discrimination test was met. All other testing procedures were identical to those used with object discrimination learning.
Spatial two-choice non-matching to position.
To assess spatial acquisition and memory, we used a two choice non-matching-to-position task. Animals were first shown a single red Lego block covering either the left or right food wells. Animals displaced the object on one side (e.g., L) to obtain the reward. A 5 s delay interval followed, after which, animals were shown two identical red Lego blocks with the reward hidden under the object on the side not rewarded previously (e.g., R). Dogs were given 10 trials per day with a 30 s intertrial interval and were tested for 40 d. During this 40 d period, when criterion was met at a 5 s delay, the delay was progressively increased to 10, 20, 30, 50, 70, and then 90 s. Two scores were derived from this learning phase of the task: maximal memory and total number of errors to reach criterion at the 5 s delay. Subsequently, animals were given 20 d of a variable delay problem. In this task, used to assess working memory, animals were exposed to delays of 20, 70, or 110 s on each day of testing. The order of the delay interval presentation was random, but each delay appeared four times per day, and 12 trials were given each day. Accuracy scores for each delay interval were calculated as a measure of spatial working memory and used for repeated measures comparisons after treatment was started.
Treatment cognitive testing
At predetermined time points during the study, animals were given tests to measure spatial attention (landmark discrimination learning), spatial memory (two-choice and three-choice), oddity learning, discrimination learning, and reversal learning (oddity learning) (Table 2).
Landmark discrimination learning.
Spatial attention was measured using a landmark discrimination task (Milgram et al., 1999, 2002b). Landmark discrimination learning was initiated after animals had been immunized twice (initial, 2 weeks) and 30 d into the treatment phase of the study. The first phase, landmark 0, involves showing animals two identical objects (wooden blocks) with a third object, the landmark, placed on top of the object that is associated with a food reward. The correct response is to select the object associated with the landmark. Animals are trained until criterion is met, by either obtaining a score of 9 of 10 on 1 d or 8 of 10 on 2 consecutive days of testing. A maximum of 40 d or 400 trials is allowed on landmark 0. In the next phase of testing, the landmark was moved at successively greater distances (1, 4, or 10 cm) away from the reward object with animals required to meet criterion before progressing to testing with a larger landmark distance. Animals that could not solve the problem at any of the longer distances within 400 trials stopped testing on this stepwise protocol. Once all animals had been tested out to a 10 cm distance on the landmark discrimination task or had reached the maximum number of trials for a single distance, all animals were given a variable landmark distance test. In this test, animals were given 12 trials per day with the landmark being placed 1, 4, or 10 cm away from the correct object. These three distances appeared for four trials per day with 12 trials per d in total, and dogs were given a total of 20 d of testing (i.e., 240 trials).
Two-choice spatial memory retesting.
Two-choice spatial memory retesting occurred at two times points: (1) after 5.5 months and on treatment and having received six immunizations, and (2) after 15.4 months of treatment and 14 immunizations. Dogs were retested for spatial memory ability using a short 5 s delay interval. As during the baseline testing procedure, a maximal memory score was derived from a total of 500 trials. Last, animals were tested on the variable delay spatial memory task to measure working memory, as described previously.
Three-choice spatial learning.
At two time points during the study, a more challenging three-choice spatial memory task was introduced. The three-choice delayed non-matching-to-position task has been described previously (Chan et al., 2002). Dogs are first shown a single red Lego block covering the left, right, or center food wells. Once the object is displaced and the reward obtained, a 5 s delay interval followed. After the delay, animals were shown two identical red Lego blocks, one covering the well seen previously and the other covering one of the two remaining food wells. The correct response was to select the object covering the novel food well. Dogs were given 50 d of testing with 12 trials per day with the location of the reward appearing four times each in the three food wells. During the 50 d of testing, if an animal reached criterion at the 5 s delay, the delay was increased to 10 s. This was continued over the 50 d of testing with gradually increasing delay intervals. Two scores were calculated from this test: total number of errors made to reach criterion (or after 50 d of testing if criterion was not met) at the 5 s delay, and maximal memory score, which represented the maximum delay interval animals could reach criterion at the 50 d of testing. After incremental delay interval increases over a 50 d period, dogs were given a variable delay procedure for 20 d. As with the two-choice spatial memory task, dogs would be exposed to a 20, 70, or 110 s delay in a single day of testing. Accuracy scores for each delay interval were calculated as a measure of spatial working memory. Animals were tested at two time points: the first was after 9.6 months of treatment and nine immunizations and the second after 19.4 months of treatment and 18 immunizations.
Oddity discrimination.
After 13.3 months of treatment and 12 immunizations, dogs were tested for oddity discrimination learning (Milgram et al., 2002a). This task involves four sets of three objects. Two objects of each set are identical and one is novel. Animals are shown the first set and all three objects simultaneously, with a food reward hidden under the novel object. Dogs are given 12 trials per day with the location of the food reward appearing once in each of the three food wells each day. Once dogs learn the first problem, the next set of objects is used. The dogs must reach criterion levels of responding from each object set before moving to the more difficult object sets. Animals that fail to learn one of the oddity problems within 40 d of testing are stopped. The similarity between the odd object and the two identical object increases with each problem set such that the last problem, oddity 4, is the most difficult to discriminate. Not all animals were able to learn all four phases of the oddity problem. The total number of errors made to reach criterion or in the 40 d of testing for each oddity problem were used to test for treatment effects.
Size discrimination and reversal learning.
After animals were treated for a period of 23.4 months and had received 22 immunizations, they were given a size discrimination and reversal problem. The procedures are identical to those used during baseline testing except the objects only differ on the basis of size (Milgram et al., 2005). Dogs were given a total of 40 d to learn the size discrimination problem. If criterion was met during this time, they were immediately placed into size reversal learning, which was identical to the discrimination task but the reward contingencies were reversed. Animals were given an additional 40 d to learn reversal. The total number of errors made to reach criterion or within 40 d of testing were used as dependent measures for both tasks. An error analysis was used for reversal learning as follows: errors were classified as stage 1 (perseverative responding: scores of 4 of 12 or less each day), stage II (chance levels of responding: 5 to 7 of 12 each day), or stage III (relearning: 8 of 12 or higher each day) (Jones and Mishkin, 1972).
Landmark discrimination retention.
The last task before the end of the study was initiated after 26.4 months of treatment and 24 immunizations. Animals were retested for landmark discrimination learning described previously. Dogs were given a total of 20 d to relearn this task. The total number of errors made to reach criterion, or the total number of errors committed during the 20 d of testing, were used as dependent measures.
Treatment groups
The total errors made for reward and object approach learning, for discrimination and reversal learning, and for acquisition of the two-choice spatial memory task at baseline were summed, and dogs were ranked according to these total error scores. Animals were placed into one of three treatment groups balancing for cognitive test scores, source, and age. These groups included (1) immunization with fibrillar Aβ1–42 and Alum (n = 9), (2) immunization with Alum only (n = 6), or (3) saline only (n = 5). The two control immunization groups did not differ significantly on measures of cognitive function throughout the study and were combined in a single control group for data analyses.
Immunization procedure
Fibrillar Aβ (provided by C. Glabe, University of California, Irvine, Irvine, CA) was prepared by adding 500 μl of PBS, pH 7.5, to 0.5 mg of peptide, and the sample was vortexed and incubated overnight at 37°C in a water bath before formulation with the adjuvant. To prepare Aβ for immunization, 0.5 mg of fibrillar Aβ (500 μl) was added to 50 μl of 2% aluminum hydroxide suspension (Accurate Chemical, Westbury, NY) and 450 μl of PBS and vortexed. Control animals either received Alum only (n = 6) or saline only (n = 5). Animals were immunized subcutaneously in the back of the neck and monitored for adverse reactions. After 2 weeks, animals were boosted with an additional injection. After the first two injections, animals received a single injection each month.
Tissue collection
At the end of the study, animals were anesthetized with sodium pentobarbital (Nembutal). Blood was taken in 10 cc red top tubes and centrifuged, and the supernatant (serum) used to assay anti-Aβ antibodies. When animals were in deep surgical stage, the brains were rapidly removed. Procedures were preformed in accordance with LRRI Institutional Animal Care and Use Committee protocols. The left hemisphere was placed in 4% paraformaldehyde at 4°C for 72–80 h before transfer to PBS, pH 7.4, with 0.02% sodium azide and stored at 4°C. The right hemisphere was coronally sectioned and flash frozen to −70°C.
Aβ immunohistochemistry and quantification
For histology experiments, the left hemisphere was embedded in a gelatin matrix, and serial coronal sections were cut at 40 μm and placed in antigen preserve solution (PBS, ethylene glycol, and polyvinylpyrrolidone) for long-term storage at −20°C (Neuroscience Associates, Knoxville, TN). We selected free-floating sections containing the dorsolateral prefrontal, entorhinal, posterior parietal, and occipital cortices, which have been described previously as establishing the pattern of Aβ deposition with age in canines (Head et al., 2000). These four regions of interest are also components of cortical circuits responsible for intact or impaired function on cognitive tasks used in the study. Aβ was detected with anti-Aβ1–17 (mouse monoclonal 6E10 antibody, 1:5000; Signet Laboratories, Dedham, MA). Briefly, the protocol consisted of pretreatment with 90% formic acid (Kitamoto et al., 1987) before overnight incubation with primary antibody, followed by anti-mouse secondary antibodies, detection with an ABC peroxidase kit, and visualization with a DAB substrate kit (both from Vector Laboratories, Burlingame, CA). Control experiments in which primary or secondary antibody was omitted resulted in negative staining. The procedure for quantifying Aβ loads has been reported previously (Head et al., 2000). Briefly, 10 images (525 × 410 μm each) were captured at a 20× objective using a high-resolution video camera and NIH Image 1.59b5 in each brain region for each animal. Sampling consisted of five images from the superficial and five from the deep cortical layers (totaling 40 images per animal). The cross-sectional area occupied by Aβ in each individual image was quantified using grayscale thresholding, which separated positive staining from background and calculated the percentage of area occupied by Aβ immunoreactivity or “Aβ load.” This entire experiment was replicated with a second set of coronal sections (at least 200 μm away from the first set) to confirm the results.
Aβ ELISA
Aβ from frozen samples of the prefrontal, entorhinal, parietal, and occipital cortex from the right hemisphere was sequentially extracted first in radioimmunoprecipitation assay (RIPA) buffer [pH 8, 50 mm Tris-HCl, 150 mm NaCl, 0.5% deoxycholate, 0.1% SDS, 1% Triton X-100, and protease inhibitor cocktail (MP Biomedicals, Costa Mesa, CA) at 1 ml buffer/150 mg wet weight tissue]. After centrifugation (1 h at 100,000 × g at 4°C), the RIPA supernatant was used to measure soluble Aβ. All pellets were resuspended in 500 μl of 70% formic acid (FA) at half the original RIPA volume and centrifuged to obtain an FA supernatant to measure insoluble Aβ. Before assay, FA fractions were thawed and then neutralized with 1.0 m Tris-base and 0.5 m NaH2P04. Brain samples were run in triplicate on ELISA plates coated with a monoclonal anti-Aβ1–16 antibody (20.1; kindly provided by Dr. William Van Nostrand, Stony Brook University, Stony Brook, NY) and detected with monoclonal HRP-conjugated anti-AβX-40 (MM32-13.1.1) and anti-AβX-42 (MM40-21.3.1) antibodies (kindly provided by Dr. Christopher Eckman, Mayo Clinic Jacksonville, Jacksonville, CA) (Das et al., 2003; Kukar et al., 2005; McGowan et al., 2005). For standards, Aβ1–40 and Aβ1–42 (Bachem California, Torrance, CA) were used after a pretreatment with hexafluoroisopropanol to prevent fibril formation. The FA fraction ELISAs from the frontal and parietal cortex were repeated with fresh tissue samples to confirm results.
Dot blot assay
Prefrontal and parietal frozen cortical tissue adjacent to samples taken for ELISA measures were cut and pulverized on dry ice using a tissue pulverizer (Research Products International, Mt. Prospect, IL). Samples were homogenized on ice at 16 μl/mg pulverized tissue weight in PBS packet (Sigma, St. Louis, MO), pH 7.4, 0.2% NaN3 with Complete Mini protease inhibitor (Roche Diagnostics, Indianapolis, IN) using 15 brief pulses with a hand-held sonicator at medium power (Fisher Scientific, Tustin, CA). After centrifugation at 100,000 × g for 1 h at 4°C, the PBS soluble fraction was recovered, and protein concentration was assayed by BCA (Pierce, Rockford, IL). All samples were brought to 1 μg/μl with PBS buffer, and 1 μl dots were blotted in triplicate onto 0.2 μm pore nitrocellulose Protran membranes (Whatman Schleicher and Schuell, Florham, NJ). Membranes were allowed to air dry for 20 min and submerged in 10 mm Tris-buffered saline with 0.1% Tween 20 (TTBS) for all washes and 5% milk/TTBS for 1 h blocking steps and overnight primary antibody incubations. A11 (rabbit polyclonal 1.8 mg/ml stock used at 1:5000) was used to detect oligomers (Kayed et al., 2003). Our secondary antibody was an HRP-conjugated IgG goat anti-rabbit used at 1:10,000 (Bio-Rad, Hercules, CA) and visualized with Supersignal Chemiluminescent Substrate (Pierce) on Hyperfilm ECL (GE Healthcare, Little Chalfont, UK). Immunoblots were quantified using NIH ImageJ software to obtain optical density (OD) measures.
Serum titers
The titers of anti-Aβ antibodies were measured as described previously with minor modifications (Cribbs et al., 2003). Briefly, wells of 96-well plates (Immulon 2HB; Thermo Fisher Scientific, Waltham, MA) were coated with 2.5 μm fibrillar Aβ42 in PBS, pH 7.2 (Sigma), and incubated overnight at 4°C. The wells were washed and then blocked with 3% nonfat dry milk for 1 h at 37°C with shaking. After washing, serial dilutions of serum from experimental and control dogs were added to the wells, and the plate was incubated for 1 h at 37°C with shaking. After washing, HRP-conjugated rabbit anti-dog IgG antibodies (Jackson ImmunoResearch, West Grove, PA) were added at 1:4000 dilution. After incubation for 1 h at 37°C with shaking, wells were washed, Ultra-TMB ELISA substrate (Pierce) was added to develop the reaction for 15 min, then 2N H2SO4 was added to the wells to stop the reaction, and the plates were analyzed on a Synergy HT Spectrophotometer (Bio-Tek Instruments, Winooski, VT) at 450 nm. Anti-Aβ antibody concentrations were calculated using the monoclonal antibody 6E10 as a standard bound to the corresponding Aβ species on the same ELISA plates and are expressed in micrograms. In addition, the sera end-point titer was defined as the maximal sera dilution in which OD for the antibodies was three times higher than the OD values of the blank wells (Table 1).
Data analysis
Our preliminary analysis of cognitive test scores (baseline and during treatment) and Aβ neuropathology established that the two control groups (Alum only and saline injected) could be combined into one control group for comparison with the fibrillar Aβ immunized animals. Subsequently, either t tests or repeated-measures ANOVA were used to compare treatment conditions on both cognitive and neurobiological outcome measures. Post hoc comparisons used the Bonferroni's correction. All analyses used SPSS for Windows (SPSS, Chicago, IL).
Results
Animals underwent an extensive baseline cognitive function assessment were subsequently matched into three treatment groups (n = 9 immunized, n = 6 Alum only, or n = 5 saline controls) and were immunized on a monthly basis throughout the duration of the study after an initial 2 week boost (total of 25 injections). Cognition was evaluated at multiple time points throughout the study, and, for each cognitive task and for the brain outcome measures, the two control groups (Alum only or saline injected) were not statistically significantly different and thus combined for all subsequent analyses. The final two groups (immunized vs control) were balanced with respect to baseline cognition (Fig. 1A). In the first year of the study, one saline-injected dog (11.5 years of age, 1.2 months of treatment) was killed because of an oronasal fistula. A second animal developed blindness (14.6 years of age, fibrillar Aβ-immunized group) and was maintained on the study but could not complete the cognitive testing protocol. A third animal in the Alum control group was killed in the second year of the treatment because of a mammary carcinoma that had spread to the lymphatic system (11.2 years of age, 1.48 years of treatment). In the last year of the study, a fourth animal in the fibrillar Aβ-immunized group was killed after poor recovery from a CSF tap (12.5 years of age, 1.78 years of treatment). However, overall, the treatment was well tolerated by all animals, and no adverse events were noted.
Figure 1.
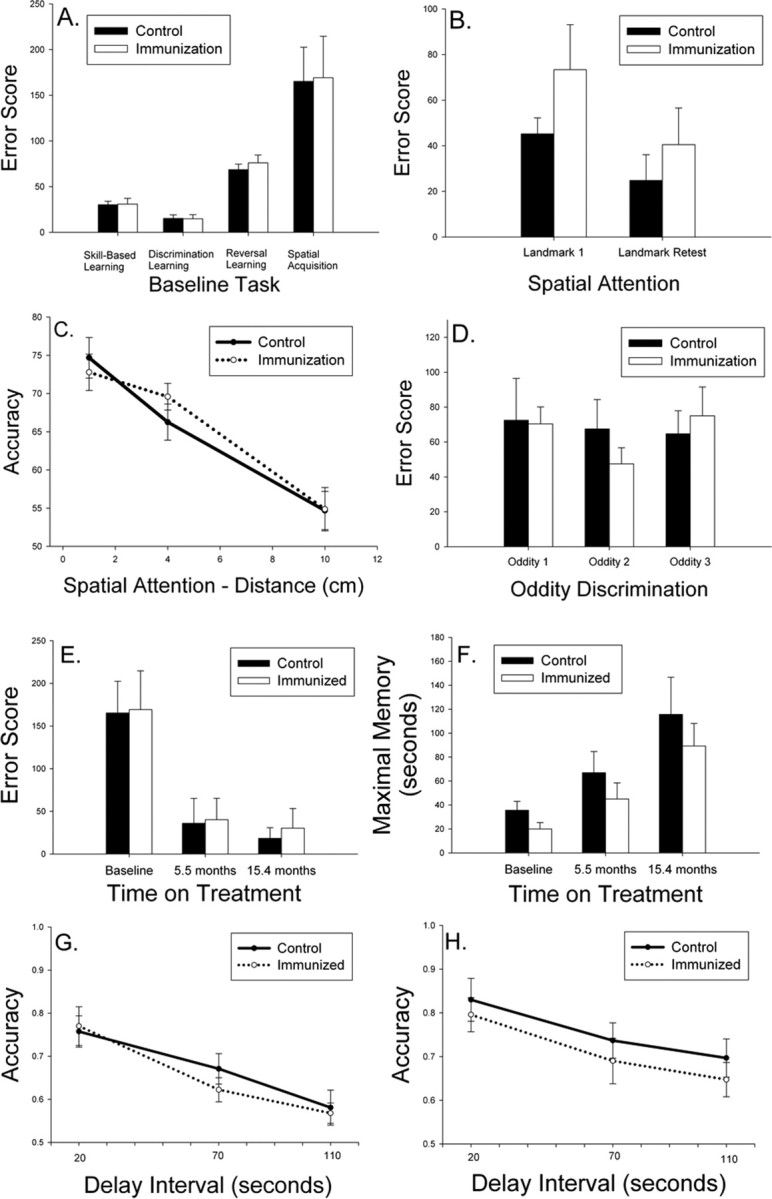
A, Aged beagles were cognitively assessed before treatment and matched into two groups with similar learning error scores. B, Spatial attention measured using a landmark discrimination task was not improved after two immunizations (Landmark 1) nor after 25 immunizations (Landmark Retest). C, Spatial attention measured using a variable distance procedure also did not reveal improvements in immunized dogs. D, On a measure of more complex learning ability, dogs receiving 12 immunizations showed no improvement on three phases of an oddity discrimination problem. E, Spatial learning of a two-choice delayed non-match-to-position task was equivalent in the two groups after up to 14 vaccinations, with all animals showing progressive improvements with repeated testing. F, The maximal memory score representing how long animals can remember spatial information also did not vary as a function of treatment. G, H, Spatial working memory assessed after eight immunizations (G) or 17 immunizations (H) also did not show significant treatment effects. Bars represent means, and error bars represent SEM.
After two injections, animals were tested for spatial attention ability using a landmark discrimination problem, which is sensitive to both age and treatment (Milgram et al., 2002b). There were no significant reductions in error scores in immunized dogs (t(16) = 1.34; p = 0.20) (Fig. 1B). Immediately before the end of the study, after 24 immunizations and 26.4 months of treatment, the remaining animals (six controls, seven fibrillar Aβ) were retested on the landmark problem. No treatment effects were observed (t(11) = 0.77; p = 0.46) (Fig. 1B), nor were treatment effects detectable compared with baseline measures. Using a variable distance landmark test at each time point, we further confirmed a lack of treatment effect (Fig. 1C). We next hypothesized that immunized animals may show improvements in a more difficult learning task, oddity discrimination, which was demonstrated previously to be sensitive to more subtle treatment effects (Cotman et al., 2002). As shown in Figure 1D, there were no significant changes in complex learning ability in treated animals after 12 immunizations (t(14) = 0.08; p = 0.94).
Spatial attention and oddity discrimination are measures of learning, and so we next assessed spatial memory because previous reports in immunized transgenic mice suggest improvements in this cognitive domain. At baseline, after six injections (5.5 months on treatment) and after 14 immunizations (15.4 months on treatment), animals were retested for two-choice spatial acquisition, maximal memory, and working memory using a delayed non-match-to-position procedure. Seven controls and eight fibrillar Aβ-immunized animals completed all three test sessions. Overall, animals in both groups progressively improved (i.e., showed a reduction in error scores) with repeated testing (F(2,26) = 16.95; p < 0.0005) (Fig. 1E). However, there was no significant main effect of treatment (F(1,13) = 0.27; p = 0.61) nor a time × treatment group interaction (F(2,26) = 0.018; p = 0.98). Next, we analyzed maximal memory ability (Fig. 1F), which was the maximum delay interval at which dogs could meet criterion levels of responding within 50 d of testing (i.e., higher delay is indicative of better memory). One animal from each group did not complete all phases of testing. Figure 1F shows that, across time, maximal memory improves (F(2,22) = 16.37; p < 0.0001), but no significant main effects of treatment (F(1,11) = 2.99; p = 0.11) nor a treatment × time interaction was observed (F(2,22) = 1.75 p = 0.20). Last, to detect improvements in spatial working memory, we used accuracy scores from a variable delay procedure. The spatial memory task was administered twice in the study while animals were being immunized. Figure 1G shows that, after 8.3 months of treatment (n = 8 control and n = 9 fibrillar Aβ-immunized animals) as the delay interval increases, there is a corresponding decrease in response accuracy (F(2,30) = 48.14; p < 0.0001). However, no treatment × delay interval interaction effects (F(2,30) = 1.24; p = 0.30) nor a main effect of treatment (F(1,15) = 0.13; p = 0.72) was observed, and immunized dogs showed similar spatial memory ability to control animals. After 17.9 months of treatment, similar effects were observed, and no treatment associated improvements in accuracy were seen (F(1,11) = 0.54; p = 0.48) (Fig. 1H).
Because of concerns about practice effects, we also used a more difficult three-choice spatial acquisition and memory task at two time points in the study: (1) after 9.6 months or 9 immunizations, and (2) 19.4 months of treatment and 18 immunizations (Fig. 2). The addition of a third possible location for the food reward reduces the ability of animals to use a body position strategy for solving the problem (Chan et al., 2002). Acquisition was tested using a short, 5 s delay for six control animals and eight fibrillar Aβ-immunized dogs. There were no significant improvements in acquisition of the task across time (F(1,12) = 0.023; p = 0.88) nor a time × treatment interaction (F(1,12) = 1.18; p = 0.30). Furthermore, there was no significant main effect of treatment on acquisition of the three-choice spatial memory task (F(1,12) = 0.55; p = 0.47) (Fig. 2A). Maximal memory was also compared across the two time points with similar negative outcomes. There was no practice effect observed over time (F(1,12) = 1.15; p = 0.31), no time × treatment interaction (F(1,12) = 0.56; p = 0.47), and no effect of treatment overall (F(1,12) = 0.04; p = 0.85) (Fig. 2B). Nonparametric analyses of maximal memory scores confirmed the lack of treatment effect. Spatial working memory using the three-choice procedure using delay intervals of 20, 70, and 110 s was also tested, and similar analyses as described for the two-choice memory test was used. After nine immunizations, seven control and nine fibrillar Aβ-immunized animals showed a decrease in accuracy with increasing delay interval (F(2,28) = 34.14; p < 0.0001), but no main treatment effects (F(1,14) = 0.57; p = 0.46) nor a group × delay interval interaction (F(2,28) = 0.83; p = 0.45) was observed (Fig. 2C). At the second time point after 18 immunizations, similar effects were observed. Six control and seven fibrillar Aβ-immunized animals showed a decrease in accuracy with increasing memory demands (F(2,22) = 7.66; p = 0.003), but no main effect of treatment (F(1,11) = 0.24; p = 0.63) nor treatment × delay interaction was observed (F(2,22) = 0.95; p = 0.40) (Fig. 2D).
Figure 2.

A, Spatial learning of a three-choice delayed non-match-to-position task was equivalent in the two treatment groups after up to 20 vaccinations with animals showing relatively stable spatial acquisition with repeated testing. B, The maximal memory score representing how long animals can remember spatial information also did not vary as a function of treatment. C, D, Spatial working memory assessed after 11 immunizations (C) or 20 immunizations (D) also did not show significant treatment effects. Bars represent means, and error bars represent SEM.
Within 5 months of the end of the study, we reassessed visual discrimination and reversal learning function (after 22 immunizations and 23.4 months of treatment) and compared scores with baseline measures. No treatment effects were observed in discrimination learning (t(11) = 0.102; p = 0.92) (Fig. 3A). After 40 d of testing, two control animals failed to learn the new problem and one immunized animal. In addition, one immunized animal stopped responding in the test apparatus and could not be continued. Subsequently for reversal learning, four control and five fibrillar Aβ-immunized dogs were compared. There was a significant reduction in reversal learning error scores in immunized dogs (t(7) = 2.37; p = 0.049) (Fig. 3B). Furthermore, reversal learning showed an overall increase in error scores over time (F(1,7) = 22.91; p = 0.002) in untreated animals, suggesting progressive decline, and a significant time × treatment effect (F(1,7) = 7.90; p = 0.026) indicated that fibrillar Aβ-immunized animals showed a maintenance of reversal learning ability (Fig. 3B). We further confirmed that frontal function, specifically, was affected by treatment by analyzing the types of errors individual animals made using previously published methods (Milgram et al., 1994). A repeated-measures univariate ANOVA suggested a learning stage × group interaction (F(2,14) = 4.00; p = 0.042), primarily driven by fibrillar Aβ-immunized dogs showing significantly fewer perseverative responses (t(7) = 2.53; p = 0.039) (Fig. 3C).
Figure 3.

A, Visual discrimination learning did not improve with treatment over time in vaccinated animals. B, Reversal learning in immunized dogs was improved after 22 immunizations compared with both control animals and over time. C, Immunized aged beagles showed fewer perseverative responses relative to control animals, suggesting an improvement in prefrontal cortex function. Bars represent means, and error bars represent SEM. *p < 0.05.
Because of the relatively small effects of the immunization treatment on cognition, we were concerned that, because of the significant Aβ deposition typically observed in animals of this age, neuropathology may have been unaffected by immunization. Animals were between 11.6 and 15.7 years (mean age control animals, 12.3 years; immunized animals, 12.7 years) at the end of the study after 28.3 months of treatment. The 4% paraformaldehyde-fixed brains of study animals were immunostained to detect diffuse Aβ plaques and to measure Aβ loads (Head et al., 1998) (representative sections shown in Fig. 4A,B). We observed dramatically reduced diffuse Aβ plaque load (detected using 6E10 antibody) in the prefrontal (∼87%), entorhinal (∼84%), and occipital (∼75%) cortical regions but not in the parietal cortex (F(3,54) = 3.61; p = 0.019) (Fig. 4C). Based on our previous work (Head et al., 2000), we estimated that, between the ages of 8 and 12 years of age (average Aβ load, 7.0%) to between 12 and 15 years of age (average Aβ load, 11.9%), an ∼1.7-fold increase in prefrontal cortex Aβ plaque load would occur. In immunized animals, the final Aβ plaque load in the prefrontal cortex was <2%, which is lower than what would have been predicted in animals at the start of the study. This suggests that not only did immunization prevent new plaque deposition but also cleared existing Aβ plaque pathology. However, as shown in Figure 4C, there can be significant individual variability in plaque loads in untreated aged dogs, particularly in the prefrontal, parietal, and occipital cortices. In most cases, immunized animals showed low individual variability (Fig. 4C). To determine whether untreated aged dogs with low plaque pathology were contributing to a lack of cognitive improvements in many of the outcome measures, we reanalyzed cognitive data and focused on prefrontal cortex and eliminated untreated dogs with a plaque load of <10%. This left five animals in the untreated group for comparison with nine animals in the immunized group. As described previously, we did not find any significant differences when comparing the two groups on the landmark test, oddity discrimination, and size discrimination learning and on the two spatial learning and memory tasks. However, the treatment effect for size reversal learning remained significant, with immunized dogs showing less decline.
Figure 4.
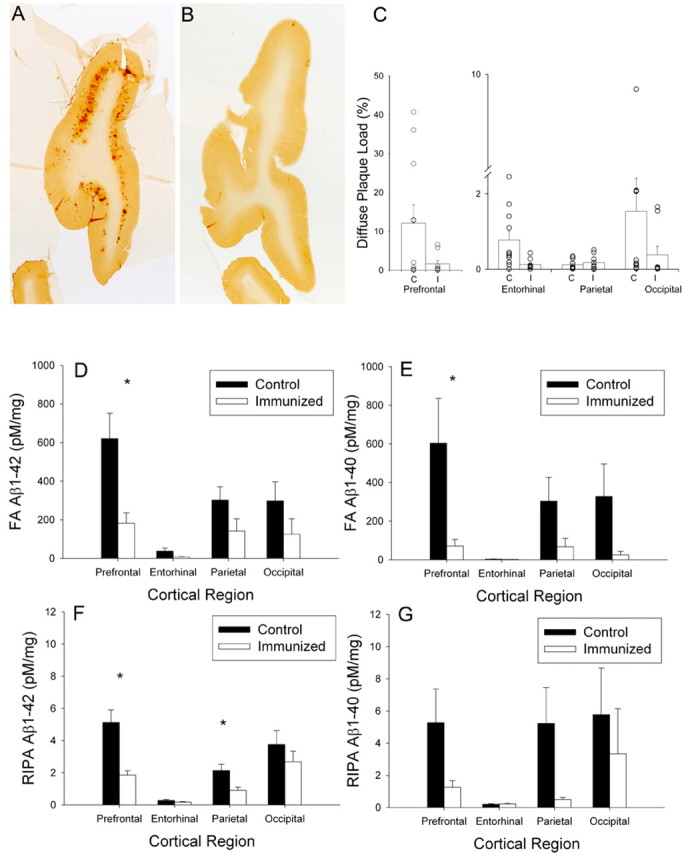
A, B, Diffuse prefrontal Aβ plaques were extensive in a control animal (A) and significantly reduced in an immunized animal (B). C, Animals chosen for this illustration had Aβ plaque loads similar to the group means of the control and immunized animals. Consistent with immunolabeling for Aβ, diffuse plaque load was reduced in the prefrontal cortex, the entorhinal cortex, and the occipital cortex but not in the parietal cortex. Each symbol represents an individual animal. D, E, Formic acid-extracted Aβ1–42 (D) and Aβ1–40 (E) were both significantly reduced in immunized dogs in multiple cortical regions. F, G, Similarly, RIPA-extracted Aβ1–42 (F) and Aβ1–40 (G) were reduced in treated dogs. Bars represent means, and error bars represent SEM. *p < 0.05.
We also evaluated the extent of Aβ angiopathy in treated animals but observed no differences from untreated dogs (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To detect inflammation in the brains of treated animals and possible microglial activation in association with remaining plaques in treated dogs, we immunostained the prefrontal cortex for human leukocyte antigen–D region-related receptors. As shown in the supplemental Methods and Results (available at www.jneurosci.org as supplemental material), we did not observe significant differences in white matter microglial activation, but remaining plaques in immunized dogs were surrounded by activated microglial cells (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
To confirm and extend our findings of reduced Aβ by plaque load, we used snap-frozen contralateral hemispheres of treatment animals. Both soluble (RIPA extracted) and insoluble (formic acid extracted) Aβ1–40 and Aβ1–42 were measured by ELISA in four cortical regions (prefrontal, parietal, occipital, and entorhinal cortex) (Table 1) (supplemental Results, available at www.jneurosci.org as supplemental material). There was a significant 63.7% reduction of formic acid-extracted Aβ1–42 overall in treated animals (F(1,14) = 5.43; p = 0.035) and a significant region × treatment group interaction (F(3,42) = 4.6; p = 0.007) (Fig. 4D), suggesting differential reductions in Aβ within specific cortical areas. Formic acid-extracted Aβ1–40 showed a similar treatment-associated reduction (78.0%; F(1,14) = 4.65; p = 0.049) that varied as a function of cortical region sampled (F(3,42) = 3.23; p = 0.032) (Fig. 4E). RIPA-extracted Aβ1–42 was significantly reduced by 50.1% overall in response to immunization (F(1,14) = 8.28; p = 0.012), but larger reductions were observed in the prefrontal (63.7%) and parietal (57.8%) cortices than in entorhinal (34.2%) or occipital (28.3%) cortices (F(3,42) = 4.61; p = 0.007). Figure 4F shows that the largest treatment effects occurred in the prefrontal cortex, with the smallest effects occurring in the entorhinal cortex. In contrast, despite trends toward decreased RIPA-extracted Aβ1–40 (67.9%), particularly in the prefrontal (76.2%) and parietal (90.7%) cortices, statistically, Aβ40 was not significantly reduced in treated animals (F(1,14) = 2.29; p = 0.15) nor differentially affected depending on the brain region sampled (F(3,42) = 1.35; p = 0.27) (Fig. 4G). This may have been attributable to large individual variability, particularly in the control group.
Less significant reductions in soluble Aβ levels compared with insoluble species suggest that the amount of soluble Aβ oligomers may have remained unchanged. Oligomers are thought to be one type of assembly state of Aβ that might be particularly toxic to neurons and synapses and linked to cognitive dysfunction (Walsh et al., 2002; Lesne et al., 2006; Haass and Selkoe, 2007). Thus, we used a new sample from the prefrontal and parietal cortex homogenized in PBS to detect soluble oligomers according to a previously published protocol (Kayed et al., 2003). Optical densities from individual animals using dot blots and the A11 antibody (Kayed et al., 2003) that detects all oligomers (not specifically Aβ42 oligomers) did not reveal a significant reduction in immunized dogs in the prefrontal cortex (t(15) < 1; p = 0.93) or parietal cortex (t(15) < 1; p = 0.51) (Fig. 5A–C).
Figure 5.

Dot blot assays for oligomers (A11) in the prefrontal cortex with individual animals shown in triplicate illustrates individual variability in the amount of oligomers detected in animals despite an equal amount of protein loaded. In the prefrontal cortex, control animals (mean ± SD OD, 1501 ± 224.75) had similar levels of A11 compared with immunized animals (mean ± SD OD, 1400.6 ± 385.43). In addition, similar measures of A11 by dot blot in the parietal cortex also revealed no difference between controls (mean ± SD OD, 1250.5 ± 386.95) and immunized dogs (mean ± SD OD, 1284.8 ± 265.08). PBS indicates control samples.
To determine whether remaining Aβ in the brain was associated with cognitive scores on the size discrimination and reversal learning tasks, we computed correlations. Table 3 shows that higher error scores on the size discrimination learning task (i.e., poorer performance) was correlated with higher levels of RIPA-extracted Aβ1–40 in the entorhinal cortex (r = 0.71; p = 0.007). In contrast, higher formic acid-extracted prefrontal Aβ1–40 was correlated with higher errors scores on the reversal learning problem. No other correlations were significant. These results suggest that reduced Aβ in a specific brain region (prefrontal cortex) is significantly associated with cognitive improvement (frontal function) mediated by these regions.
Table 3.
Correlation between brain Aβ and cognition
| Region/species | Formic acid extracted |
RIPA extracted |
||
|---|---|---|---|---|
| Size discrimination | Size reversal | Size discrimination | Size reversal | |
| Prefrontal Aβ1–42 | −0.313; p = 0.299 | 0.635; p = 0.066 | −0.167; p = 0.585 | 0.567; p = 0.111 |
| Prefrontal Aβ1–40 | −0.374; p = 0.208 | 0.714; p = 0.031* | −0.248; p = 0.414 | 0.627; p = 0.071 |
| Entorhinal Aβ1–42 | −0.372; p = 0.211 | 0.319; p = 0.402 | −0.392; p = 0.185 | 0.659; p = 0.053 |
| Entorhinal Aβ1–40 | −0.291; p = 0.334 | 0.165; p = 0.672 | 0.706; p = 0.007* | −0.241; p = 0.533 |
| Parietal Aβ1–42 | −0.256; p = 0.399 | 0.592; p = 0.093 | −0.148; p = 0.630 | 0.565; p = 0.113 |
| Parietal Aβ1–40 | −0.017; p = 0.955 | 0.541; p = 0.133 | −0.218; p = 0.474 | 0.437; p = 0.240 |
| Occipital Aβ1–42 | −0.098; p = 0.751 | 0.476; p = 0.196 | 0.015; p = 0.960 | 0.502; p = 0.168 |
| Occipital Aβ1–40 | −0.177; p = 0.563 | 0.588; p = 0.096 | 0.076; p = 0.804 | 0.326; p = 0.392 |
*p < 0.05.
We next tested the hypothesis that serum anti-fibrillar Aβ titers would predict the extent of Aβ remaining in the brain and specifically that higher antibody titers against Aβ would be associated with lower Aβ in the brain. Antibody titers were log transformed, and a Spearman's rank correlation was used to measure the association. Table 4 shows that higher antibody titers against Aβ were associated with lower brain Aβ level. Interestingly, this effect primarily occurred for prefrontal and parietal cortices, and serum titers were less predictive of Aβ in the entorhinal or occipital cortex.
Table 4.
Spearman's rank correlations between serum anti-Aβ antibodies and brain Aβ
| Region/species | FA extracted | RIPA extracted |
|---|---|---|
| Prefrontal Aβ1–42 | −0.634; p = 0.008* | −0.697; p = 0.003* |
| Prefrontal Aβ1–40 | −0.528; p = 0.036* | −0.457; p = 0.075 |
| Entorhinal Aβ1–42 | −0.443; p = 0.086 | −0.332; p = 0.209 |
| Entorhinal Aβ1–40 | −0.203; p = 0.45 | 0.144; p = 0.596 |
| Parietal Aβ1–42 | −0.524; p = 0.037* | −0.576; p = 0.02* |
| Parietal Aβ1–40 | −0.372; p = 0.156 | −0.215; p = 0.424 |
| Occipital Aβ1–42 | −0.377; p = 0.150 | −0.295; p = 0.267 |
| Occipital Aβ1–40 | −0.418; p = 0.107 | −0.190; p = 0.480 |
*p < 0.05.
Discussion
We present data from a long-term therapeutic anti-fibrillar Aβ active immunization study, using aged canines, that naturally accumulate human-type Aβ in the CNS and cognitive deficits. A maintenance of executive function was observed, similar to the AN1792 study (Hock et al., 2003), but there were no improvements in complex learning, spatial memory, or attention. It is unlikely that floor or ceiling effects account for the limited treatment effect; the same series of cognitive tasks are sensitive to age (Milgram et al., 1994, 1999; Head et al., 1995, 1998; Chan et al., 2002; Tapp et al., 2003a; Studzinski et al., 2006) and to interventions (e.g., antioxidant diet and behavioral enrichment) (Cotman et al., 2002; Milgram et al., 2002b, 2004, 2005; Siwak et al., 2005). The relatively limited effects of Aβ immunization on cognition contrasts with the significant reduction in Aβ plaque load and extractable Aβ1–40/Aβ1–42. Immunotherapy reduced Aβ pathology to a greater extent in the prefrontal cortex relative to other cortical regions. The extent of remaining prefrontal Aβ was correlated with frontal-dependent cognition (size reversal learning) and serum antibody titers best predicted treatment-associated Aβ reduction in this same brain region. We did not observe any changes in Aβ angiopathy in treated animals, which suggests that increased angiopathy in human clinical trials (Masliah et al., 2005) may reflect a transient active phase of plaque removal (Nicoll, 2007). One of the simplest interpretations of these data are that reducing preexisting brain Aβ is insufficient to restore neuronal and cognitive function. However, immunotherapy may help to maintain cognition. We also found that the total amount of oligomers in canine brain was unaffected by treatment. Given the possible link between toxic assembly states of Aβ, such as oligomers, and cognition, this may be one reason for the lack of improvements in several cognitive domains (Lue et al., 1999; Walsh et al., 2002; Walsh and Selkoe, 2004).
Cognitive outcomes in response to immunization in canines contrast with reports in transgenic mice. Aged transgenic mice, with preexisting pathology, show improved learning and memory in response to immunization (Dodart et al., 2002; Kotilinek et al., 2002; Wilcock et al., 2004a; Hartman et al., 2005; Oddo et al., 2006). However, Aβ reduction may be absent or not as extensive as that reported in younger mice (Das et al., 2001; Zhou et al., 2005). In contrast, a recent study using a large sample size showed that, overall, active immunization of PDAPP mice (mice overexpressing mutant human amyloid precursor protein) did not lead to improved spatial learning (Chen et al., 2007). If animals showing very low levels of brain Aβ after immunization were compared with nontransgenic controls, the two groups performed similarly, suggesting improved spatial learning. Significant differences in the responses of aged transgenic mice and canines to Aβ clearance may be attributable to several factors: (1) lack of an overexpression system, (2) lack of mutant human amyloid precursor protein, (3) lack of effect on oligomer levels (Klyubin et al., 2005), and (4) differences in the length of time canine neurons are exposed to Aβ during the aging process (Head et al., 2000).
The results of the study in aged dogs are consistent with studies in Alzheimer's disease patients. The first clinical trial in AD patients used the AN1792 vaccine, which initially consisted of fibrillar Aβ42 as the antigen and QS-21 as the adjuvant. Subsequently, the vaccine was reformulated to include Polysorbate 80. In the Swiss cohort of the AN1792 patients, there was a maintenance of function on a global test of cognition and on a hippocampal-dependent task in individuals who developed antibodies capable of binding to plaques (Hock et al., 2003). In a second, larger study, no differences between antibody responders and placebo groups were observed on several cognitive and disability scales (Gilman et al., 2005). However, a composite score of a neuropsychological test battery indicated “less worsening” of decline in antibody responders after 12 months and an improvement in the memory domain (Gilman et al., 2005). A small number of patients enrolled in the AN1792 study have come to autopsy and show Aβ plaque reduction without any effect on the extent of neurofibrillary tangles or cerebral amyloid angiopathy (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). Interestingly, in the case report by Masliah et al. (2005), the frontal cortex showed the largest response to immunotherapy, which is similar to the canine.
There are several differences between the canine study and the human clinical trial. First, we observed no adverse autoimmune responses in canines, which suggest that a Th2 adjuvant (e.g., Alum) may be a safe alternative. Second, the majority of AD patients in the initial clinical trial received between two and three immunizations, and long-term effects of chronic immunization have not been evaluated. Our canine study suggests that, even with longer treatment periods, there is no additional benefit to cognitive outcome measures. Third, the antibody titers reported in clinical trial participants (Hock et al., 2002) were relatively low compared with those seen in the canine (Head et al., 2006), and all immunized canines responded to treatment. Higher antibody titers in treated aged canines may be attributable to a larger dose of Aβ peptide than that used in humans (Nicoll et al., 2003; Orgogozo et al., 2003).
Learning and memory deficits observed in aged canines may be attributable to chronic exposure of the brain to Aβ and possible downstream molecular cascades that can lead to neuronal dysfunction and death. Similar to the human brain, canine Aβ accumulates over a period of many years. This is sufficient time to allow for spontaneous posttranslational modifications to extracellular Aβ such as isomerization, racemization, and oxidation, similar to human brain aging and AD (Fonseca et al., 1999; Azizeh et al., 2000; Head et al., 2001; Kalback et al., 2002). One of the interesting outcomes of the current experiment is that Aβ immunization is potent enough to clear modified forms of Aβ thought to be more difficult to degrade typically observed in human and canine brain but not in transgenic mouse brain (Kalback et al., 2002). The limited functional benefit suggests that prevention of Aβ accumulation by initiating treatment in middle-aged animals may be more efficacious as has been suggested in transgenic mouse models (Janus et al., 2000; Morgan et al., 2000; Das et al., 2001; Sigurdsson et al., 2004; Asuni et al., 2006).
There is another possible interpretation of the current study. In humans, Aβ accumulation in diffuse plaques and in the cerebral vasculature may begin 10–20 years before the development of clinical signs of impairment (Price and Morris, 1999; Price, 2003). It is only when there is extensive Aβ pathology, neuritic plaques, neurofibrillary tangles, and neuron loss that cognitive decline is observed (Morris et al., 2001; Price et al., 2001). Thus, Aβ alone may be insufficient to cause cognitive decline in human brain and may explain why there were limited improvements in cognition if Aβ accumulation in the canine brain is also “clinically silent.” However, the extent of Aβ is correlated with cognitive decline in aged dogs (Cummings et al., 1996b; Head et al., 1998), and animals with more extensive Aβ pathology have more severe cognitive deficits. In the current study, reducing prefrontal Aβ impacted frontal function, suggesting that reducing Aβ may result in cognitive benefits. In addition, aged canines exhibit several signs of pathology such as neuron loss in the hippocampus (Siwak-Tapp et al., 2008) and cortical atrophy in association with Aβ and cognition in vulnerable brain regions (Tapp et al., 2004). The lack of neurofibrillary tangle pathology in canines may also not entirely explain the limited cognitive benefits of Aβ reduction. In several autopsy studies of immunized patients with AD, there was a lack of impact on neurofibrillary tangle pathology, suggesting that this was not a mechanism underlying cognitive maintenance observed in the human clinical trial (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). In addition, all of the previous animal immunotherapy studies were primarily in transgenic mice that do not develop tauopathy and led to significant behavioral improvements (Schenk et al., 1999; Janus et al., 2000; Morgan et al., 2000; Das et al., 2003).
If immunotherapy as a treatment is pursued, then our work in canines suggests another possible approach. We previously published the results of a longitudinal study in which aged beagles were fed an antioxidant-enriched diet, provided with behavioral enrichment, or given a combination of both treatments. Treated animals showed significant cognitive improvements, with the combination being the most efficacious (Cotman et al., 2002; Milgram et al., 2002b, 2005). Antioxidant-fed but not behaviorally enriched animals showed decreased brain Aβ (Pop et al., 2003). Thus, additional and/or combinatorial interventions that may both reduce Aβ (immunization) or reduce Aβ oligomers (Lee et al., 2006; Chauhan, 2007) and in parallel restore neuron health (e.g., antioxidant-enriched diet or behavioral enrichment) (Milgram et al., 2005) after chronic Aβ exposure may be a promising approach for future treatment studies.
Footnotes
This work was supported by National Institutes of Health (NIH)/National Institute on Aging Grants AG20242 and AG20241, NIH/National Institute of Neurological Disorders and Stroke Grant NS50895, and NIH/Alzheimer's Disease Research Center Grant P50 AG16573.
References
- Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Vaccination of Alzheimer's model mice with Abeta derivative in alum adjuvant reduces Abeta burden without microhemorrhages. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizeh BY, Head E, Ibrahim MA, Torp R, Tenner AJ, Kim RC, Lott IT, Cotman CW. Molecular dating of senile plaques in aged Down's syndrome and canine brains. Exp Neurol. 2000;163:111–122. doi: 10.1006/exnr.2000.7359. [DOI] [PubMed] [Google Scholar]
- Chan AD, Nippak PM, Murphey H, Ikeda-Douglas CJ, Muggenburg B, Head E, Cotman CW, Milgram NW. Visuospatial impairments in aged canines (Canis familiaris): the role of cognitive-behavioral flexibility. Behav Neurosci. 2002;116:443–454. [PubMed] [Google Scholar]
- Chauhan NB. Intracerebroventricular passive immunization with anti-oligoAbeta antibody in TgCRND8. J Neurosci Res. 2007;85:451–463. doi: 10.1002/jnr.21110. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen KS, Kobayashi D, Barbour R, Motter R, Games D, Martin SJ, Morris RG. Active β-amyloid immunization restores spatial learning in PDAPP mice displaying very low levels of β-amyloid. J Neurosci. 2007;27:2654–2662. doi: 10.1523/JNEUROSCI.3710-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Head E, Muggenburg BA, Zicker S, Milgram NW. Brain aging in the canine: a diet enriched in antioxidants reduces cognitive dysfunction. Neurobiol Aging. 2002;23:809–818. doi: 10.1016/s0197-4580(02)00073-8. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Head E, Ruehl WW, Milgram NW, Cotman CW. The canine as an animal model of human aging and dementia. Neurobiol Aging. 1996a;17:259–268. doi: 10.1016/0197-4580(95)02060-8. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW. Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem. 1996b;66:11–23. doi: 10.1006/nlme.1996.0039. [DOI] [PubMed] [Google Scholar]
- Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Abeta1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging. 2001;22:721–727. doi: 10.1016/s0197-4580(01)00245-7. [DOI] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-β immunization effectively reduces amyloid deposition in FcRγ−/− knock-out mice. J Neurosci. 2003;23:8532–8538. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Head E, Velasquez P, Cotman CW, Tenner AJ. The presence of isoaspartic acid in b-amyloid plaques indicates plaque age. Exp Neurol. 1999;157:277–288. doi: 10.1006/exnr.1999.7058. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Boada Rovira M, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-β antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E, Mehta R, Hartley J, Kameka AM, Cummings BJ, Cotman CW, Ruehl WW, Milgram NW. Spatial learning and memory as a function of age in the dog. Behav Neurosci. 1995;109:851–858. doi: 10.1037//0735-7044.109.5.851. [DOI] [PubMed] [Google Scholar]
- Head E, Callahan H, Muggenburg BA, Cotman CW, Milgram NW. Visual-discrimination learning ability and beta-amyloid accumulation in the dog. Neurobiol Aging. 1998;19:415–425. doi: 10.1016/s0197-4580(98)00084-0. [DOI] [PubMed] [Google Scholar]
- Head E, McCleary R, Hahn FF, Milgram NW, Cotman CW. Region-specific age at onset of beta-amyloid in dogs. Neurobiol Aging. 2000;21:89–96. doi: 10.1016/s0197-4580(00)00093-2. [DOI] [PubMed] [Google Scholar]
- Head E, Garzon-Rodriguez W, Johnson JK, Lott IT, Cotman CW, Glabe C. Oxidation of Ab and plaque biogenesis in Alzheimer's disease and Down syndrome. Neurobiol Dis. 2001;8:792–806. doi: 10.1006/nbdi.2001.0431. [DOI] [PubMed] [Google Scholar]
- Head E, Barrett EG, Murphy MP, Das P, Nistor M, Sarsoza F, Glabe CC, Kayed R, Milton S, Vasilevko V, Milgram NW, Agadjanyan MG, Cribbs DH, Cotman CW. Immunization with fibrillar Abeta(1–42) in young and aged canines: Antibody generation and characteristics, and effects on CSF and brain Abeta. Vaccine. 2006;24:2824–2834. doi: 10.1016/j.vaccine.2005.12.058. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer's disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10:299–305. doi: 10.1016/0169-328x(91)90088-f. [DOI] [PubMed] [Google Scholar]
- Jones B, Mishkin M. Limbic lesions and the problem of stimulus-reinforcement associations. Exp Neurol. 1972;36:362–377. doi: 10.1016/0014-4886(72)90030-1. [DOI] [PubMed] [Google Scholar]
- Kalback W, Watson MD, Kokjohn TA, Kuo YM, Weiss N, Luehrs DC, Lopez J, Brune D, Sisodia SS, Staufenbiel M, Emmerling M, Roher AE. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer's disease senile plaques. Biochemistry. 2002;41:922–928. doi: 10.1021/bi015685+. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol. 2004;82:497–505. doi: 10.1111/j.0818-9641.2004.01286.x. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgram NW, Head E, Weiner E, Thomas E. Cognitive functions and aging in the dog: acquisition of nonspatial visual tasks. Behav Neurosci. 1994;108:57–68. doi: 10.1037//0735-7044.108.1.57. [DOI] [PubMed] [Google Scholar]
- Milgram NW, Adams B, Callahan H, Head E, Mackay W, Thirlwell C, Cotman CW. Landmark discrimination learning in the dog. Learn Mem. 1999;6:54–61. [PMC free article] [PubMed] [Google Scholar]
- Milgram NW, Zicker SC, Head E, Muggenburg BA, Murphey H, Ikeda-Douglas C, Cotman CW. Dietary enrichment counteracts age-associated cognitive dysfunction in canines. Neurobiol Aging. 2002a;23:737–745. doi: 10.1016/s0197-4580(02)00020-9. [DOI] [PubMed] [Google Scholar]
- Milgram NW, Head E, Muggenburg BA, Holowachuk D, Murphey H, Estrada J, Ikeda-Douglas CJ, Zicker SC, Cotman CW. Landmark discrimination learning in the dog: effects of age, an antioxidant fortified diet, and cognitive strategy. Neurosci Biobehav Rev. 2002b;26:679–695. doi: 10.1016/s0149-7634(02)00039-8. [DOI] [PubMed] [Google Scholar]
- Milgram NW, Head E, Zicker SC, Ikeda-Douglas C, Murphey H, Muggenberg BA, Siwak CT, Dwight Tapp P, Lowry SR, Cotman CW. Long-term treatment with antioxidants and a program of behavioral enrichment reduces age-dependent impairment in discrimination and reversal learning in beagle dogs. Exp Gerontol. 2004;39:753–765. doi: 10.1016/j.exger.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Milgram NW, Head E, Zicker SC, Ikeda-Douglas CJ, Murphey H, Muggenburg B, Siwak C, Tapp D, Cotman CW. Learning ability in aged beagle dogs is preserved by behavioral enrichment and dietary fortification: a two-year longitudinal study. Neurobiol Aging. 2005;26:77–90. doi: 10.1016/j.neurobiolaging.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Morley JE, Farr SA, Flood JF. Antibody to amyloid beta protein alleviates impaired acquisition, retention, and memory processing in SAMP8 mice. Neurobiol Learn Mem. 2002;78:125–138. doi: 10.1006/nlme.2001.4047. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- Nicoll J. Potential mechanisms of Abeta plaque clearance in patients immunized with Abeta. In: Fisher A, Hanin I, Poewe W, Windisch M, editors. Alzheimer's and Parkinson's diseases: progress and new perspectives; 8th International Conference AD/PD; Salzburg, Austria: Karger; 2007. p. 14. [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pop V, Head E, Nistor M, Milgram NW, Muggenburg BA, Cotman CW. Reduced Ab deposition with long-term antioxidant diet treatment in aged canines. Soc Neurosci Abstr. 2003;29:525–4. [Google Scholar]
- Price JL. Aging, preclinical Alzheimer disease, and early detection. Alzheimer Dis Assoc Disord. 2003;17(Suppl 2):S60–S62. doi: 10.1097/00002093-200304002-00009. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandervert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-b attentuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Bell DS, Podlisny MB, Price DL, Cork LC. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer's disease. Science. 1987;235:873–877. doi: 10.1126/science.3544219. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siwak CT, Tapp PD, Head E, Zicker SC, Murphey HL, Muggenburg BA, Ikeda-Douglas CJ, Cotman CW, Milgram NW. Chronic antioxidant and mitochondrial cofactor administration improves discrimination learning in aged but not young dogs. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:461–469. doi: 10.1016/j.pnpbp.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Siwak-Tapp CT, Head E, Muggenburg BA, Milgram NW, Cotman CW. Region specific neuron loss in the aged canine hippocampus is reduced by enrichment. Neurobiol Aging. 2008;29:39–50. doi: 10.1016/j.neurobiolaging.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studzinski CM, Christie LA, Araujo JA, Burnham WM, Head E, Cotman CW, Milgram NW. Visuospatial function in the beagle dog: an early marker of cognitive decline in a model of human aging and dementia. Neurobiol Learn Mem. 2006;86:197–204. doi: 10.1016/j.nlm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Tapp PD, Siwak CT, Estrada J, Holowachuk D, Milgram NW. Effects of age on measures of complex working memory span in the beagle dog (Canis familiaris) using two versions of a spatial list learning paradigm. Learn Mem. 2003a;10:148–160. doi: 10.1101/lm.56503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapp PD, Siwak CT, Estrada J, Muggenburg BA, Head E, Cotman CW, Milgram NW. Size and reversal learning in the beagle dog as a measure of executive function and inhibitory control in aging. Learn Mem. 2003b;10:64–73. doi: 10.1101/lm.54403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapp PD, Siwak CT, Gao FQ, Chiou JY, Black SE, Head E, Muggenburg BA, Cotman CW, Milgram NW, Su MY. Frontal lobe volume, function, and β-amyloid pathology in a canine model of aging. J Neurosci. 2004;24:8205–8213. doi: 10.1523/JNEUROSCI.1339-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004a;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Kayed R, Hernandez I, Webster SD, Yazan O, Cribbs DH, Glabe CG, Tenner AJ. Novel Abeta peptide immunogens modulate plaque pathology and inflammation in a murine model of Alzheimer's disease. J Neuroinflammation. 2005;2:28. doi: 10.1186/1742-2094-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]