

Abstract
Botulinum neurotoxins (designated BoNT/A–BoNT/G) are bacterial enzymes that block neurotransmitter release by cleaving essential components of the vesicle fusion machinery. BoNT/A, which cleaves SNAP-25 (synaptosomal-associated protein of 25 kDa), is extensively exploited in clinical medicine to treat neuromuscular pathologies, facial wrinkles, and various types of pain. It is widely assumed that BoNT/A remains at the synaptic terminal and its effects are confined to the injection site. Here we demonstrate that catalytically active BoNT/A is retrogradely transported by central neurons and motoneurons and is then transcytosed to afferent synapses, in which it cleaves SNAP-25. SNAP-25 cleavage by BoNT/A was observed in the contralateral hemisphere after unilateral BoNT/A delivery to the hippocampus. Appearance of cleaved SNAP-25 resulted in blockade of hippocampal activity in the untreated hemisphere. Injections of BoNT/A into the optic tectum led to the appearance of BoNT/A-truncated SNAP-25 in synaptic terminals within the retina. Cleaved SNAP-25 also appeared in the facial nucleus after injection of the toxin into rat whisker muscles. Experiments excluded passive spread of the toxin and demonstrated axonal migration and neuronal transcytosis of BoNT/A. These findings reveal a novel pathway of BoNT/A trafficking in neurons and have important implications for the clinical uses of this neurotoxin.
Keywords: synaptic transmission, SNAP-25, retrograde axonal transport, transcytosis, hippocampus, visual system
Introduction
Botulinum neurotoxins (BoNT/A–BoNT/G) are the most potent poisons known (Johnson, 1999; Rossetto et al., 2006). They disrupt neurotransmission via their proteolytic activity directed specifically on SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment receptor) proteins, which are essential for vesicle fusion (Schiavo et al., 2000; Turton et al., 2002; Sudhof, 2004; Rossetto et al., 2006). Specifically, BoNT/A and BoNT/E target SNAP-25 (synaptosomal-associated protein of 25 kDa), which resides in the plasma membrane facing the nerve cell cytosol (Montecucco et al., 2005). BoNT/A and BoNT/E cleave SNAP-25 at different sites near the C terminus (Schiavo et al., 2000; Davletov et al., 2005). Because of its long-lasting effects, BoNT/A is widely exploited to treat several human neuromuscular disorders characterized by hyperfunction of cholinergic terminals, such as dystonia and spasticity (Johnson, 1999; Davletov et al., 2005; Montecucco and Molgo, 2005). BoNT/A is also used in pharmacocosmesis to remove facial wrinkles and in the management of various types of pain (Davletov et al., 2005; Montecucco and Molgo, 2005; Dolly and Aoki, 2006). The range of clinical applications of BoNT/A is continuously increasing (Foster et al., 2006), and it is therefore important to elucidate the mechanisms involved in binding and intracellular trafficking of BoNT/A in neurons (Verderio et al., 2006).
Recently, BoNT/A was shown to enter neurons by binding to the synaptic vesicle protein SV2 (Dong et al., 2006; Mahrhold et al., 2006). BoNT/A is then internalized by vesicle endocytosis and translocated into the cytosol, in which it exerts its proteolytic activity (Schiavo et al., 2000; Rossetto et al., 2006). It is commonly believed that BoNT/A effects remain localized to synapses near the injection site. Contrary to this idea, here we demonstrate that BoNT/A undergoes retrograde axonal transport and is transcytosed to afferent neurons, in which it cleaves its substrate SNAP-25. These findings have important implications for understanding the mechanisms of action of BoNT/A.
Materials and Methods
Adult C57BL/6N mice and Sprague Dawley rats bred in our animal facility were used in this study. The animals were kept on a 12 h light/dark cycle and had access to food and water ad libitum. All experiments were performed in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC) and were approved by the Ministry of Health.
BoNT injections.
BoNT/A and BoNT/E were prepared and tested as described previously (Schiavo and Montecucco, 1995; Costantin et al., 2005; Caleo et al., 2007). Intrahippocampal injections (0.2 μl of a 10 nm solution; BoNT/A, n = 48 mice, BoNT/E, n = 27 mice) were performed with a glass micropipette connected to an injector. Coordinates (in millimeters with respect to bregma) were as follows: anteroposterior (A-P), −2.0; mediolateral (M-L), 1.5; 1.7 below dura. Injections of BoNT/A (3 nm, 0.5 μl; n = 45 animals) were made into the superior colliculus (SC) of Sprague Dawley rats at the following coordinates (in mm with respect to lambda): A-P, −0.5; M-L, 1; 2.6 below dura (Tropea et al., 2003). The axonal transport blocker colchicine (10 μg; Sigma, St. Louis, MO) was injected either into the SC (n = 3) or intravitreally (n = 4) 16–18 h before BoNT/A. Control experiments demonstrated that this dose of colchicine was effective in blocking transport of a retrograde tracer (cholera toxin B subunit) from the tectum to the retina.
For intraocular delivery of BoNT/E (25 nm), a glass micropipette was inserted at the ora serrata, and the injection volume (0.5 μl) was slowly released into the vitreous (Caleo et al., 1999). For analysis of transport in motoneurons, injections of BoNT/A (3 nm, 0.3 μl; n = 14 rats) were performed with a microsyringe on the right side of the snout at the center of the whisker pad (i.e., between rows B and C of the vibrissae) (Franchi, 2002; Franchi and Veronesi, 2004).
Immunoblotting.
Immunoblotting was performed as described previously (Caleo et al., 2007). Proteins were extracted with lysis buffer (1% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS, 10% glycerol, 20 mm TrisHCl, pH 8, 150 mm NaCl, 1 mm EDTA, 50 mm NaF, 1 mm Na2MoO4, 0.5 mm Na3VO4, 5 mm Na4P2O7, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.01 mm PMSF), and the total concentration of the samples was assessed with a protein assay kit (Bio-Rad, Hercules, CA) using a bovine serum albumin-based standard curve. Protein extracts were separated by electrophoresis and blotted, and filters were incubated with primary antibodies overnight at 4°C (anti-BoNT/E-cleaved SNAP-25, 1:50 dilution; anti-BoNT/A-cleaved SNAP-25, 1:500 dilution). Blots were then reacted with HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and developed by ECL (GE Healthcare, Little Chalfont, UK). Filters were also probed with anti-β-tubulin monoclonal antibody (1:500 dilution; Sigma), which serves as an internal standard for protein quantification.
Immunohistochemistry.
Animals were deeply anesthetized with chloral hydrate and perfused through the heart with freshly prepared 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Tissues were dissected and postfixed for 2 h at 4°C. Brain sections (40 μm thick) were cut with a freezing microtome. Eye cups were embedded in OCT compound (Leica Microsystems, Wetzlar, Germany), and retinal vertical sections (20 μm thick) were cut with a cryostat. For immunostaining, sections were blocked with 10% normal horse serum in PBS containing 0.25% Triton X-100 and then incubated overnight at room temperature with the anti-BoNT/A-cleaved SNAP-25 antibody (1:200 dilution). On the following day, sections were incubated with AlexaFluor568-conjugated secondary antibody (1:400; Invitrogen, Carlsbad, CA) for 2 h at room temperature. Sections were then washed in PBS and mounted with anti-fading agent (Vectashield; Vector Laboratories, Burlingame, CA).
For double-labeling experiments, sections were first reacted with anti-BoNT/A-cleaved SNAP-25, followed by AlexaFluor568-conjugated secondary antibodies. Sections were then incubated with one of the following antibodies: mouse monoclonal anti-neuronal nuclei (NeuN) (1:500; Millipore Bioscience Research Reagents, Temecula, CA), goat polyclonal anti-choline acetyltransferase (1:250; Millipore Bioscience Research Reagents), and goat polyclonal anti-vesicular acetylcholine transporter (1:1000; Millipore Bioscience Research Reagents). Bound primary antibodies were revealed by AlexaFluor488-conjugated secondary antibodies (Invitrogen).
Double labeling for BoNT/A-cleaved SNAP-25 and SV2C was performed as follows. Retinal sections were incubated first with the anti-cleaved SNAP-25 antibody that was revealed by AlexaFluor568-conjugated secondary antibodies. Sections were then fixed in paraformaldehyde for 30 min at room temperature, thoroughly washed in PBS, and incubated with rabbit polyclonal anti-SV2C antibody (a kind gift from Dr. Thomas Sudhof, Southwestern Medical Center, Dallas, TX). SV2C staining was revealed by AlexaFluor488-conjugated secondary antibodies.
Extracellular recordings of spiking activity.
Adult mice were unilaterally injected with either BoNT/A or BoNT/E into the dorsal hippocampus. They were recorded either 1 or 3 d after toxin administration (n = 4 animals per point). Three animals injected with vehicle solution were also recorded. Recordings were performed under urethane anesthesia as described previously (Costantin et al., 2005). Mice were anesthetized and placed in a stereotaxic frame. Body temperature was continuously monitored and maintained at 37°C by a thermostat-controlled electric blanket. After exposure of the cerebral surface, a micropipette (tip resistance, 2 MΩ) filled with 3 m NaCl was inserted into the brain to reach the dorsal hippocampus. Two to five penetrations per hemisphere were made to map spike activity in CA1. Starting coordinates were as follows (in mm from bregma): A-P, 2; M-L, 1.5. Spontaneous discharge of spikes of high amplitude was encountered as the electrode passed through the dense CA1 cell layer. The CA1 band was typically encountered at a depth of 1100–1300 μm from the pial surface. Signals were amplified 25,000-fold, bandpass filtered (500–5000 Hz), and conveyed to a computer for storage and analysis.
Results
Characterization of the antibodies against cleaved SNAP-25
Cleaved SNAP-25 was detected using polyclonal antibodies raised against either the BoNT/A- or the BoNT/E-truncated C-terminal peptide of SNAP-25. Western blot experiments demonstrated that they recognize specifically the cleaved form of SNAP-25 and not the whole protein (Fig. 1 A,B).
Figure 1.

Characterization of the antibodies against cleaved SNAP-25. A, Immunoblotting for BoNT/E-cleaved SNAP-25 [cl. SNAP-25(E)] on protein extracts from the mouse hippocampus. A 22 kDa band is detected in the sample injected with BoNT/E but not in a control uninjected hippocampus (control). β-Tubulin, Internal standard. B, Immunoblotting for BoNT/A-cleaved SNAP-25 [cl. SNAP-25(A)] on protein extracts from the mouse hippocampus. A 24 kDa band is detected in the sample injected with BoNT/A but not in a control uninjected hippocampus (control). β-Tubulin, Internal standard. C, Immunoblotting for BoNT/A-cleaved SNAP-25 [cl. SNAP-25(A)] on hippocampal extracts after injection of different doses of toxin. D, Quantification of the levels of BoNT/A-damaged SNAP-25 versus amounts of injected toxin. Values are normalized to the data obtained with 90 pg of BoNT/A. Error bars indicate SE and, when not seen, are within the symbol.
The antibody against BoNT/E-truncated SNAP-25 has been characterized previously (Costantin et al., 2005; Antonucci et al., 2007; Caleo et al., 2007). Here, a dose–response curve was done to determine the sensitivity of the anti-BoNT/A-cleaved SNAP-25 antibody and to establish how levels of cleaved SNAP-25 relate to the amounts of injected toxin. A series of adult mice were injected into the dorsal hippocampus with 0.2 μl of BoNT/A at different concentrations (3 nm, 0.3 nm, 0.03 nm, 3 pm, and 0.3 pm). Hippocampal tissues were harvested 3 d later, lysed, and subjected to Western blot analysis. The results from these experiments are reported in Figure 1, C and D. SNAP-25 cleavage was still detectable after intrahippocampal injection of 0.09 pg of BoNT/A (i.e., 0.2 μl of a 3 pm solution).
Different duration of action of BoNT/A and BoNT/E in the brain
Next, we compared the duration of action of BoNT/A and BoNT/E in the mouse brain. Adult mice received unilateral injections of either BoNT/A or BoNT/E (10 nm) into the dorsal hippocampus, and the injected hippocampi were dissected at different intervals to analyze the time course of SNAP-25 cleavage. We found that BoNT/E-truncated SNAP-25 was maximal 1–10 d after injection, decreased at 14 d, and was no longer detectable at 21 d (Fig. 2 A). BoNT/A effects were much more persistent. BoNT/A-cleaved SNAP-25 remained high for up to 120 d and was reduced at 180 d (Fig. 2 B). These data confirm for brain synapses the well known long duration of action of BoNT/A (Davletov et al., 2005; Montecucco and Molgo, 2005).
Figure 2.

Retrograde propagation of BoNT/A effects in the limbic system. A, Immunoblotting for BoNT/E-cleaved SNAP-25 [cl. S-25(E)] on protein extracts from the injected hippocampus at different times after BoNT/E injection. BoNT/E effects are extinguished by 21 d. Fifteen micrograms of protein were loaded per lane. β-Tubulin, Internal standard. B, Persistence of BoNT/A-truncated SNAP-25 in the injected hippocampus. Levels of BoNT/A-cleaved SNAP-25 [cl.S-25(A)] remain high for up to 120 d after injection and decrease at 180 d. C, Unilateral BoNT/E administration results in SNAP-25 cleavage in the injected (ipsi) but not contralateral (contra) hemisphere. D, Appearance of BoNT/A-truncated SNAP-25 on the side contralateral to the injection starting from 3 d after toxin delivery. E, Hippocampal coronal section from a mouse unilaterally injected with BoNT/A 3 d earlier. Staining for cleaved SNAP-25 (red) is evident in both hemispheres. Green, Neuronal counterstaining (NeuN antibody). Arrows indicate stratum oriens (s.o.) and radiatum (s.r.). Scale bar, 500 μm. F, Section through the entorhinal cortex ipsilateral to the side of intrahippocampal BoNT/A injection. Labeling for cleaved SNAP-25 (red) is evident in superficial layers. Neuronal counterstaining is in green. Note that other areas of the cortex are not stained, ruling out passive spread. Dorsal is up and lateral is to the left. Scale bar, 100 μm.
Spread of BoNT/A effects in the limbic system
We also analyzed SNAP-25 cleavage in the hippocampus contralateral to the injection of either BoNT/E or BoNT/A. BoNT/E-truncated SNAP-25 was restricted to the injected hemisphere at all time points examined (Fig. 2 C), in keeping with our previous reports (Costantin et al., 2005; Antonucci et al., 2007; Caleo et al., 2007). In animals treated with BoNT/A, proteolysis of SNAP-25 was confined to the ipsilateral hippocampus 1 d after toxin administration. Unexpectedly, robust expression of BoNT/A-cleaved SNAP-25 was detected on the contralateral side starting from 3 d after injection (Fig. 2 D).
Immunohistochemical analysis confirmed the interhemispheric spread of BoNT/A effects, because 3 d after injection there was strong labeling for cleaved SNAP-25 in both hippocampi (Fig. 2 E). On the contralateral side, staining was particularly prominent in stratum oriens and radiatum of the CA1 and CA3 sectors. Some staining was also visible in the hilus (Fig. 2 E). Notably, a small spot of the entorhinal cortex on the injected hemisphere was also positive for cleaved SNAP-25. Staining was found within the neuropil of superficial layers II–III (Fig. 2 F). Because neurons in layers II–III of entorhinal cortex project to the hippocampus, these data suggest a retrograde propagation of BoNT/A effects. Other parts of the cerebral cortex (such as parietal areas that are closer to the injection site in the dorsal hippocampus) were completely devoid of staining. The entorhinal cortex on the uninjected side was as well not labeled. Thus, BoNT/A-cleaved SNAP-25 appears exclusively in neuronal populations that are directly connected to the injection site.
Appearance of BoNT/A-truncated SNAP-25 in the untreated hippocampus results in functional consequences
To assess whether appearance of BoNT/A-damaged SNAP-25 in the contralateral, uninjected hemisphere has functional consequences, we made in vivo recordings of hippocampal activity. As reported previously for BoNT/E (Costantin et al., 2005; Antonucci et al., 2007), we found that unilateral BoNT/A infusion into the hippocampus produced within 1 d a nearly complete blockade of spontaneous spiking activity from CA1 pyramidal cells on the injected side (Fig. 3 A). Three days after BoNT/A injection, activity was strongly depressed in both the injected side and the contralateral hippocampus (Fig. 3 B). This is in keeping with the appearance of BoNT/A-truncated SNAP-25 in the untreated hemisphere 3 d after BoNT/A (Fig. 2 D). Silencing of the contralateral side was not secondary to blockade of activity in the injected hippocampus. Indeed, recordings performed 3 d after unilateral injection of BoNT/E (that does not spread contralaterally) revealed blockade of activity in the injected side and vigorous discharges in the untreated hemisphere (Fig. 3 C).
Figure 3.

Unilateral, intrahippocampal infusion of BoNT/A results in bilateral blockade of activity within 3 d. A, B, Representative examples of extracellular recordings of spiking activity from CA1, performed 1 d (A) and 3 d (B) after unilateral administration of BoNT/A into the dorsal hippocampus. Note that spontaneous discharges in the contralateral side are strongly suppressed 3 d after BoNT/A. Ipsi, Injected side; contra, contralateral side. C, Extracellular recordings from CA1 performed 3 d after unilateral administration of BoNT/E. Activity is blocked only in the treated side.
Spread of BoNT/A effects in the visual system
To obtain additional evidence for a retrograde spread of BoNT/A, we turned to the visual system. It is well known that the SC receives massive unidirectional projections from the contralateral retina and ipsilateral primary visual cortex. BoNT/A was injected into the SC of adult rats, and SNAP-25 cleavage was assessed either 1 or 3 d later. Western blot experiments performed at 1 d demonstrated high levels of BoNT/A-truncated SNAP-25 in the injected SC but no SNAP-25 proteolysis in either the retina or visual cortex (Fig. 4 A). Notably, truncated SNAP-25 appeared in the contralateral retina and ipsilateral striate cortex 3 d after injection (Fig. 4 A). Levels of cleaved SNAP-25 in the retina and cortex were much smaller than those found in the injection site (Fig. 4 A). BoNT/A-cleaved SNAP-25 was detectable in neither the ipsilateral retina nor the contralateral primary visual cortex (Fig. 4 B). Retrograde appearance of BoNT/A-truncated SNAP-25 in the retina was prevented by infusion of the microtubule depolymerizing agent colchicine, pointing to a role for microtubule-based axonal transport. Colchicine (10 μg) was administered either into the SC or intravitreally 16–18 h before tectal injection of BoNT/A (Fig. 4 C). Together, these data rule out a systemic spread of the toxin (via blood or CSF).
Figure 4.

Retrograde propagation of BoNT/A effects in the visual system. A, Immunoblotting for cleaved SNAP-25 [cl. SNAP-25(A)] on protein extracts from the injected SC, the contralateral retina (ret), and the ipsilateral visual cortex (ctx). Spread of BoNT/A effects to the retina and cortex is evident 3 d after administration of the toxin. Fifteen micrograms of protein were loaded per lane. β-Tubulin, Internal standard. B, Immunoblotting demonstrates no cleaved SNAP-25 [cl. S-25(A)] in either the ipsilateral retina (ret i) or contralateral striate cortex (ctx c). C, Immunoblotting for cleaved SNAP-25 [cl. S-25(A)] on protein extracts from the injected SC and contralateral retina (ret). Intravitreal injection of colchicine 16–18 h before BoNT/A (colchicine+BoNT/A) prevents retrograde spread of toxin effects. D, Immunostaining for BoNT/A-truncated SNAP-25 (red) in a coronal section of primary visual cortex ipsilateral to the tectal injection. Staining for neurons is in green. Immunoreactivity for cleaved SNAP-25 is concentrated in layer V. Cortical layers are indicated on the right. Scale bar, 100 μm. E, Transverse section through the retina contralateral to the injection site demonstrates labeling for cleaved SNAP-25 (red) in the inner plexiform layer (IPL); GCL, ganglion cell layer. Green, Nuclear staining. Scale bar, 20 μm.
To determine the localization of cleaved SNAP-25 in the visual cortex and retina, immunostaining experiments were performed. Immunolabeling of cortical sections demonstrated that cleaved SNAP-25 was selectively localized within the neuropil of layer V of primary visual cortex (Fig. 4 D). This layer is known to contain the pyramidal cells projecting to the SC. Other layers of striate cortex (Fig. 4 D) as well as more medial cortical areas overlying the SC were not stained. This provides additional evidence against passive spread of the toxin and/or its cleaved substrate.
In the retina, labeling was found in punctate structures within the inner plexiform layer. Staining was particularly prominent in ON and OFF sublaminas that contain synaptic inputs to retinal ganglion cells (Fig. 4 E), the retinal neurons projecting to the SC. Colocalization analysis with neurochemical markers was performed to determine the type of synapses containing cleaved SNAP-25. We found that cleaved SNAP-25 exhibited a high degree of colocalization with markers of cholinergic terminals [choline acetyltransferase (Fig. 5 A) and vesicular acetylcholine transporter (data not shown)]. The pattern of staining for BoNT/A-damaged SNAP-25 in the retina (Fig. 4 E) is strikingly similar to that observed for the synaptic vesicle protein SV2C (Wang et al., 2003). Indeed, we found that most of the cleaved SNAP-25-stained profiles were also positive for SV2C (Fig. 5 B). We conclude that BoNT/A-truncated SNAP-25 appears mainly in cholinergic, SV2C-positive synapses of the retina 3 d after toxin injection into the SC.
Figure 5.

Cleaved SNAP-25 is mainly localized within cholinergic, SV2C-positive terminals in the retina. A, Confocal images of a single optical section through the inner plexiform layer showing colocalization of cleaved SNAP-25 (red) with choline acetyltransferase (ChAT; green). B, Confocal images of a single optical section through the inner plexiform layer showing colocalization of cleaved SNAP-25 (red) with SV2C (green). The ganglion cell layer is down. Scale bar, 10 μm.
Functional proof that active BoNT/A is retrogradely transported from the superior colliculus to the retina
These findings demonstrate that BoNT/A effects propagate from the site of delivery to distant synapses via a retrograde route. Specifically, cleaved SNAP-25 appears in synapses contacting neuronal cell bodies whose terminals were exposed to the toxin, indicative of retrograde axonal transport followed by transcytosis. Because only the distribution of cleaved SNAP-25 was examined, it is at least theoretically possible that the cleaved substrate is transported and transcytosed rather than the toxin itself. To formally prove a retrograde transfer of BoNT/A, we designed an experiment (Fig. 6 A) in which a group of rats (n = 14) were injected with BoNT/A into the SC. Four of these rats were dissected 3 d later and showed the expected SNAP-25 cleavage in the retina (Fig. 6 C, left lanes). In the remaining rats, we sectioned the optic nerve to prevent additional transport from the SC, and we injected BoNT/E intraocularly (Fig. 6 A). BoNT/E removes the BoNT/A-truncated C-terminal peptide of SNAP-25 (Fig. 6 B). Accordingly, delivery of BoNT/E into the eye led to the disappearance or strong reduction of BoNT/A-truncated SNAP-25 within 2 d (Fig. 6 C, middle lanes). We reasoned that this reduction should be permanent if cleaved SNAP-25 is transported, because there is no way of generating new truncated substrate in the retina. Conversely, if active BoNT/A is transported, its action would reappear when BoNT/E effects have vanished (Keller et al., 1999). Indeed, high amounts of BoNT/A-cleaved SNAP-25 reappeared in the retina at the completion of BoNT/E effects (i.e., 25 d after BoNT/E) (Fig. 6 C, right lines). Quantification of the immunoblotting experiments indicated a very significant reduction of BoNT/A-truncated SNAP-25 2 d after BoNT/E (one-way ANOVA, p < 0.01 followed by Tukey's test, p < 0.05) and a return of high levels of the cleaved substrate 25 d after BoNT/E (Fig. 6 D). The return of BoNT/A-truncated SNAP-25 indicates that the BoNT/A protease was transported to the retina during the first 3 d after injection and remained catalytically active in retinal terminals. Thus, a significant fraction of active BoNT/A is retrogradely transported from the optic tectum to the retina, in which it cleaves SNAP-25.
Figure 6.
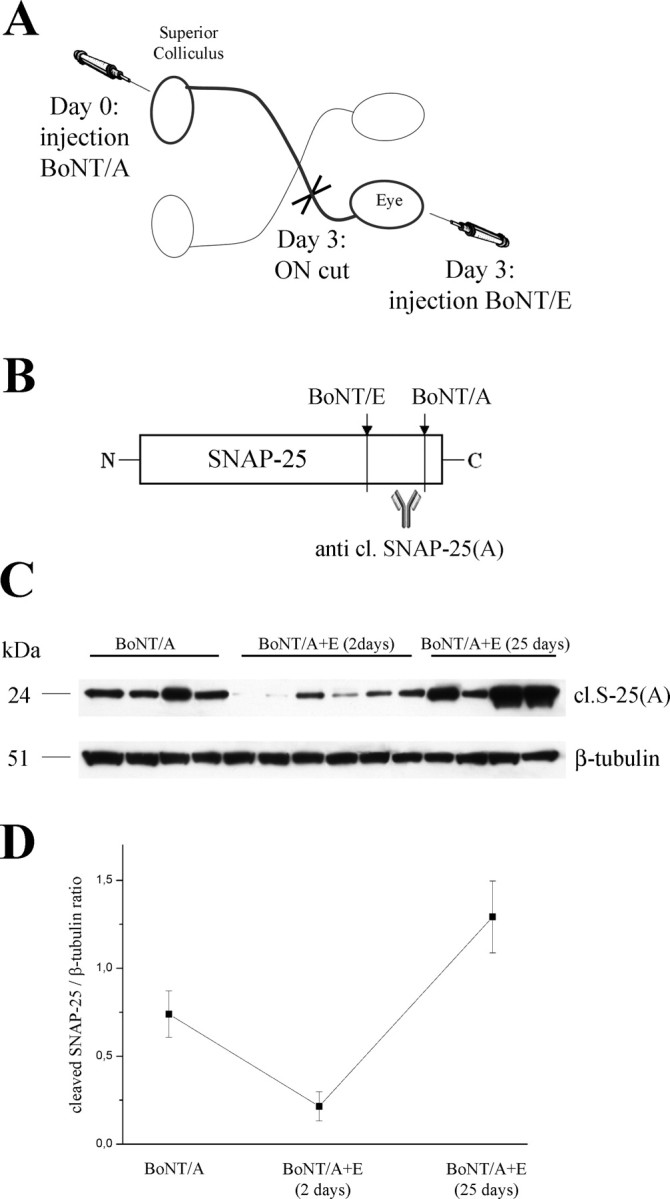
Retrograde transport of active BoNT/A in the visual system. A, Experimental design. Adult rats were injected with BoNT/A into the SC. At day 3, the optic nerve (ON) was sectioned, and BoNT/E was delivered into the vitreous. Retinal tissue samples were dissected either 2 or 25 d after BoNT/E. B, Schematic representation of BoNT/A and BoNT/E cleavage sites. BoNT/E proteolysis removes the BoNT/A-truncated C-terminal peptide of SNAP-25. C, Immunoblotting for BoNT/A-cleaved SNAP-25 on retinal protein extracts in animals that received BoNT/A into the SC. Samples were taken 3 d after BoNT/A (BoNT/A, left lanes), and either 2 d (middle lanes) or 25 d (right lanes) after ON cut and intraocular injection of BoNT/E. The recovery of high levels of BoNT/A-altered SNAP-25 at the completion of BoNT/E effects indicates persistent activity of BoNT/A within the retina. Fifty micrograms of protein were loaded per lane. β-Tubulin, Internal standard. D, Quantification of the immunoblotting experiments. BoNT/A-damaged SNAP-25 is high 3 d after BoNT/A, decreases 2 d after intravitreal BoNT/E, and returns high 25 d after BoNT/E.
Retrograde propagation of BoNT/A effects in facial motoneurons
The data reported above demonstrate retrograde transfer of BoNT/A in central neurons. Because the natural target of BoNT/A is the neuromuscular junction, it was important to examine transport of BoNT/A in motoneurons. For this analysis, we selected facial motoneurons projecting to the whisker muscles. This model system was chosen because facial motoneurons lack direct proprioceptive and sensory innervation (Paxinos, 2004), thus excluding any possible supply of BoNT/A and/or cleaved SNAP-25 by anterograde axonal transport. Adult rats were injected with BoNT/A into the whisker pad. One day after injection, the injected vibrissae were atonic and positioned backward. The facial nucleus ipsilateral to the injection was microdissected 3 d after BoNT/A. Western blot analysis demonstrated significant amounts of BoNT/A-truncated SNAP-25 in the facial nucleus (Fig. 7 A). Immunostaining with the anti-BoNT/A-cleaved SNAP-25 antibody confirmed this finding. Cleaved SNAP-25-immunopositive profiles were detected in the neuropil surrounding the soma of facial motoneurons (Fig. 7 B).
Figure 7.

Retrograde spread of BoNT/A effects in facial motoneurons. A, Immunoblotting showing expression of cleaved SNAP-25 in the facial nucleus 3 d after BoNT/A injection into the whisker pad. Nor, Tissue from a normal uninjected animal. One hundred micrograms of protein were loaded per lane. β-Tubulin, Internal standard. B, Confocal image of facial motoneurons (NeuN staining, green) surrounded by cleaved SNAP-25-immunopositive profiles (red). Scale bar, 20 μm.
Discussion
In this paper, we have examined the distribution of cleaved SNAP-25 after injection of BoNT/A into central and peripheral targets. We found that BoNT/A-truncated SNAP-25 appears not only at the injection site but also in distant regions that project to the infusion area. This retrograde spread was blocked by colchicine, pointing to an involvement of microtubule-dependent axonal transport. Cleaved SNAP-25 appeared at distant synaptic sites, indicative of transcytosis. The finding of a persistent BoNT/A catalytic activity inside distant synaptic terminals proved that active BoNT/A (rather than the cleaved substrate) is retrogradely transported and transcytosed. Finally, electrophysiological recordings showed functional consequences of the spread toxin. We conclude that BoNT/A does not stay confined to the injection site but spreads in physiologically significant amounts to distant synapses via a retrograde axonal route.
Retrograde transport and transcytosis of BoNT/A
We used detection of cleaved SNAP-25 as an assay of BoNT/A trafficking. This is the most sensitive test to monitor the presence of active BoNT/A in vivo, because a single toxin molecule can proteolyse a large number of SNAP-25 target molecules, providing a dramatic amplifying effect. We found high amounts of BoNT/A-truncated SNAP-25 in the infusion area, consistent with the well known local actions of BoNT/A (Johnson, 1999; Montecucco and Molgo, 2005). In addition, there was significant SNAP-25 cleavage in distant regions that send axonal projections to the toxin injection site. No SNAP-25 fragments were detectable in nonconnected areas, providing clear evidence that BoNT/A does not spread in a passive way from the site of injection in the CNS. A similar conclusion, but with a lower spatial resolution, was achieved for the peripheral nervous system (Eleopra et al., 2004).
In the case of intrahippocampal infusions, staining was specifically found in the contralateral hippocampus and superficial layers of the ipsilateral entorhinal cortex. The two hippocampi are known to be interconnected by a large set of commissural connections, originating predominantly from CA3 and CA1 sectors (Paxinos, 2004). Spread of SNAP-25 cleavage to the contralateral, uninjected hippocampus was accompanied by significant physiological changes, as shown by the blockade of spontaneous firing of pyramidal cells. In the entorhinal cortex ipsilateral to the injected hippocampus, staining for cleaved SNAP-25 was restricted to the superficial layers. These layers contain the projection neurons innervating the hippocampal dentate gyrus, thus suggesting a retrograde axonal route.
Previous data in the literature indicated the possibility that BoNT/A might undergo a limited extent of retrograde axonal transport from the muscle to the spinal cord. Radiolabeled BoNT/A injected into the cat gastrocnemius muscle was detected in the sciatic nerve and spinal cord 24–38 h later. The authors concluded that BoNT/A reaches the spinal cord after intramuscular injection (Habermann, 1974; Wiegand et al., 1976). However, it remained unclear whether the radioactivity corresponded to intact, catalytically active BoNT/A, also because the iodination procedure used is known to affect toxicity. Moreover, no evidence for transcytosis was provided (Habermann, 1974; Wiegand et al., 1976).
Our experiments in the visual system provide the first evidence that, in addition to the local effects at the intoxicated synapses, a significant fraction of active BoNT/A is transported to the cell soma and released in intact form to second-order neurons. Thus, not only can tetanus neurotoxin undergo transcytosis within the CNS (Schwab et al., 1979), but BoNT/A can also. After tectal injections of the toxin, cleaved SNAP-25 appeared in cholinergic synapses in the retina. These data indicate retrograde transport to retinal ganglion cells followed by transcytosis into starbust amacrine cells but leave open the question of whether the toxin itself or the cleaved product circulates in the brain. Accordingly, we provided a functional proof that active BoNT/A is indeed retrogradely transported and transcytosed. Rats were injected with BoNT/A into the SC and, 3 d later, when BoNT/A-truncated SNAP-25 is detectable in the retina, the optic nerve was transected to prevent additional transport from the tectum. BoNT/E was also injected intraocularly to convert the BoNT/A-cleaved SNAP-25 into a BoNT/E-altered form, which is no longer recognized by the anti-BoNT/A-cleaved SNAP-25 antibody. We observed a complete recovery of the BoNT/A-truncated SNAP-25 at the completion of the BoNT/E effects, indicating that active BoNT/A was transported to the eye within the first 3 d after injection and retained its enzymatic activity in retinal synapses.
Our findings shed a new light on the trafficking of BoNT/A in neurons. Although it is currently held that all internalized BoNT/A is released into the neuronal cytosol, the present data indicate that a significant fraction of BoNT/A is loaded onto the retrograde transport machinery, escapes degradation in the cell soma, and is released at synaptic sites and eventually taken up by afferent neurons, in which it cleaves SNAP-25. The precise molecular mechanisms as well as the cellular compartments involved in this process remain to be determined.
Retrograde transport and transcytosis have been described previously for tetanus neurotoxin and some trophic factors (von Bartheld, 2004; Deinhardt and Schiavo, 2005; Rind et al., 2005; Deinhardt et al., 2006). For example, rat hypoglossal motoneurons retrogradely transport glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor and subsequently transfer these factors across the synaptic cleft to afferent synapses (Rind et al., 2005). As demonstrated for tetanus neurotoxin, transcytosis may be restricted to specific synaptic inputs. Indeed, it is well known that, after retrograde transport from the muscle to the spinal cord, tetanus toxin is selectively transcytosed to inhibitory interneurons, in which it blocks GABA exocytosis (Lalli et al., 2003; von Bartheld, 2004).
Some specificity in transcytosis appear to exist also for BoNT/A, because we have shown that profiles immunopositive for BoNT/A-truncated SNAP-25 in the retina are predominantly (though not exclusively) colocalized with markers of cholinergic terminals. These data suggest a preferential uptake of BoNT/A by cholinergic amacrine cells. It is worth noting that BoNT/A-cleaved SNAP-25 also highly colocalized with the synaptic vesicle protein SV2C in the retina. Among the SV2 isoforms acting as protein receptors for BoNT/A, SV2C exhibits the most robust BoNT/A binding activity (Dong et al., 2006; Mahrhold et al., 2006). This might explain the preferential uptake of BoNT/A by cholinergic retinal terminals.
Comparison with previous studies and clinical implications
Our studies document retrograde spread of BoNT/A effects to the facial nucleus in the brain after injection of the toxin into the whisker muscles. The total amount of BoNT/A injected into the whisker pad (135 pg) was based on previous studies aimed at blocking transmission at the neuromuscular junction (Franchi, 2002; Franchi and Veronesi, 2004). Previous reports showing spinal spread of radiolabeled toxin after intramuscular injection used much higher amounts of BoNT/A in cats [i.e., >100 ng (Habermann, 1974; Wiegand et al., 1976)]. Central effects of BoNT/A were reported after the injection of 9 ng of BoNT/A into the lateral rectus muscle of the cat (Moreno-Lopez et al., 1997a; Pastor et al., 1997). In clinical practice, doses of BoNT/A vary widely depending on type of the disease and size of the muscles to be treated. Doses of BoNT/A ranging from 2 to 22 ng are typically reported for the treatment of torticollis spasmodicus and head/neck dystonia (Garner et al., 1993; Wohlfarth et al., 2001). Comparison of these doses with those used by us suggests that retrograde transport may be a common occurrence after intramuscular delivery of BoNT/A. The fraction of BoNT/A that spreads retrogradely in different systems is likely to depend on several factors, including amount and concentration of injected toxin and levels of expression of relevant receptors in the specific pathways under consideration.
Several papers have described changes at the level of the CNS in man and animals treated intramuscularly with BoNT/A (Garner et al., 1993; Giladi, 1997; Moreno-Lopez et al., 1997a,b; Kanovsky et al., 1998; Wohlfarth et al., 2001; Abbruzzese and Berardelli, 2006; Kim et al., 2006). For example, delivery of BoNT/A into the lateral rectus muscle of cats dramatically reduces the firing rates of abducens motoneurons projecting to the injected muscle (Moreno-Lopez et al., 1997a,b). These electrophysiological changes are accompanied by ultrastructural alterations in synaptic boutons impinging on motoneurons. These boutons show an increase in the total number of synaptic vesicles, an observation consistent with impairments in neuroexocytosis (Pastor et al., 1997).
Physiological studies in humans also demonstrate central effects of the toxin that are unlikely explained by local neuromuscular effects alone. For example, changes have been described at the level of spinal cord circuitry (Priori et al., 1995; Wohlfarth et al., 2001). An alteration of cortical excitability has also been reported in dystonic patients treated intramuscularly with BoNT/A (Kanovsky et al., 1998; Gilio et al., 2000).
These central effects of BoNT/A have been usually ascribed to (1) plastic rearrangements subsequent to denervation or (2) alterations in sensory input (Priori et al., 1995; Giladi, 1997; Abbruzzese and Berardelli, 2006). The data presented in our study indicate that they may be consequences of a direct central action of BoNT/A. Indeed, we have shown for the first time that BoNT/A applied in the periphery can affect central circuits via retrograde transport and transcytosis. Our findings provide the first evidence for a mechanism by which BoNT/A can gain access to the CNS after peripheral administration.
Footnotes
This work was supported by Telethon Grant GGP04086 (M.C.) and Fondo per gli Investimenti della Ricerca di Base Project “CHEM-PROFARMA-NET.” We are grateful to Lamberto Maffei, Cesare Montecucco, Yuri Bozzi, Lucia Galli-Resta, and Enrica Strettoi for comments on this manuscript. Chiara Cerri and Alessio Lancioni participated in some experiments.
References
- Abbruzzese G, Berardelli A. Neurophysiological effects of botulinum toxin type A. Neurotox Res. 2006;9:109–114. doi: 10.1007/BF03033927. [DOI] [PubMed] [Google Scholar]
- Antonucci F, Di Garbo A, Novelli E, Manno I, Sartucci F, Bozzi Y, Caleo M. Botulinum neurotoxin E (BoNT/E) reduces CA1 neuron loss and granule cell dispersion, with no effects on chronic seizures, in a mouse model of temporal lobe epilepsy. Exp Neurol. 2007 doi: 10.1016/j.expneurol.2007.11.012. in press. [DOI] [PubMed] [Google Scholar]
- Caleo M, Lodovichi C, Maffei L. Effects of nerve growth factor on visual cortical plasticity require afferent electrical activity. Eur J Neurosci. 1999;11:2979–2984. doi: 10.1046/j.1460-9568.1999.00737.x. [DOI] [PubMed] [Google Scholar]
- Caleo M, Restani L, Gianfranceschi L, Costantin L, Rossi C, Rossetto O, Montecucco C, Maffei L. Transient synaptic silencing of developing striate cortex has persistent effects on visual function and plasticity. J Neurosci. 2007;27:4530–4540. doi: 10.1523/JNEUROSCI.0772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantin L, Bozzi Y, Richichi C, Viegi A, Antonucci F, Funicello M, Gobbi M, Mennini T, Rossetto O, Montecucco C, Maffei L, Vezzani A, Caleo M. Antiepileptic effects of botulinum neurotoxin E. J Neurosci. 2005;25:1943–1951. doi: 10.1523/JNEUROSCI.4402-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davletov B, Bajohrs M, Binz T. Beyond BOTOX: advantages and limitations of individual botulinum neurotoxins. Trends Neurosci. 2005;28:446–452. doi: 10.1016/j.tins.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Schiavo G. Endocytosis and retrograde axonal traffic in motor neurons. Biochem Soc Symp. 2005:139–150. doi: 10.1042/bss0720139. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Berninghausen O, Willison HJ, Hopkins CR, Schiavo G. Tetanus toxin is internalized by a sequential clathrin-dependent mechanism initiated within lipid microdomains and independent of epsin1. J Cell Biol. 2006;174:459–471. doi: 10.1083/jcb.200508170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolly JO, Aoki KR. The structure and mode of action of different botulinum toxins. Eur J Neurol. 2006;13(Suppl 4):1–9. doi: 10.1111/j.1468-1331.2006.01648.x. [DOI] [PubMed] [Google Scholar]
- Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, Chapman ER. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592–596. doi: 10.1126/science.1123654. [DOI] [PubMed] [Google Scholar]
- Eleopra R, Tugnoli V, Quatrale R, Rossetto O, Montecucco C. Different types of botulinum toxin in humans. Mov Disord. 2004;19(Suppl 8):S53–S59. doi: 10.1002/mds.20010. [DOI] [PubMed] [Google Scholar]
- Foster KA, Bigalke H, Aoki KR. Botulinum neurotoxin: from laboratory to bedside. Neurotox Res. 2006;9:133–140. doi: 10.1007/BF03033931. [DOI] [PubMed] [Google Scholar]
- Franchi G. Time course of motor cortex reorganization following botulinum toxin injection into the vibrissal pad of the adult rat. Eur J Neurosci. 2002;16:1333–1348. doi: 10.1046/j.1460-9568.2002.02195.x. [DOI] [PubMed] [Google Scholar]
- Franchi G, Veronesi C. Time course for the reappearance of vibrissal motor representation following botulinum toxin injection into the vibrissal pad of the adult rat. Eur J Neurosci. 2004;20:1873–1884. doi: 10.1111/j.1460-9568.2004.03653.x. [DOI] [PubMed] [Google Scholar]
- Garner CG, Straube A, Witt TN, Gasser T, Oertel WH. Time course of distant effects of local injections of botulinum toxin. Mov Disord. 1993;8:33–37. doi: 10.1002/mds.870080106. [DOI] [PubMed] [Google Scholar]
- Giladi N. The mechanism of action of botulinum toxin type A in focal dystonia is most probably through its dual effect on efferent (motor) and afferent pathways at the injected site. J Neurol Sci. 1997;152:132–135. doi: 10.1016/s0022-510x(97)00151-2. [DOI] [PubMed] [Google Scholar]
- Gilio F, Curra A, Lorenzano C, Modugno N, Manfredi M, Berardelli A. Effects of botulinum toxin type A on intracortical inhibition in patients with dystonia. Ann Neurol. 2000;48:20–26. [PubMed] [Google Scholar]
- Habermann E. 125I-labeled neurotoxin from Clostridium botulinum A: preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn Schmiedebergs Arch Pharmacol. 1974;281:47–56. doi: 10.1007/BF00500611. [DOI] [PubMed] [Google Scholar]
- Johnson EA. Clostridial toxins as therapeutic agents: benefits of nature's most toxic proteins. Annu Rev Microbiol. 1999;53:551–575. doi: 10.1146/annurev.micro.53.1.551. [DOI] [PubMed] [Google Scholar]
- Kanovsky P, Streitova H, Dufek J, Znojil V, Daniel P, Rektor I. Change in lateralization of the P22/N30 cortical component of median nerve somatosensory evoked potentials in patients with cervical dystonia after successful treatment with botulinum toxin A. Mov Disord. 1998;13:108–117. doi: 10.1002/mds.870130122. [DOI] [PubMed] [Google Scholar]
- Keller JE, Neale EA, Oyler G, Adler M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999;456:137–142. doi: 10.1016/s0014-5793(99)00948-5. [DOI] [PubMed] [Google Scholar]
- Kim DY, Oh BM, Paik NJ. Central effect of botulinum toxin type A in humans. Int J Neurosci. 2006;116:667–680. doi: 10.1080/00207450600674525. [DOI] [PubMed] [Google Scholar]
- Lalli G, Bohnert S, Deinhardt K, Verastegui C, Schiavo G. The journey of tetanus and botulinum neurotoxins in neurons. Trends Microbiol. 2003;11:431–437. doi: 10.1016/s0966-842x(03)00210-5. [DOI] [PubMed] [Google Scholar]
- Mahrhold S, Rummel A, Bigalke H, Davletov B, Binz T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006;580:2011–2014. doi: 10.1016/j.febslet.2006.02.074. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Molgo J. Botulinal neurotoxins: revival of an old killer. Curr Opin Pharmacol. 2005;5:274–279. doi: 10.1016/j.coph.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem Sci. 2005;30:367–372. doi: 10.1016/j.tibs.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Moreno-Lopez B, de la Cruz RR, Pastor AM, Delgado-Garcia JM. Effects of botulinum neurotoxin type A on abducens motoneurons in the cat: alterations of the discharge pattern. Neuroscience. 1997a;81:437–455. doi: 10.1016/s0306-4522(97)00199-1. [DOI] [PubMed] [Google Scholar]
- Moreno-Lopez B, Pastor AM, de la Cruz RR, Delgado-Garcia JM. Dose-dependent, central effects of botulinum neurotoxin type A: a pilot study in the alert behaving cat. Neurology. 1997b;48:456–464. doi: 10.1212/wnl.48.2.456. [DOI] [PubMed] [Google Scholar]
- Pastor AM, Moreno-Lopez B, De La Cruz RR, Delgado-Garcia JM. Effects of botulinum neurotoxin type A on abducens motoneurons in the cat: ultrastructural and synaptic alterations. Neuroscience. 1997;81:457–478. doi: 10.1016/s0306-4522(97)00200-5. [DOI] [PubMed] [Google Scholar]
- Paxinos G. The rat nervous system. Oxford: Elsevier; 2004. [Google Scholar]
- Priori A, Berardelli A, Mercuri B, Manfredi M. Physiological effects produced by botulinum toxin treatment of upper limb dystonia. Changes in reciprocal inhibition between forearm muscles. Brain. 1995;118:801–807. doi: 10.1093/brain/118.3.801. [DOI] [PubMed] [Google Scholar]
- Rind HB, Butowt R, von Bartheld CS. Synaptic targeting of retrogradely transported trophic factors in motoneurons: comparison of glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor, and cardiotrophin-1 with tetanus toxin. J Neurosci. 2005;25:539–549. doi: 10.1523/JNEUROSCI.4322-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto O, Morbiato L, Caccin P, Rigoni M, Montecucco C. Presynaptic enzymatic neurotoxins. J Neurochem. 2006;97:1534–1545. doi: 10.1111/j.1471-4159.2006.03965.x. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Montecucco C. Tetanus and botulism neurotoxins: isolation and assay. Methods Enzymol. 1995;248:643–652. doi: 10.1016/0076-6879(95)48041-2. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Suda K, Thoenen H. Selective retrograde transsynaptic transfer of a protein, tetanus toxin, subsequent to its retrograde axonal transport. J Cell Biol. 1979;82:798–810. doi: 10.1083/jcb.82.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tropea D, Caleo M, Maffei L. Synergistic effects of brain-derived neurotrophic factor and chondroitinase ABC on retinal fiber sprouting after denervation of the superior colliculus in adult rats. J Neurosci. 2003;23:7034–7044. doi: 10.1523/JNEUROSCI.23-18-07034.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turton K, Chaddock JA, Acharya KR. Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem Sci. 2002;27:552–558. doi: 10.1016/s0968-0004(02)02177-1. [DOI] [PubMed] [Google Scholar]
- Verderio C, Rossetto O, Grumelli C, Frassoni C, Montecucco C, Matteoli M. Entering neurons: botulinum toxins and synaptic vesicle recycling. EMBO Rep. 2006;7:995–999. doi: 10.1038/sj.embor.7400796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS. Axonal transport and neuronal transcytosis of trophic factors, tracers, and pathogens. J Neurobiol. 2004;58:295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- Wang MM, Janz R, Belizaire R, Frishman LJ, Sherry DM. Differential distribution and developmental expression of synaptic vesicle protein 2 isoforms in the mouse retina. J Comp Neurol. 2003;460:106–122. doi: 10.1002/cne.10636. [DOI] [PubMed] [Google Scholar]
- Wiegand H, Erdmann G, Wellhoner HH. 125I-labelled botulinum A neurotoxin: pharmacokinetics in cats after intramuscular injection. Naunyn Schmiedebergs Arch Pharmacol. 1976;292:161–165. doi: 10.1007/BF00498587. [DOI] [PubMed] [Google Scholar]
- Wohlfarth K, Schubert M, Rothe B, Elek J, Dengler R. Remote F-wave changes after local botulinum toxin application. Clin Neurophysiol. 2001;112:636–640. doi: 10.1016/s1388-2457(01)00478-3. [DOI] [PubMed] [Google Scholar]