

Abstract
The K+ M-current (I M, Kv7) is an important regulator of cortical excitability, and mutations in these channels cause a seizure disorder in humans. The neuropeptide somatostatin (SST), which has antiepileptic properties, augments I M in hippocampal CA1 pyramidal neurons. We used SST receptor knock-out mice and subtype-selective ligands to investigate the receptor subtype that couples to I M and mediates the antiepileptic effects of SST. Using pentylenetetrazole as a chemoconvulsant, SST2, SST3, and SST4 receptor knock-out mice all had shorter latencies to different seizure stages and increased seizure severity when compared with wild-type mice. However, the most robust differences were observed in the SST4 knock-outs. When seizures were induced by systemic injection of kainate, only SST4 knock-outs showed an increase in seizure sensitivity. We next examined the action of SST and subtype-selective SST agonists on electrophysiological parameters in hippocampal slices of wild-type and receptor knock-out mice. SST2 and SST4 appear to mediate the majority of SST inhibition of epileptiform activity in CA1. SST lacked presynaptic effects in mouse CA1, in contrast to our previous findings in rat. SST increased I M in CA1 pyramidal neurons of wild-type and SST2 knock-out mice, but not SST4 knock-out mice. Using M-channel blockers, we found that SST4 coupling to M-channels is critical to its inhibition of epileptiform activity. This is the first demonstration of an endogenous enhancer of I M that is important in controlling seizure activity. SST4 receptors could therefore be an important novel target for developing new antiepileptic and antiepileptogenic drugs.
Keywords: somatostatin, Kv7 channels, KCNQ, epilepsy, knock-out mice, electrophysiology
Introduction
The neuropeptide somatostatin (SST) is an important regulator of hippocampal excitability. SST has strong antiseizure actions in many rodent models (Vezzani and Hoyer, 1999), with the hippocampus indicated as the major site of action (Mazarati and Telegdy, 1992). At the cellular level, SST has inhibitory actions in rat CA1 hippocampus, including inhibition of excitatory neurotransmission (Boehm and Betz, 1997; Tallent and Siggins, 1997), and augmentation of two distinct K+ current, the voltage-sensitive M-current (I M) (Moore et al., 1988) and a voltage-insensitive leak current (Schweitzer et al., 1998).
The Kv7 (KCNQ) family of K+ channels comprise M-channels. Mutations in two members of this family, Kv7.2 and Kv7.3, cause the epilepsy syndrome benign familial neonatal convulsions (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998). Patients with this disorder develop seizures shortly after birth that spontaneously remit within a few months. However, these patients also have a tenfold higher likelihood of developing temporal lobe epilepsy as adults (Singh et al., 2003), suggesting these channels remain important throughout the lifespan. We showed previously that I M is critical in preventing the transition from preseizure interictal epileptiform bursting to ictal seizure-like events in hippocampal slices from both adult and immature rats (Qiu et al., 2007). Furthermore, the drug retigabine, that robustly increases I M, has potent antiepileptic actions in pharmacoresistant animal models (Armand et al., 1999, 2000). After successful completion of Phase II clinical trials (Porter et al., 2007b), retigabine is currently in Phase III clinical trials for treatment of refractory partial seizures (Porter et al., 2007a,b).
The contribution of I M to the antiepileptic actions of SST is unknown. The neuropeptide nociceptin/OFQ also couples to I M in CA1 and CA3 hippocampus, however, its antiepileptic actions, at least in vitro, appear to be independent of this current (Tallent et al., 2001). Furthermore, whether endogenous regulators of M-channels would have similar antiepileptic actions as retigabine has not been determined. Targeting an upstream regulator of M-channels could be advantageous, because Kv7 channels are widely distributed throughout the brain and periphery, increasing the possibility of unwanted side effects.
The SST family of receptors has 5 members, SST1–SST5, all of which are Gi/Go-coupled receptors. SST1–SST4 are present in the brain, and SST2, SST3, and SST4 are expressed in cortex and hippocampus (Dournaud et al., 1996; Handel et al., 1999; Schreff et al., 2000), although expression of SST1 in forebrain is still somewhat controversial (Hervieu and Emson, 1998; Schulz et al., 2000). The receptor subtype mediating inhibitory effects of SST in hippocampus is unknown. The goal of this study was to address these deficiencies using SST2, SST3, and SST4 knock-out (KO) mice and selective pharmacological tools. We show here that SST4 is the major player in mediating the antiepileptic actions of SST, although SST2 and SST3 also contribute. Furthermore, we demonstrate that the major mechanism through which SST4 acts is augmentation of I M. Thus, targeting SST4, which has limited distribution in the brain, could lead to development of novel antiepileptic drugs.
Materials and Methods
Generation of SST3 and SST4 knock-out mice.
We isolated genomic DNA clones containing the coding region for SST3 and SST4 from a 129Sv genomic phage library (Lambda Fix; Stratagene, La Jolla, CA) (Schwabe et al., 1996). Targeting vectors were assembled around a neomycin cassette (pAB5) (Zeyda et al., 2001) consisting of the SV40 enhancer, the tk promoter, and the coding region for neomycin. For the SST3 targeting vector, we deleted a 427 bp SacI–SalI fragment from the middle of the coding region and replaced it with the neomycin cassette, flanked 5′ by a 1.7 kb SacI fragment, and 3′ by a 6.5 kb SalI-BglII fragment (see Fig. 1 A). For the SST4 targeting vector a 592 bp SacI-HincII fragment covering most of the coding region for SST4 was deleted and replaced by the neo cassette, flanked 5′ by a 6.7 kb SacI fragment and 3′ by a 1.1 kb HincII-EcoRI fragment (see Fig. 1 B).
Figure 1.
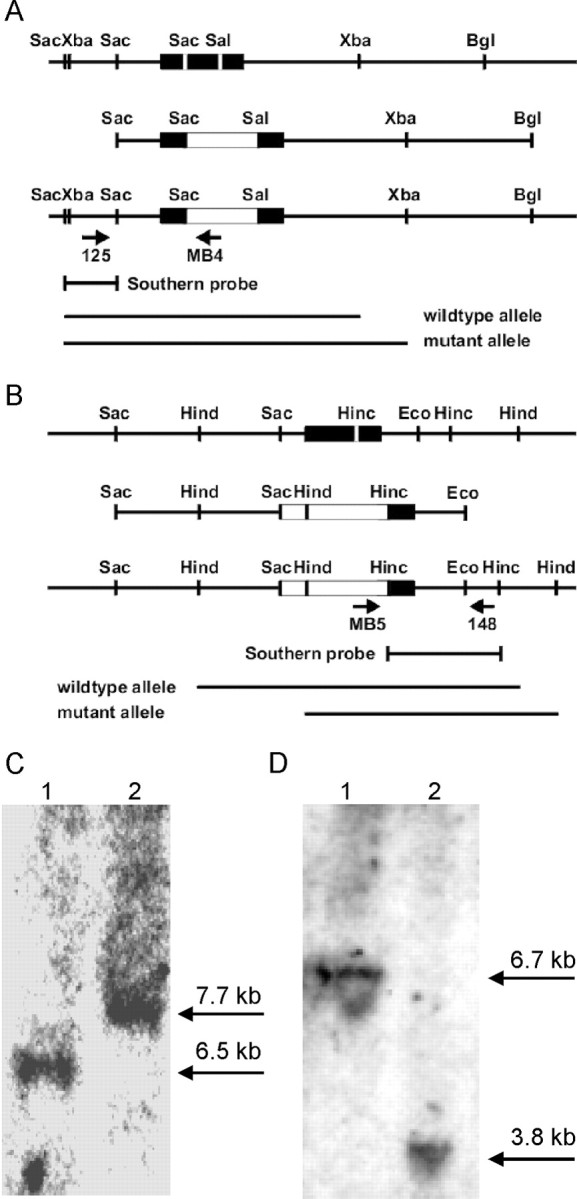
Targeted disruption of the murine SST3 and SST4 genes. A, Targeting strategy for disruption of the SST3 gene. A 427 bp SacI–SalI fragment from the middle of the coding region for SST3 is deleted and replaced by a neo cassette (white box). The neo cassette is flanked 5′ by 1.7 kb and 3′ by 6.5 kb of genomic sequence from the SST3 locus. B, A 592 bp SacI-HincII fragment covering most of the coding region for SST4 is deleted and replaced by a neo cassette (white box). The neo cassette is flanked 5′ by 6.7 kb and 3′ by a 1.1 kb of genomic sequence from the SST4 locus. C, Southern blot analysis of mouse tail DNA from wild-type (1) and SST3 KO mice (2). DNA was digested with XbaI and subjected to hybridization using a genomic probe (XbaI-SacI fragment). The expected increase of fragment size in the mutant (from 6.5 to 7.7 kb) is depicted schematically and shown in the autoradiograph. D, Southern blot analysis of mouse tail DNA from wild-type (1) and SST4 KO mice (1). DNA was digested with HindIII and subjected to hybridization using a genomic probe (HincII fragment). The expected decrease of fragment size in the mutant (from 6.7 to 3.8 kb) is depicted schematically and shown in the autoradiograph. MB4, 5′ TCCACACCCTAACTGACACAC; MB5, 5′ ATCCAGGAAACCAGCAGCGGCTAT.
We introduced the targeting plasmids by electroporation into J1 embryonic stem cells as described previously (Li et al., 1992; Zeyda et al., 2001). Colonies were screened for homologous integration by PCR, using primers 5′ of the start of the targeting vector (F3: 5′ CCAGATTCGCTAGACCCAGCTCAA) and from the neo cassette (R3: 5′ TCCACACCCTAACTGACACAC) for SST3, and 3′ of the start of the targeting vector (R4: 5′ CCTTCCCCCTCATAGCATAGCTCG) and from the neo cassette (F4: 5′ ATCCAGGAAACCAGCAGCGGCTAT) for SST4.
Clones with the expected rearrangement at the targeted locus were injected into C57BL/6J blastocysts. We mated chimeric males transmitting the mutation through the germline to 129Sv wild-type females (129SvEv-Tac; Taconic, Germantown, NY) to keep the mutation in an isogenic background. Genotyping was done by PCR on tail DNA as described (Zeyda et al., 2001). Presence or absence of the mutant and wild-type alleles was determined using the following PCR primers: wild-type-F3, ctacacggccgcactgggcttctt; mutant-F3, ATCCAGGAAACCAGCA GCGGCTAT; R3, TTCCATCACTGGACCCTGGTACCA; wild-type F4, cactagg ctcgtgctaatggtggt; mutant-F4, ATCCAGGAAACCAGCAGCGGCTAT; and R4, GCTTGCAGCCAGGTTCTGCTTGCA.
We analyzed DNA from wild-type and mutant littermates by Southern blotting for the expected rearrangement at the mutated locus(Fig. 1 C,D). PCR analysis of reverse-transcribed mRNA from whole brain of wild-type and homozygous mutant mice was performed as described previously (Sharifi et al., 2001).
Animal use was in accordance with National Institutes of Health policy and was approved by the Drexel University College of Medicine Institutional Animal Care and Use Committee. Male and female mice were 5–8 weeks old for in vitro studies and 6–10 weeks old for in vivo studies. We used four strains of mice in this study: somatostatin receptor subtype 2 (SST2) knock-outs, SST3 knock-outs, SST4 knock-outs, and wild-type mice. SST2 knock-out mice were provided by Merck Pharmaceuticals (Whitehouse Station, NJ) and have been characterized phenotypically (Viollet et al., 2000; Dutar et al., 2002). We did limited studies on SST2/SST4 double knock-outs, which were bigenic crosses between the SST2 and SST4 knock-outs. SST receptor knock-out mice were bred in-house and C57BL/6J wild-type mice were purchased from Jackson Laboratories (Bar Harbor, ME). For in vivo seizure studies and electrophysiological studies, we backcrossed receptor knock-out mice at least 10 generations to C57BL/6J to increase genetic homogeneity (Silva et al., 1997).
Seizure models.
Pentylenetetrazole (PTZ) was dissolved in saline and injected intraperitoneally. The dosage was 50 mg/kg. Animals were observed and videotaped for 30 min after injection. We scaled seizure behavior as follows (modified from Racine, 1972): stage 1, hypoactivity; stage 2, tail extension or limb jerk; stage 3, whole-body clonus; stage 4, rolling, running, jumping, and tonic-clonic. The experimenter was blind to the phenotype of the mice.
In the second seizure model, we dissolved kainic acid in saline, adjusted pH to 7.2, and injected subcutaneously. Dosage required to reliably elicit seizures in wild-type mice was established for each group of mice, after which wild-type and knock-out mice were tested side by side. Dosage was 20 mg/kg for wild-type and SST3 and SST4 knock-out mice, and 30 mg/kg for SST2 knock-out mice, which were tested at a later time. We observed and videotaped mice for 120 min after injection. Seizure behavior was scaled as the following: 1, hypoactivity; 2, myoclonic jerks of head and neck, forelimb, or hindlimb clonus; 3, partial rearing and rearing; 4, rearing and falling; 5, generalized tonic-clonic convulsions with loss of postural tone, rolling, and jumping. In both models, animals did not always progress sequentially through each seizure stage, but could skip stages.
In vitro electrophysiology.
We made hippocampal slices from mice as described previously (Tallent et al., 2001). Briefly, mice were anesthetized with halothane (4%), decapitated and the brains rapidly removed. We cut transverse hippocampal slices (350–400 μm) on a vibraslicer (Vibrotome, St. Louis, MO) or tissue chopper (Vibratome) and placed them in artificial CSF (ACSF), gassed with 95% O2/5% CO2 (carbogen), of the following composition (in mm): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 1.5 MgSO4, 2 CaCl2, 24 NaHCO3, and 10 glucose. After 20 min of incubation with their upper surfaces exposed to warmed, humidified carbogen, the slices were submerged and superfused with ACSF (31°C) at a constant rate (3–4 ml/min) for the remainder of the experiment. The inner chamber had a total volume of 1 ml; at the superfusion rates used, 90% replacement of the chamber solution could be obtained within 1–1.5 min. We added drugs and peptides to the bath from stock solutions at known concentrations. Mg2+-free ACSF was the same composition as above except no MgSO4 was added and 1.0 mm of KCl was used. SST-14 (1–4 μm), octreotide (0.5–1 μm), J-2156 [(1′S,2S)-4-amino-N-(1′-carbamoyl-2′-penylethyl)-2-(4″-methyl-1″-naphthalenesulfonylamino)butanamide] (0.5–1 μm), and ACQ090 (1 μm) were all aliquotted in DMSO, to keep the vehicle consistent between all of the drugs, and added to the bath at 1:1000 dilution, so that the final concentration of DMSO was 0.1%. Vehicle alone had no affect on burst or membrane characteristics.
Extracellular recording.
We recorded extracellular epileptiform bursts by conventional means in the CA1 pyramidal layer using glass extracellular pipettes (1–3 MΩ tip resistance when filled with 3 m NaCl) and a Molecular Devices (Union City, CA) Axoclamp 2B or Multiclamp amplifier (Tallent and Siggins, 1997). Recordings were filtered at 3–10 kHz, digitized, and analyzed using pClamp software (Molecular Devices). For recording spontaneous extracellular bursts, we superfused Mg2+-free ACSF until a stable burst rate was achieved, usually <30 min. Bursting events were acquired via computer and continuously monitored using Axoscope (Molecular Devices). We measured bursting over a 1–2 min period to calculate frequencies, and used as the control rate the period immediately before the beginning of drug superfusion. We calculated maximal drug effects (change in burst rate), which occurred 5–7 min after beginning superfusion for all drugs tested. All drug effects were reversible such that after washout burst rate returned to 93–100% of control values (data not shown).
Intracellular recording.
We used voltage-clamp techniques with sharp intracellular micropipettes (3 m KCl, 50–80 MΩ) as described previously (Tallent and Siggins, 1997; Tallent et al., 2001) to record I M. TTX (1 μm) was used to block sodium channels during recordings. We recorded from CA1 pyramidal neurons; after stabilization in current-clamp, discontinuous voltage clamp recordings were made using an Axoclamp-2B amplifier (Molecular Devices) and stored on a PC for data analysis using pClamp software (Molecular Devices). Neurons were held near −40 mV and hyperpolarizing 1 s voltage steps were applied to measure M-current deactivation. Deactivation kinetics and amplitudes were analyzed using a single-exponential fit of the region of the current spanning the two capacitance transients. SST effects were measured by subtracting control current traces from currents recorded in the presence of the peptide, to obtain the net SST-induced current.
We recorded EPSCs in CA1 pyramidal neurons using whole-cell patch clamp and visualized neurons using infrared microscopy. EPSCs were evoked by orthodromic stimulation (0.05 ms stimulus duration; 0.1 Hz frequency) of Schaeffer collaterals with a bipolar tungsten electrode placed in the stratum radiatum. We superfused bicuculline (30 μm) to block GABAA receptors to isolate EPSCs. Trials were recorded on a computer and continuously monitored with Axoscope software.
Drugs.
SST was from Bachem (Bubendorf, Switzerland). Octreotide, ACQ090, SRA880 [(3R,4aR,10aR)-1,2,3,4,4a,5,10,10a-octahydro-6-methoxy-1-methyl-benz(g)quinoline-3-carboxylic-acid-4-(4-nitro-phenyl)-piperazine-amide, hydrogen malonate] (Novartis, Basel, Switzerland), J-2156 (Juvantia Pharmaceuticals, Turku, Finland), and l-796,778 and l-803,087 (Merck Pharmaceuticals) were gifts. All other chemicals were from Sigma (St. Louis, MO). We chose drug concentrations based on receptor affinity profiles (Rohrer et al., 1998; Ramirez et al., 2002) and previous brain slice studies (Cammalleri et al., 2004; Meis et al., 2005).
Statistics.
We performed statistical analysis using two-factor ANOVA with or without replication, Student's t test (paired for within group and unpaired for between group comparison), or χ2, as indicated, using Microsoft (Redmond, WA) Excel or SPSS. Data are reported as mean ± SEM and considered statistically significant at p < 0.05.
Results
Generation of SST3 and SST4 knock-out mice
The generation of SST3 and SST4 knock-out mice are detailed in Figure 1. SST receptor genes are intronless genes. Integration of a selection cassette into the coding region disrupts the processing of mRNA and the assembly of the receptor protein. The targeting vectors for SST3 carry a 427 bp and 592 bp deletion, respectively, in the coding region (Fig. 1 A). We mated chimeric mice transmitting the mutated alleles through the germline to 129/Sv wild-type mice, resulting in the generation of knock-out strains 129/Sv-sst3tm1ute and 129/Sv-sst4tm1ute. Heterozygous offspring were mated to generate homozygous null mutant mice. Southern blot analyses of DNA from wild-type and mutant littermates show the expected rearrangements for the mutant locus (Fig. 1 C). An XbaI digest probed with a SacI-XbaI fragment shows the absence of a 6.5 kb wild-type fragment and the presence of a 7.7 kb fragment in the sst3 mutant because of the insertion of the neo cassette (Fig. 1 C). A HindIII digest probed with a HincII fragment reveals the absence of the 6.7 kb wild-type fragment and the presence of a 3.8 kb fragment in the SST4 mutant because of the additional HindIII site from the neo cassette (Fig. 1 D). We performed PCR analysis of reverse-transcribed mRNA from whole brain of wild-type and SST3 and SST4 homozygous mutant mice as described previously (Sharifi et al., 2001) and showed the absence of the respective mRNA in the mutants (data not shown).
Mutant mice were born at the frequency expected for a recessive mutation. Mice lacking SST3 or SST4 are viable, appear healthy, and are fertile.
SST receptor knock-out mice had shorter latencies and developed more severe seizures in PTZ seizure model
In the PTZ model, wild-type (n = 26), SST2 (n = 15), SST3 (n = 23), and SST4 (n = 23) knock-out mice almost all exhibited stage 1 and stage 2 seizures, with no difference between the groups in the proportion that reached these mild seizures stages (p > 0.05, χ2) (for a summary of these results, see Table 1). However, significantly more SST3 and SST4 knock-out mice than wild-type mice progressed to the more severe seizures (i.e., stage 3 and stage 4, p < 0.05). For wild-type mice, 48 and 33% developed stage 3 and stage 4 seizures, respectively. For SST4 knock-outs, 100 and 87% developed stage 3 and stage 4 seizures, whereas 89 and 67% of SST3 knock-outs developed stage 3 and 4 seizures, respectively. In contrast, there was no significant difference between wild-type and SST2 knock-out mice in the proportion that progressed to stage 3 and 4 seizures (p > 0.05), although a larger proportion of SST2 knock-outs progressed to these stages (78 and 56% for SST2 knock-outs, respectively, vs 48 and 33% for wild types) (Table 1). Also, four of 23 SST4 knock-out mice progressed to status epilepticus and died during seizures (p < 0.05), whereas this did not occur in any of the wild-type or SST2 or SST3 knock-out mice.
Table 1.
PTZ-induced seizure stages in SST receptor knock-out mice
| Wild type | SST2 KO | SST3 KO | SST4 KO | |
|---|---|---|---|---|
| Stage 1 | 89% | 100% | 94% | 100% |
| Stage 2 | 81% | 89% | 94% | 96% |
| Stage 3 | 48% | 78% | 89%* | 100%* |
| Stage 4 | 33% | 56% | 67%* | 87%* |
| Death | 0% | 0% | 0% | 17%* |
The percentage of mice from each strain that exhibited each seizures stage is shown. Asterisks indicate significant difference from wild type (p < 0.05; χ2).
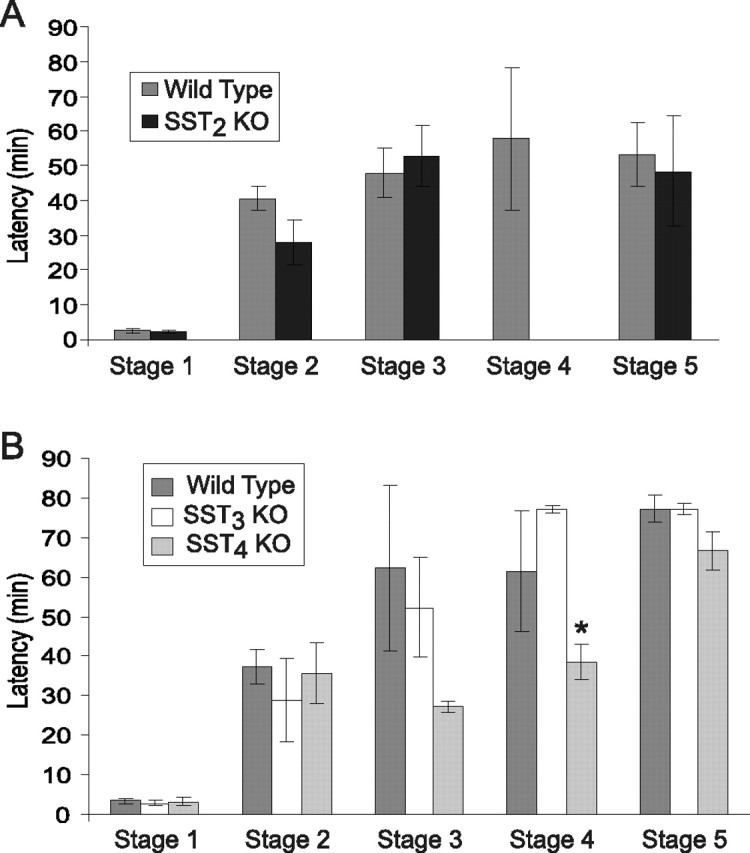
We also measured the latency to reach each seizure stage (Fig. 2). SST2, SST3, and SST4 knock-out mice all had significantly shorter latencies to stage 1 and stage 2 seizures compared with wild-type mice (ANOVA, p < 0.05), which had latencies almost twofold of the knock-outs. SST4 knock-out mice had the shortest latency of all of the knock-out strains to both stage 1 and stage 2 seizures. For stage 3, only SST4 knock-out mice showed significantly shorter latencies compared with wild-type controls (p < 0.05). SST2 and SST3 knock-out mice failed to show significant difference in latency to stage 3, although they had a trend toward shorter latencies (p > 0.05). There were no significant difference among all strains for the latency to stage 4 (p > 0.05). However, this comparison is confounded by the caveat that, as previously noted, only 33% of wild-type mice entered stage 4 seizures, whereas a much higher proportion of the SST receptor knock-out mice reached this seizure stage (Table 1). Thus, these wild-type mice may represent a particularly vulnerable subpopulation.
Figure 2.

Latency from time of injection to different seizure stages induced by PTZ in wild-type and SST receptor knock-out mice. Error bars indicate SEM. Asterisk indicates significant difference from wild-type mice. For a description of seizures stages, see Materials and Methods.
SST4 knock-out mice developed more severe seizures in the kainate model
For kainate-induced seizures, a similar number of animals from wild-type mice (n = 9) and SST2 knock-out mice (n = 8) reached each seizure stage, with no significant difference detected between groups (χ2, p > 0.05) (Table 2). The latencies to each seizure stage were also not significantly different between wild-types and SST2 knock-outs (p < 0.05) (Fig. 3 A).
Table 2.
Comparison of wild-type and SST2 knock-out mice in proportion reaching each seizure stage after kainate treatment
| Wild type | SST2 KO | |
|---|---|---|
| Stage 1 | 100% | 100% |
| Stage 2 | 67% | 88% |
| Stage 3 | 67% | 100% |
| Stage 4 | 33% | 0% |
| Stage 5 | 78% | 50% |
| Death | 0% | 0% |
No significant difference was observed between wild type and SST2 knock-outs for any of the seizure stages (χ 2).
Figure 3.
Latency from time of kainate injection to different seizure stages in wild-type and SST receptor knock-out mice. A, Comparison of wild-type and SST2 knock-out mice. B, Latency to kainate-induced seizure stages in wild-type and SST3 and SST4 knock-out mice. Error bars indicate SEM. Asterisk indicates significant difference from wild-type mice.
SST4 knock-out mice developed more severe seizures with kainate induction (Table 3), similar to our findings in the PTZ model. The majority of SST4 knock-out mice (seven of nine) exhibited stage 4 seizures, compared with only 3 of 11 wild-type and 2 of 8 SST3 knock-out mice. The difference between SST4 and wild type was significant (p < 0.05). In addition, the mortality rate was significantly higher in SST4 knock-out group than that in wild-type and SST3 knock-out groups (p < 0.05). Three of nine SST4 knock-out mice died during the seizure episode, whereas none from other groups died. Unlike in the PTZ model, SST3 knock-out mice did not develop more severe seizures than wild-type controls (Table 3).
Table 3.
Proportion of wild types and SST3 and SST4 knock-outs reaching each seizure stage after kainate treatment
| Wild type | SST3 KO | SST4 KO | |
|---|---|---|---|
| Stage 1 | 100% | 100% | 100% |
| Stage 2 | 82% | 50% | 56% |
| Stage 3 | 27% | 50% | 22% |
| Stage 4 | 27% | 25% | 78%* |
| Stage 5 | 18% | 25% | 56% |
| Death | 0% | 0% | 33%* |
No significant differences were observed in the proportion of SST3 knock-out mice that reached each seizure stage compared with wild-type mice. For SST4 knock-out mice, a greater proportion reached stage 4 seizures than wild-type mice. In addition, seizure-induced mortality was observed in a significantly greater proportion of SST4 knock-outs than wild-type mice.
When latencies were measured, SST3 knock-outs were found to have a similar latency to reach each seizures stage as wild-type mice. SST4 knock-outs, however, had a significantly shorter latency to reach stage 4 compared with wild-type controls (Fig. 3 B) (ANOVA, p < 0.05). There was no difference between SST4 knock-outs and wild-type mice in latencies to any of the other seizure stages (Fig. 3 B) (p > 0.05).
SST had diminished effect on inhibition of spontaneous epileptiform bursting in CA1 of SST receptor knock-out mice
In extracellular recordings from CA1 in hippocampal slices, there was no significant difference of bursting frequencies among wild-type and knock-out mice (data not shown). In wild-type slices (n = 11), 1 μm SST (a maximal concentration) (data not shown) decreased bursting frequency from 0.38 ± 0.07 to 0.16 ± 0.04 (58 ± 4.6% inhibition). However, this same concentration of SST had a significantly reduced effect in SST knock-out mice (Fig. 4) (ANOVA, p < 0.05). Inhibition by SST was 23% in slices from SST2 knock-out mice (n = 7), 34% in SST3 knock-out mice (n = 9), and 25% in SST4 knock-out mice (n = 8), respectively (p < 0.05 for control vs SST in all three knock-out strains, paired t test). In CA1 of slices from SST2/SST4 double knock-outs, SST did not inhibit epileptiform bursting (105 ± 2% of control burst rate; n = 7, p > 0.05).
Figure 4.
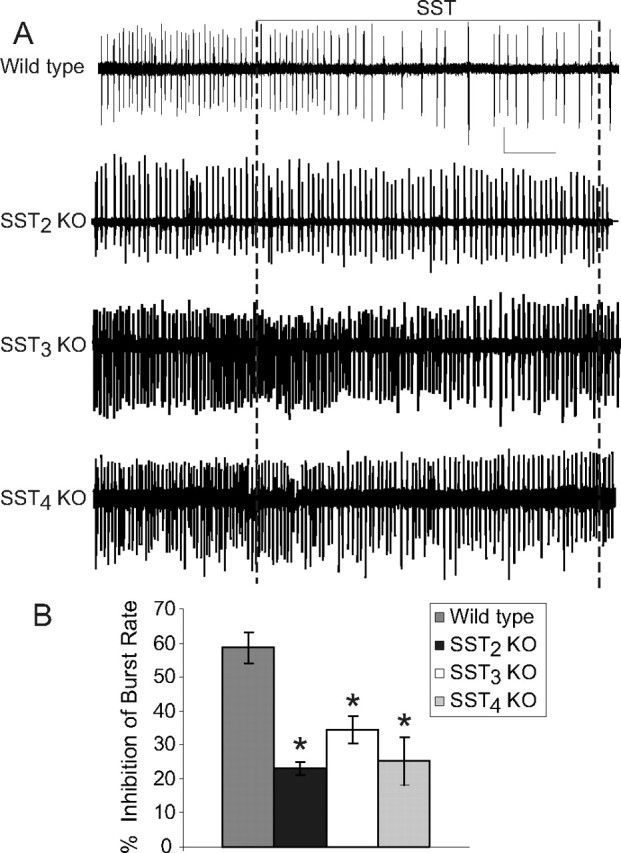
Inhibition of epileptiform bursting in CA1 of SST receptor knock-out mice. A, Representative voltage recordings for each strain. Calibration: wild type, SST2 KO, 0.25 mV; SST3 and SST4 wild type, 0.2 mV; 1 min. B, Mean inhibition of epileptiform burst rate by 1 μm SST for each mouse strain. Asterisks indicate a significant difference from wild-type mice.
Pharmacological validation of receptor knock-out studies
We used SST receptor agonists or antagonists to determine the inhibitory effect of individual receptor subtypes in wild-type mice and to validate the results from knock-out mice. Octreotide is the most commonly used SST2 agonist (Vezzani et al., 1991; Hoyer et al., 1995; Perez et al., 1995; Schoeffter et al., 1995; Hicks et al., 1998). Application of 1 μm octreotide resulted in 33.9% inhibition of epileptiform bursting in CA1 of wild-type mice (Table. 4), and a reduced but significant (12 ± 2%) inhibition in SST2 knock-out mice (p < 0.05; n = 5). J-2156 is a newly available high affinity nonpeptide SST4 agonist (Engstrom et al., 2006). Superfusion of 1 μm J-2156 resulted in a 21.8 ± 3% inhibition of bursting in wild-type mice (p < 0.05) (Table 4) and had no effect on SST4 knock-out mice (p > 0.05; n = 5). Another SST4-selective nonpeptide agonist, l-803,087 (1 μm), did not significantly reduce epileptiform burst rate in wild-type mice (Table 4).
Table 4.
Inhibition of bursting by subtype-selective ligands
| Ligand | Receptor(s) activated | % inhibition of burst rate | Number of slices |
|---|---|---|---|
| SST | All | 58.6 ± 4% | 11 |
| Octreotide | SST2 > SST3 | 33.9 ± 4%* | 9 |
| l-796,778 | SST3 | 5.2 ± 3.4* , ** | 5 |
| J-2156 | SST4 | 21.8 ± 3%* | 10 |
| l-803,087 | SST4 | 2.1 ± 5.7%* , ** | 8 |
| Octreotide + J-2156 | SST2 + SST4 | 47.0 ± 5 | 6 |
| SST + SRA880 a | All but SST1 | 47.0 ± 2 | 7 |
| SST + ACQ090 b | All but SST3 | 46 ± 5 | 6 |
Concentration is 1 μm for all drugs except when octreotide and J-2156 are coapplied (0.5 μm each).
aSST1-selective antagonist.
bSST3-selective antagonist.
*Significantly different from SST alone (p < 0.05; unpaired t test);
**no significant inhibition of burst rate (p > 0.05; paired t test).
The SST3-selective nonpeptide agonist l-769,778 (Rohrer et al., 1998) did not modulate epileptiform bursting in CA1 of wild-type slices at 1 μm (p > 0.05; n = 5) (Table 4). At 10 μm concentration, l-796,778 irreversibly increased burst rate in two slices while having no effect in the other two (120 ± 28% of control, n = 4, p > 0.05). We also examined the effect of a selective SST3 antagonist, ACQ090, in wild-type mice. ACQ090 (1 μm) was applied 15 min before application of 1 μm SST. ACQ090 alone significantly increased burst rate by 18% (p < 0.05), but inhibition by exogenous SST in the presence of ACQ090 was not significantly different from SST alone (p > 0.05; n = 6) (Table 4).
We also coapplied octreotide and J-2156 together to determine whether activation of both SST2 and SST4 resulted in a similar degree of inhibition as SST. We found that 0.5 μm octreotide and 0.5 μm J-2156 together reduced epileptiform bursting by 47 ± 5% (n = 6). This was not significantly different from 1 μm SST (p > 0.05).
Because SST1 has been suggested previously to contribute to inhibition of bursting by SST in mouse hippocampus (Cammalleri et al., 2004), we also examined whether a SST1-selective antagonist could affect the inhibitory action of SST. When applied alone, the SST1 antagonist SRA-880 (1 μm) modestly increased burst rate (112 ± 2% of control; n = 7). However, SST inhibition of epileptiform bursting was not affected by coapplication of SRA-880 (47 ± 2% inhibition), indicating that SST1 does not play a major role in regulating epileptiform bursting in CA1.
Contribution of K+ M-current to inhibition of epileptiform bursting by SST
A major mechanism of action of SST in CA1 is an increase in the K+ M-current (I M) (Moore et al., 1988; Schweitzer et al., 1990), a voltage sensitive K+ current important in regulating epileptiform activity in hippocampus (Qiu et al., 2007). To determine the contribution of I M in SST inhibition of epileptiform bursting, we blocked the current using the selective M-channel blocker linopirdine. As we have reported previously for rat hippocampus (Qiu et al., 2007), after I M blockade, the duration of the interictal bursts increase. The effect of linopirdine on burst duration was not significantly different between wild type (216 ± 12% of control), SST2 (230 ± 51%), and SST4 knock-outs (225 ± 32%). In wild-type mice, when linopirdine (20 μm) was superfused beginning 20 min earlier, 1 μm SST reduced epileptiform bursting by 28 ± 3% (n = 9), compared with 58 ± 5% inhibition by SST alone (Fig. 5). Thus, with I M blocked, SST efficacy in inhibiting epileptiform bursting is reduced by ∼50%. We did similar experiments in SST2 and SST4 knock-out mice. In SST2 knock-out mice, linopirdine completely prevents inhibition of bursting by SST (4.2 ± 3.6%), compared with 23 ± 4% for SST alone in slices from the same mice (Fig. 5) (n = 6). In slices from SST4 knock-out mice, no significant difference was found in SST inhibition of epileptiform burst rate in the presence of linopirdine (27 ± 1%; n = 5) when compared with SST alone (23 ± 5%; n = 7). These results suggest that the major inhibitory SST receptor remaining in SST2 knock-out mice, likely SST4, mediates its action by increasing I M. However, the major SST receptor remaining in SST4 knock-out mice, likely SST2, does not appear to act via I M in mediating the inhibitory effects of SST on epileptiform bursting.
Figure 5.

Contribution of I M to SST mediated inhibition of epileptiform bursting. A, Representative traces showing SST inhibition of bursting when I M is blocked with linopirdine (Linop) in wild-type and SST2 and SST4 knock-out mice. In wild-type and SST4 knock-out mice, SST inhibits burst rate to a similar degree, but no inhibition is present in SST2 knock-out mice. Experiments with and without linopirdine were done in different slices to avoid confounds caused by repeated applications of SST. Calibration: 0.5 mV, 10 s. B, Averaged data from the different mouse strains showing inhibition of epileptiform burst rate with and without linopirdine. With I M blocked, SST inhibition is partially inhibited in wild-type mice, absent in SST2 knock-outs, and unaffected in SST4 knock-outs.
SST4 mediates SST-induced increase in I M
To directly assess coupling of SST2 and SST4 to I M, we did voltage-clamp studies in wild-type and knock-out mice using sharp intracellular electrodes. Resting membrane potential (RMP) and input resistance (R in) were not different between wild type (RMP, −66 ± 1.2 mV; R in, 86 ± 4.9 MΩ), SST2 (−66 ± 1.0 mV; 74 ± 3.8 MΩ), and SST4 knock-outs (−67 ± 1.0 mV; 77 ± 0.7.0 MΩ). Control I M amplitudes measured from a holding potential of −40 mV with a step to −60 mV were similar to a previous report on C57BL/6J mice (Otto et al., 2006). We did not observe a significant difference in amplitudes between wild-type (57 ± 12 pA), SST2 (77 ± 10 pA), and SST4 knock-out mice (56 ± 6 pA; one-way ANOVA, p > 0.05). The kinetics of deactivation (τ) measured with the same step protocol were also not significantly different between wild-type (162 ± 30 ms), SST2 (139 ± 42 ms), and SST4 knock-outs (139 ± 22 ms; one-way ANOVA, p > 0.05) and were also comparable with the previous report in C57BL/6J mice (Otto et al., 2006).
We examined whether SST augmented I M on mouse hippocampus, as has been reported in rat (Moore et al., 1988). Superfusion of 1 μm SST increased I M measured using hyperpolarizing voltage steps from a holding potential near −40 mV (Fig. 6 A,B). The SST-induced increase at voltage steps to −60 mV measured 128 ± 20 pA. This SST effect is similar in magnitude to that reported in rat (Schweitzer et al., 1993; Moore et al., 1994). SST also increased the holding current at −40 mV by 190 pA.
Figure 6.
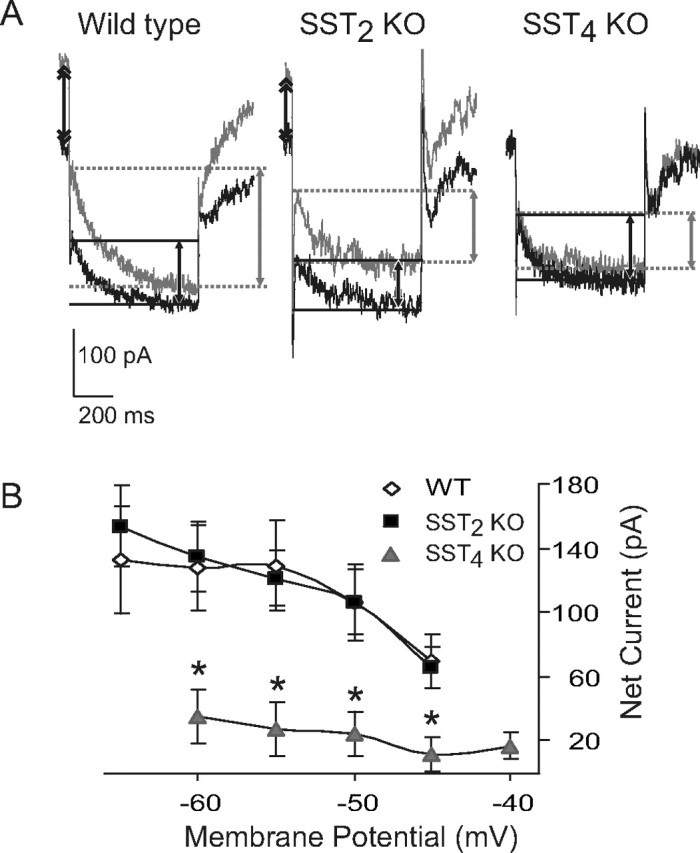
SST4 mediates SST increase in M-current. A, Representative current traces from CA1 hippocampal neurons of wild-type, SST2 and SST4 knock-out mice evoked by stepping from a holding voltage of −40 to −60 mV for 1 s. Note the increase in outward holding current (double arrows) and the inward “relaxation” (single arrows) in the presence of 1 μm SST (gray traces) in wild type and SST2 knock-outs, but not SST4 knock-outs. B, Summary of current-voltage data for SST-induced current in CA1 neurons from wild-type and SST2 and SST4 knock-out mice. Neurons were held at approximately −40 mV and 5–10 mV hyperpolarizing voltage steps given. Currents in presence of 1 μm SST were subtracted from control currents to obtain the net SST-induced current. Net SST current is similar in wild type and SST2 knock-outs, whereas in SST4 knock-outs SST-induced current is mostly absent. Error bars indicate SEM. Asterisk indicates significant difference from wild type.
We next examined SST actions on I M in SST2 and SST4 knock-out mice. SST (1 μm) significantly enhanced I M amplitude at each individual command step in SST2 knock-out mice (p < 0.05) (Fig. 6 A,B). There was no significant difference in the SST effect between wild type and SST2 knock-outs (ANOVA, p < 0.05). SST increased the holding current at −40 mV by 216 pA, which was not significantly different from wild-type mice. In contrast to wild-type and SST2 knock-out mice, application of 1 μm SST in SST4 knock-out mice did not significantly increase I M recorded from CA1 pyramidal neurons (p > 0.05) (Fig. 6 B). There was a significant difference in the SST effect on I M between wild type and SST4 knock-outs (p < 0.05). For example, with a voltage step to −60 mV, the net increase of I M by SST was 128 ± 20 pA in wild-type controls, 135 ± 23 pA in SST2 knock-out mice, and 35 ± 8 pA in SST4 knock-out mice (Fig. 6 B). SST also did not affect the holding current recorded at −40 mV in SST4 knock-outs (−36 ± 44 pA).
SST does not decrease EPSCs in pyramidal neurons of mouse CA1
SST inhibition of EPSCs in rat has been reported to be mediated by SST2 (Boehm and Betz, 1997). We explored whether this mechanism could be involved in SST2-mediated inhibitory effects in mice. We examined the action of SST on EPSCs recorded in wild-type mouse CA1 pyramidal neurons using whole-cell patch clamp and evoked by stimulating Schaeffer collaterals. After addition of 1 μm SST, mean EPSC amplitude remained 95 ± 5% of control (Fig. 7 A) (n = 8; p > 0.05). We verified SST effects in rat CA1 neurons (n = 4), and found that 1 μm SST decreased EPSCs to 66 ± 3% of control (Fig. 6 B) (p < 0.05), similar to our previous report (Tallent and Siggins, 1997). Thus, unlike in rat, SST does not reduce EPSC amplitude in mouse CA1 pyramidal neurons, indicating this mechanism does not account for SST2-mediated inhibition of epileptiform bursting.
Figure 7.

SST does not affect CA1 EPSCs in mice. A, Representative EPSCs generated by stimulating Schaeffer collaterals and recorded using whole-cell patch clamp. SST (1 μm) inhibits EPSC in recordings from rat (left), but not mice (right). B, Graph showing mean SST inhibition of CA1 EPSC amplitude in rats and mice. In rats but not mice, SST (1 μm) significantly reduced EPSC amplitude. Error bars indicate SEM. Asterisk indicates significant difference from control.
Discussion
Our results demonstrate a major role for SST4 receptors in SST function in hippocampus. Interestingly, in all three of the SST receptor knock-out mice studied, SST2, SST3, and SST4, we observed a decreased latency to stage 1 and stage 2 seizures in the PTZ model. Stage 3 and stage 4 seizures also occurred significantly more in SST3 and SST4 knock-out mice, but not in SST2 knock-out mice. SST4 knock-outs showed the most robust response to PTZ, and were the only strain where seizure-induced death occasionally occurred. In wild-type animals, PTZ is considered a rather mild chemoconvulsant with little or no mortality observed in most studies (Sarkisian, 2001).
We used a second seizure model, subcutaneous injection of kainate, to confirm the importance of SST4 in seizure modulation. SST2 and SST3 knock-outs did not exhibit more severe seizures in this model. A previous study using intrahippocampal kainate injections showed a decreased vulnerability to seizures in SST2 knock-outs (Moneta et al., 2002), but we did not observe this with systemic IP injections. SST4 knock-outs exhibited both a significantly higher proportion and a shorter latency to stage 4 seizures in the kainate model than wild-type mice. Thus, although the results in SST4 knock-out mice were consistent between the two models, SST2 and SST3 knock-outs demonstrated increased seizure susceptibility only in the PTZ model. This could reflect the different mechanism of seizure generation by these two drugs, because PTZ is a GABA receptor blocker whereas kainate activates glutamate receptors. These discrepancies could also be related to differences in seizure types generated by the two chemoconvulsants. PTZ induces generalized cortical seizures, with hippocampal involvement late (Starzl et al., 1953; Brevard et al., 2006). Kainate is used to model of partial seizures, with critical involvement of hippocampus from early stages (Sarkisian, 2001). Thus, the increased vulnerability in SST4 knock-outs to kainate-induced seizure could reflect its relative importance in controlling hippocampal excitability compared with SST2 and SST3. This agrees with anatomical studies showing that SST4 is the predominate SST receptor in CA1 hippocampus (Videau et al., 2003).
Both SST2 and SST4 contribute to SST inhibition of bursting in CA1 in hippocampal slices. SST actions on CA1 bursting are attenuated to a similar degree in SST2 and SST4 knock-outs, and no SST inhibition of epileptiform bursting remains in the SST2/SST4 double knock-out. We confirmed these findings with pharmacological studies in wild-type mice, suggesting that functionally these knock-out mice have few compensatory changes in expression of the remaining receptors. We demonstrate that SST2 and SST4-selective ligands inhibit bursting to a lesser degree than SST itself. With coapplication, the degree of inhibition by SST2 and SST4 agonists is similar to SST. Thus, the majority of SST inhibition of epileptiform bursting in CA1 is mediated by SST2 and SST4. A previous study suggested that SST1 can mediate SST inhibition of bursting in hippocampal slices (Cammalleri et al., 2004); however, the SST1 antagonist used in previous work, SRA-880, did not attenuate SST-mediated inhibition of bursting in our model.
The role of SST3 in hippocampus is less clear. SST3 knock-out mice had shorter latencies to stage 1 and stage 2 seizures induced by PTZ, and a reduced response to SST in CA1 in vitro. These results suggest that SST3 may contribute to the antiepileptic actions of SST. However, unlike with the SST2 and SST4 knock-outs, we were unable to confirm these findings pharmacologically. The SST3-selective agonist did not regulate epileptiform bursting in wild-type mice, nor did the SST3 antagonist reduce inhibition of bursting by SST. Because testing of these SST3 ligands in situ has been limited, one explanation is that these compounds do not appropriately interact with SST3 in our hippocampal slice preparation, as seems to be the case with the SST4 agonist l-803,087. However, additional data support the conclusion that SST3 does not mediate acute electrophysiological actions of SST. As discussed above, SST does not inhibit bursting in the SST2/SST4 double knock-out, and coapplication of SST2 and SST4 agonists largely recapitulates the SST effect. Thus, the preponderance of evidence suggests that SST3 does not contribute to acute electrophysiological actions of SST in hippocampus. SST3 is located on neuronal cilia that are believed to be involved in neuronal signaling (Fuchs and Schwark, 2004). SST3 could therefore modulate hippocampal excitability by regulating signaling pathways.
SST modulation of I M in rat CA1 neurons has been well characterized (Moore et al., 1988; Schweitzer et al., 1990, 1993). We demonstrate here that SST likewise augments I M in mouse CA1 neurons. This action of SST is absent in SST4 knock-out mice, suggesting that this receptor is critical in coupling to I M. SST mediated increase in I M is intact in SST2 knock-outs, indicating SST2 does not couple to this channel.
Mutations in M-channel subunits underlie a human epilepsy, benign familial neonatal convulsions (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998), and we showed previously that I M is critical in preventing generation of ictal events in hippocampus in both immature and adult rats (Qiu et al., 2007). Retigabine, a drug that directly augments M-channels, reduces epileptiform activity in several in vitro hippocampal models (Armand et al., 2000; Dost and Rundfeldt, 2000), and is in clinical trials for use in epilepsy (Porter et al., 2007b). After I M blockade, SST inhibition of bursting is reduced by ∼50%. In SST2 knock-outs, where the major SST receptor mediating SST inhibition of bursting would be SST4, I M blockade prevents inhibition of epileptiform bursting by SST. In contrast, I M blockade does not alter SST inhibition of bursting in SST4 knock-outs, where SST2 likely mediates the majority of SST actions. These results suggest that the major mechanism through which SST4 inhibits epileptiform activity in CA1 is by activation of I M, and that SST2-mediated inhibition of bursting is independent of I M.
Although receptor-mediated inhibition of I M has been well studied (Delmas and Brown, 2005), mechanisms through which G-protein-coupled receptors augment I M have not received the same focus. Previous studies suggested arachidonic acid metabolites mediate SST augmentation of I M in hippocampal CA1 pyramidal neurons via activation of PLA2 (Schweitzer et al., 1990, 1993). Because inhibition of I M by muscarinic receptors has been demonstrated to be via activation of PLC (Suh and Hille, 2007), this suggests reciprocal modulation of M-channels by phospholipase pathways.
SST receptors have distinct patterns of expression on hippocampal principle neurons in rats and mice. SST2 receptors are present on soma and proximal dendrites (Dournaud et al., 1996; Allen et al., 2003). SST4 is expressed mostly in medial and proximal dendrites of principle neurons (Schreff et al., 2000). SST3 has a pattern of expression unique to G-protein-coupled receptors, in that it is exclusively localized on neuronal cilia (Handel et al., 1999). The function of neuronal cilia is unknown, although they have been speculated to play a role in signaling (Fuchs and Schwark, 2004; Whitfield, 2004). Thus, principle neurons of hippocampus are virtually coated with SST receptors, suggesting that SST is an important signaling molecule in this region. Interestingly, SST terminals are located at the distal dendrites of pyramidal neurons in CA1, CA3, and dentate (Freund and Buzsaki, 1996). This would suggest that SST must diffuse quite far from its release site to activate SST2 and SST3. SST4 however, is localized closer to SST terminals, and therefore would likely require smaller amounts of SST release to be activated.
Our results show that SST2, SST3, and SST4 all appear to regulate cortical excitability, with SST4 playing the major role in regulating seizure events. Our in vitro studies implicate SST2 and SST4 as mediating the acute electrophysiological actions of SST in CA1 hippocampus. SST4 regulates hippocampal excitability via enhancement of I M, whereas the mechanism through which SST2 acts is unknown. In rat hippocampal neurons, inhibition of EPSCs by SST has been proposed to be mediated by SST2, because the SST2-selective ligands seglitide and octreotide can mediate this response (Boehm and Betz, 1997). However, in mice, EPSCs generated by stimulation of Schaeffer collaterals were not sensitive to SST, even though we confirmed this effect in rat slices.
Another possible mechanism through which SST2 may act is inhibition of Ca2+ channels, because SST has this action in dentate granule cells (Baratta et al., 2002). Ca2+ channels contribute to dendritic depolarization during seizure events (Traub and Wong, 1983; Traub et al., 1993). The mechanism of action of SST3 is also unclear, but may involve activation of longer-term signaling pathways because this receptor does not appear to mediate acute electrophysiological actions of SST in CA1 hippocampus.
Our results suggest that new drugs acting on SST4 could have diverse clinical applications, because M-channels are under investigation as therapeutic target of not only epilepsy, but other conditions such as neuropathic pain (Porter et al., 2007a). Because of more restricted localization of SST4 in both brain and periphery (Cooper et al., 2000; Schreff et al., 2000; Porter et al., 2007a), targeting this receptor could lead to drugs with fewer unwanted side effects than direct activators of M-channels.
Footnotes
This work was supported by National Institutes of Health Grants NS38633 and NS048241 to M.K.T. and MH58543 to L.d.L., and an Epilepsy Foundation predoctoral fellowship to C.Q. We thank Ms. Emily Einstein and Ms. Carlyn Patterson for excellent technical assistance. We would also like to thank the following people for generously providing compounds: Dr. Daniel Hoyer and Dr. Herbert Schmid (Novartis) for octreotide, ACQ090, and SRA880; Dr. M. Engstrom (Juvantia Pharmaceuticals) for J-2156; and Dr. Susan Rohrer (Merck) for L-796,778 and L-803,087. We also thank Dr. James Schaeffer (Merck) for providing the SST2 knock-out mice.
References
- Allen JP, Hathway GJ, Clarke NJ, Jowett MI, Topps S, Kendrick KM, Humphrey PPA, Wilkinson LS, Emson PC. Somatostatin receptor 2 knock-out/LacZ knockin mice show impaired motor coordination and reveal sites of somatostatin action within the striatum. Eur J Neurosci. 2003;17:1881–1895. doi: 10.1046/j.1460-9568.2003.02629.x. [DOI] [PubMed] [Google Scholar]
- Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by 4-aminopyridine in rat entorhinal cortex hippocampal slices. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:33–39. doi: 10.1007/pl00005320. [DOI] [PubMed] [Google Scholar]
- Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by low magnesium in rat entorhinal cortex hippocampal slices. Epilepsia. 2000;41:28–33. doi: 10.1111/j.1528-1157.2000.tb01501.x. [DOI] [PubMed] [Google Scholar]
- Baratta MV, Lamp T, Tallent MK. Somatostatin depresses long-term potentiation and Ca2+ signaling in mouse dentate gyrus. J Neurophysiol. 2002;88:3078–3086. doi: 10.1152/jn.00398.2002. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. J Neurosci. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brevard ME, Kulkarni P, King JA, Ferris CF. Imaging the neural substrates involved in the genesis of pentylenetetrazol-induced seizures. Epilepsia. 2006;47:745–754. doi: 10.1111/j.1528-1167.2006.00502.x. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Cervia D, Langenegger D, Liu Y, Monte MD, Hoyer D, Bagnoli P. Somatostatin receptors differentially affect spontaneous epileptiform activity in mouse hippocampal slices. Eur J Neurosci. 2004;20:2711–2721. doi: 10.1111/j.1460-9568.2004.03741.x. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–4919. doi: 10.1073/pnas.090092797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Dost R, Rundfeldt C. The anticonvulsant retigabine potently suppresses epileptiform discharges in the low Ca++ and low Mg++ model in the hippocampal slice preparation. Epilepsy Res. 2000;38:53–66. doi: 10.1016/s0920-1211(99)00065-0. [DOI] [PubMed] [Google Scholar]
- Dournaud P, Gu YZ, Schonbrunn A, Mazella J, Tannenbaum GS, Beaudet A. Localization of the somatostatin receptor SST2A in rat brain using a specific anti-peptide antibody. J Neurosci. 1996;16:4468–4478. doi: 10.1523/JNEUROSCI.16-14-04468.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutar P, Vaillend C, Viollet C, Billard JM, Potier B, Carlo AS, Ungerer A, Epelbaum J. Spatial learning and synaptic hippocampal plasticity in type 2 somatostatin receptor knock-out mice. Neuroscience. 2002;112:455–466. doi: 10.1016/s0306-4522(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Engstrom M, Savola JM, Wurster S. Differential efficacies of somatostatin receptor agonists for G-protein activation and desensitization of somatostatin receptor subtype 4-mediated responses. J Pharmacol Exp Ther. 2006;316:1262–1268. doi: 10.1124/jpet.105.094128. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fuchs JL, Schwark HD. Neuronal primary cilia: a review. Cell Biol Int. 2004;28:111–118. doi: 10.1016/j.cellbi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Hollt V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89:909–926. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- Hervieu G, Emson PC. The localization of somatostatin receptor 1 (sst1) immunoreactivity in the rat brain using an N-terminal specific antibody. Neuroscience. 1998;85:1263–1284. doi: 10.1016/s0306-4522(98)00024-4. [DOI] [PubMed] [Google Scholar]
- Hicks GA, Feniuk W, Humphrey PP. Outward current produced by somatostatin (SRIF) in rat anterior cingulate pyramidal cells in vitro. Br J Pharmacol. 1998;124:252–258. doi: 10.1038/sj.bjp.0701824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer D, Perez J, Schoeffter P, Langenegger D, Schuepbach E, Kaupmann K, Luebbert H, Bruns C, Reubi JC. Pharmacological identity between somatostatin SS-2 binding sites and SSTR-1 receptors. Eur J Pharmacol Mol Pharmacol. 1995;289:151–161. doi: 10.1016/0922-4106(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Telegdy G. Effects of somatostatin and anti-somatostatin serum on picrotoxin-kindled seizures. Neuropharmacology. 1992;31:793–797. doi: 10.1016/0028-3908(92)90043-o. [DOI] [PubMed] [Google Scholar]
- Meis S, Sosulina L, Schulz S, Hollt V, Pape HC. Mechanisms of somatostatin-evoked responses in neurons of the rat lateral amygdala. Eur J Neurosci. 2005;21:755–762. doi: 10.1111/j.1460-9568.2005.03922.x. [DOI] [PubMed] [Google Scholar]
- Moneta D, Richichi C, Aliprandi M, Dournaud P, Dutar P, Billard JM, Carlo AS, Viollet C, Hannon JP, Fehlmann D, Nunn C, Hoyer D, Epelbaum J, Vezzani A. Somatostatin receptor subtypes 2 and 4 affect seizure susceptibility and hippocampal excitatory neurotransmission in mice. Eur J Neurosci. 2002;16:843–849. doi: 10.1046/j.1460-9568.2002.02146.x. [DOI] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Joels M, Siggins GR. Somatostatin augments the M-current in hippocampal neurons. Science. 1988;239:278–280. doi: 10.1126/science.2892268. [DOI] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Schweitzer P, Siggins GR. Voltage-dependent effects of opioid peptides on hippocampal CA3 pyramidal neurons in vitro . J Neurosci. 1994;14:809–820. doi: 10.1523/JNEUROSCI.14-02-00809.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–2059. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J, Vezzani A, Civenni G, Tutka P, Rizzi M, Schuepbach E, Hoyer D. Functional effects of D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Trp-NH2 and differential changes in somatostatin receptor messenger RNAs, binding sites and somatostatin release in kainic acid-treated rats. Neuroscience. 1995;65:1087–1097. doi: 10.1016/0306-4522(94)00535-d. [DOI] [PubMed] [Google Scholar]
- Porter RJ, Nohria V, Rundfeldt C. Retigabine. Neurotherapeutics. 2007a;4:149–154. doi: 10.1016/j.nurt.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RJ, Partiot A, Sachdeo R, Nohria V, Alves WM. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology. 2007b;68:1197–1204. doi: 10.1212/01.wnl.0000259034.45049.00. [DOI] [PubMed] [Google Scholar]
- Qiu C, Johnson BN, Tallent MK. K(+) M-current regulates the transition to seizures in immature and adult hippocampus. Epilepsia. 2007;48:2047–2058. doi: 10.1111/j.1528-1167.2007.01193.x. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ramirez JL, Mouchantaf R, Kumar U, Otero Corchon V, Rubinstein M, Low MJ, Patel YC. Brain somatostatin receptors are up-regulated in somatostatin-deficient mice. Mol Endocrinol. 2002;16:1951–1963. doi: 10.1210/me.2002-0068. [DOI] [PubMed] [Google Scholar]
- Rohrer SP, Birzin ET, Mosley RT, Berk SC, Hutchins SM, Shen DM, Xiong Y, Hayes EC, Parmar RM, Foor F, Mitra SW, Degrado SJ, Shu M, Klopp JM, Cai SJ, Blake A, Chan WW, Pasternak A, Yang L, Patchett AA, et al. Rapid identification of subtype-selective agonists of the somatostatin receptor through combinatorial chemistry. Science. 1998;282:737–740. doi: 10.1126/science.282.5389.737. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR. Overview of the current animal models for human seizure and epileptic disorders. Epilepsy Behav. 2001;2:201–216. doi: 10.1006/ebeh.2001.0193. [DOI] [PubMed] [Google Scholar]
- Schoeffter P, Perez J, Langenegger D, Schuepbach E, Bobirnac I, Luebbert H, Bruns C, Hoyer D. Characterization and distribution of somatostatin SS-1 and SRIF-1 binding sites in rat brain: Identity with SSTR-2 receptors. Eur J Pharmacol Mol Pharmacol. 1995;289:163–173. doi: 10.1016/0922-4106(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Schreff M, Schulz S, Handel M, Keilhoff G, Braun H, Pereira G, Klutzny M, Schmidt H, Wolf G, Hollt V. Distribution, targeting, and internalization of the sst4 somatostatin receptor in rat brain. J Neurosci. 2000;20:3785–3797. doi: 10.1523/JNEUROSCI.20-10-03785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Handel M, Schreff M, Schmidt H, Hollt V. Localization of five somatostatin receptors in the rat central nervous system using subtype-specific antibodies. J Physiol Paris. 2000;94:259–264. doi: 10.1016/s0928-4257(00)00212-6. [DOI] [PubMed] [Google Scholar]
- Schwabe W, Brennan MB, Hochgeschwender U. Isolation and characterization of the mouse (Mus musculus) somatostatin receptor type-4-encoding gene (mSSTR4) Gene. 1996;168:233–235. doi: 10.1016/0378-1119(95)00748-2. [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Madamba S, Siggins GR. Arachidonic acid metabolites as mediators of somatostatin-induced increase of neuronal M-current. Nature. 1990;346:464–467. doi: 10.1038/346464a0. [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Madamba S, Champagnat J, Siggins GR. Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J Neurosci. 1993;13:2033–2049. doi: 10.1523/JNEUROSCI.13-05-02033.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer P, Madamba SG, Siggins GR. Somatostatin increases a voltage-insensitive K+ conductance in rat CA1 hippocampal neurons. J Neurophysiol. 1998;79:1230–1238. doi: 10.1152/jn.1998.79.3.1230. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knock-out mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Simpson EM, Takahashi JS, Lipp H, Nakanishi S, Wehner JM, Giese KP, Tully T, Abel T, Chapman PF, Fox K, Grant S, Itohara S, Lathe R, Mayford M, McNamara JO, Morris RJ, Picciotto M, Roder M, Shin H, et al. Mutant mice and neuroscience: recommendations concerning genetic background. Banbury Conference on genetic background in mice. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, Anderson VE, Sanguinetti MC, Leppert MF. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–2737. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- Starzl TE, Niemer WT, Dell M, Forgrave PR. Cortical and subcortical electrical activity in experimental seizures induced by metrazol. J Neuropathol Exp Neurol. 1953;12:262–276. doi: 10.1097/00005072-195307000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of KCNQ channels by manipulation of phosphoinositides. J Physiol. 2007;582:911–916. doi: 10.1113/jphysiol.2007.132647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin depresses excitatory but not inhibitory neurotransmission in rat CA1 hippocampus. J Neurophysiol. 1997;78:3008–3018. doi: 10.1152/jn.1997.78.6.3008. [DOI] [PubMed] [Google Scholar]
- Tallent MK, Madamba SG, Siggins GR. Nociceptin reduces epileptiform events in CA3 hippocampus via presynaptic and postsynaptic mechanisms. J Neurosci. 2001;21:6940–6948. doi: 10.1523/JNEUROSCI.21-17-06940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK. Synchronized burst discharge in disinhibited hippocampal slice. II. Model of cellular mechanism. J Neurophysiol. 1983;49:459–471. doi: 10.1152/jn.1983.49.2.459. [DOI] [PubMed] [Google Scholar]
- Traub RD, Miles R, Jefferys JG. Synaptic and intrinsic conductances shape picrotoxin-induced synchronized after-discharges in the guinea-pig hippocampal slice. J Physiol (Lond) 1993;461:525–547. doi: 10.1113/jphysiol.1993.sp019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Hoyer D. Brain somatostatin: a candidate inhibitory role in seizures and epileptogenesis. Eur J Neurosci. 1999;11:3767–3776. doi: 10.1046/j.1460-9568.1999.00838.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Serafini R, Stasi MA, Vigano G, Rizzi M, Samanin R. A peptidase-resistant cyclic octapeptide analogue of somatostatin (SMS 201–995) modulates seizures induced by quinolic acid and kainic acid differently in the rat hippocampus. Neuropharmacology. 1991;30:345–352. doi: 10.1016/0028-3908(91)90059-k. [DOI] [PubMed] [Google Scholar]
- Videau C, Hochgeschwender U, Kreienkamp HJ, Brennan MB, Viollet C, Richter D, Epelbaum J. Characterisation of [125I]-Tyr0DTrp8-somatostatin binding in sst1- to sst4- and SRIF-gene-invalidated mouse brain. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:562–571. doi: 10.1007/s00210-003-0758-8. [DOI] [PubMed] [Google Scholar]
- Viollet C, Vaillend C, Videau C, Bluet-Pajot MT, Ungerer A, L'Heritier A, Kopp C, Potier B, Billard J, Schaeffer J, Smith RG, Rohrer SP, Wilkinson H, Zheng H, Epelbaum J. Involvement of sst2 somatostatin receptor in locomotor, exploratory activity and emotional reactivity in mice. Eur J Neurosci. 2000;12:3761–3770. doi: 10.1046/j.1460-9568.2000.00249.x. [DOI] [PubMed] [Google Scholar]
- Whitfield JF. The neuronal primary cilium—an extrasynaptic signaling device. Cell Signal. 2004;16:763–767. doi: 10.1016/j.cellsig.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Zeyda T, Diehl N, Paylor R, Brennan MB, Hochgeschwender U. Impairment in motor learning of somatostatin null mutant mice. Brain Res. 2001;906:107–114. doi: 10.1016/s0006-8993(01)02563-x. [DOI] [PubMed] [Google Scholar]