

Abstract
A hallmark of rapid eye movement (REM) sleep is a potent suppression of postural muscle tone. Motor control in REM sleep is unique because it is characterized by flurries of intermittent muscle twitches that punctuate muscle atonia. Because somatic motoneurons are bombarded by strychnine-sensitive IPSPs during REM sleep, it is assumed that glycinergic inhibition underlies REM atonia. However, it has never been determined whether glycinergic inhibition of motoneurons is indeed responsible for triggering the loss of postural muscle tone during REM sleep. Therefore, we used reverse microdialysis, electrophysiology, and pharmacological and histological methods to determine whether glycinergic and/or GABAA-mediated neurotransmission at the trigeminal motor pool mediates masseter muscle atonia during REM sleep in rats. By antagonizing glycine and GABAA receptors on trigeminal motoneurons, we unmasked a tonic glycinergic/GABAergic drive at the trigeminal motor pool during waking and non-rapid eye movement (NREM) sleep. Blockade of this drive potently increased masseter muscle tone during both waking and NREM sleep. This glycinergic/GABAergic drive was immediately switched-off and converted into a phasic glycinergic drive during REM sleep. Blockade of this phasic drive potently provoked muscle twitch activity in REM sleep; however, it did not prevent or reverse REM atonia. Muscle atonia in REM even persisted when glycine and GABAA receptors were simultaneously antagonized and trigeminal motoneurons were directly activated by glutamatergic excitation, indicating that a powerful, yet unidentified, inhibitory mechanism overrides motoneuron excitation during REM sleep. Our data refute the prevailing hypothesis that REM atonia is caused by glycinergic inhibition. The inhibitory mechanism mediating REM atonia therefore requires reevaluation.
Keywords: sleep, REM, atonia, glycine, GABA, trigeminal, motoneuron, NREM sleep, REM sleep behavior disorder
Introduction
A defining feature of mammalian rapid eye movement (REM) sleep is a potent suppression of postural muscle tone. Compared with waking and non-rapid eye movement (NREM) sleep, motor control in REM sleep is unique because it is characterized by flurries of periodic muscle twitches that punctuate REM sleep atonia. Determining the neural mechanisms mediating muscle atonia is of major clinical importance because abnormal motor control during REM sleep underlies some of the major sleep disorders, including REM sleep behavior disorder (RBD), narcolepsy, obstructive sleep apnea (OSA) and bruxism.
There is considerable controversy concerning the neural mechanisms generating muscle atonia in REM sleep. The prevailing hypothesis is that glycinergic inhibition of somatic motoneurons is responsible for loss of postural muscle tone in REM sleep (Chase and Morales, 2005). This hypothesis stems from observations that trigeminal and lumbar motoneurons are hyperpolarized by large amplitude IPSPs that are reduced, but not eliminated by antagonism of glycine receptors (Soja et al., 1987; Chase et al., 1989).
Studies of respiratory motor control in sleep, however, have shown that neither glycinergic nor GABAA-mediated inhibition at the hypoglossal motor pool mediates the typical suppression of inspiratory genioglossus muscle activity during natural or carbachol-induced REM sleep (Kubin et al., 1993; Morrison et al., 2003). Although inspiratory muscle suppression in REM sleep is not caused by glycinergic/GABAergic inhibition of respiratory motoneurons, it has never been determined whether the REM-specific IPSPs recorded from nonrespiratory motoneurons (e.g., trigeminal) are in fact responsible for triggering REM atonia in postural muscles.
We aimed to determine whether masseter muscle atonia during REM sleep is caused by glycinergic and/or GABAA-mediated inhibition of trigeminal motoneurons in freely behaving rats. The trigeminal-masseteric motor system was used because trigeminal motoneurons innervate masseter muscles, which are postural muscles that are affected by, and contribute to, the pathogenesis of several sleep disorders including RBD, OSA, cataplexy/narcolepsy, and bruxism (Guilleminault et al., 1974; Horner, 1996; Schenck and Mahowald, 2002; Kato et al., 2003). Importantly, the glycinergic and GABAergic control of trigeminal motoneurons has also been well documented during natural REM (Soja et al., 1987; Chase et al., 1989).
We used reverse-microdialysis, electrophysiology, neuropharmacological and histological techniques to determine whether changes in glycinergic and GABAA-mediated neurotransmission at the trigeminal motor pool are responsible for loss of masseter muscle tone during REM sleep. First, we identified the presence of a tonic glycinergic and GABAergic tone at the trigeminal motor pool during waking and NREM sleep by antagonizing glycine and GABAA receptors on trigeminal motoneurons. Then, we demonstrated that this tonic inhibitory tone is transformed into a phasic glycinergic drive in REM sleep. Blockade of glycinergic and GABAA-mediated inhibition at the trigeminal motor pool during REM sleep triggered a potent increase in periodic muscle twitches; however, it did not prevent REM atonia. Our data therefore confirm the presence of a phasic glycinergic drive during REM sleep; however, they do not support the prevailing hypothesis that glycinergic inhibition of motoneurons is responsible for postural muscle atonia in REM sleep.
Materials and Methods
Animals
Rats were housed individually and maintained on a 12 h light/dark cycle (lights on at 7:00 A.M. and off at 7:00 P.M.) and both food and water were available ad libitum. All procedures and experimental protocols were approved by the University of Toronto's animal care committee and were in accordance with the Canadian Council on Animal Care.
Surgical preparation for sleep and microdialysis studies
Studies were performed using 19 male Sprague Dawley rats (average weight: 342 ± 10 g). To implant electroencephalographic (EEG) and electromyographic (EMG) electrodes and a microdialysis probe, sterile surgery was performed under anesthesia induced with intraperitoneal ketamine (85 mg/kg) and xylazine (15 mg/kg), and maintained with additional anesthesia given by inhalation (isoflurane, 0.5–2%). Effective depth of anesthesia was determined by the abolishment of the pedal withdrawal and blink reflexes. Body temperature was monitored with a rectal probe (CWE, Ardmore, PA) and maintained at 37 ± 1°C.
Three insulated, multistranded stainless-steel wire EMG electrodes (Cooner Wire, Chatsworth, CA) were implanted into the left and right masseter muscles. The wires were tunneled subcutaneously to an incision along the dorsal surface of the cranium. Three EMG electrodes were also inserted into the nuchal muscle. Four stainless-steel screws (JI Morris, Southbridge, MA), attached to insulated 34 gauge wire (Cooner Wire) were implanted in the skull for recording cortical EEG activity; their coordinates were 2 mm rostral and 2 mm to the left and right of bregma, and 3 mm caudal and 2 mm to the left and right of bregma. An additional reference electrode was also secured onto the skull; its position was 9.4 mm caudal and 0.5 mm lateral to bregma.
To implant a microdialysis probe into the left trigeminal motor nucleus a ∼2 mm burr hole was made at 9.4 mm caudal and 1.8 mm lateral to bregma (Paxinos and Watson, 1998). A microdialysis guide probe (CMA, St. Laurent, Quebec, Canada) was then lowered 8.2 mm below the skull surface by stereotaxic manipulation. Dental cement (1234; Lang Dental, Wheeling, IL) secured the probe in place and after the cement was dry, EEG and EMG electrodes were connected to pins (Allied Electronics, Fort Worth, TX) and inserted into a custom-made head plug (Allied Electronics) that was affixed to the skull with dental cement.
After surgery, rats were given an intraperitoneal injection of 0.03 mg/kg buprenorphine and kept warmed by a heating pad. They were also given a dietary supplement (i.e., Nutri-Cal) and soft food for the subsequent 2 d. Rats recovered for at least 7–10 d before experimental testing began.
Experimental procedures for sleep and microdialysis studies
Recording environment.
During experiments, animals were housed in Raturn system (BioAnalytical Systems, West Lafayette, IN), which is a movement-responsive caging system eliminating the need for a commutator or liquid swivel. This caging system was housed inside a sound-attenuated, ventilated, and illuminated (lights on: 110 lux) chamber.
Electrophysiological recordings.
EEG and EMG activities were recorded by attaching a lightweight cable to the plug on the rat's head, which was connected to a Super-Z head-stage amplifier and BMA-400 AC/DC Bioamplifier (CWE). The EEG signal was amplified 1000 times and bandpass filtered between 1 and 100 Hz. EMG signals were amplified between 500 and 1000 times and bandpass filtered between 30 Hz and 30 kHz. All electrophysiological signals were digitized at 500 Hz (Spike 2 software, 1401 interface; Cambridge Electronic Design, Cambridge, UK) and monitored and stored on a computer.
Microdialysis probe.
A microdialysis probe was used to perfuse candidate drugs into the trigeminal motor nucleus. The microdialysis probe (34 kDa cutoff; membrane length and diameter, 1 mm by 250 μm; CMA) was placed into the left trigeminal motor nucleus. The microdialysis probe was connected to Teflon tubing (inside diameter, 0.12 mm; Eicom, Kyoto, Japan), which was connected to a 1 ml gastight syringe via a liquid switch (BioAnalytical Systems). The probe was continually perfused with filtered (0.2 μm nylon; Fisher Scientific, Ottawa, Ontario, Canada) artificial CSF (aCSF) [containing (in mm) 125 NaCl, 5 KCl, 1.25 KH2PO4, 24 NaHCO3, 2.5 CaCl2, 1.25 MgSO2, 20 d-glucose] at a flow rate of 2 μl/min using a syringe pump (BioAnalytical Systems).
Drug preparation.
Strychnine [strychnine hydrochloride; formula weight (FW), 370.9; Sigma, Oakville, Ontario, Canada], a glycine receptor antagonist, bicuculline [(−)-bicuculline methochloride; FW, 444.87; Tocris Bioscience, Ellisville, MO)], a GABAA receptor antagonist, glycine (FW, 75.07; Tocris Bioscience), and muscimol (molecular weight, 121.31; Tocris Bioscience) a GABAA receptor agonist, were prepared in aCSF. Each drug was made immediately before each experiment. The AMPA receptor agonist (α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; FW, 186.17; Tocris Bioscience) was made in advance and stored in stock solutions at −20°C. All drugs were vortexed and filtered (0.22 μm polyvinylidene difluoride; Fisher Scientific) before use.
Experimental protocols
Each experiment took 2 d to complete. On the first day, at 8:00–10:00 A.M., animals were placed into the sleep recording chamber and given at least 1 h to habituate before they were connected to the electrical tether. They were then given a minimum of 3 h to habituate to this before recordings began. Baseline recordings (without the microdialysis probe in place) were established on day 1 of experiments, between 1:00 and 4:00 P.M. The microdialysis probe was inserted between 5:00 and 7:00 P.M. and aCSF perfused throughout the night. Probes were inserted the night before experiments began because previous studies demonstrate that probe insertion induces spontaneous neurotransmitter release and local neuronal activation (Di Chiara, 1990; Kodama et al., 1998).
On the second day of experimentation, perfusion of candidate drugs began at 8:00–9:00 A.M. Drug treatments were randomized and no >3 drug treatments were given to any one animal. Each drug was applied onto the trigeminal nucleus for 2–4 h; this typically allowed sufficient time for the animal to transition through 3 complete sleep cycles (i.e., wake to NREM to REM sleep). An aCSF washout period of at least 2 h followed every drug treatment.
Study 1: is glycinergic inhibition of somatic motoneurons responsible for REM atonia?
To answer this question, we antagonized glycine receptors by perfusing 0.1 mm strychnine into the left trigeminal motor pool while monitoring left masseter muscle EMG activity during sleep and waking. We used this concentration of strychnine because previous in vitro and in vivo studies show that it antagonizes glycinergic neurotransmission on somatic motoneurons (Song and Huang, 1990; Jonas et al., 1998; Morrison et al., 2002).
Study 2: is GABAA-mediated inhibition of motoneurons responsible for REM atonia?
We antagonized GABAA receptors by applying 0.1 mm bicuculline into the left trigeminal motor pool while monitoring left masseter muscle EMG activity across the sleep cycle. This dosage of bicuculline was previously shown to antagonize GABAA-mediated neurotransmission on somatic motoneurons in vitro and at the hypoglossal motor pool in vivo (Jonas et al., 1998; Liu et al., 2003; Pagnotta et al., 2005).
Study 3: does REM atonia require concurrent glycinergic and GABAA-mediated inhibition of motoneurons?
Because trigeminal motoneurons are inhibited by both glycinergic and GABAergic inputs during REM sleep (Soja et al., 1987), and because GABA and glycine are coreleased onto motoneurons (Jonas et al., 1998; O'Brien and Berger, 1999), we simultaneously antagonized both glycine and GABAA receptors by perfusing 0.1 mm strychnine and 0.1 mm bicuculline onto the trigeminal motor pool during sleep–wake behaviors.
Study 4: is REM atonia mediated by increased inhibition and reduced excitation of motoneurons?
We hypothesize that motor atonia during REM sleep is mediated by concomitant inhibition and disfacilitation (i.e., reduced excitation) of motoneurons during REM sleep. To test this hypothesis, we antagonized both glycine and GABAA receptors (using 0.1 mm strychnine and bicuculline) while simultaneously activating trigeminal motoneurons with 0.1 mm AMPA. This dose of AMPA provokes a robust increase in masseter muscle tone during waking and NREM sleep when applied to the trigeminal motor pool in rats (Burgess et al., 2005) and also activates genioglossus muscle activity when perfused into the hypoglossal motor pool in anesthetized rats (Aoki et al., 2006).
Study 5: demonstration that doses of strychnine and bicuculline antagonize glycine and GABAA receptors.
We microdialyzed 1 mm glycine and 1 μm muscimol (GABAA receptor agonist) into the left trigeminal motor pool before and while simultaneously applying 0.1 mm strychnine and 0.1 mm bicuculline. We used these doses of glycine and GABAA receptor agonists because they suppress genioglossus muscle EMG activity when applied to the hypoglossal motor pool in anesthetized rats (Morrison et al., 2002; Liu et al., 2003). All manipulations were made during waking when masseter muscle tone was maximal so the inhibitory effects of glycine and muscimol would induce the greatest degree of suppression. After a steady-state suppression of masseter tone was observed, we began perfusing glycine/muscimol and strychnine/bicuculline.
Verification of microdialysis probe location
Two procedures were used to demonstrate that microdialysis probes were both functional and located in the left trigeminal motor pool. At the end of each experiment, 0.1 mm AMPA was perfused into the left trigeminal motor pool, which induced a rapid and potent increase in basal levels of left masseter muscle tone without affecting either the right masseter or neck EMG activity. This result verified that trigeminal motoneurons were viable and able to respond to glutamatergic activation, that microdialysis probes were functional at the end of each experiment and that probes were located in the trigeminal motor nucleus. We also used postmortem histological analysis to demonstrate that microdialysis probes were physically located in the left trigeminal nucleus.
Under deep anesthesia (ketamine, 85 mg/kg; xylazine, 15 mg/kg, i.p.), rats were decapitated, and their brains removed and placed in chilled 4% paraformaldehyde (in 0.1 m PBS) for 24 h. Brains were cryoprotected in 30% sucrose (in 0.1 m PBS) for 48 h; they were then frozen in dry-ice and transversely sectioned in 40 μm slices using a microtome (Leica, Richmond Hill, ON). Brain sections were mounted, dried and stained with Neutral Red. Tissue sections were viewed using a light microscope (Olympus, Center Valley, PA) and the location of probe lesion tracts were plotted on standardized brain maps (Paxinos and Watson, 1998).
Data analysis
Behavioral state.
We classified four behavioral states. Alert wake (AW) was characterized by high-frequency, low-voltage EEG signals coupled with high levels of EMG activity (i.e., chewing, grooming, drinking). Quiet wake (QW) was characterized by high-frequency, low-voltage EEG signals, but in the absence of overt motor activity. NREM sleep was characterized by high-amplitude, low-frequency EEG signals and minimal EMG activity. REM sleep was characterized by low-amplitude, high-frequency theta-like EEG activity and REM atonia interspersed by periodic muscle twitches. Sleep states were visually identified and analyzed in 5 s epochs using the Sleepscore version 1.01 script (Cambridge Electronic Design).
EMG analysis.
Raw EMG signals were full-wave rectified, integrated and quantified in arbitrary units. Average EMG activity for left and right masseter and neck muscle activity was quantified in 5 s epochs for each behavioral state. When candidate drugs were applied onto the left trigeminal motor pool, EMG data were not analyzed for the first 30 min of perfusion because the latency from the syringe pump to the microdialysis probe is ∼15 min.
At least three episodes of each behavioral state (i.e., AW, QW, NREM, and REM) were analyzed for each experimental condition. In each rat the average EMG activity was calculated for each behavioral state and for each drug perfused into the trigeminal motor pool.
EMG analysis in REM sleep.
REM sleep consists of both tonic and phasic motor events. The stereotypical periods of motor atonia occur during tonic REM sleep and the periodic muscle twitches that punctuate REM atonia occur during phasic REM sleep (i.e., during rapid eye movements) (Aserinsky and Kleitman, 1953; Jouvet, 1967). Because a major goal of this study was to determine the role for glycinergic and GABAergic neurotransmission in modulating motor activity in REM sleep, we developed an objective method for identifying and quantifying the phasic (i.e., muscle twitches) and tonic (REM atonia) periods of REM sleep. To quantify motor atonia during tonic REM sleep, we determined the 99th percentile of EMG activity during the first 5 s of each REM period because muscle twitches are conspicuously absent during this time (Morrison et al., 2003; Lu et al., 2005). The muscle twitches that define phasic REM sleep were classified as motor events (lasting >20 ms) that exceeded the 99th percentile of EMG activity during the first 5 s of REM; conversely, REM sleep atonia was classified as any period in which muscle activity was equal to or less than the 99th percentile of EMG activity during the first 5 s of REM. In each rat, REM atonia and muscle twitches were quantified for each REM episode during baseline conditions and for each drug perfused into the trigeminal motor pool.
EEG spectral analysis.
Spectral analysis was performed using EEG Band Detect version 1.06 in Spike 2. The EEG was windowed using a Hamming function and subjected to a fast Fourier transform to yield the power spectrum. The power within four frequency bands was recorded as absolute power and as a percentage of the total power of the signal that was calculated over each 5 s epoch. The band limits used were delta (δ), 0.48–4 Hz; theta (θ), 4.25–8 Hz; alpha (α), 8.25–15 Hz; and beta (β), 15.25–35 Hz.
Statistical analyses
All statistical analyses used SigmaStat (SPSS, Chicago, IL) and applied a critical two-tailed α value of p < 0.05. Differences in basal EMG tone during waking, NREM, and tonic REM sleep under baseline (i.e., ACSF) and drug treatments (i.e., 0.1 mm strychnine or 0.1 mm bicuculline or 0.1 mm strychnine/bicuculline or strychnine/bicuculline/AMPA) were determined using ANOVA with repeated measures (RM-ANOVA) and post hoc comparisons were performed using a Student–Newman–Keuls (SNK) test. Comparisons between treatments for frequency, amplitude and duration of muscle twitches per REM episode were made using ANOVA (i.e., drug treatments versus aCSF) and post hoc comparisons were performed using a SNK test. All data are expressed as mean ± SEM (SEM).
Results
Inserting microdialysis probes into the trigeminal motor pool transiently increased masseter tone, but had no effect on the sleep–wake cycle
Inserting a microdialysis probe into the left motor pool caused a transient yet robust activation of the left masseter muscle (relative to aCSF; paired t test, p = 0.011) without affecting the activity of either right masseter (paired t test, p = 0.261) or neck muscle (paired t test, p = 0.637) activities. This finding demonstrates that probe insertion only activates motoneurons in the left trigeminal motor pool. We also found that microdialysis of 0.1 mm AMPA into the left trigeminal motor pool at the end of each experiment increased left masseter muscle activity (paired t test, p = 0.035) (Fig. 1 A) without altering levels of either the right masseter (paired t test, p = 0.563) (Fig. 1 A) or neck muscle EMG activity (paired t test, p = 0.219). This procedure verified that probes were located within the left trigeminal nucleus and that microdialysis probes were functional. It also shows that motoneurons were viable and able to respond to changes in excitation neurotransmission.
Figure 1.
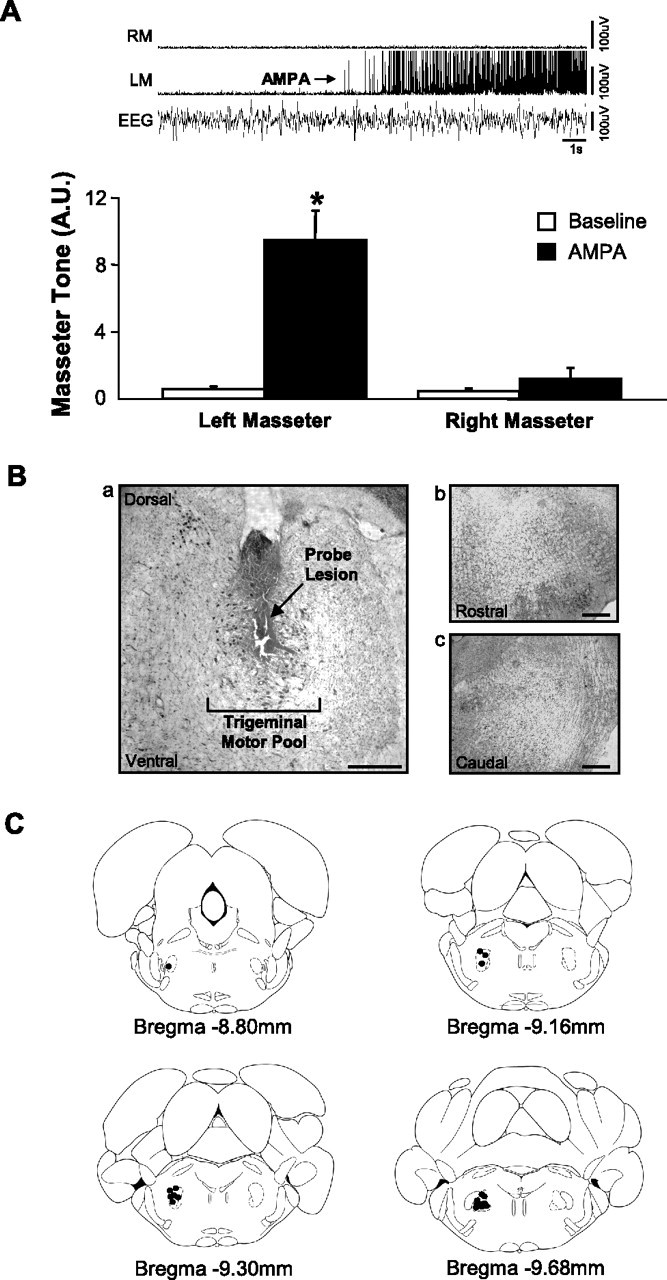
Microdialysis probes were located in the left trigeminal motor pool. A, Group data showing that AMPA perfusion increased left masseter (LM) but not right masseter (RM) tone, indicating that the probe was located in the left motor pool. Top, A representative trace showing LM and RM EMG tone before and during AMPA perfusion into the left trigeminal nucleus. The AMPA effect on LM tone is indicated by an arrow. Ba, Example of a lesion site left by a microdialysis probe in the left trigeminal motor pool. Bb, Bc, Sections immediately flanking the rostral and caudal borders of the trigeminal nucleus; no lesion was found in these areas, demonstrating that the probe was located exclusively within the motor pool. Scale bars, 500 μm. C, Filled circles represent the location of probe lesions in the left trigeminal nuclei in the 19 rats used in this study. The asterisk indicates a significant change (p < 0.05) in activity from baseline levels. A.U., Arbitrary units. All values are mean ± SEM.
In all 19 rats, we used postmortem histology to confirm that microdialysis probes were either within or immediately adjacent to the left trigeminal motor pool (Fig. 1 B,C).
We also found that placing a probe and perfusing glycinergic and GABAergic antagonists into the trigeminal nucleus had no effect on the amount of time spent in each sleep–wake state when baseline and antagonist treatment (i.e., strychnine and bicuculline) were compared (RM-ANOVA, p = 0.374). There was also no change in the ratio of high to low frequencies in EEG powers (i.e., %β/%δ) before and after antagonist treatment (RM-ANOVA, p = 0.872), indicating that these interventions did not substantially effect sleep–wake regulation.
Masseter muscle atonia in REM sleep is not mediated by glycinergic inhibition of trigeminal motoneurons
If a functional glycinergic drive underlies loss of muscle tone in REM sleep, then antagonism of glycine receptors on motoneurons should reverse REM sleep atonia. In 11 rats, we perfused 0.1 mm strychnine into the left trigeminal motor pool while monitoring masseter EMG activity during sleep and waking. Strychnine perfusion had a potent stimulatory effect on basal levels of masseter tone during both QW and NREM sleep. Compared with left masseter EMG activity under baseline conditions, antagonism of glycine receptors increased basal masseter tone during QW by 51 ± 8% (p < 0.05 post hoc test) and during NREM sleep by 29 ± 4% (p < 0.05 post hoc test) (Fig. 2 A,B). This stimulatory effect on masseter tone was tonically sustained throughout periods of QW and NREM sleep, demonstrating the presence of a tonic glycinergic drive during these states. We also found that strychnine increased left masseter tone by the same magnitude at the beginning (43 ± 15% above baseline; paired t test, p = 0.013) and end (41 ± 14% above baseline; paired t test, p = 0.012) of the 2.3 ± 0.1 h perfusion period (paired t test, p = 0.899) (Fig. 3), indicating that glycine receptor antagonism was not subject to time-dependent changes.
Figure 2.
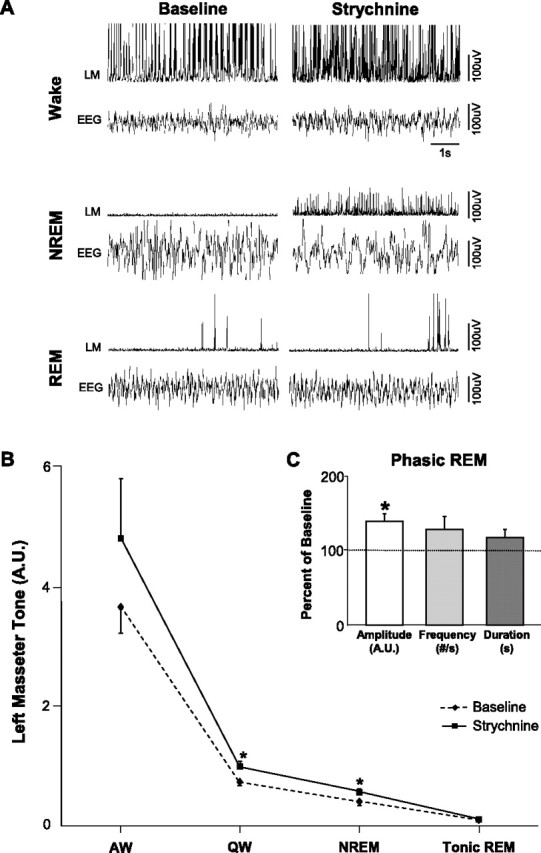
There is an endogenous glycinergic drive at the trigeminal motor pool in waking and NREM sleep, but not during tonic REM sleep. A, Raw EMG and EEG traces from one rat showing that strychnine perfusion into the left trigeminal motor pool increased left masseter muscle (LM) tone during waking and NREM sleep; this intervention did not affect the atonia of tonic REM sleep. However, it did increase the left masseter muscle twitch amplitude during phasic REM. B, Group data (n = 11) demonstrating that strychnine increased LM tone in QW and NREM sleep, without changing levels of motor tone during tonic REM sleep. Although this intervention consistently increased LM tone in AW, it was not statistically significant. C, Group data (n = 101 REM periods) demonstrating that strychnine perfusion into the left trigeminal motor pool increased left masseter muscle twitch amplitude during phasic REM sleep, without significantly effecting either muscle twitch frequency or duration. *p < 0.05. A.U., Arbitrary units; #/s, number muscle twitches per second. All values are mean ± SEM.
Figure 3.

The magnitude of masseter muscle activation by glycinergic and GABAergic antagonists was unaffected by time. Group data (n = 11) demonstrating that strychnine, bicuculline, or strychnine/bicuculline perfusion into the left trigeminal motor pool increased left masseter EMG tone by the same magnitude at the beginning (white bars) and end (gray bars) of respective drug application periods. This finding demonstrates there was no time-dependent changes in antagonist efficacy. All measurements were taken during NREM sleep and values are mean ± SEM.
Although we identified a tonic glycinergic drive at the trigeminal motor pool during both waking and NREM sleep, we could not confirm the presence of such drive during tonic REM sleep. Compared with baseline conditions, strychnine perfusion had no effect on basal levels of masseter tone during tonic REM sleep (p = 0.958 post hoc test) (Fig. 2 A,B). Indeed, we demonstrate that the tonic glycinergic drive present in NREM sleep is rapidly lost on entrance into REM sleep, but is reinstated during post-REM waking (Fig. 4 B–D). We analyzed levels of masseter EMG activity in the 30 s periods immediately proceeding (i.e., NREM) and following (i.e., post-REM waking) each REM episode. In the 101 REM periods analyzed (mean REM duration: 80 ± 11 s), we found that strychnine increased left masseter tone above baseline levels in all pre-REM (p < 0.001 post hoc test) and post-REM (p < 0.001 post hoc test) periods. However, there was rapid and complete loss of strychnine's stimulatory effects during periods of tonic REM sleep (p = 0.860 post hoc test; i.e., not different from baseline levels), which resulted in the persistence of masseter atonia despite continued glycine receptor antagonism (Fig. 4 B–D).
Figure 4.

The tonic glycinergic drive present at the trigeminal motor pool in NREM sleep is abruptly switched off in REM sleep. A, Typical EMG and EEG traces showing how left masseter muscle (LM) activity changes during the transitions into and out of REM sleep during baseline (i.e., aCSF at the trigeminal nucleus). B, Another trace from the same rat showing that strychnine perfusion increases LM activity during the NREM and waking periods preceding and after REM onset (compared with traces in A), but that masseter atonia persists even though glycine receptors are blocked. C, A typical raw trace showing the abrupt loss of masseter tone on entrance into REM sleep, despite strychnine perfusion at the trigeminal nucleus. D, Group data showing that compared with baseline, strychnine perfusion significantly increased LM activity in NREM and post-REM waking (QW), but not during REM sleep. A.U., Arbitrary units. All values are mean ± SEM.
Because motoneurons are inhibited by glycinergic inputs during the muscle twitches of phasic REM sleep (Chase and Morales, 1983), we also determined whether strychnine affected muscle twitches. Antagonism of glycine receptors significantly increased the amplitude of muscle twitches by 27 ± 9% above baseline levels during phasic REM sleep (p = 0.018). However, neither twitch duration (p = 0.455) nor frequency (p = 0.219) were affected by strychnine perfusion (Fig. 2 C). Because strychnine increased the magnitude of masseter twitches, this not only identifies the presence of a phasic glycinergic drive at the trigeminal motor pool during REM sleep, but that 0.1 mm strychnine antagonized and reversed the inhibitory effects of this drive.
GABAA-mediated inhibition of trigeminal motoneurons does not cause masseter atonia in REM sleep
Although we identified a phasic glycinergic drive during REM sleep, it is not responsible for mediating REM atonia. Because GABA is the most common inhibitory neurotransmitter in the brain (Bloom and Iversen, 1971) and because motoneurons are influenced by GABAergic processes in REM sleep (Soja et al., 1987), we hypothesized that motor atonia of REM sleep may be mediated by GABAergic inhibition. Therefore, we antagonized GABAA receptors on trigeminal motoneurons using 0.1 mm bicuculline while monitoring masseter EMG activity during sleep and waking (n = 11).
Bicuculline perfusion into the left trigeminal nucleus induced a robust facilitation of masseter tone during both waking and NREM sleep. Compared with left masseter EMG activity under baseline conditions, antagonism of GABAA receptors increased basal masseter tone in AW by 106 ± 30% (p = 0.009 post hoc test), in QW by 183 ± 30% (p < 0.05 post hoc test), and during NREM sleep by 180 ± 30% (p < 0.05 post hoc test) (Fig. 5 A,B). This stimulatory effect on masseter tone was tonically sustained throughout individual periods of waking and NREM sleep and its stimulatory effects were maintained at constant levels across the 2.7 ± 0.2 h perfusion period (paired t test, p = 0.657) (Fig. 3). These findings not only identify a tonic GABAergic tone at the trigeminal nucleus during waking and NREM sleep, but that 0.1 mm bicuculline antagonized GABAergic inhibition.
Figure 5.
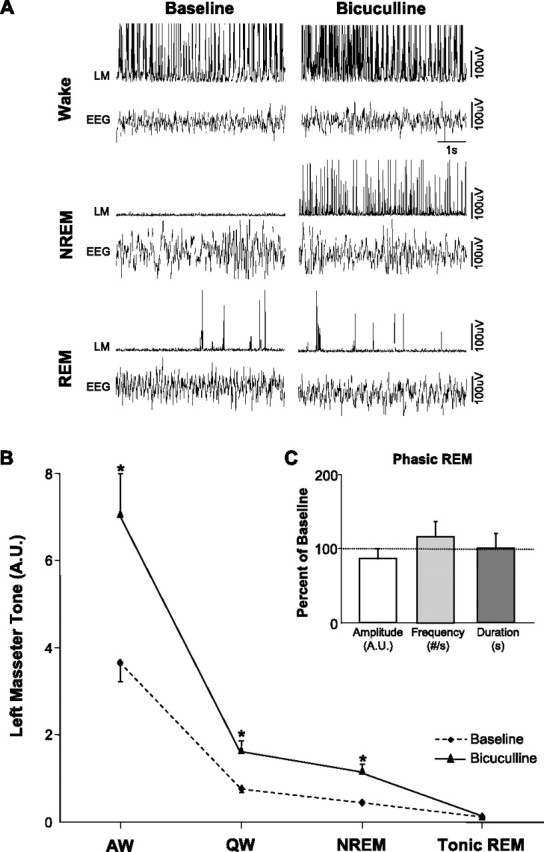
Trigeminal motoneurons are inhibited by a tonic GABAergic drive during waking and NREM sleep, but not in REM sleep. A, Raw traces showing that bicuculline perfusion into the left trigeminal motor pool increases left masseter muscle (LM) tone during waking and NREM sleep; this intervention did not affect either basal masseter tone in tonic REM sleep or masseter muscle twitch activity in phasic REM. B, Group data (n = 11) showing that bicuculline perfusion potently increased LM tone in AW, QW, and NREM sleep, without reversing muscle atonia in tonic REM sleep. *p < 0.05. C, Group data (n = 107 REM periods) demonstrating that bicuculline perfusion did not affect masseter muscle twitch amplitude, frequency, or duration during phasic REM sleep. A.U., Arbitrary units; #/s, number muscle twitches per second. All values are mean ± SEM.
Bicuculline perfusion had no effects on basal levels of masseter tone during tonic REM sleep (p = 0.958) (Fig. 5 A,B). We found that the stimulatory effects of bicuculline during NREM sleep were rapidly lost on entrance into REM sleep (Fig. 6 C). In the 107 REM periods analyzed (mean REM duration, 74 ± 11 s), we found that bicuculline increased masseter tone (relative to baseline levels), in all pre-REM (p < 0.001 post hoc test) and post-REM (p < 0.001 post hoc test) periods; however, there was rapid and complete loss of the stimulatory effects of bicuculline during each REM episode (p = 0.622 post hoc test) (Fig. 6 B,D).
Figure 6.

Masseter muscle atonia in REM sleep is not mediated by GABAergic inhibition of trigeminal motoneurons. A, Raw traces demonstrating how left masseter muscle (LM) activity changes during the transition into and out of REM sleep during baseline (i.e., aCSF). B, An example trace from the same rat showing that bicuculline perfusion at the left trigeminal motor pool increases LM activity during the NREM and waking periods preceding and after REM onset (compared with baseline), but that masseter atonia is unaffected by blocking GABAA receptors. C, A representative trace showing the abrupt loss of masseter tone on entrance into REM sleep despite bicuculline perfusion at the trigeminal nucleus. D, Group data showing that compared with baseline, bicuculline perfusion significantly increased LM activity in NREM and post-REM waking (QW), but not during REM sleep. A.U., Arbitrary units. All values are mean ± SEM.
Unlike strychnine perfusion, which facilitated muscle twitch activity during REM sleep, we found that bicuculline had no significant effect (compared with aCSF baseline) on the amplitude (p = 0.271), duration (p = 0.651), or frequency (p = 0.219) of masseter muscle twitches during REM sleep (Fig. 5 C).
Coantagonism of glycine and GABAA receptors on trigeminal motoneurons does not reverse the masseter atonia in REM sleep
We found that neither glycine nor GABAA receptor antagonism reversed masseter muscle atonia during REM sleep. However, because motoneurons are concurrently inhibited by GABA and glycine during natural REM sleep (Soja et al., 1987) and because both transmitters are coreleased onto motoneurons (O'Brien and Berger, 1999), especially during pharmacologically induced REM-like sleep (Kodama et al., 2003), we hypothesized that both transmitters may be required to induce REM atonia. Therefore, we simultaneously antagonized glycine and GABAA receptors in the left trigeminal motor pool by perfusing 0.1 mm strychnine and 0.1 mm bicuculline (n = 11).
Coantagonism of glycine and GABAA receptors resulted in a potent activation of masseter tone during both waking and NREM sleep. Perfusion of strychnine and bicuculline increased masseter tone in AW by 215 ± 35% (p < 0.001 post hoc test), in QW by 502 ± 95% (p < 0.05 post hoc test), and during NREM sleep by 643 ± 121% (p < 0.05 post hoc test) (Fig. 7 A,B). This stimulatory effect was tonically sustained throughout individual episodes of waking and NREM sleep, and its stimulatory effects were maintained at constant levels across the 2.6 ± 0.1 h perfusion period. Compared with baseline, left masseter tone increased during the first NREM period by 500 ± 130% (paired t test; p = 0.002) and during the last NREM period by 491 ± 170% (paired t test; p = 0.016); there was no difference in the magnitude of these responses (paired t test; p = 0.930) (Fig. 3).
Figure 7.
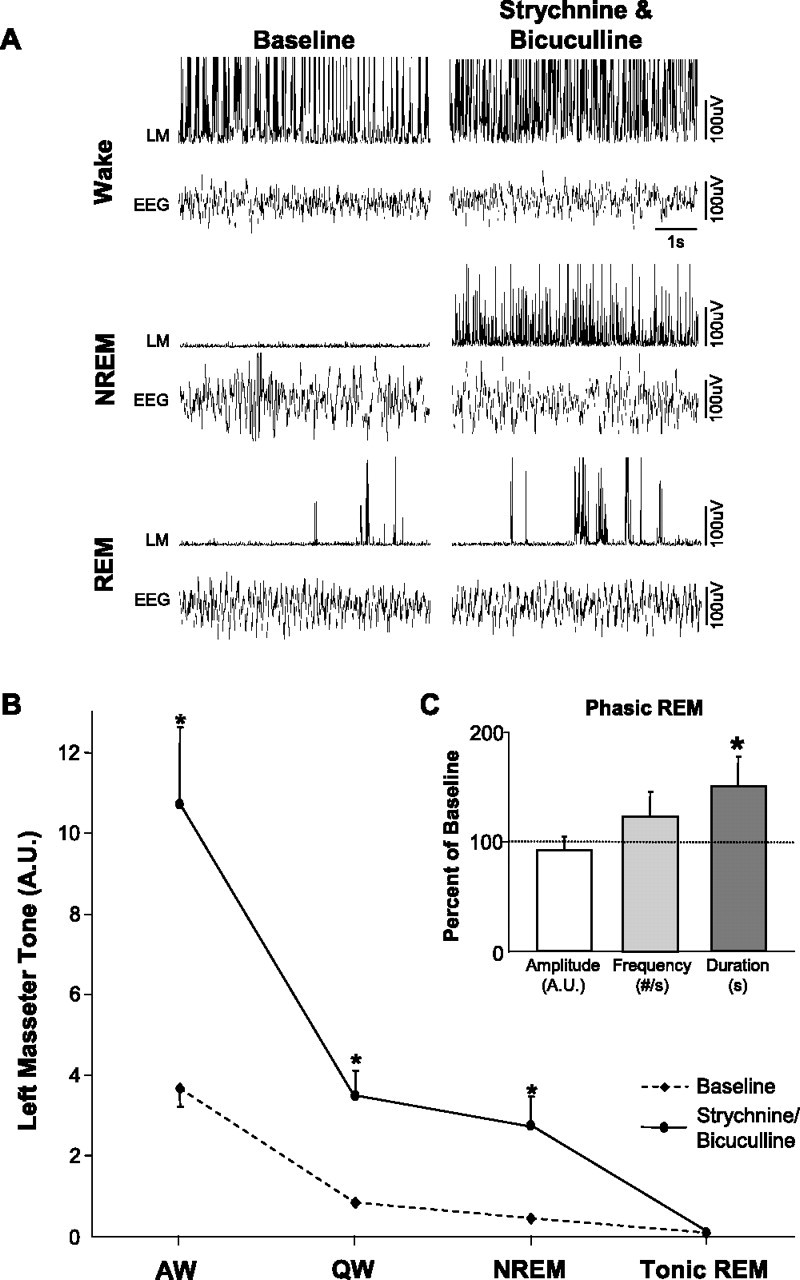
A tonic glycinergic/GABAergic drive at the trigeminal motor pool functions to suppress masseter tone in waking and NREM sleep, but not during tonic REM sleep. A, Typical EMG and EEG traces illustrating that strychnine/bicuculline perfusion into the left trigeminal motor pool increases left masseter muscle (LM) tone during waking and NREM sleep; however, it has no effect on masseter atonia in tonic REM sleep despite provoking masseter muscle twitch activity in phasic REM. B, Group data (n = 11) showing that strychnine/bicuculline perfusion increases LM tone in AW and QW and in NREM sleep, without changing levels of motor tone during tonic REM sleep. C, Group data showing strychnine/bicuculline perfusion into the left trigeminal nucleus increases masseter muscle twitch duration in phasic REM sleep, without significantly affecting either twitch amplitude or frequency. *p < 0.05. A.U., Arbitrary units; #/s, number muscle twitches per second. All values are mean ± SEM.
Although combined glycine and GABAA receptor antagonism had potent excitatory effects on basal masseter tone during waking and NREM sleep, this stimulatory effect was immediately lost on entrance into REM sleep, but immediately regained during post-REM waking. Dual strychnine/bicuculline perfusion tonically increased basal masseter tone (relative to baseline levels) during all pre-REM (p < 0.001 post hoc test) and post-REM (p < 0.001 post hoc test) periods; however, there was rapid and complete loss of this stimulatory effect during tonic REM sleep (p = 0.588 post hoc test) (Fig. 8 B–D).
Figure 8.

Masseter atonia in REM sleep is not mediated by concurrent glycinergic and GABAergic inhibition of trigeminal motoneurons. A, Traces showing how left masseter muscle (LM) activity changes during the transition into and out of REM sleep during baseline. B, A trace from the same rat illustrating that strychnine/bicuculline perfusion at the left trigeminal motor pool increases LM activity during the NREM and waking periods preceding and after REM onset, but that masseter atonia is unaffected by this intervention. C, An example showing the sudden loss of masseter tone on entrance into REM sleep despite strychnine/bicuculline perfusion at the trigeminal nucleus. D, Group data showing that strychnine/bicuculline perfusion increases basal LM tone in NREM and post-REM waking (QW), but not during REM sleep. A.U., Arbitrary units. All values are mean ± SEM.
Although coantagonism of glycine and GABAA receptors could not reverse REM sleep motor atonia, it significantly potentiated muscle twitch activity during phasic REM sleep (Fig. 7 C). Strychnine and bicuculline perfusion potently increased the mean duration of individual muscle twitches by 52 ± 2.7% (p = 0.036); however, it had no effect on either muscle twitch amplitude (p = 0.330) or frequency (p = 0.219).
REM atonia persists even when glycine and GABAA receptors are antagonized and trigeminal motoneurons are directly activated by AMPA
Because masseter muscle atonia during REM sleep could not be reversed by blockade of glycine and GABAA receptors, we hypothesized that REM atonia may be mediated by a disfacilitation of excitatory inputs (Fenik et al., 2005; Chan et al., 2006). We therefore antagonized both glycine and GABAA receptors (using 0.1 mm strychnine and bicuculline) while simultaneously activating trigeminal motoneurons with a potent dose of AMPA (0.1 mm; n = 5).
We found this intervention had powerful stimulatory effects on masseter tone during waking and NREM sleep, increasing left masseter EMG activity above baseline levels by 1539 ± 502 (p = 0.003 post hoc test) and 983 ± 280% (p = 0.031 post hoc test) (Fig. 9 A), respectively. This effect was also significantly greater than the effect that combined strychnine and bicuculline perfusion (without AMPA) had on levels of masseter tone during NREM sleep (paired t test; p < 0.001). However, this stimulatory effect was immediately abolished at the transition from NREM sleep into REM sleep, with REM atonia persisting despite simultaneous antagonism of both glycine and GABAA receptors and activation of AMPA receptors (n = 11 REM periods) (Fig. 9 B). Compared with baseline levels, perfusion of strychnine, bicuculline and AMPA into the left trigeminal motor pool did not prevent or reverse masseter atonia in REM sleep (p = 0.795 post hoc test) (Fig. 9 C).
Figure 9.
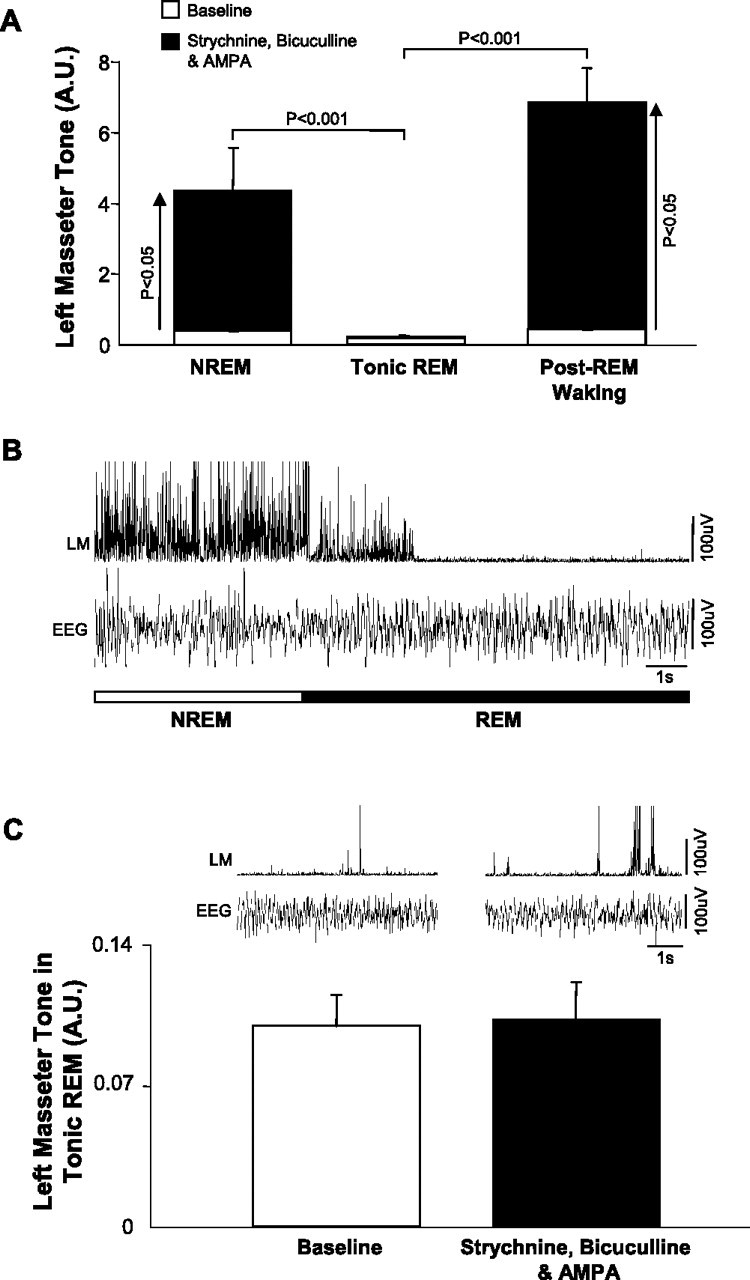
REM atonia persists even when glycine and GABAA receptors are antagonized and trigeminal motoneurons are directly activated by AMPA. A, Group data showing that simultaneous strychnine/bicuculline and AMPA perfusion into the left trigeminal nucleus potently increases basal LM tone in NREM and post-REM waking (QW), but not during tonic REM sleep. B, A typical example trace showing the sudden loss of left masseter (LM) tone on entrance into REM sleep despite glycine and GABAA receptor antagonism and direct activation of trigeminal motoneurons by AMPA. Note that basal LM tone in NREM is facilitated by this intervention, but this response is rapidly lost in REM sleep. C, Left top trace, LM activity during baseline. Right top trace, LM activity during strychnine/bicuculline/AMPA perfusion. This intervention had no effect on atonia but did increase the duration of masseter twitches in phasic REM (Fig. 6 C). Group data (n = 11 REM periods) shows that masseter atonia is unaffected by strychnine/bicuculline/AMPA perfusion. A.U., Arbitrary units. All values are mean ± SEM.
Strychnine and bicuculline antagonize the effects of glycine and GABAA receptor agonism at the trigeminal motor pool
Although previous in vitro and in vivo studies have demonstrated that 0.1 mm strychnine and 0.1 mm bicuculline antagonize glycine and GABAA receptors (Song and Huang, 1990; Jonas et al., 1998; Morrison et al., 2003; Pagnotta et al., 2005), we aimed to verify that these doses blocked glycinergic and GABAergic inhibition on trigeminal motoneurons. To do this, we exogenously applied 1 mm glycine and 1 μm muscimol into the trigeminal nucleus before and while simultaneously microdialyzing 0.1 mm strychnine and bicuculline in three rats.
We found that glycine and muscimol perfusion induced a significant suppression of waking levels of left masseter muscle tone, reducing it to 58 ± 2% of baseline levels (p = 0.025 post hoc test). This suppression was rapidly reversed when 0.1 mm strychnine and bicuculline were concurrently perfused with glycine and muscimol (p = 0.009 post hoc test) (Fig. 10). Not only did strychnine and bicuculline block the inhibitory effects of the agonists, it increased masseter tone above waking levels during baseline conditions (p = 0.04 post hoc test; 39 ± 9%) (Fig. 10). This observation not only demonstrates that this concentration of antagonists effectively reversed glycine and GABAA receptor agonism, but that it also antagonized the endogenous inhibitory drive present in waking.
Figure 10.

Strychnine and bicuculline antagonize the effects of glycine and GABAA receptor agonism at the trigeminal motor pool. Group data (n = 3) showing that agonism of glycine and GABAA receptors by glycine and muscimol application (light gray bar) significantly reduces LM activity to 58 ± 2% of baseline levels (i.e., aCSF; white bar). This suppression was reversed when strychnine and bicuculline were concurrently perfused with glycine and muscimol (dark gray bars). A.U., Arbitrary units. All values are mean ± SEM.
Discussion
A tonic glycinergic and GABAergic drive at the trigeminal motor pool suppresses masseter motor tone in waking
Intracellular studies demonstrate that trigeminal motoneurons are hyperpolarized by IPSPs in waking cats (Chase and Morales, 2005). We confirm the presence of an endogenous glycinergic and GABAergic tone that contributes to levels of trigeminal motoneuron excitability and masseter muscle activity during waking. We show that antagonism of glycine and GABAA receptors on trigeminal motoneurons results in a sustained increase in basal masseter tone throughout wakefulness, indicating that a tonic inhibitory drive functions to suppress waking muscle tone. A similar glycinergic/GABAergic tone at the hypoglossal motor pool has been shown to modulate inspiratory genioglossus muscle activity in waking rats (Morrison et al., 2003).
A waking inhibitory tone may function to gate the glutamatergic and monoaminergic drives that activate somatic motoneurons during wakefulness (Chan et al., 2006; Mir et al., 2006). Stiff-person syndrome and hyperekplexia are neurological disorders caused by either dysregulation of GABAA receptor homeostasis (Gilbert et al., 2006) or mutations in the α1 subunit of the glycine receptor (Shiang et al., 1993), with both mutations impairing glycinergic and/or GABAergic inhibition of motoneurons (Becker et al., 2002). A major symptom of these disorders is muscle hypertonia in waking (Kurczynski, 1983; Shiang et al., 1993). We identify a wake-related glycinergic/GABAergic tone at the trigeminal nucleus, that when blocked, produces muscle hypertonia. Together, these observations suggest that a waking inhibitory tone functions to dampen muscle tone and the inability to transduce this natural inhibitory drive (i.e., glycine and/or GABAA receptors mutations) underlies the muscle rigidity of stiff-person syndrome and hyperekplexia.
Somatic motoneurons are inhibited by a tonic glycinergic and GABAergic drive during NREM sleep
Antagonism of glycine and GABAA receptors at the trigeminal motor pool potently increased masseter muscle tone throughout NREM sleep, thus unmasking the presence of a tonic inhibition during this state. Inhibitory tone is maximal during NREM sleep because blocking it resulted in a 643% increase in masseter tone compared with the 215 and 502% increases evoked in alert and quiet waking, respectively. We suggest that a progressively strengthened glycinergic/GABAergic tone at the trigeminal motor pool functions to state-dependently suppress masseter tone from alert waking into NREM sleep. This perspective is congruent with intracellular recordings in cats demonstrating that trigeminal and lumbar motoneurons are progressively hyperpolarized from alert wake into NREM sleep (Chase et al., 1980).
Basal muscle tone is not only suppressed in NREM sleep by a pervasive inhibitory tone, it is also suppressed by withdrawal of glutamatergic and noradrenergic excitation of motoneurons (Burgess et al., 2005; Chan et al., 2006; Mir et al., 2006). Therefore, both passive disfacilitation and active inhibition of motoneurons are responsible for suppressing basal muscle tone in NREM sleep.
A phasic inhibitory drive functions to oppose muscle twitches during REM sleep
We demonstrate that the functional glycinergic and GABAA-mediated drive present at the trigeminal nucleus in waking and NREM sleep is immediately switched off and converted to a phasic glycinergic drive during REM sleep. Antagonism of this drive potently facilitated muscle twitch activity without affecting REM sleep muscle atonia. This is in striking contrast to the tonic inhibitory drive of waking and NREM sleep, which when blocked resulted in a sustained increase in basal masseter tone. The phasic inhibitory drive of REM sleep is mediated by glycine because strychnine application onto trigeminal motoneurons potentiated muscle twitches in REM sleep, whereas bicuculline had no effect. These findings mirror observations from intracellular studies showing that motoneurons are maximally hyperpolarized by glycinergic IPSPs during periods of REM sleep with muscle twitches (Lopez-Rodriguez et al., 1990, 1992). During REM sleep, trigeminal and spinal motoneurons are concurrently inhibited and excited by glycinergic and glutamatergic inputs (Chase and Morales, 1982, 1983; Glenn and Dement, 1985; Pedroarena et al., 1994; Soja et al., 1995); therefore, we suggest that a phasic inhibitory drive during REM sleep functions to oppose the glutamatergic inputs that trigger muscle twitches.
Dysfunction of the REM-related inhibitory drive may explain the primary symptom of RBD, a neurological condition of unknown origin whose presenting clinical symptom is excessive phasic muscle activity during REM sleep (Schenck et al., 1988). It may also explain why point-mutations in the α1 subunit of the glycine receptor, which causes hyperekplexia, also leads to myoclonic twitching in sleep (de Groen and Kamphuisen, 1978). These data suggest that the phasic inhibitory drive during REM sleep functions to oppose muscle twitch activity and loss of this drive could underlie the excessive myoclonic activity reported in RBD and hyperekplexia.
Glycinergic and GABAA-mediated inhibition of somatic motoneurons is not responsible for mediating REM sleep atonia
Our most fundamental observation is that REM sleep atonia could not be reversed by either glycine or GABAA receptor antagonism. This finding contradicts the prevailing hypothesis that REM atonia is mediated by glycinergic inhibition of motoneurons (Chase and Morales, 2005). Chase and Morales (2005) established this hypothesis because they found that lumbar and trigeminal motoneurons are hyperpolarized by large amplitude IPSPs that are reduced (but not eliminated) by antagonism of glycine receptors (Soja et al., 1987; Chase et al., 1989). Although we found that strychnine at the trigeminal motor pool unmasked a tonic inhibitory drive and increased basal masseter tone during waking and NREM sleep, it had no effect on REM atonia. Antagonism of either GABAA receptors alone or both glycine and GABAA receptors also had no effect on REM sleep muscle atonia despite causing robust increases in muscle activity during waking, NREM, and phasic REM sleep.
One concern is whether REM atonia could not be reversed because glycine and GABAA receptors were not completely antagonized. However, we used strychnine and bicuculline at concentrations that were previously shown to eliminate glycine- and GABAA-mediated inhibition of motoneurons (Song and Huang, 1990; Jonas et al., 1998; Morrison et al., 2003; Pagnotta et al., 2005). We also demonstrate that dosages of these antagonists blocked the suppressive effects that high doses of glycine and muscimol at the trigeminal motor pool had on masseter muscle tone (Fig. 10).
However, the most concrete demonstration that glycine and GABAA receptors were antagonized stems from the fact that strychnine and bicuculline provoked masseter twitch activity in REM sleep. Because IPSPs that hyperpolarize trigeminal motoneurons are at their maximal frequency and amplitude during periods of REM sleep when muscle twitches occur (Pedroarena et al., 1994), and because we show that antagonists potently increased muscle twitches, we conclude that motoneurons were indeed antagonized. Therefore, the persistence of muscle atonia during strychnine and bicuculline application indicates that REM atonia is not mediated by glycinergic or GABAA-mediated inhibition.
This conclusion is congruent with the findings that suppression of respiratory-related genioglossus muscle activity during either natural or carbachol-induced REM sleep could not be reversed by antagonism of glycinergic or GABAA-mediated inhibition at the hypoglossal motonucleus in rats (Kubin et al., 1993; Morrison et al., 2003). We conclude that glycinergic and GABAA-mediated inhibition of motoneurons is not the mechanism mediating the loss of postural muscle tone during REM sleep.
What is the root mechanism responsible for REM atonia?
Although motoneurons are hyperpolarized during REM sleep (Nakamura et al., 1978) and glycine and GABA are released onto them during REM-like atonia (Kodama et al., 2003), we demonstrate that glycinergic and GABAA-mediated inhibition of trigeminal motoneurons does not mediate REM sleep atonia. Multiple lines of evidence indicate that muscle atonia in REM (and cataplexy) may result from disfacilitation of noradrenergic inputs onto motoneurons (Aston-Jones and Bloom, 1981; Mignot et al., 1993; Wu et al., 1999; Lai et al., 2001; Fenik et al., 2005). However, glutamatergic, noradrenergic and serotonergic activation of motoneurons during REM sleep fails to reverse muscle atonia (Jelev et al., 2001; Chan et al., 2006; Mir et al., 2006). Our current study demonstrates that loss of postural muscle tone in REM sleep persists even when glycine and GABAA receptors are antagonized and motoneurons are directly activated by glutamatergic excitation, indicating that a powerful, yet unidentified, inhibitory mechanism overrides motoneuron excitation during REM sleep.
Although REM atonia is not triggered by either glycine- or GABAA-mediated inhibition, it is possible that GABAB receptors could be involved. Previous studies demonstrate that activation of GABAB receptors on hypoglossal motoneurons suppresses their activity in vitro (O'Brien and Berger, 1999) and reduces genioglossus motor tone in anesthetized rats (Okabe et al., 1994). However, it is unknown whether GABAB receptors are endogenously active during natural sleep–wake motor behaviors.
Another possible source of motoneuron inhibition during REM sleep may be from cholinergic neurons in the pedunculopontine tegmental nucleus. Neurons in this region are maximally active during REM sleep (el Mansari et al., 1989; Steriade et al., 1990) and they project to and inhibit motoneurons (Bellingham and Berger, 1996; Liu et al., 2005). The role that cholinergic neurons play in triggering REM atonia has not been determined, but clearly merits consideration.
In summary, we conclude that REM atonia is induced by inhibition of somatic motoneurons; however, neither glycinergic nor GABAA-mediated inhibition are responsible. The biochemical substrate mediating REM atonia remains unknown and therefore requires identification.
Footnotes
This work was supported by Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council (NSERC) grants to J.H.P. P.L.B. received salary funding from NSERC. We are grateful to Drs. Jerry Siegel and Richard Horner for their helpful comments and Christian Burgess for technical assistance, and we are indebted to Dr. James Duffin for his expert assistance and advice.
References
- Aoki CR, Liu H, Downey GP, Mitchell J, Horner RL. Cyclic nucleotides modulate genioglossus and hypoglossal responses to excitatory inputs in rats. Am J Respir Crit Care Med. 2006;173:555–565. doi: 10.1164/rccm.200509-1469OC. [DOI] [PubMed] [Google Scholar]
- Aserinsky E, Kleitman N. Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science. 1953;118:273–274. doi: 10.1126/science.118.3062.273. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker L, von Wegerer J, Schenkel J, Zeilhofer HU, Swandulla D, Weiher H. Disease-specific human glycine receptor alpha1 subunit causes hyperekplexia phenotype and impaired glycine- and GABAA-receptor transmission in transgenic mice. J Neurosci. 2002;22:2505–2512. doi: 10.1523/JNEUROSCI.22-07-02505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Berger AJ. Presynaptic depression of excitatory synaptic inputs to rat hypoglossal motoneurons by muscarinic M2 receptors. J Neurophysiol. 1996;76:3758–3770. doi: 10.1152/jn.1996.76.6.3758. [DOI] [PubMed] [Google Scholar]
- Bloom FE, Iversen LL. Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature. 1971;229:628–630. doi: 10.1038/229628a0. [DOI] [PubMed] [Google Scholar]
- Burgess CR, Lai YY, Siegel JM, Peever JH. Glutamatergic control of somatic motoneurons in freely behaving rats. Sleep. 2005;28:A13. [Google Scholar]
- Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med. 2006;174:1264–1273. doi: 10.1164/rccm.200605-597OC. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. Phasic changes in motoneuron membrane potential during REM periods of active sleep. Neurosci Lett. 1982;34:177–182. doi: 10.1016/0304-3940(82)90172-0. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. Subthreshold excitatory activity and motoneuron discharge during REM periods of active sleep. Science. 1983;221:1195–1198. doi: 10.1126/science.6310749. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. Control of motoneurons during sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Ed 3. Philadelphia: W. B. Saunders; 2005. pp. 154–168. [Google Scholar]
- Chase MH, Chandler SH, Nakamura Y. Intracellular determination of membrane potential of trigeminal motoneurons during sleep and wakefulness. J Neurophysiol. 1980;44:349–358. doi: 10.1152/jn.1980.44.2.349. [DOI] [PubMed] [Google Scholar]
- Chase MH, Soja PJ, Morales FR. Evidence that glycine mediates the postsynaptic potentials that inhibit lumbar motoneurons during the atonia of active sleep. J Neurosci. 1989;9:743–751. doi: 10.1523/JNEUROSCI.09-03-00743.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groen JH, Kamphuisen HA. Periodic nocturnal myoclonus in a patient with hyperexplexia (startle disease) J Neurol Sci. 1978;38:207–213. doi: 10.1016/0022-510x(78)90067-9. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. In vivo brain dialysis of neurotransmitters. Trends Pharmacol Sci. 1990;11:116–121. doi: 10.1016/0165-6147(90)90197-g. [DOI] [PubMed] [Google Scholar]
- el Mansari M, Sakai K, Jouvet M. Unitary characteristics of presumptive cholinergic tegmental neurons during the sleep-waking cycle in freely moving cats. Exp Brain Res. 1989;76:519–529. doi: 10.1007/BF00248908. [DOI] [PubMed] [Google Scholar]
- Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med. 2005;172:1322–1330. doi: 10.1164/rccm.200412-1750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SL, Zhang L, Forster ML, Anderson JR, Iwase T, Soliven B, Donahue LR, Sweet HO, Bronson RT, Davisson MT, Wollmann RL, Lahn BT. Trak1 mutation disrupts GABAA receptor homeostasis in hypertonic mice. Nat Genet. 2006;38:245–250. doi: 10.1038/ng1715. [DOI] [PubMed] [Google Scholar]
- Glenn LL, Dement WC. Membrane potential and input resistance in alpha motoneurons of hindlimb extensors during isolated and clustered episodes of phasic events in REM sleep. Brain Res. 1985;339:79–86. doi: 10.1016/0006-8993(85)90623-7. [DOI] [PubMed] [Google Scholar]
- Guilleminault C, Wilson RA, Dement WC. A study on cataplexy. Arch Neurol. 1974;31:255–261. doi: 10.1001/archneur.1974.00490400069008. [DOI] [PubMed] [Google Scholar]
- Horner RL. Motor control of the pharyngeal musculature and implications for the pathogenesis of obstructive sleep apnea. Sleep. 1996;19:827–853. doi: 10.1093/sleep/19.10.827. [DOI] [PubMed] [Google Scholar]
- Jelev A, Sood S, Liu H, Nolan P, Horner RL. Microdialysis perfusion of 5-HT into hypoglossal motor nucleus differentially modulates genioglossus activity across natural sleep-wake states in rats. J Physiol (Lond) 2001;532:467–481. doi: 10.1111/j.1469-7793.2001.0467f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandkuhler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Jouvet M. The states of sleep. Sci Am. 1967;216:62–68. doi: 10.1038/scientificamerican0267-62. [DOI] [PubMed] [Google Scholar]
- Kato T, Dal-Fabbro C, Lavigne GJ. Current knowledge on awake and sleep bruxism: overview. Alpha Omegan. 2003;96:24–32. [PubMed] [Google Scholar]
- Kodama T, Lai YY, Siegel JM. Enhanced glutamate release during REM sleep in the rostromedial medulla as measured by in vivo microdialysis. Brain Res. 1998;780:178–181. [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Lai YY, Siegel JM. Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J Neurosci. 2003;23:1548–1554. doi: 10.1523/JNEUROSCI.23-04-01548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L, Kimura H, Tojima H, Davies RO, Pack AI. Suppression of hypoglossal motoneurons during the carbachol-induced atonia of REM sleep is not caused by fast synaptic inhibition. Brain Res. 1993;611:300–312. doi: 10.1016/0006-8993(93)90517-q. [DOI] [PubMed] [Google Scholar]
- Kurczynski TW. Hyperekplexia. Arch Neurol. 1983;40:246–248. doi: 10.1001/archneur.1983.04050040076015. [DOI] [PubMed] [Google Scholar]
- Lai YY, Kodama T, Siegel JM. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci. 2001;21:7384–7391. doi: 10.1523/JNEUROSCI.21-18-07384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sood S, Liu H, Nolan P, Morrison JL, Horner RL. Suppression of genioglossus muscle tone and activity during reflex hypercapnic stimulation by GABAA mechanisms at the hypoglossal motor nucleus in vivo. Neuroscience. 2003;116:249–259. doi: 10.1016/s0306-4522(02)00564-x. [DOI] [PubMed] [Google Scholar]
- Liu X, Sood S, Liu H, Horner RL. Opposing muscarinic and nicotinic modulation of hypoglossal motor output to genioglossus muscle in rats in vivo. J Physiol (Lond) 2005;565:965–980. doi: 10.1113/jphysiol.2005.084657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Rodriguez F, Morales FR, Soja PJ, Chase MH. Suppression of the PGO-related lumbar motoneuron IPSP by strychnine. Brain Res. 1990;535:331–334. doi: 10.1016/0006-8993(90)91619-r. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez F, Chase MH, Morales FR. PGO-related potentials in lumbar motoneurons during active sleep. J Neurophysiol. 1992;68:109–116. doi: 10.1152/jn.1992.68.1.109. [DOI] [PubMed] [Google Scholar]
- Lu JW, Mann GL, Ross RJ, Morrison AR, Kubin L. Differential effect of sleep-wake states on lingual and dorsal neck muscle activity in rats. Respir Physiol Neurobiol. 2005;147:191–203. doi: 10.1016/j.resp.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology (Berl) 1993;113:76–82. doi: 10.1007/BF02244337. [DOI] [PubMed] [Google Scholar]
- Mir S, Yee N, Brooks PL, Peever JH. Noradrenergic control of motoneurons during natural sleep in rats. Sleep. 2006;29:A27. [Google Scholar]
- Morrison JL, Sood S, Liu X, Liu H, Park E, Nolan P, Horner RL. Glycine at hypoglossal motor nucleus: genioglossus activity, CO(2) responses, and the additive effects of GABA. J Appl Physiol. 2002;93:1786–1796. doi: 10.1152/japplphysiol.00464.2002. [DOI] [PubMed] [Google Scholar]
- Morrison JL, Sood S, Liu H, Park E, Liu X, Nolan P, Horner RL. Role of inhibitory amino acids in control of hypoglossal motor outflow to genioglossus muscle in naturally sleeping rats. J Physiol (Lond) 2003;552:975–991. doi: 10.1113/jphysiol.2003.052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Goldberg LJ, Chandler SH, Chase MH. Intracellular analysis of trigeminal motoneuron activity during sleep in the cat. Science. 1978;199:204–207. doi: 10.1126/science.202025. [DOI] [PubMed] [Google Scholar]
- O'Brien JA, Berger AJ. Cotransmission of GABA and glycine to brain stem motoneurons. J Neurophysiol. 1999;82:1638–1641. doi: 10.1152/jn.1999.82.3.1638. [DOI] [PubMed] [Google Scholar]
- Okabe S, Woch G, Kubin L. Role of GABAB receptors in the control of hypoglossal motoneurons in vivo. NeuroReport. 1994;5:2573–2576. doi: 10.1097/00001756-199412000-00042. [DOI] [PubMed] [Google Scholar]
- Pagnotta SE, Lape R, Quitadamo C, Nistri A. Pre- and postsynaptic modulation of glycinergic and gabaergic transmission by muscarinic receptors on rat hypoglossal motoneurons in vitro. Neuroscience. 2005;130:783–795. doi: 10.1016/j.neuroscience.2004.09.046. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pedroarena C, Castillo P, Chase MH, Morales FR. The control of jaw-opener motoneurons during active sleep. Brain Res. 1994;653:31–38. doi: 10.1016/0006-8993(94)90368-9. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Mahowald MW. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120–138. doi: 10.1093/sleep/25.2.120. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Hurwitz TD, Mahowald MW. REM sleep behavior disorder. Am J Psychiatry. 1988;145:652. doi: 10.1176/ajp.145.5.652a. [DOI] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Soja PJ, Finch DM, Chase MH. Effect of inhibitory amino acid antagonists on masseteric reflex suppression during active sleep. Exp Neurol. 1987;96:178–193. doi: 10.1016/0014-4886(87)90179-8. [DOI] [PubMed] [Google Scholar]
- Soja PJ, Lopez-Rodriguez F, Morales FR, Chase MH. Effects of excitatory amino acid antagonists on the phasic depolarizing events that occur in lumbar motoneurons during REM periods of active sleep. J Neurosci. 1995;15:4068–4076. doi: 10.1523/JNEUROSCI.15-05-04068.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YM, Huang LY. Modulation of glycine receptor chloride channels by cAMP-dependent protein kinase in spinal trigeminal neurons. Nature. 1990;348:242–245. doi: 10.1038/348242a0. [DOI] [PubMed] [Google Scholar]
- Steriade M, Datta S, Pare D, Oakson G, Curro Dossi RC. Neuronal activities in brain-stem cholinergic nuclei related to tonic activation processes in thalamocortical systems. J Neurosci. 1990;10:2541–2559. doi: 10.1523/JNEUROSCI.10-08-02541.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience. 1999;91:1389–1399. doi: 10.1016/s0306-4522(98)00600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]