

Abstract
The treatment of alcoholism requires the proper management of ethanol withdrawal symptoms, such as anxiety, to prevent further alcohol use and abuse. In this study, we investigated the potential role of brain chromatin remodeling, caused by histone modifications, in alcoholism. We found that the anxiolytic effects produced by acute alcohol were associated with a decrease in histone deacetylase (HDAC) activity and increases in acetylation of histones (H3 and H4), levels of CREB (cAMP-responsive element binding) binding protein (CBP), and neuropeptide Y (NPY) expression in the amygdaloid brain regions of rats. However, the anxiety-like behaviors during withdrawal after chronic alcohol exposure were associated with an increase in HDAC activity and decreases in acetylation of H3 and H4, and levels of both CBP and NPY in the amygdala. Blocking the observed increase in HDAC activity during alcohol withdrawal with the HDAC inhibitor, trichostatin A, rescued the deficits in H3 and H4 acetylation and NPY expression (mRNA and protein levels) in the amygdala (central and medial nucleus of amygdala) and prevented the development of alcohol withdrawal-related anxiety in rats as measured by the elevated plus maze and light/dark box exploration tests. These results reveal a novel role for amygdaloid chromatin remodeling in the process of alcohol addiction and further suggest that HDAC inhibitors may be potential therapeutic agents in treating alcohol withdrawal symptoms.
Keywords: alcoholism, anxiety, amygdala, histone acetylation, HDAC inhibitors, NPY
Introduction
Excessive consumption of ethanol is a major health concern worldwide (National Institute on Alcohol Abuse and Alcoholism, 1993). The cessation of chronic ethanol consumption is often accompanied by several withdrawal symptoms indicative of a hyperexcitable state, such as increased anxiety, risk of convulsions, and tremors (Roelofs, 1985; Koob, 2003). Anxiety is a common early symptom of ethanol withdrawal and serves as an important factor in the negative reinforcement leading to excessive alcohol drinking (Weiss and Rosenberg, 1985; Kushner et al., 1990; Schuckit and Hesselbrock, 1994; Koob, 2003; Pandey, 2003). Amygdaloid structures, particularly the central (CeA) and medial nucleus of amygdala (MeA), serve as major neural substrates for anxiety and alcohol-drinking behaviors (Davis, 1997; McBride, 2002; Koob, 2003; Pandey et al., 2006).
Epigenetic mechanisms, such as DNA methylation and histone modifications (acetylation, methylation, and phosphorylation), regulate gene transcription (Grunstein, 1997; Turner, 2002; Egger et al., 2004; Hsieh and Gage, 2005; Verdone et al., 2005). Studies have shown a role for changes in chromatin structure in the regulation of gene expression attributable to covalent histone modifications via acetylation and deacetylation (Grunstein, 1997; Hsieh and Gage, 2005; Verdone et al., 2005). The fundamental unit of chromatin is the nucleosome, containing 147 bp of DNA wrapped around the histone octamer core, consisting of a central heterotetramer of histones H3 and H4 along with two heterodimers of histones H2A and H2B. Two groups of enzymes, known as histone acetyltransferases (HATs) and histone deacetylases (HDACs) are involved in chromatin remodeling (Grunstein, 1997; Hsieh and Gage, 2005). Previous studies have shown that the phosphorylated form of cAMP-responsive element binding (CREB) protein (p-CREB) regulated neuronal plasticity via recruitment of CREB binding protein (CBP), which has intrinsic HAT activity, to regulate chromatin structure (Silva et al., 1998; Korzus et al., 2004; Hsieh and Gage, 2005). HATs can induce lysine acetylation at the N-terminal of histones, which can cause the relaxation of the nucleosomes and thereby promote gene expression. However, HDACs can remove acetyl groups, allowing histones in the deacetylated state to pack the DNA into a more condensed chromatin, blocking access to transcriptional activators and decreasing gene transcription (Grunstein, 1997; Turner, 2002; Hsieh and Gage, 2005). Thus, HATs and HDACs appear to be important molecular targets in remodeling chromatin structure via histone covalent modifications eventually changing the expression of various genes, such as neuropeptide Y (NPY).
NPY is a CREB-related gene that acts as a potent endogenous anxiolytic compound and deletion of the NPY gene has been shown to promote alcohol intake (Heilig et al., 1989; Heilig and Widerlov, 1995; Thiele et al., 1998; Pandey, 2004). Using various animal models, we have demonstrated that decreased functioning of CREB and NPY in the CeA and MeA, but not in the basolateral amygdala (BLA), may be involved in alcohol preference and dependence (Pandey et al., 2003, 2005). However, the contribution of histone modifications related to epigenetic mechanisms that may be associated with the process of alcohol addiction is currently unknown. Therefore, for the first time we investigated the involvement of chromatin remodeling in the neuromechanisms of alcoholism.
Materials and Methods
Acute ethanol exposure.
All experiments were conducted in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee. Adult male Sprague Dawley rats (270–300 g) were used in this study. For acute ethanol exposure studies, rats were injected with ethanol (1 g/kg dose; ethanol was diluted to 0.2 g/ml in n-saline and was injected as 5 μl/g of body weight) or n-saline (5 μl/g of body weight) intraperitoneally and 1 h after the injections, anxiety-like behaviors were measured using the elevated plus maze (EPM) and light/dark box (LDB) exploration tests, described below. Immediately after anxiety measurements, brains were used for immunohistochemistry, in situ reverse transcription (RT)-PCR, or for measuring HDAC activity, as described below.
Chronic ethanol exposure and TSA treatment.
Male adult Sprague Dawley rats were housed individually and offered 80 ml of the nutritionally complete Lieber-DeCarli control diet (Lieber-DeCarli Diet 82; Bio-Serv, Frenchtown, NJ) as their source of food and fluid. Rats were fed with the Lieber-DeCarli ethanol and control-diets, as described previously by us (Pandey et al., 1999, 2001, 2003, 2008). One group of rats was gradually habituated to ethanol (within 7 d) and maintained on the ethanol-containing (9% v/v) Lieber-DeCarli liquid diet for 15 or 16 d (ethanol diet-fed group). Another group of rats continued to feed with the control liquid diet for 15 or 16 d (pair-fed control group). The mean ± SEM consumption of control liquid diet in control diet-fed rats (n = 36) was 55.17 ± 0.28 ml/d and mean ± SEM consumption of ethanol-diet in ethanol diet-fed rats (n = 53) was 53.22 ± 0.28 ml/d. Ethanol diet-fed rats were withdrawn for 0 and 24 h. The ethanol diet-fed rats (n = 36) were given control diet during withdrawal and consumed 54.33 ± 0.98 ml of diet within 24 h of ethanol withdrawal. The control diet-fed, ethanol diet-fed (0 h withdrawal), and ethanol withdrawn (ethanol-fed rats with 24 h of withdrawal) rats were injected with trichostatin A (TSA) [2 mg/kg, i.p.; TSA was dissolved in DMSO and then diluted (1:5 dilution) with PBS] or vehicle (DMSO, 1:5 dilution with PBS) 2 h before measuring anxiety-like behaviors by the EPM and LDB tests. We chose 24 h of withdrawal because we had shown previously that peak anxiety occurs at this time point of withdrawal after 15 d of ethanol treatment (Pandey et al., 1999). The TSA dose we used in our studies (2 mg/kg) was based on previous studies showing that this dose was able to increase histone acetylation in the brain (Korzus et al., 2004; Kumar et al., 2005). This dose of TSA appeared to be nontoxic, as higher doses (7.5 mg/kg; chronic treatment) of TSA have been shown to produce no neurotoxic effects or cell death in the brain in mice (Camelo et al., 2005). All rats were used for measurement of anxiety-like behaviors, as described below, and their brains were processed for neurochemical studies. We also collected blood at the time of brain collection and measured blood ethanol levels in ethanol diet-fed rats using an Analox Alcohol Analyzer (Analox Instruments, Lunenburg, MA).
EPM test.
The test procedure was the same as that described by us previously (Pandey et al., 2003, 2005, 2006). Each rat was observed for exploration to open and closed arms during a 5 min test period and the number of entries made to each type of arm (open or closed) was recorded. The results were expressed as the mean ± SEM of the percentage of open-arm entries and the mean percentage of time spent on the open arms (open-arm activity). The general activity of the rats was measured by the total number of closed arm entries as described previously (File, 1993; Pandey et al., 2006).
LDB exploration test.
The LDB exploration test procedure was the same as that described by other investigators (Jonkman et al., 2005; Slawecki, 2005). The light/dark box was located in a dark room and consisted of a dark compartment without illumination and a light compartment with illumination (0.25 Amp; light-emitting diode light). Both compartments were connected through an opening. On the day of testing, rats were removed from their home cages and individually taken to the test room in a cage with bedding. The cage was placed on the floor and each rat was allowed a 5 min pretest habituation period in the room before testing. After the habituation period, the rat was gently placed in the dark compartment with its head facing away from the opening. The rat was observed for a 5 min test period, and the time spent in each compartment of the LDB was recorded. The compartments of the LDB were connected to an infrared beam and animal activity was computer monitored. The percentage of time spent in either the dark compartment or light compartment was calculated for each animal. The box was wiped with a wet paper towel and dried after each rat testing.
Gold immunolabeling of acetylated histones, CBP, and NPY proteins.
Rats were anesthetized with pentobarbital (50 mg/kg) and then perfused intracardially with n-saline (100 ml), followed by 400 ml of 4% ice-cold paraformaldehyde fixative. Brains were removed and placed in fixative for 20 h at 4°C. After fixation, brains were soaked in 10%, followed by 20%, and then 30% sucrose (prepared in 0.1 m phosphate buffer, pH 7.4). Brains were then frozen, sectioned (20 μm) using a cryostat, and used for gold immunolabeling, as described previously (Pandey et al., 2001, 2003, 2005, 2006). The antibodies for acetylated histones H3 (Lys 9) and H4 (Lys 8) (Millipore, Billerica, MA), CBP (SC-583; Santa Cruz Biotechnology, Santa Cruz, CA), and for NPY (Immunostar, Hudson, WI) were used. The quantification of gold-immunolabeled proteins was performed using the Image Analysis System (Loats Associates, Westminster, MD) connected to a light microscope that calculated the number of immunogold particles/100 μm2 area of a defined amygdaloid structure at high magnification (100×). The threshold of each image was set up in such a way that an area without staining gave zero counts. Under this condition, immunogold particles in the defined areas of three adjacent brain sections of each rat were counted and values were averaged for each rat.
In situ RT-PCR for NPY mRNA measurement.
Rat brain sections were used to determine the mRNA levels of NPY using in situ RT-PCR as we reported previously (Pandey et al., 2003, 2004, 2005). The free-floating brain sections (40 μm thickness) were treated with proteinase K (1 μg/ml in 1× PBS containing 0.05% Triton X-100) for 15 min at 37°C. After washing with 1× PBS, the sections were subjected to DNase digestion. The sections were again washed with 1× PBS and transferred to PCR tubes containing 100 μl of reverse transcription reaction mixture (Applied Biosystems, Foster City, CA) and reverse transcribed with reverse transcriptase enzyme in the presence of oligo(dt)16. For the negative sections, reverse transcriptase enzyme was not added. PCR was performed with Taq DNA polymerase enzyme and 100 pmol of each NPY primer (5′-TAGGTAACAAACGAATGGGG-3′ and 5′-AGGATGAGATGAGATGTGGG-3′) and 1 mm of each NTP, with the exception of dTTP which was replaced by digoxigenin (DIG)-11-dUTP [PCR conditions: 94°C for 3 min; 94°C for 45 s; 60°C for 45 s; 72°C for 45 s (total of 25 cycles); and then 72°C for 7 min]. After PCR, sections were mounted on slides and NPY-positive cell bodies were detected by an alkaline phosphatase conjugated anti-DIG antibody and the subsequent staining of the complex with the specific substrate nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolylphosphate (Roche, Indianapolis, IN). The optical density (OD) of positive cell bodies was calculated with an Image Analyzer (Loats Associates). The OD of the negative brain sections was subtracted from the OD of the positive brain sections. The mean OD of NPY-positive cell bodies in the amygdaloid structures of three adjacent brain sections from each rat were calculated and then values were averaged for each rat. The results were represented as mean OD/100 pixels of area for NPY mRNA levels.
HDAC activity in the amygdala.
Immediately after anxiety behaviors were measured, rats were anesthetized with pentobarbital (50 mg/kg), brains were removed, and amygdaloid structures, primarily the CeA and MeA, were dissected out and and frozen. The cytosolic and nuclear fractions were prepared using a nuclear extraction kit from Sigma (St. Louis, MO). The two fractions from each rat amygdala were mixed and referred to as total cell lysates. HDAC activity (class I and II HDACs) was determined according to the manufacturer's instructions for the colorimetric HDAC activity assay kit (BioVision Research, Mountain View, CA) by measuring the deacetylation of acetylated lysine side chains. The OD of the samples was measured using an ELISA plate reader at 405 nm (Spectra MR; Dynex Technologies, Chantilly, VA). The results were calculated as OD per milligram of protein and then converted to percentage of control.
Statistical analysis.
The differences between two groups were evaluated using the Student's t test. The differences between more than two groups were evaluated by the one-way ANOVA test. Post hoc multiple comparisons were performed using Tukey's test. The data normality was also tested and passed.
Results
Inhibition of HDAC activity and increased histone acetylation and NPY expression in the amygdala by acute ethanol exposure
To gain insight into chromatin remodeling during the process of alcoholism, we first examined the effects of acute ethanol exposure on anxiety-like behaviors, HDAC activity, H3 and H4 acetylation, CBP levels, and NPY expression in the amygdala of rats. We injected ethanol (1 g/kg; i.p.) or n-saline and measured anxiety-like behaviors 1 h after injection [mean ± SEM (n = 10) blood ethanol levels, 92 ± 2.6 mg% at the time of brain collection] using the EPM test in one batch of rats. We found that acute ethanol significantly (p < 0.001) increased the percentage of open arm entries and the percentage of time spent on open arms (Fig. 1A), as measured by the EPM test. The general activity, as measured by total number of closed arm entries of EPM was similar among the groups (Fig. 1A). In another batch of rats [mean ± SEM (n = 12) blood ethanol levels, 91 ± 3.2 mg% at the time of brain collection], the anxiety-like behaviors were measured by the LDB exploration test. We found that acute ethanol significantly (p < 0.01) increased the time spent in the light compartment and significantly (p < 0.01) decreased the time spent in the dark compartment of the LDB (Fig. 1B).
Figure 1.
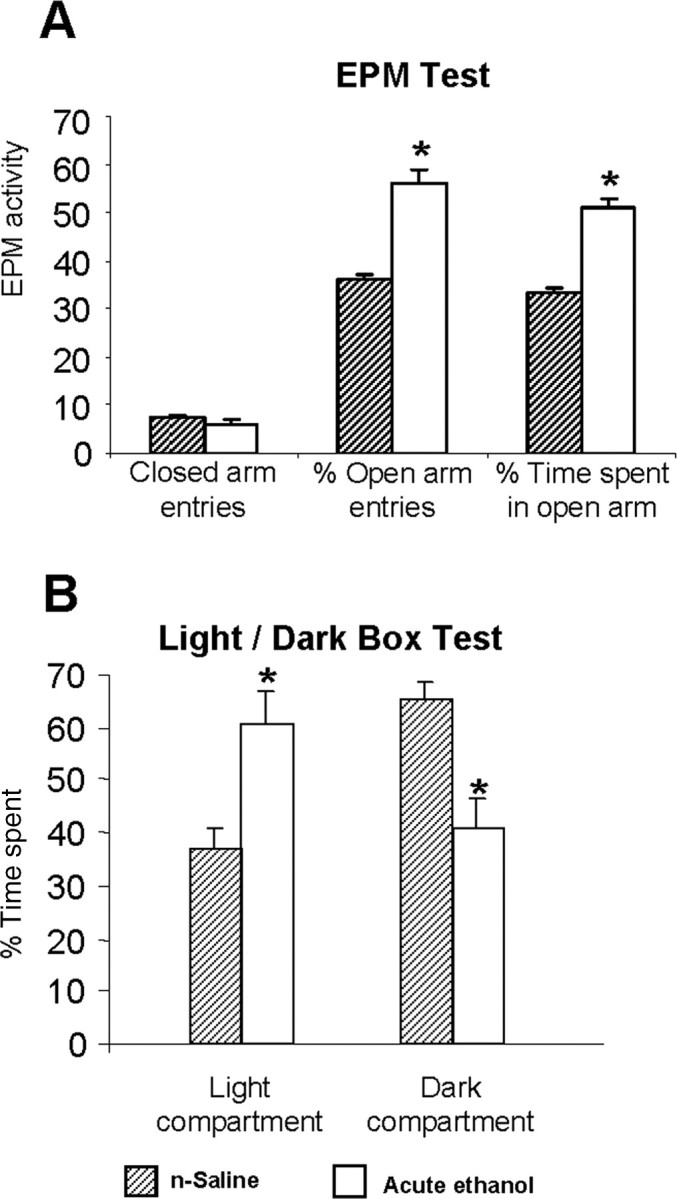
A, B, The effect of acute ethanol exposure (1 h after 1 g/kg; i.p.) on open-arm and closed-arm activity in the EPM test (A) and on LDB exploration test (B) for anxiety-like behaviors. Values are the mean ± SEM of 11–12 rats in each group. *Significantly different from n-saline-treated rats (p < 0.01–0.001).
We also measured total HDACs (class I and II) activity in the amygdala 1 h after acute ethanol injection. We found that acute ethanol significantly (p < 0.01) inhibited the HDAC activity in total cell lysates of amygdala (Fig. 2A). In addition, we found that acute ethanol significantly increased (p < 0.001) the acetylation of both H3 and H4 and increased (p < 0.001) protein levels of CBP in the CeA and MeA, but not in the BLA of rats (Fig. 2B,C). Acute ethanol exposure also significantly increased (p < 0.001) the mRNA and protein levels of NPY in the CeA and MeA, but not in the BLA of rats (Fig. 3A,B). These results suggest that acute ethanol exposure significantly increased acetylation of H3 and H4 and increased NPY expression in selected regions of the amygdala, which may be related to either the ethanol-induced inhibition of HDAC activity or to the observed increase in CBP levels in the amygdala. These results also suggest the possibility that the anxiolytic effects of ethanol may be related to the inhibition of HDAC activity in the amygdala of rats.
Figure 2.
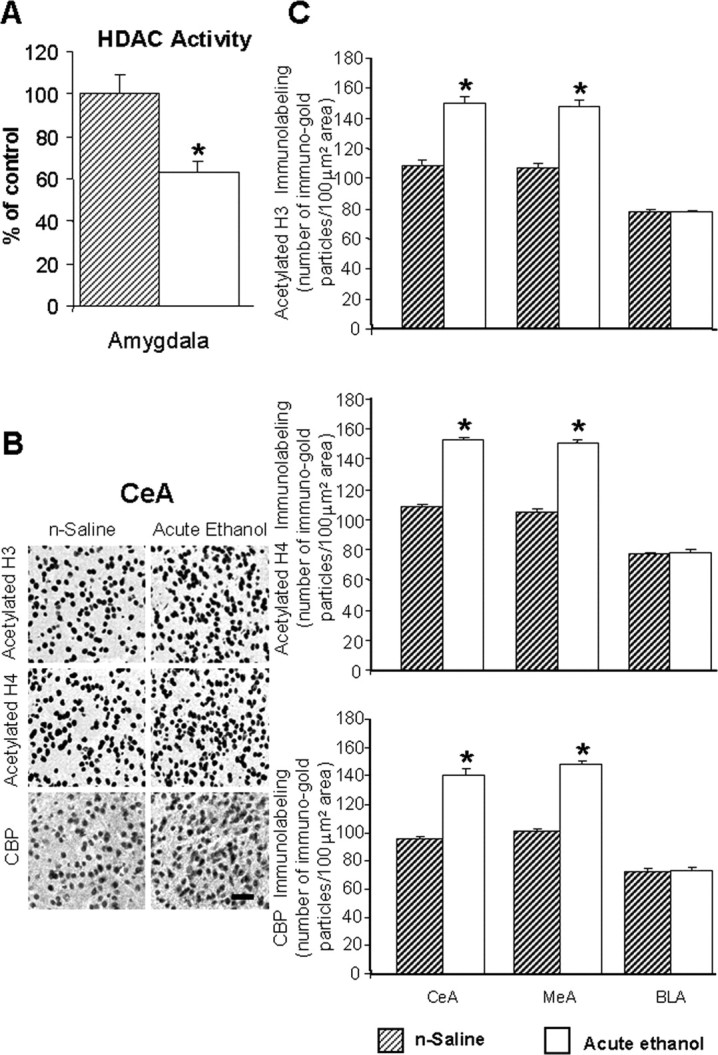
A, Acute ethanol exposure inhibited HDAC activity in the amygdala of rats [injected with ethanol (1 h after 1 g/kg, i.p.) or n-saline]. The HDAC activity was determined in the cell lysates of amygdala by measuring the deacetylation of acetylated lysine side chains. The values are the mean ± SEM of seven rats per group. *Significantly different from n-saline-treated rats (p < 0.01; Student's t test). B, Low-magnification photomicrographs of acetylated histones H3 (Lys 9) and H4 (Lys 8) and CBP gold-immunolabeling (protein levels) in central amygdaloid (CeA) structures of n-saline or acute-ethanol-treated rats. Scale bar, 40 μm. C, Effect of acute ethanol treatment on protein levels of acetylated H3 and H4, and of CBP in various amygdaloid (CeA, MeA, and BLA) structures of rats. Values are the mean ± SEM of seven to nine rats per group. *Significantly different from the n-saline-treated rats (p < 0.001; Student's t test).
Figure 3.

A, Low-magnification photomicrographs of NPY mRNA (in situ RT-PCR) and NPY gold-immunolabeling (protein) in CeA of n-saline and acute ethanol-treated rats. Scale bar, 40 μm. B, Effect of acute ethanol treatment on mRNA and protein levels of NPY in CeA, MeA, and BLA of rats. Values are the mean ± SEM of five rats per group. *Significantly different from the n-saline-treated rats (p < 0.001; Student's t test).
HDAC inhibitor treatment prevents anxiety and rescues the deficits in both histone acetylation and NPY expression during ethanol withdrawal
Next, we examined the effects of chronic ethanol treatment and its withdrawal (24 h), with or without TSA treatment, on anxiety-like behaviors, HDAC activity, CBP levels, acetylation of histones H3 and H4, and NPY expression in the amygdaloid structures of rats. Rats were chronically fed with either a liquid control-diet or ethanol-diet (containing 9% v/v ethanol). The ethanol-fed groups [blood ethanol levels (mean ± SEM) in ethanol-fed rats were 177 ± 11 mg% (n = 17) at the time of brain collection] were withdrawn for 0 and 24 h. The control diet-fed, ethanol diet-fed (0 h withdrawal), and ethanol-withdrawn (24 h) rats were injected one time with either TSA (2 mg/kg, i.p.) or vehicle, 2 h before commencement of the anxiety measurements using the EPM test and LDB exploration test. There were no significant differences observed in the body weights (mean ± SEM) of rats among the various groups: control diet-fed rats plus vehicle, 295 ± 2.8 g (n = 18); control diet-fed rats plus TSA, 294 ± 3.5 g (n = 18); ethanol diet-fed rats (0 h withdrawal) plus vehicle, 292 ± 2.7 g (n = 17); ethanol-withdrawn (24 h) rats plus vehicle, 299 ± 1.3 g (n = 18); and ethanol-withdrawn rats plus TSA, 296 ± 2.5 g (n = 18). As shown in Figure 4A, ethanol withdrawal (24 h) after chronic ethanol exposure produced a significant reduction in open-arm activity [percentage entries (F(4,39) = 28.83; p < 0.001) and percentage of time (F(4,39) = 19.63; p < 0.001) spent on open arms] in the EPM test. In another batch of rats, we found that ethanol-withdrawn rats spent significantly less time in the light compartment (F(4,40) = 15.96; p < 0.001) and more time in the dark compartment (F(4,40) = 15.98; p < 0.001) during a 5 min exploration period in the LDB test (Fig. 4B). Interestingly, treatment with TSA during ethanol withdrawal protected against the development of anxiety-like behaviors in ethanol-withdrawn rats during both tests for anxiety (Fig. 4A,B).
Figure 4.
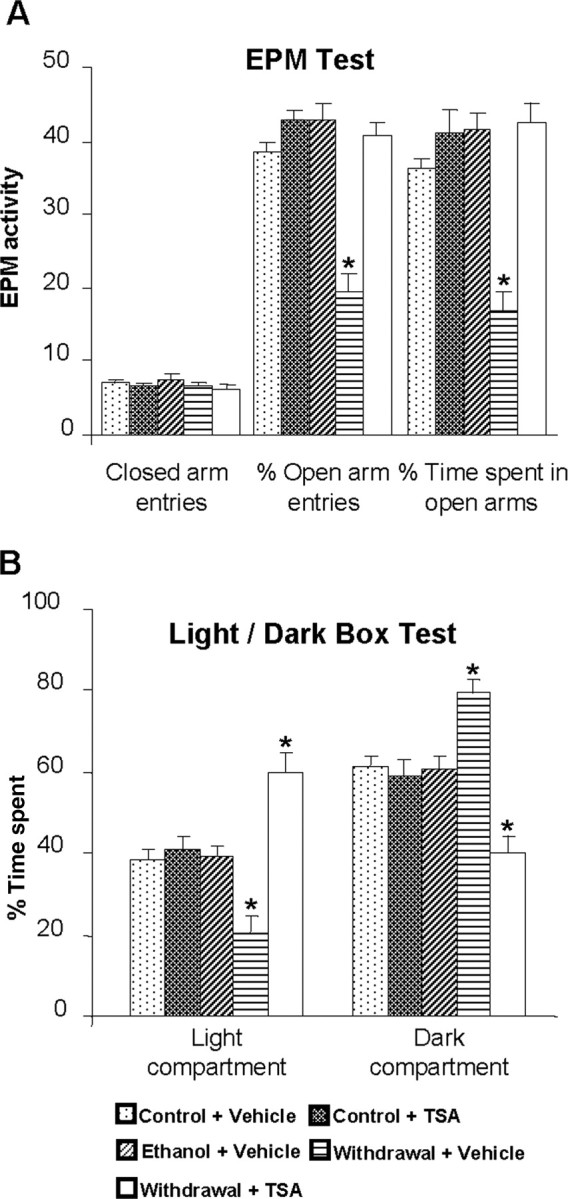
A, Effect of TSA treatment on open-arm and closed-arm activities in the EPM test for anxiety-like behaviors during ethanol withdrawal. The control diet-fed, ethanol diet-fed (0 h withdrawal), and ethanol diet-fed (24 h withdrawal) rats were injected with TSA (2 mg/kg, i.p.) or vehicle once, 2 h before measuring anxiety-like behaviors. The groups are represented as follows: control diet-fed + vehicle = control + vehicle; control diet-fed + TSA = control + TSA; ethanol diet-fed (0 h withdrawal) + vehicle = ethanol + vehicle; ethanol diet-fed (24 h withdrawal) + vehicle = withdrawal + vehicle; ethanol diet-fed (24 h withdrawal) + TSA = withdrawal + TSA. Values are the mean ± SEM of eight to nine rats per group. *Significantly different from control diet-fed rats treated with vehicle (p < 0.001; ANOVA followed by Tukey's test). B, Effect of TSA treatment on LDB exploration test for anxiety-like behaviors during ethanol withdrawal after chronic ethanol exposure. Values are the mean ± SEM of nine rats per group. *Significantly different from control diet-fed rats treated with vehicle (p < 0.01–0.001; ANOVA followed by Tukey's test).
Ethanol withdrawal after chronic ethanol exposure significantly decreased the acetylation of histones H3 (p < 0.001) and H4 (p < 0.001) (Fig. 5B,C), and also decreased the mRNA (p < 0.001) and protein (p < 0.001) levels (Fig. 6A,B) of NPY in the CeA and MeA, but not in BLA. Deficits in histone acetylation (Fig. 5B,C) and NPY expression (Fig. 6A,B) were not present in the amygdala of rats chronically treated with ethanol, subsequently these rats did not display anxiety-like behaviors (Fig. 4A,B). Moreover, TSA treatment given to ethanol-withdrawn rats significantly restored the reductions in histone acetylation and in the mRNA and protein levels of NPY in the CeA and MeA. TSA treatment given to control rats had no significant effect on anxiety levels, as measured by either anxiety test (Fig. 4A,B), and also had no effects on the acetylation states of H3 and H4 or on NPY expression in the CeA, MeA, or BLA (Figs. 5B,C, 6A,B). These results suggest that a histone acetylation-associated reduction in NPY expression may be involved in ethanol withdrawal-related anxiety behaviors.
Figure 5.
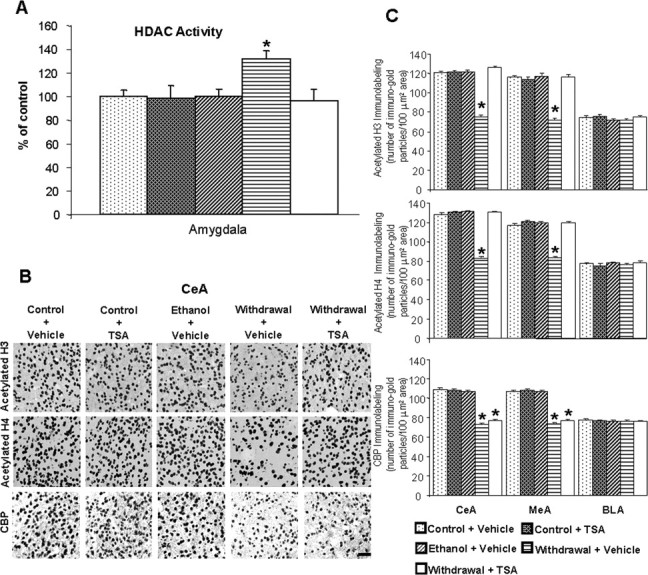
A, Effect of ethanol withdrawal after chronic ethanol treatment and effect of TSA treatment on HDAC activity in the amygdala of rats. The HDAC activity was determined in the cell lysates of amygdala by measuring the deacetylation of acetylated lysine side chains. Values are the mean ± SEM of seven rats per group. *Significantly different from control diet-fed rats treated with vehicle (p < 0.05; ANOVA followed by Tukey's test). B, Low-magnification photomicrographs of acetylated histones H3 (Lys 9) and H4 (Lys 8), and CBP gold-immunolabeling (protein levels) in the CeA of control diet-fed, ethanol-fed, and ethanol-withdrawn rats treated with TSA or vehicle. Scale bar, 40 μm. The groups are represented as follows: control diet-fed + vehicle = control + vehicle; control diet-fed + TSA = control + TSA; ethanol diet-fed (0 h withdrawal) + vehicle = ethanol + vehicle; ethanol diet-fed (24 h withdrawal) + vehicle = withdrawal + vehicle; ethanol diet-fed (24 h withdrawal) + TSA = withdrawal + TSA. C, Effect of TSA treatment on protein levels of acetylated H3 and H4, and CBP in the CeA, MeA, and BLA of rats during ethanol withdrawal after chronic ethanol exposure. Values are the mean ± SEM of five to six rats per group. *Significantly different from the control diet-fed rats treated with vehicle (p < 0.001; ANOVA followed by Tukey's test).
Figure 6.

A, Low-magnification photomicrographs of NPY mRNA and NPY gold-immunolabeling (protein) in the CeA of control diet-fed, ethanol diet-fed, and ethanol-withdrawn rats treated with TSA or vehicle. Scale bar, 40 μm. The groups are represented as follows: control diet-fed + vehicle = control + vehicle; control diet-fed + TSA = control + TSA; ethanol diet-fed (0 h withdrawal) + vehicle = ethanol + vehicle; ethanol diet-fed (24 h withdrawal) + vehicle = withdrawal + vehicle; ethanol diet-fed (24 h withdrawal) + TSA = withdrawal + TSA. B, Effect of TSA treatment on protein and mRNA levels of NPY in the CeA, MeA, and BLA of rats during ethanol withdrawal after chronic ethanol exposure. Values are the mean ± SEM of five to six rats per group. *Significantly different from the control diet-fed rats treated with vehicle (p < 0.001; ANOVA followed by Tukey's test).
HDAC activity and CBP levels in the amygdala during ethanol withdrawal
To examine whether or not decreased histone acetylation may be related to either the decreased functioning of CBP or increased functioning of HDACs, we determined both the protein levels of CBP and HDAC activity in the amygdala during ethanol treatment and its withdrawal. We found that chronic ethanol treatment had no effect on either the protein levels of CBP or HDAC activity in the amygdala. However, the protein levels of CBP significantly decreased (p < 0.001) in the CeA and MeA during ethanol withdrawal (24 h), which was not affected by TSA treatment (Fig. 5A–C). HDAC activity was significantly (p < 0.05) increased in the amygdala during ethanol withdrawal and was normalized by TSA treatment (Fig. 5A). These results suggest that activation of HDACs and inhibition of the intrinsic HAT activity of CBP may contribute to the decreased acetylation of H3 and H4, thereby leading to a decrease in NPY gene expression in the CeA and MeA during ethanol withdrawal. Furthermore, TSA treatment during ethanol withdrawal inhibited the increased HDAC activity and concomitantly increased both histone acetylation and NPY expression in the CeA and MeA of ethanol-withdrawn rats (Fig. 5A–C). Therefore, the actions of TSA in ethanol-withdrawn rats may lead to the prevention of anxiety-like behaviors, as demonstrated by both the EPM test and LDB exploration test.
Discussion
The present investigation revealed a novel epigenetic mechanism suggesting that increased histone acetylation and decreased HDAC activity in the amygdala may be involved in the anxiolytic effects of acute ethanol and conversely, decreased histone acetylation and increased HDAC activity in the amygdala may be involved in the development of anxiety during withdrawal after chronic ethanol exposure. The cessation of chronic ethanol consumption has been shown to lead to the development of withdrawal symptoms both in human alcoholics and animal models (Weiss and Rosenberg, 1985; Wilson, 1988; Lal et al., 1993; Rassnick et al., 1993; Koob, 2003; Pandey, 2003; Pandey et al., 2003, 2008). Anxiety-like behaviors, which appear during the early phase of withdrawal, play crucial roles in the maintenance of alcohol drinking behaviors in both humans and animals (Hershon, 1977; Wilson, 1988; Koob, 2003; Pandey, 2003; Funk et al., 2006). Thus, strategies to treat alcoholism should be directed toward the prevention of withdrawal symptoms that occur after cessation of drinking. The evidence presented here suggests that decreased histone acetylation, attributable to increased HDAC activity in the amygdala during alcohol withdrawal, may be associated with withdrawal symptoms, such as anxiety, and that acute ethanol exposure may produce anxiolytic effects because of its ability to increase histone acetylation through HDAC inhibition. We also shown here that treatment with TSA, an HDAC inhibitor, prevented the development of anxiety-like behaviors during ethanol withdrawal in rats. In addition, TSA blocked the observed increase in HDAC activity, rescued deficits in the acetylation states of H3 and H4, and restored the decrease in NPY expression (mRNA and protein levels) in the CeA and MeA (Fig. 7).
Figure 7.

Proposed model for chromatin remodeling in the central and medial nucleus of amygdala during acute and chronic ethanol exposure. The present investigation reveals that increased histone acetylation caused by either inhibition of HDACs or activation of HATs (increased CBP levels) may lead to increased expression of NPY in the amygdala that may be operative in the anxiolytic effects of acute ethanol exposure in rats. However, ethanol withdrawal-related anxiety might be related to decreased histone acetylation caused by activation of HDACs or inhibition of HATs (decreased CBP levels), and may also be responsible for the observed decrease in NPY expression in the amygdala. TSA treatment during ethanol withdrawal attenuated anxiety-like behaviors and rescued the reductions in both histone acetylation and NPY expression in the CeA and MeA during ethanol withdrawal. Thus, HDAC-induced chromatin remodeling in the amygdala may be crucial in the dynamic process that occurs during the development of alcoholism.
Three distinct families of HDACs have been described. TSA is a potent inhibitor of both class I (isoforms 1, 2, 3, 8, and 11) and class II HDACs (isoforms 4, 5, 6, 7, 9 and 10), but not class III HDACs (Sir2), which require a cofactor, nicotinamide-adenine dinucleotide, for enzymatic activity (Marks et al., 2003; Blander and Guarente, 2004; Villar-Garea and Esteller, 2004; Dokmanovic and Marks, 2005). Here, we found that HDAC (HDACs I and II) activity was inhibited by acute ethanol and conversely, was activated in the amygdala during ethanol withdrawal after chronic ethanol exposure. We also found that TSA both inhibited HDAC activity and elicited an anxiolytic response in ethanol-withdrawn rats. However, acute TSA treatment had no effects on anxiety-like behaviors, HDAC activity, or histone acetylation in the amygdala of control rats. It may be possible that the levels of one or more isoforms of HDACs in the amygdala were increased during ethanol withdrawal, leading to increased HDAC activity and thereby increasing the effectiveness of TSA in ethanol-withdrawn rats. Future studies are needed to investigate the involvement of specific isoforms of class I and II HDACs and Sir2 in ethanol-induced chromatin remodeling and gene expression in the amygdala. A particular class II HDACs, HDAC 4 isoform, has been shown to be involved in neuronal cell death and cocaine addiction (Bolger and Yao, 2005; Colvis et al., 2005; Kumar et al., 2005). In addition, HDAC 2 and 5 isoforms of class I and II HDACs, respectively, have been implicated in social defeat behaviors and in the mechanisms of action of antidepressant drugs (Cassel et al., 2006; Newton and Duman, 2006; Tsankova et al., 2006, 2007). Several HDAC inhibitors have been used in the treatment of cancer (Villar-Garea and Esteller, 2004) and have recently been used to investigate the epigenetic mechanisms underlying synaptic plasticity within the framework of the CNS (Colvis et al., 2005; Starkman and Pandey, 2007; Tsankova et al., 2007). Treatment with HDAC inhibitors, such as sodium butyrate, has been shown to increase the acetylation of histones H3 and H4, induce sprouting of dendrites in the hippocampus, and reinstate learning behaviors in a mouse model (Fischer et al., 2007). HDAC inhibitors have also been used in establishing a role for histone acetylation in the molecular mechanisms of cocaine addiction, schizophrenia and Huntington's disease (Ferrante et al., 2003; Colvis et al., 2005; Kumar et al., 2005; Sharma, 2005). Data obtained in the present study indicates that HDAC inhibitors might also be beneficial in treating alcoholism.
Regulation of gene expression requires activation of gene transcription factors, such as CREB, via phosphorylation (serine-133) by cAMP-dependent protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinases, as well as by mitogen activated protein kinases, in addition to the subsequent recruitment of multifunctional coactivators such as CBP and p300 to the transcriptional machinery (Silva et al., 1998; Impey et al., 1999; Soderling, 1999; Korzus et al., 2004; Hsieh and Gage, 2005). Several investigators have shown that the intrinsic HAT activity of CBP had profound effects on both chromatin remodeling in the brain and behavior. For example, a haplodeficiency of the CBP gene caused deficits in histone acetylation in the hippocampus and impaired memory consolidation. Both of these deficits in CBP-deficient mice were reinstated by treatment with TSA or suberoylanilide hydroxamic acid (Alarcon et al., 2004; Korzus et al., 2004). Also, transgenic mice expressing a truncated form of CBP displayed impairments in hippocampal-dependent long-term memory formation (Wood et al., 2005). Furthermore, mutations in the CBP gene produced Rubinstein-Taybi syndrome in humans, characterized by severe mental retardation and abnormal gross anatomy (Rubinstein and Taybi, 1963; Hennekam et al., 1992; Petrij et al., 1995). We reported previously that acute ethanol increased CREB phosphorylation in the CeA and MeA but not in BLA of rats and mice (Pandey et al., 2004, 2005, 2008). Conversely, ethanol withdrawal after chronic ethanol exposure decreased CREB phosphorylation, but not total CREB protein levels, in the CeA and MeA of rats (Pandey et al., 2003, 2008). Combining these findings with our current findings on CBP suggests the possibility that decreased functioning of CREB may lead to decreased HAT activity (decreased CBP levels), which may attenuate the acetylation states of histones H3 and H4 in the amygdala during ethanol withdrawal. Acute ethanol challenge may increase HAT activity because of increased CREB function, thereby increasing histone acetylation in the subregions of the amygdala. Challenge with an HDAC inhibitor prevented the development of anxiety and reversed the deficit in histone acetylation, but not the deficit in CBP levels, suggesting the possibility that neuroadaptations in the amygdala during chronic ethanol exposure may involve both HATs and HDACs during the dynamic process of chromatin remodeling.
We investigated the changes in NPY expression by TSA treatment during ethanol withdrawal because NPY in the amygdala had previously been implicated in anxiety and alcohol drinking behaviors (Heilig and Widerlov, 1995; Thiele et al., 1998; Pandey et al., 2003, 2005). Earlier, we found that both CREB phosphorylation and NPY expression were decreased in both the CeA and MeA, but not in the BLA of rats, during withdrawal and that restoring CREB phosphorylation in the CeA, using an activator of PKA, blocked the development of anxiety-like behaviors and restored NPY expression in the CeA (Pandey et al., 2003; Zhang and Pandey, 2003). Here, we found that TSA treatment normalized the reductions in both histone acetylation and NPY expression in the CeA and MeA during ethanol withdrawal, suggesting that histone modifications may regulate NPY expression in the amygdala during withdrawal. These results also suggest the possibility that the anxiolytic effects, observed with TSA treatment, may be related to increased NPY expression in the CeA and MeA of ethanol-withdrawn rats. Previous studies using drugs to block the translation of mRNA into protein have shown that consolidation of fear memories required protein synthesis in the amygdala (Nader et al., 2000; Schafe and LeDoux, 2000). Our data on acute ethanol exposure indicate an increase in NPY protein levels, whereas during withdrawal there is an observed decrease in NPY protein levels, suggesting the possibility that changes in protein synthesis in the amygdala may also be involved in the anxiolytic and anxiogenic effects of ethanol treatment and withdrawal, respectively. Recently, we observed that expression of other CREB target genes such as brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated protein (Arc) were increased during acute ethanol exposure and decreased during ethanol withdrawal after chronic ethanol exposure in CeA and MeA of rats (Pandey et al., 2008). The decreased functioning of BDNF-Arc signaling in the CeA may also be associated with ethanol withdrawal-related anxiety and with alcohol drinking behaviors (Pandey et al., 2006, 2008). Future studies will be aimed at examining the changes in these CREB-related genes attributable to chromatin remodeling in the amygdala and other brain regions during ethanol exposure and withdrawal.
In conclusion, our results provide the first evidence that chromatin remodeling in the amygdala is an important mechanism that may be involved in the anxiolytic and anxiogenic effects of ethanol exposure and withdrawal, respectively (Fig. 7). Furthermore, a deficiency in NPY expression in the CeA and MeA during ethanol withdrawal after chronic ethanol exposure can be rescued by TSA treatment, suggesting that chromatin remodeling might regulate NPY expression. Together, our results suggest a role for HDAC inhibitors as potential therapeutic agents in treating alcoholism and may also pave the way for increased research on the epigenetic mechanisms that occur in the brain during alcohol tolerance and dependence.
Footnotes
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants AA-010005, AA-013341, AA-016690, and AA-015626 and by the Department of Veterans Affairs (Merit Review Grant; Research Career Scientist award) to S.C.P. We thank Kaushik Misra for his technical help during the early stages of this study.
References
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Cassel S, Carouge D, Gensburger C, Anglard P, Burgun C, Dietrich JB, Aunis D, Zwiller J. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol Pharmacol. 2006;70:487–492. doi: 10.1124/mol.106.022301. [DOI] [PubMed] [Google Scholar]
- Colvis CM, Pollock JD, Goodman RH, Impey S, Dunn J, Mandel G, Champagne FA, Mayford M, Korzus E, Kumar A, Renthal W, Theobald DE, Nestler EJ. Epigenetic mechanisms and gene networks in the nervous system. J Neurosci. 2005;25:10379–10389. doi: 10.1523/JNEUROSCI.4119-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. Neurobiology of fear responses: the role of the amygdala. J Neuropsych Clin Neurosci. 1997;9:382–402. doi: 10.1176/jnp.9.3.382. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File SE. The interplay of learning and anxiety in the elevated plus-maze. Behav Brain Res. 1993;58:199–202. doi: 10.1016/0166-4328(93)90103-w. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Funk CK, O'Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Heilig M, Widerlov E. Neurobiology and clinical aspects of neuropeptide Y. Crit Rev Neurobiol. 1995;9:115–136. [PubMed] [Google Scholar]
- Heilig M, Soderpalm B, Engel JA, Widerlov E. Centrally administered neuropeptide Y (NPY) produces anxiolytic-like effects in animal anxiety models. Psychopharmacology. 1989;98:524–529. doi: 10.1007/BF00441953. [DOI] [PubMed] [Google Scholar]
- Hennekam RC, Baselier AC, Beyaert E, Bos A, Blok JB, Jansma HB, Thorbecke-Nilsen VV, Veerman H. Psychological and speech studies in Rubinstein-Taybi syndrome. Am J Ment Retard. 1992;96:645–660. [PubMed] [Google Scholar]
- Hershon HI. Alcohol withdrawal symptoms and drinking behavior. J Stud Alcohol. 1977;38:953–971. doi: 10.15288/jsa.1977.38.953. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Gage FH. Chromatin remodeling in neural development and plasticity. Curr Opin Cell Biol. 2005;17:664–671. doi: 10.1016/j.ceb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Jonkman S, Henry B, Semenova S, Markou A. Mild anxiogeneic effects of nicotine withdrawal in mice. Eur J Pharmacol. 2005;516:40–45. doi: 10.1016/j.ejphar.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, Russo SJ, LaPlant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Kushner MG, Sher KJ, Beitman BD. The relation between alcohol problems and the anxiety disorders. Am J Psychiatry. 1990;147:685–695. doi: 10.1176/ajp.147.6.685. [DOI] [PubMed] [Google Scholar]
- Lal H, Prather PL, Rezazadeh SM. Potential role of 5HT1C and/or 5HT2 receptors in the mianserin-induced prevention of anxiogenic behaviors occurring during ethanol withdrawal. Alcohol Clin Exp Res. 1993;17:411–417. doi: 10.1111/j.1530-0277.1993.tb00785.x. [DOI] [PubMed] [Google Scholar]
- Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3:344–351. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- McBride WJ. Central nucleus of the amygdala and the effects of alcohol and alcohol-drinking behavior in rodents. Pharmacol Biochem Behav. 2002;71:509–515. doi: 10.1016/s0091-3057(01)00680-3. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- National Institute on Alcohol Abuse and Alcoholism. Eighth special report to the US congress on alcohol and health. Bethesda, MD: DHHS ADM 281-91-003; 1993. [Google Scholar]
- Newton SS, Duman RS. Chromatin remodeling: a novel mechanism of psychotropic drug action. Mol Pharmacol. 2006;70:440–443. doi: 10.1124/mol.106.027078. [DOI] [PubMed] [Google Scholar]
- Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci. 2003;24:456–460. doi: 10.1016/S0165-6147(03)00226-8. [DOI] [PubMed] [Google Scholar]
- Pandey SC. The gene transcription factor cyclic AMP-responsive element binding protein: role in positive and negative affective states of alcohol addiction. Pharmacol Ther. 2004;104:47–58. doi: 10.1016/j.pharmthera.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Zhang D, Mittal N, Nayyar D. Potential role of the gene transcription factor cyclic AMP-responsive element binding protein in ethanol withdrawal-related anxiety. J Pharmacol Exp Ther. 1999;288:866–878. [PubMed] [Google Scholar]
- Pandey SC, Roy A, Mittal N. Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the CREB gene transcription factor in rat cortex. J Pharmacol Exp Ther. 2001;296:857–868. [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H. The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res. 2003;27:396–409. doi: 10.1097/01.ALC.0000056616.81971.49. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, Xu T. Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci. 2004;24:5022–5030. doi: 10.1523/JNEUROSCI.5557-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP responsive-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest. 2005;115:2762–2773. doi: 10.1172/JCI24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26:8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K. Effector immediate-early gene Arc in the amygdala plays a critical role in alcoholism. J Neurosci. 2008;28:2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Henekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- Roelofs SM. Hyperventilation, anxiety, craving for alcohol: a subacute alcohol withdrawal syndrome. Alcohol. 1985;2:501–505. doi: 10.1016/0741-8329(85)90123-5. [DOI] [PubMed] [Google Scholar]
- Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities: a possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20(RC96) doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA, Hesselbrock V. Alcohol dependence and anxiety disorders: what is the relationship? Am J Psychiatry. 1994;151:1723–1734. doi: 10.1176/ajp.151.12.1723. [DOI] [PubMed] [Google Scholar]
- Sharma RP. Schizophrenia, epigenetics, and ligand-activated nuclear receptors. Schizophr Res. 2005;72:79–90. doi: 10.1016/j.schres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ. Comparison of anxiety-like behavior in adolescent and adult Sprague-Dawley rats. Behav Neurosci. 2005;119:1477–1483. doi: 10.1037/0735-7044.119.6.1477. [DOI] [PubMed] [Google Scholar]
- Soderling TR. The Ca2+/calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Starkman BG, Pandey SC. Brain-derived neurotrophic factor and mental illness: an epigenetic approach. Proc Nat Acad Sci India Part B. 2007;77:105–113. [Google Scholar]
- Thiele TE, Marsh DJ, Marie LS, Berstein IL, Palmiter RD. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature. 1998;396:366–369. doi: 10.1038/24614. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83:344–353. doi: 10.1139/o05-041. [DOI] [PubMed] [Google Scholar]
- Villar-Garea A, Esteller M. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer. 2004;112:171–178. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]
- Weiss KJ, Rosenberg DJ. Prevalence of anxiety disorder among alcoholics. J Clin Psychiatry. 1985;46:3–5. [PubMed] [Google Scholar]
- Wilson GT. Alcohol and anxiety. Behav Res Ther. 1988;26:369–381. doi: 10.1016/0005-7967(88)90070-8. [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pandey SC. Effects of PKA modulation on the expression of neuropeptide Y in rat amygdaloid structures during ethanol withdrawal. Peptides. 2003;24:1397–1402. doi: 10.1016/j.peptides.2003.08.008. [DOI] [PubMed] [Google Scholar]