

Abstract
Systemic inflammatory events, such as infection, increase the risk of stroke and are associated with worse outcome, but the mediators of this clinically important effect are unknown. Our aim here was to elucidate mechanisms contributing to the detrimental effects of systemic inflammation on mild ischemic brain injury in mice. Systemic inflammation was induced in mice by peripheral interleukin-1β (IL-1β) challenge and focal cerebral ischemia by transient middle cerebral artery occlusion (MCAo). Systemic inflammation caused an alteration in the kinetics of blood–brain barrier (BBB) disruption through conversion of a transient to a sustained disruption of the tight junction protein, claudin-5, and also markedly exacerbated disruption to the cerebrovascular basal lamina protein, collagen-IV. These alterations were associated with a systemic inflammation-induced increase in neurovascular gelatinolytic activity that was mediated by a fivefold increase in neutrophil-derived matrix metalloproteinase-9 (MMP-9) in the brains of IL-1β-challenged mice after MCAo. Specific inhibition of MMP-9 abrogated the effects of systemic inflammation on the sustained but not the acute disruption of claudin-5, which was associated with phosphorylation of cerebrovascular myosin light chain. MMP-9 inhibition also attenuated the deleterious impact of systemic inflammation on brain damage, edema, neurological deficit, and incidence of hemorrhagic transformation. These data indicate that a transformation from transient to sustained BBB disruption caused by enhanced neutrophil-derived neurovascular MMP-9 activity is a critical mechanism underlying the exacerbation of ischemic brain injury by systemic inflammation. These mechanisms may contribute to the poor clinical outcome in stroke patients presenting with antecedent infection.
Keywords: blood–brain barrier, cerebral ischemia, claudin-5, interleukin-1, MCAo, tight junction
Introduction
Stroke is the second most common cause of mortality and the leading cause of adult neurological disability worldwide (Meairs et al., 2006). There is growing evidence that the susceptibility to stroke and subsequent prognosis are influenced by systemic inflammatory processes (McColl et al., 2007b; Emsley and Hopkins, 2008). Acute infection, mainly of bacterial origin and affecting the respiratory or urinary tracts, increases the risk of stroke, particularly in the first few days after infection (Smeeth et al., 2004; Clayton et al., 2008), and poorer outcome in stroke patients presenting with antecedent infection has been reported (Palasik et al., 2005; Zeller et al., 2005). Other established risk factors, such as atherosclerosis, obesity, type 2 diabetes, periodontal disease, and rheumatoid arthritis are also linked to an elevated systemic inflammatory status (Müller-Ladner et al., 2005; Wellen and Hotamisligil, 2005; Hansson and Libby, 2006; Moutsopoulos and Madianos, 2006). Systemic inflammation has now been shown to markedly exacerbate brain damage in experimental models of cerebral ischemia (McColl et al., 2007a; Spencer et al., 2007).
The mechanisms underlying the impact of systemic inflammation on the outcome to ischemic brain injury are poorly understood, although we recently demonstrated a critical role for neutrophils in an experimental paradigm (McColl et al., 2007a). Neutrophils contain a number of proteolytic enzymes stored in intracellular granules, notably the serine proteases, cathepsin G and elastase, and the gelatinase, matrix metalloproteinase-9 (MMP-9) (Borregaard and Cowland, 1997). MMP-9, in particular, is strongly implicated as a pathological mediator during the acute phase after stroke. Putative mechanisms include the proteolysis of neurovascular substrates such as tight junction proteins essential for blood–brain barrier (BBB) integrity and components of the extracellular matrix integral to cerebrovascular and neuronal viability (Zhao et al., 2007). In contrast to the classic biphasic response, which involves acute and delayed periods of BBB opening (Belayev et al., 1996; Rosenberg et al., 1998), we previously showed that a peripheral inflammatory challenge results in an unremitting increase in BBB permeability after experimental stroke (McColl et al., 2007a). Although the molecular basis for this is unknown, it is conceivable that alterations in neurovascular proteolytic activity may be important in mediating effects of systemic inflammation on stroke damage.
To test this hypothesis and to identify the molecular effectors of the detrimental effects of systemic inflammation on ischemic brain injury, we have used a previously validated model of systemic inflammation induced by peripheral administration of the cytokine, interleukin-1β (IL-1β) (McColl et al., 2007a). We have shown previously that the effects of IL-1β challenge on stroke in mice are indistinguishable from those induced by the bacterial endotoxin, lipopolysaccharide (McColl et al., 2007a), and that effects of the latter are inhibited by the IL-1 receptor antagonist. Moreover, localized peripheral and systemic actions of IL-1β contribute to conditions that predispose to stroke, such as infection (Dinarello, 1992), atherosclerosis (Kirii et al., 2003), and type 2 diabetes (Larsen et al., 2007). We show that systemic inflammatory challenge causes sustained BBB disruption and poorer outcome after MCAo via an MMP-9-mediated pathway.
Materials and Methods
Mice.
All experiments were performed on 10–12-week-old (25–30 g) C57BL/6J mice (Harlan) under appropriate UK Home Office personal and project licenses and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986).
Focal cerebral ischemia.
Focal ischemia was induced by transient (30 min) middle cerebral artery occlusion (MCAo) as described previously (McColl et al., 2007a). Briefly, core body temperature was regulated at 37 ± 0.5°C. Under halothane anesthesia (30% O2, 70% N2O), a 6-0 nylon monofilament (Dermalon) with 2 mm tip (180 μm diameter) coated in thermo-melting glue (Jet Melt) was introduced into the external carotid artery and advanced along the internal carotid artery (ICA) until occluding the origin of the MCA. After 30 min, the filament was withdrawn to establish reperfusion. Sham-operated mice underwent the same procedure except the filament was advanced along the ICA and then immediately withdrawn.
Drug administration.
All treatments were administered in a blinded manner by intraperitoneal injection in a volume of 4 ml/kg. Systemic inflammation was induced by injection of recombinant IL-1β (10 IU; National Institute for Biological Standards and Controls, UK) diluted in vehicle (0.5% endotoxin-free bovine serum albumin in sterile PBS) 30 min before the onset of MCAo. To block MMP-9, we used the specific inhibitor, SB-3CT. SB-3CT is a highly selective mechanism-based gelatinase inhibitor with particular specificity for MMP-9 and has previously been shown to specifically inhibit MMP-9-driven pathways in disease models in vivo (Gu et al., 2005; Bonfil et al., 2006). We verified this selectivity in vitro; SB-3CT inhibited MMP-9- but not MMP-2-mediated gelatinase activity in medium from IL-1β-treated glial cells. SB-3CT (20 mg/kg diluted in 0.1% DMSO in sterile saline; Biomol International) was administered 30 min before the induction of MCAo and 4 h after the onset of reperfusion.
Neutrophil depletion.
To deplete neutrophils, mice received three injections of rabbit anti-PMN IgG (2 mg/kg; Accurate Scientific) or rabbit IgG as control at the same dose diluted in sterile saline. One injection was given per day for 3 d before MCAo.
Assessment of neurological deficit.
Neurological status was assessed blinded to drug treatment and according to a neurological grading score of increasing severity of deficit (Bederson et al., 1986): 0, no observable deficit; 1, torso flexion to right; 2, spontaneous circling to right; 3, leaning/falling to right; 4, no spontaneous movement.
Tissue processing.
For measurement of ischemic damage and brain edema and for in situ zymography, mice were perfused transcardially with 0.9% saline then brains removed and snap-frozen in isopentane on dry ice. For immunohistochemistry, mice were perfused with 0.9% saline followed by 4% paraformaldehyde. Brains were removed, postfixed, cryoprotected (15% sucrose), and frozen in isopentane on dry ice. For all experiments, sections (20 μm) were cut on a cryostat (Leica Microsystems). For immunoblotting and gel zymography, mice were perfused with 0.9% saline, and striatal and cortical brain samples were rapidly removed and frozen in dry ice. Frozen samples were homogenized on ice in buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.02% NaN, 1% Triton X-100) and then centrifuged (14,000 × g for 30 min at 4°C). Protein concentration was determined in supernatants by BCA protein assay (Pierce) and all samples equilibrated to a concentration of 2 mg/ml total protein.
Assessment of ischemic brain damage and edema.
Ischemic damage and edema were measured blinded to drug treatment as described previously (McColl et al., 2007a). Adjacent sections were stained with hematoxylin and eosin to detect hemorrhagic transformation (HT).
Immunohistochemistry.
Antibodies used were as follows: goat anti-MMP-9 (1:500; R&D Systems), rabbit anti-neutrophil SJC (1:100; kindly provided by Drs. Daniel Anthony and Sandra Campbell, University of Oxford, Oxford, UK), mouse anti-neuronal-specific nuclear protein (neuN) to label neurons (1:50; Millipore), rabbit anti-GFAP to label astrocytes (1:500; Dako), rabbit anti-iba1 to label microglia (1:500; Wako), rabbit anti-claudin-5 (1:200; Zymed Laboratories), rabbit anticollagen-IV (1:500; Abcam), rabbit anti-phosho-myosin light chain (MLC) (1:50; Cell Signaling Technology). Endogenous peroxidase activity was blocked (this step omitted for double-labeling immunofluorescence) with 0.3% H2O2 in methanol and nonspecific binding sites blocked with 10% normal serum (Vector Laboratories). Sections were incubated in primary antibody (diluted in 5% normal serum in PBS) overnight at 4°C. For peroxidase-based staining, sections were then incubated with appropriate biotinylated secondary antibody (1:200 in PBS; Vector Laboratories) before incubation in Vectastain ABC solution (Vector Laboratories) and development of staining by diaminobenzidine reaction (Vector Laboratories). Sections were lightly counterstained with cresyl violet. For double-labeling immunofluorescence, after primary antibody incubation, sections were incubated with Alexa fluorochrome-conjugated secondary antibodies (1:1000 in PBS; Invitrogen) and mounted with ProLong Gold containing 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) counterstain (Invitrogen).
Quantification of MMP-9 immunostaining.
Cerebral MMP-9 immunoreactivity was quantified blinded to treatment. The number of MMP-9-immunopositive cells was counted in three fields of the cortex (somatosensory, insular, piriform) or striatum (dorsal, lateral, ventral) at two coronal levels (0.2 mm and −0.5 mm relative to bregma). The mean was calculated from the six fields in the cortex or striatum and adjusted to express as mean number of cells/mm2.
Immunoblotting.
Antibodies used were as follows: rabbit anti-claudin-5 (1:500; Zymed Laboratories), goat anti-occludin (1:1000; Santa Cruz Biotechnology), rabbit anti-collagen-IV (1:1000; Abcam), rabbit anti-laminin (1:100; Sigma-Aldrich). Homogenized samples (containing 50 μg protein) were separated by SDS-PAGE and proteins transferred to polyvinylidene difluoride membrane. Membranes were blocked [10% nonfat milk in 0.01% PBS-Tween20 (PBS-T)] and incubated in primary antibody (diluted in 1% bovine serum albumin in PBS-T) overnight at 4°C. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (in 5% milk in PBS-T). Blots were developed using an Enhanced Chemiluminescent Detection Kit (GE Healthcare) and protein bands visualized on x-ray film (Biomax MR1; Kodak). Semiquantitative densitometry was performed on digitized images using Northern Eclipse software.
Gel zymography.
Protein samples were purified for gelatinases as described previously (Zhang and Gottschall, 1997). Briefly, samples (containing 1.5 mg protein) were incubated with gelatin-Sepharose beads (GE Healthcare) for 1 h at 4°C with gentle agitation. After centrifugation (14,000 × g for 2 min at 4°C), the gelatin-Sepharose pellet containing bound gelatinases was washed (in homogenization buffer without detergent) and the purified gelatinases then separated from beads by incubation with elution buffer (10% DMSO in wash buffer) for 30 min at 4°C with constant agitation followed by centrifugation. Purified samples (25 μl) were separated on SDS-polyacrylamide gels impregnated with 1.5 mg/ml gelatin under nonreducing, nondenaturing conditions. SDS was removed to renature enzymes by incubating gels in wash buffer (50 mm Tris-HCl, pH 7.6, 5 mm CaCl2, 1 μm ZnCl2, 2.5% Triton X-100). Gels were then incubated in reaction buffer (wash buffer without detergent) for 72 h at 37°C to stimulate gelatinolysis. Gels were stained with Coomassie Brilliant Blue R-250 (0.1%) diluted in 40% methanol and 10% acetic acid for 1 h and destained in a solution containing 10% methanol and 1% acetic acid until clear gelatinolytic bands appeared. Gels were dried and semiquantitative densitometry performed on digitized images using Northern Eclipse software.
In situ zymography.
Snap-frozen unfixed sections were incubated with reaction buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 1 μm ZnCl2, 0.02% NaN) containing 20 μg/ml FITC-conjugated DQ gelatin (Invitrogen) overnight at 37°C. Gelatinases cleave the gelatin resulting in uncaging of the conjugated FITC and in increase in fluorescence corresponding to the extent of proteolytic activity. Sections were washed and mounted with ProLong Gold (Invitrogen). Some sections were coincubated with the broad-spectrum MMP inhibitor, GM-6001 (Calbiochem). For colocalization of gelatinolytic activity, immunofluorescence was performed as described above immediately after zymography.
Microscopy.
Brightfield images were collected on a Zeiss Axioskop upright microscope and captured using Zeiss Axiovision software. Widefield fluorescence images were collected on an Olympus BX51 upright microscope and captured using MetaVue software (Molecular Devices). Confocal images were collected using a Nikon Eclipse 90i upright confocal microscope. Fluorescent images were processed using ImageJ software.
Statistical analysis.
Parametric data were analyzed using Student's t test for single comparisons or one-way ANOVA followed by Student's t test with Bonferroni correction for multiple comparisons. Correlation coefficients were computed using Pearson correlation. Nonparametric data were analyzed using Kruskal–Wallis test followed by Dunn's test for multiple comparisons. Categorical data (neurological deficit scores) were analyzed using generalized Fisher's exact test with Bonferroni correction. Statistical significance was assumed at p ≤ 0.05.
Results
Systemic inflammation alters the kinetics of BBB tight-junction disruption
We showed previously that a systemic inflammatory challenge exacerbates BBB permeability and brain edema after MCAo (McColl et al., 2007a). In view of the fundamental role of tight junctions in regulating BBB permeability, we examined the effects of systemic inflammation on the tight junction proteins, claudin-5 (Fig. 1A–C) and occludin (Fig. 1D), the major interendothelial junctional proteins limiting paracellular permeability at the BBB. MCAo induced a 50% reduction in claudin-5 protein compared with sham-operated mice after 4 h reperfusion in both vehicle and IL-1β-challenged mice (Fig. 1A). After 8 h reperfusion, a similar MCAo-induced reduction in claudin-5 to that at 4 h was observed in IL-1β-challenged mice. In contrast, claudin-5 expression in vehicle-treated mice was comparable with that in sham-operated mice. Claudin-5 expression was significantly reduced in IL-1β-challenged mice compared with vehicle-treated mice at this time point (p < 0.05). After 24 h reperfusion there was further disruption to claudin-5 in IL-1β-challenged mice with levels reduced to 30% of sham-operated mice. Claudin-5 was also significantly reduced at 24 h compared with vehicle-treated mice (p < 0.05) which showed comparable levels to sham-operated animals.
Figure 1.

Systemic inflammation causes sustained claudin-5 disruption after MCAo. Systemic inflammation was induced by intraperitoneal IL-1β injection, and the effects on the BBB tight junction proteins, claudin-5 and occludin, were assessed in cortical tissue ipsilateral to MCAo by immunoblot and immunofluorescence at indicated times after MCAo. A, MCAo induced a 50% reduction in claudin-5 levels in both vehicle (veh)- and IL-1β-treated mice compared with sham-operated mice after 4 h reperfusion. This disruption to claudin-5 resolved in vehicle-treated mice by 8 h after MCAo and was maintained at 24 h in which claudin-5 levels were similar to sham-operated mice. There was a sustained disruption to claudin-5 in IL-1β-treated mice 8 and 24 h after MCAo resulting in a 70% reduction compared with sham-operated mice at 24 h. The sustained loss of claudin-5 in IL-1β-challenged mice resulted in a significant reduction compared with vehicle-treated mice 8 and 24 h after MCAo. B, Claudin-5 immunofluorescence (green channel) with DAPI counterstain (blue channel) demonstrated dense networks of cerebrovascular immunoreactivity in sham-operated mice. Marked reductions in immunoreactivity were evident 4 h after MCAO in vehicle-treated mice and 4, 8, and 24 h after MCAo in IL-1β-challenged mice, consistent with the immunoblot data. C, Confocal immunofluorescence revealed the disruption by systemic inflammation to the highly organized arrangement of claudin-5 in the interendothelial tight junctions of cortical blood vessels in the ipsilateral hemisphere 24 h after MCAo. D, There were no significant effects of systemic inflammation or MCAo on occludin levels. All immunofluorescence images are from the cortex ipsilateral to MCAo. *p < 0.05 versus vehicle; Student's t test. Scale bars: B, 100 μm; C, 10 μm.
Disruption of claudin-5 was further examined by immunofluorescence (Fig. 1B). Immunoreactivity was restricted to the cerebral vasculature, and in sham-operated mice dense networks of penetrating arterioles and microvessels were immunoreactive (Fig. 1B). In parallel with the immunoblot data, MCAo induced a marked loss of immunoreactivity after 4 h reperfusion in vehicle and IL-1β-challenged mice. A sustained loss of immunoreactivity was evident in IL-1β-challenged mice as reperfusion progressed. In contrast, the disruption was transient in vehicle-treated mice with the pattern of immunostaining similar to sham-operated mice 8 and 24 h after MCAo (Fig. 1B). Confocal microscopy highlighted the density and precise interendothelial location of claudin-5 in individual vessels. A highly regular and organized arrangement of claudin-5 immunoreactivity was evident in sham-operated and vehicle-treated mice 24 h after MCAo, whereas marked disorganization and fragmentation was seen in IL-1β-challenged mice (Fig. 1C).
In contrast to claudin-5, MCAo did not cause disruption of occludin in vehicle- or IL-1β-challenged mice at any time point (Fig. 1D).
Systemic inflammation exacerbates cerebrovascular basal lamina disruption
The integrity of the BBB is dependent on adequate structural and biochemical support from the basement membrane that closely apposes the abluminal surface of the endothelial monolayer (del Zoppo and Milner, 2006). Therefore, we next determined whether systemic inflammation affects the disruption of the key basal lamina components, collagen-IV and laminin, after MCAo (Fig. 2). There was no effect of MCAo on collagen-IV levels in vehicle-treated mice at any time point assessed. After 4 h reperfusion, IL-1β challenge induced a modest reduction in collagen-IV levels relative to sham-operated mice, but there was no significant difference compared with vehicle-treated mice (Fig. 2A). There was, however, a progressive reduction in collagen-IV levels as reperfusion continued to 8 and 24 h after MCAo in IL-1β-challenged mice, and at these time points there was a significant reduction compared with vehicle-treated mice (p < 0.05). In addition to the progressive loss in 160 kDa collagen-IV, we also observed the appearance of a smaller band of ∼80 kDa that was consistently present in all samples from IL-1β-challenged mice but only rarely (2 of 6) in vehicle-treated mice 8 and 24 h after MCAo. This band was not present in sham-operated mice or 4 h after MCAo suggesting that it may represent a collagen-IV cleavage product and that systemic inflammation aggravates proteolytic degradation of the full-length species.
Figure 2.
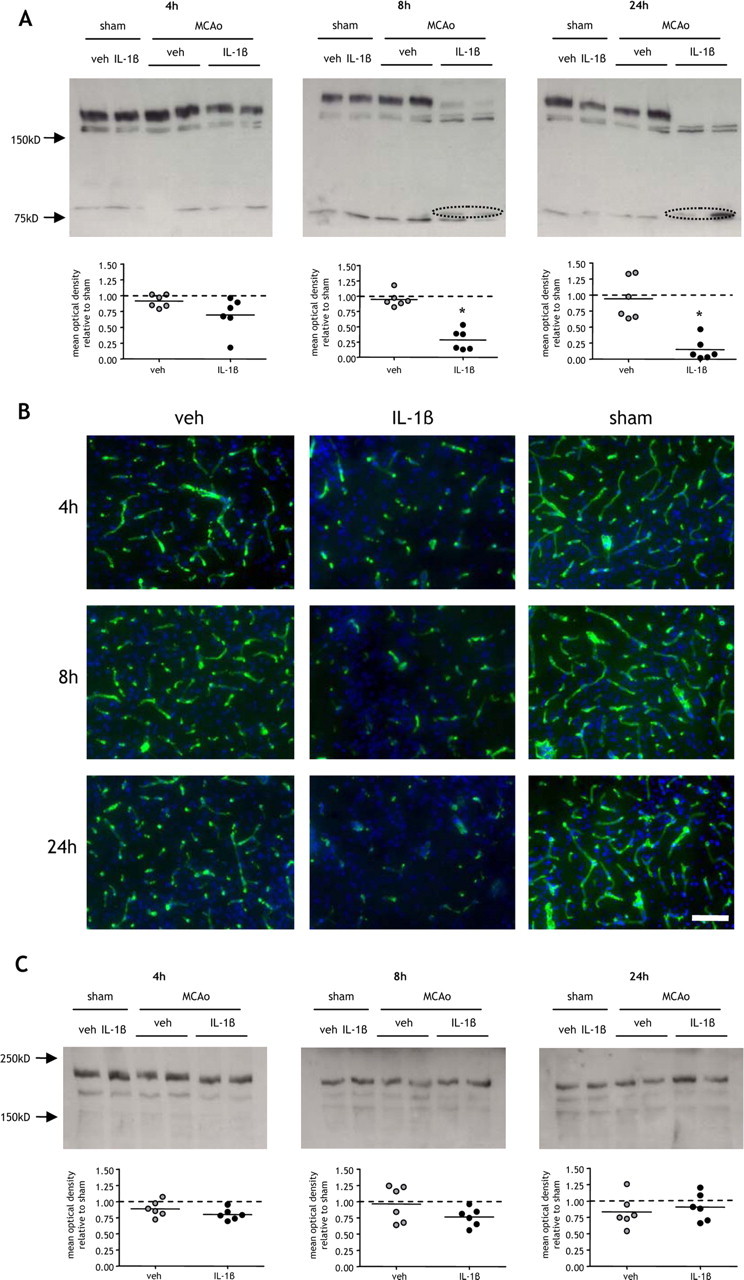
Systemic inflammation aggravates cerebrovascular collagen-IV disruption after MCAo. Systemic inflammation was induced by intraperitoneal IL-1β injection and the effects on the cerebrovascular basement lamina proteins, collagen-IV, and laminin, were assessed in cortical tissue ipsilateral to MCAo by immunoblot and immunofluorescence at indicated times after MCAo. A, Collagen-IV levels in vehicle (veh)-treated mice were similar in sham-operated mice and after MCAo at all time points examined. IL-1β challenge caused a progressive decline in collagen-IV as reperfusion progressed and resulted in an 80% reduction compared with sham levels 24 h after MCAo. There was a significant reduction in collagen-IV levels in IL-1β-challenged mice compared with vehicle-treated mice 8 and 24 h after MCAo, and a putative cleavage collagen-IV cleavage product (circled) was evident in IL-1β-challenged mice at these time points. B, Collagen-IV immunofluorescence (green channel) with DAPI counterstain (blue channel) demonstrated extensive and dense networks of cerebrovascular immunoreactivity in sham-operated mice and a similar pattern was evident in vehicle-treated mice after MCAo. IL-1β challenge induced a marked reduction in collagen-IV immunoreactivity 4, 8, and 24 h after MCAo. C, Systemic inflammation or MCAo did not significantly affect laminin levels. All immunofluorescence images are from the cortex ipsilateral to MCAo. *p < 0.05 versus vehicle; Student's t test. Scale bar, 50 μm.
The pattern of collagen-IV immunostaining on brain sections was consistent with the immunoblotting results. Immunoreactivity was associated exclusively with the cerebral vasculature, and dense networks of collagen-IV-labeled vessels were observed in sham-operated mice (Fig. 2B). A modest reduction in immunoreactivity was observed in vehicle-treated mice in response to MCAo, but the duration of reperfusion did not demonstrably affect the extent of immunoreactivity. In contrast, as reperfusion progressed in IL-1β-challenged mice, we observed a progressive reduction in collagen-IV immunoreactivity resulting in a marked loss 24 h after MCAo (Fig. 2B).
The major laminin immunoreactive band was observed at 200 kDa, likely corresponding to the α4 isoform enriched in the vascular basement membrane. There were no significant changes in laminin levels in response to MCAo or IL-1β challenge (Fig. 2C). Immunohistochemistry on brain sections confirmed the preservation of vessel-associated laminin; however, we did observe scattered loss of neuronal-associated laminin 24 h after MCAo (data not shown) consistent with a previous study (Gu et al., 2005).
Systemic inflammation exacerbates neurovascular gelatinolytic activity
Several mechanisms may contribute to BBB disruption after stroke. Foremost among these is the proteolytic disruption of BBB substrates, including tight junction and basal lamina proteins, associated with neurovascular MMP activity. We next determined whether systemic inflammation affects gelatinase (MMP-9 and MMP-2)-derived proteolytic activity in the brain after MCAo using gel and in situ zymography (Fig. 3). MMP-9 activity was undetectable at any time point in sham-operated mice. A band corresponding to murine pro-MMP-9 (105 kDa) was evident in vehicle and IL-1β-challenged mice 8 and 24 h after MCAo. There was negligible induction of MMP-9 activity 4 h after MCAo and no difference between vehicle and IL-1β-challenged mice (Fig. 3A). After 8 and 24 h reperfusion, IL-1β-challenge resulted in a significant increase in MMP-9 activity (p < 0.01) (Fig. 3A). The activated form of murine MMP-9 (97 kDa) was also detectable at these time points. In contrast to MMP-9 activity, no gelatinolysis at the 72 kDa level (corresponding to MMP-2) was detected after sham surgery or MCAo suggesting that MMP-2 was not induced by peripheral inflammatory challenge or ischemia. This is consistent with neutrophils representing the major source of MMP-9 in the postischemic brain because these cells do not produce MMP-2 (Van den Steen et al., 2002). Furthermore, we did not detect induction of MMP-3 by immunohistochemistry (data not shown), another MMP that has been implicated in BBB disruption.
Figure 3.
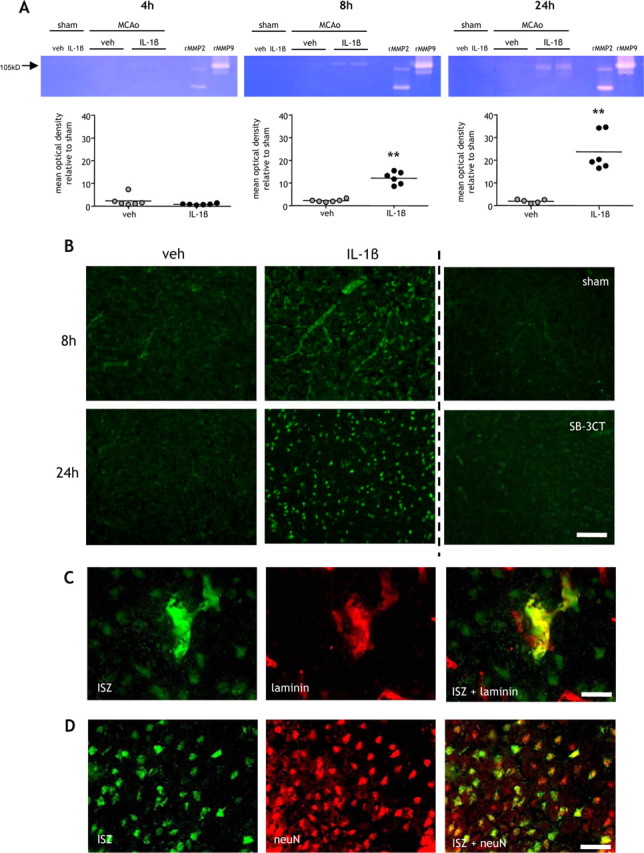
Systemic inflammation exacerbates neurovascular gelatinolytic activity. A, B, Systemic inflammation was induced by intraperitoneal IL-1β injection and the extent of gelatinolytic activity determined by gel zymography in purified cortical brain samples (A) and by in situ zymography on brain sections at indicated times after MCAo (B). A, Gelatinolytic activity was minimal 4 h after MCAo, and there was no difference between vehicle (veh)- and IL-1β-treated mice. Clear gelatinolytic bands corresponding to pro-MMP-9 (105 kDa) and active MMP-9 (97 kDa) were evident 8 and 24 h after MCAo in vehicle- and IL-1β-treated mice but not after sham occlusion. The extent of gelatinolytic activity (105 kDa) was significantly greater in IL-1β-treated mice 8 and 24 h after MCAo. There was no detectable gelatinolytic activity corresponding to MMP-2 (72 kDa). B, In situ zymography demonstrated that increased gelatinolytic activity in IL-1β-challenged mice was primarily localized to the cerebral vasculature and neurons 8 and 24 h after MCAo. In situ gelatinolytic activity was minimal in sham-operated mice and attenuated by coincubation with the MMP-9 inhibitor, SB-3CT. C, Immunofluorescence was combined with in situ zymography to confirm the localization of gelatinolytic activity. D, Gelatinolytic activity colocalized with the vascular basement membrane protein, laminin, and the neuronal marker neuN. **p < 0.01 versus vehicle; Student's t test. Scale bars: B, 100 μm; C, 15 μm; D, 25 μm.
To further assess the spatiotemporal induction of MMP activity, we used in situ zymography. Gelatinolytic activity was not detectable after 4 h reperfusion in vehicle or IL-1β-challenged mice (data not shown). An induction of activity was observed 8 and 24 h after MCAo and was markedly greater in IL-1β-treated mice compared with vehicle-treated (Fig. 3B). After 8 h reperfusion, IL-1β treatment resulted in intense and extensive proteolytic activity throughout cortical tissue, which was primarily localized to the microvasculature and to a lesser extent surrounding neuronal cell bodies. Peri-neuronal activity increased further 24 h after MCAo. Addition of the specific MMP-9 inhibitor, SB-3CT, to the incubation medium abolished gelatinolytic activity indicating that MMP-9 was the gelatinase responsible for proteolysis (Fig. 3B). Colocalization with laminin (Fig. 3C) and neuN (Fig. 3D) confirmed the vascular and neuronal localization of in situ gelatinolytic activity. These observations are consistent with the gel zymography data which show specific induction of MMP-9 and together suggest that systemic inflammation exacerbates neutrophil MMP-9-driven proteolysis of neurovascular substrates.
Infiltrating neutrophils are the major source of increased MMP-9 caused by systemic inflammation
Cells resident in the brain and infiltrating immune cells can express MMP-9 in the postischemic brain suggesting that systemic inflammation could enhance MMP-9 activity by stimulating resident brain cells via immune-brain signaling or via increased trafficking of MMP-9-laden cells into the brain. We next investigated the cellular source(s) responsible by examining the spatiotemporal profile of cerebral MMP-9 immunoreactivity after MCAo (Fig. 4). MMP-9 immunoreactivity was evident in the striatum and cortex of the hemisphere ipsilateral to MCAo but not in the contralateral hemisphere or in sham-operated mice. MMP-9 was localized to cells within blood vessels, including penetrating vessels (Fig. 4A) and microvessels (Fig. 4B), and to cells throughout the parenchyma (Fig. 4C). MMP-9 immunoreactivity was more abundant and more extensively distributed in IL-1β-treated mice when compared with vehicle-treated, particularly after 8 or 24 h reperfusion (Fig. 4D). Quantification of cellular MMP-9 in the striatum showed a general decline in the number of MMP-9+ cells as reperfusion progressed (Fig. 4E). The number of MMP-9+ cells in the striatum was significantly greater in IL-1β-challenged mice 8 h after MCAo (p < 0.05). In contrast, the number of MMP-9+ cells in the cortex increased as reperfusion progressed and IL-1β challenge resulted in a significant increase in MMP-9 immunoreactivity at all time points after MCAo (4 and 8 h, p < 0.05; 24 h, p < 0.01).
Figure 4.

Systemic inflammation increases cerebral MMP-9 immunoreactivity after MCAo. Systemic inflammation was induced by intraperitoneal IL-1β injection and MMP-9 immunostaining assessed at indicated times after MCAo. A–C, MMP-9 immunostaining was evident in cells adherent to cortical penetrating vessels (A) and microvessels (B) and in cells throughout the parenchyma (C) in the hemisphere ipsilateral to MCAo. There was minimal immunoreactivity in the contralateral hemisphere and in sham-operated mice. D, Representative brain sections illustrate the marked increase in MMP-9-immunoreactive cells in the cortex of IL-1β-treated mice 8 and 24 h after MCAo. E, Quantification of cellular MMP-9 immunoreactivity. Systemic inflammation significantly increased the number of MMP-9+ cells in the cortex 4, 8, and 24 h after MCAo and in the striatum 8 h after MCAo. *p < 0.05, **p < 0.01 versus vehicle (veh); Student's t test. Scale bars: (in C) A–C, 25 μm; D, 100 μm.
The distribution and localization of MMP-9 immunostaining was strikingly similar to the pattern of neutrophil infiltration we reported previously (McColl et al., 2007a). To determine whether neutrophils were the major source of MMP-9 in this paradigm, we performed double-labeling immunofluorescence (Fig. 5). We observed strong colocalization between MMP-9 and the neutrophil-specific marker, SJC, but not with neuronal (neuN), microglial/macrophage (Iba1), or astrocytic (GFAP) markers (Fig. 5A). We also observed strong colocalization between MMP-9 and the vascular basement membrane protein, laminin (Fig. 5A). The vascular (and cellular) MMP-9 immunoreactivity was lost in neutropenic mice but not in mice treated with an isotype control (Fig. 5B), suggesting that the vascular MMP-9 was neutrophil-derived and confirming neutrophils as the key source of MMP-9 in the ischemic brain in our model. This was further substantiated by a strong correlation between the number of MMP-9+ cells and neutrophils (r2 = 0.91; p < 0.001) (Fig. 5C). These data suggest that systemic inflammation exacerbates cerebrovascular and parenchymal MMP-9 levels and activity by increasing the trafficking of peripheral immune cells, particularly neutrophils, into the brain.
Figure 5.
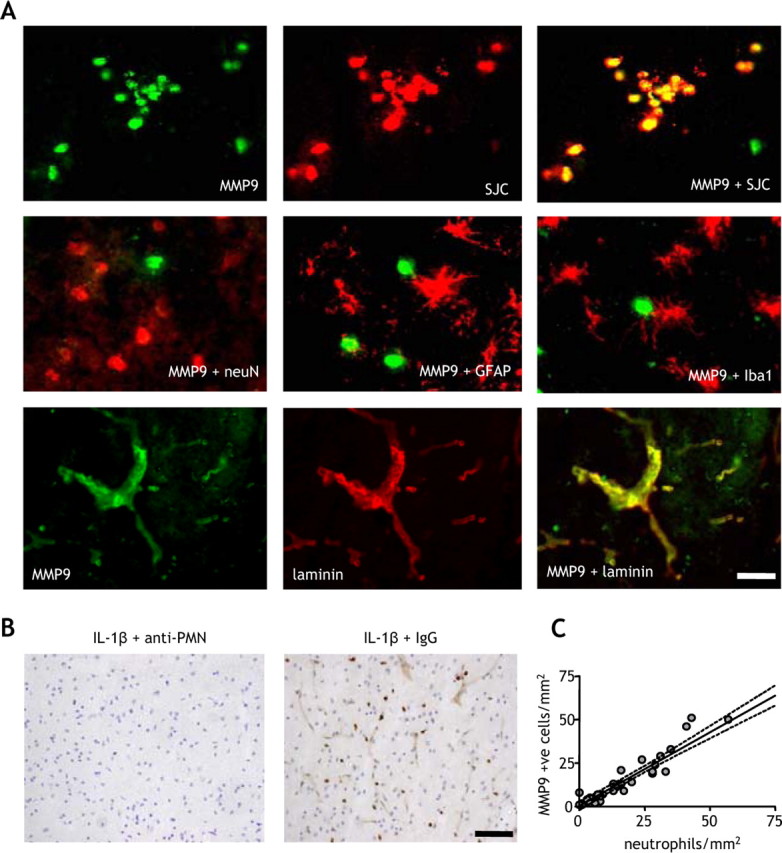
Infiltrating neutrophils are the primary source of MMP-9 in the ischemic brain. The cellular localization of MMP-9 immunoreactivity in the ischemic brain was examined by double-labeling immunofluorescence. A, There was strong and extensive colocalization between MMP-9 and the specific neutrophil marker, SJC, but not with neuronal (neuN), astrocytic (GFAP), or microglial (Iba1) markers. There was also strong colocalization of MMP-9 with the vascular basement membrane protein, laminin. B, Depletion of neutrophils using an anti-PMN antibody abolished the cellular and vascular MMP-9 immunoreactivity in the brain, but there was no effect with an isotype control antibody. C, There was a strong correlation between the number of MMP+ cells and the number of neutrophils in the cortex of vehicle- and IL-1β-treated mice. R2 = 0.91; p < 0.01, Pearson correlation. All double-labeling immunofluorescence images are from the cortex ipsilateral to MCAo of IL-1β-treated mice 24 h after MCAo. Scale bars: A, 25 μm; B, 100 μm.
Inhibition of MMP-9 abrogates the delayed and sustained disruption to BBB substrates caused by systemic inflammation
The temporal profile of exacerbated MMP-9 activity was consistent with the sustained disruption to claudin-5 after MCAo caused by systemic inflammation. We next determined directly whether MMP-9 was responsible for the systemic inflammation-induced alteration in the kinetics of BBB disruption after MCAo. MMP-9 inhibition did not significantly affect the reduction in claudin-5 levels in vehicle- or IL-1β-treated mice 4 h after MCAo (Fig. 6A). The claudin-5 disruption resolved by 8 h after MCAo in vehicle-treated mice regardless of whether MMP-9 was inhibited or not. This resolution in vehicle-treated mice was maintained at 24 h after MCAo and again was independent of MMP-9 inhibition. In contrast, the sustained reduction in claudin-5 levels in IL-1β-challenged mice 8 and 24 h after MCAo was significantly (p < 0.05) attenuated by MMP-9 inhibition (Fig. 6A).
Figure 6.

Sustained aggravation of postischemic BBB disruption caused by systemic inflammation is dependent on MMP-9. A, B, Systemic inflammation was induced by intraperitoneal IL-1β injection, and the effects of MMP-9 inhibition using the specific inhibitor, SB-3CT, on the BBB tight junction protein, claudin-5 (A), and the cerebrovascular basal lamina protein, collagen-IV (B), were assessed in cortical tissue ipsilateral to MCAo by immunoblot at indicated times after MCAo. A, MCAo induced a 75% reduction in claudin-5 levels after 4 h reperfusion in vehicle (veh)- and IL-1β-treated mice, and there was no effect of MMP-9 inhibition at this time point. Claudin-5 levels were restored to sham occlusion levels 8 and 24 h after MCAo, and there was no significant effect of MMP-9 inhibition. Peripheral inflammatory challenge caused a sustained reduction in claudin-5 8 and 24 h after MCAo, and these effects were significantly attenuated by inhibition of MMP-9. B, Collagen-IV levels were similar after sham occlusion and MCAo in vehicle-treated mice at all time points examined. Peripheral inflammatory challenge induced a significant reduction in collagen-IV 8 and 24 h after MCAo, and these effects were significantly attenuated by inhibition of MMP-9. *p < 0.05 versus vehicle, #p < 0.05 versus IL-1β, Kruskal–Wallis test followed by Dunn's multiple-comparison test.
MMP-9 inhibition had no effect on collagen-IV levels in vehicle-treated mice, and levels were similar to sham-operated mice at all time points examined (Fig. 6B). There was a trend toward reduced collagen-IV levels 4 h after MCAo in IL-1β-challenged mice, and MMP-9 inhibition restored levels to those in sham-operated mice. MMP-9 inhibition significantly attenuated the reduction in collagen-IV 8 and 24 h after MCAo in IL-1β-challenged mice (p < 0.05). At the later time points, MMP-9 inhibition prevented the appearance of the 80 kDa immunoreactive band, a potential collagen-IV cleavage product.
Together, these data suggest that systemic inflammation can drive a sustained MMP-9-dependent disruption of the BBB that supersedes and compounds a previous MMP-9-independent disruption that can resolve in the absence of systemic inflammation.
Phosphorylation of cerebrovascular myosin light chain occurs independently of systemic inflammation 4 h after MCAo
The data above suggest an MMP-9-independent disruption to claudin-5, 4 h after MCAo. The resolution of claudin-5 disruption in vehicle-treated mice by 8 h also suggested a protease-independent mechanism, because cleavage of claudin-5 would be expected to induce a prolonged effect. Therefore, we next assessed whether MCAo triggered brain endothelial MLC phosphorylation, a critical step in brain endothelial cytoskeletal reorganization and tight junction disruption in vitro (Afonso et al., 2007). Using an antibody that specifically detects phosphorylation of serine19 of MLC, we observed an increase in phosphorylated MLC 4 h after MCAo in vehicle- and IL-1β-treated mice compared with sham-operated mice (supplemental figure, available at www.jneurosci.org as supplemental material). Increased MLC phosphorylation was observed in microvessels of diameter ∼10 μm deep in the cerebral cortex. These vessels do not have a surrounding smooth muscle layer indicating that the observed response was attributable to phosphorylation of endothelial MLC. Phosphorylated MLC was also apparent in large cortical surface arteries which do contain smooth muscle (supplemental figure, available at www.jneurosci.org as supplemental material), but there was no induction compared with sham-operated mice, perhaps reflecting the constitutive function of these vessels in regulating cortical perfusion.
Systemic inflammation exacerbates ischemic brain injury via an MMP-9 dependent mechanism
In view of the important role of BBB dysfunction in ischemic brain damage, the transformation from transient to sustained tight-junction disruption caused by systemic inflammation and mediated by MMP-9 is likely to have a marked effect on outcome after stroke. Therefore, we next determined whether the systemic inflammation-induced MMP-9-dependent sustained disruption to BBB substrates is an important mechanism underlying the exacerbation of brain damage after mild focal ischemia we demonstrated previously (McColl et al., 2007b) (Fig. 7). Peripheral IL-1β challenge significantly exacerbated (2-fold) the extent of ischemic brain damage (p < 0.05) (Fig. 7A), brain edema (p < 0.05) (Fig. 7B), and the severity of neurological deficit (p < 0.05) (Fig. 7C) and also caused HT of infarcts in two of six mice. Inhibition of MMP-9 significantly attenuated the effects of peripheral IL-1β on damage (p < 0.05), edema (p < 0.05), and neurological deficit (p < 0.05), and prevented HT (Fig. 7D). The abrogation of brain injury was primarily the result of an attenuation of damage in cortical tissue (Fig. 7E), and there was no effect of MMP-9 inhibition in vehicle-treated mice. These results are consistent with the predominance of inflammatory processes in cortical tissue and the mainly anoxic/excitotoxic modes of cell death operating in the striatal core. Inhibition of MMP-9 also attenuated the effect of peripheral inflammatory challenge on cortical neutrophil accumulation (Fig. 7F) and gelatinolytic activity (Fig. 7F).
Figure 7.

Inhibition of MMP-9 ameliorates the exacerbation of ischemic brain damage by systemic inflammation. Systemic inflammation was induced by intraperitoneal IL-1β injection, and the effects of MMP-9 inhibition using the specific inhibitor, SB-3CT, on the extent of ischemic brain damage, brain edema, and the severity of neurological deficit were determined 24 h after MCAo. A–C, Systemic inflammation significantly exacerbated the volume of ischemic damage (A), the extent of brain edema (B), and the neurological deficit (C). Inhibition of MMP-9 significantly attenuated the deleterious effects of systemic inflammation on brain damage (A), edema (B), and neurological deficit (C). D, Representative cresyl violet-stained brain sections illustrate the exacerbation of brain damage by systemic inflammation and the attenuation of this effect by SB-3CT. E, SB-3CT prevented the hemorrhagic transformation (arrowheads) caused by systemic inflammation. F, G, Inhibition of MMP-9 also attenuated the potentiation of neutrophil infiltration (F) and gelatinolytic activity (G) caused by peripheral inflammatory challenge. A, B, F: *p < 0.05 versus vehicle (veh), #p < 0.05 versus IL-1β, one-way ANOVA followed by Student's t test with Bonferroni correction; C: *p < 0.05 versus vehicle, #p = 0.05 versus IL-1β, generalized Fisher's exact test with Bonferroni correction. Scale bars, 100 μm.
Discussion
The present data provide an explanation for the deleterious impact of systemic inflammatory events, such as infection, on ischemic brain injury, and highlight the importance of considering the underlying immune status as a key modifier of stroke outcome. We demonstrate that systemic inflammation aggravates neurovascular proteolytic activity and exacerbates and alters the kinetics of disruption to key components of the BBB after mild focal cerebral ischemia. A rapid and transient claudin-5 disruption occurs after ischemia independently of systemic inflammation and is associated with phosphorylation of cerebrovascular MLC. Systemic inflammation transforms this transient disruption to a sustained reduction in claudin-5. We identify infiltrating neutrophil-derived MMP-9 as the perpetrator responsible for this sustained BBB disruption because inhibition of MMP-9 reversed the sustained but not the hyperacute transient disruption. These data imply that systemic inflammation alters the kinetics of BBB tight-junction disruption via a peripherally derived MMP-9-dependent phase of disruption that prevents the resolution of the initial tight junction alterations induced independently of MMP-9. The impact of these alterations on brain injury is underscored by our finding that inhibition of MMP-9 prevents the exacerbation of brain damage, edema, and neurological deficit caused by systemic inflammatory challenge.
There is growing evidence that inflammatory events outside the brain impinge on susceptibility, outcome, and prognosis in a variety of neurological conditions, including cerebral ischemia (McColl et al., 2007b). Acute peripheral inflammatory challenge worsens outcome after focal cerebral ischemia (McColl et al., 2007a) and increases hippocampal neuronal death after global cerebral ischemia (Spencer et al., 2007). These effects are independent of core body and brain temperature (Parry-Jones et al., 2008). Systemic inflammation also sensitizes the brain to neonatal hypoxic and hemorrhagic injury (Lehnardt et al., 2003; Xue and Del Bigio, 2005) and exacerbates excitotoxic (Favrais et al., 2007) and traumatic brain injury (Utagawa et al., 2008). The clinical relevance of these findings is underlined by epidemiological and clinical studies demonstrating that systemic infection is a risk factor for stroke and that preceding infection is associated with less favorable outcome (Emsley and Hopkins, 2008). Furthermore, systemic infection acquired after stroke is a common complication and an important determinant of outcome in animals and humans (Prass et al., 2003; Chamorro et al., 2007). Collectively, these studies suggest that the systemic inflammatory profile in the peri-ischemic period is an important variable that can modulate stroke outcome. This concept parallels the disease-modifying effects of systemic inflammation on chronic neurodegeneration (Perry et al., 2007) and highlights an important generic role for innate immune–brain interactions in acute and chronic neurological dysfunction.
The mechanisms underlying the detrimental effects of systemic inflammation on ischemic brain injury are not well understood. In the present study, systemic inflammation altered the kinetics of BBB tight-junction disruption by converting a transient reduction in claudin-5 to a sustained disruption that did not resolve. Brain endothelial cells are enriched in claudin-5 which is the critical structural and functional element for correct formation of tight junction strands and the limited paracellular permeability of the BBB (Nitta et al., 2003; Piontek et al., 2008). The present data provide a potential explanation for our previous results showing that systemic inflammation induces a sustained increase in BBB permeability and an exacerbation of brain edema (McColl et al., 2007a). In vehicle-challenged mice, claudin-5 disruption and BBB permeability were rapid and transient. A similar acute and resolving episode of BBB permeability has been described in previous studies reporting a biphasic profile of BBB opening after stroke in the absence of systemic inflammation (Belayev et al., 1996; Rosenberg et al., 1998). In view of these studies, our current data suggest that this early BBB opening does not resolve but is protracted when systemic inflammation is coincident with stroke. Although prolonged, the conversion from transient to sustained BBB disruption occurs early and before the development of increased brain damage caused by peripheral IL-1β challenge (McColl et al., 2007a) which supports a causative role for these BBB alterations in the exacerbation of injury. The consequences of unremitting loss of BBB integrity may be manifold including excessive edema, raised intracranial pressure and increased infiltration of hematogenous immune cells, all of which are detrimental to stroke outcome. In support of this, systemic inflammation significantly increased neutrophil accumulation in the brain (McColl et al., 2007a).
An important finding in our current study is the identification of MMP-9 as a key mediator underlying the sustained disruption to cerebrovascular claudin-5 caused by systemic inflammation. In contrast, MMP-9 was not responsible for the rapid disruption to claudin-5 which preceded the MMP-9-dependent phase in IL-1β-challenged mice or the transient disruption in vehicle-treated mice. Accordingly, our data implicate MMP-9-independent (early) and MMP-9-dependent (delayed) phases of tight-junction disruption and suggest that MMP-9 perturbs claudin-5 expression in a temporally distinct manner after stroke when there is coincident systemic inflammation. Inhibition of MMP-9 restored collagen-IV levels to baseline 4 h after MCAo confirming that the absence of an effect on claudin-5 at this time point was not attributable to lack of biological activity. MMP-9 has also been proposed to disrupt other components of the endothelial tight-junction-cytoskeletal framework, including claudin-3, ZO-1, and occludin, and MMP-9 is associated with BBB breakdown in a variety of neuroinflammatory conditions, including ischemia-reperfusion, trauma, and infection (Rosenberg et al., 1998; Wang et al., 2000; Asahi et al., 2001; Alvarez and Teale, 2007). Although we do not exclude that other mediators could contribute to the sustained BBB disruption caused by systemic inflammation, the present data do support an important role for MMP-9.
The time-dependent effect of MMP-9 inhibition on BBB disruption is consistent with the temporal profile of enhanced neurovascular gelatinolytic activity associated with systemic inflammation. A significant increase in gelatinolytic activity in IL-1β-challenged mice was not observed until 8 h after MCAo and is explained by our data showing that infiltrating neutrophils are the primary source of MMP-9. We demonstrated previously that systemic inflammation exacerbates neutrophil infiltration after MCAo (McColl et al., 2007a) with similar kinetics to the increase in gelatinolytic activity observed here. These alterations precede the appearance of cortical damage in IL-1β-challenged mice (McColl et al., 2007a), further supporting a causative role for enhanced MMP-9 activity in the poorer outcome caused by systemic inflammation. Because resident brain cells and infiltrating inflammatory cells can both express MMP-9, our results indicate that systemic inflammation aggravates a peripheral inflammatory pathway rather than stimulating MMP-9 expression directly in the CNS. MMP-9 is stored as a zymogen in neutrophil granules and on degranulation extracellular cleavage of the prodomain exposes the zinc-binding catalytic site (Opdenakker et al., 2001). Detection of cleaved MMP-9 only after systemic inflammatory challenge suggests that systemic inflammation both elevates levels of MMP-9 and enhances the conversion to the cleaved, active form. This could result from sensitization of neutrophils to degranulation stimuli encountered at the neurovascular compartment of the ischemic brain resulting in enhanced release and activation. This suggests that peripheral inflammatory events may alter the responsiveness of invading immune cells to locally produced signals at the blood–brain interface. In support of this, neutrophil and total leukocyte counts and the extent of leukocyte-platelet adhesion and activation are elevated in ischemic stroke patients with preceding infection (Emsley et al., 2003; Zeller et al., 2005).
The acute (4 h) disruption of claudin-5 in the present study occurs independently of systemic inflammatory challenge and MMP-9 activity. We did not detect an induction of MMP-2 or MMP3 (stromelysin-1) (data not shown) both of which have been implicated in claudin-5 degradation under neuroinflammatory conditions (Gurney et al., 2006; Yang et al., 2007). In contrast, our results suggest that a protease-independent mechanism may operate during this phase because we observed an increase in phosphorylation of endothelial MLC 4 h after MCAo that was independent of peripheral inflammatory challenge. Further studies will investigate this in more detail. MLC phosphorylation in endothelial cells induces transient cytoskeletal reorganization, cell retraction, and conformational changes in tight junction proteins that can conceal immunoreactive epitopes and inhibition of MLC kinase prevents hypoxia-induced opening of the BBB in vitro (Wysolmerski and Lagunoff, 1990; Haorah et al., 2005; Kuhlmann et al., 2007). This cascade could explain the severe but transient loss of immunoreactive claudin-5 in vehicle-treated mice and the hyper-acute MMP-9-independent phase of disruption in IL-1β-challenged mice. An important effect of systemic inflammation is to limit resolution of this initial BBB disruption. The BBB is therefore a point of convergence for systemically and CNS-initiated inflammatory processes (Fig. 8).
Figure 8.

Temporal involvement of MMP-9 in BBB disruption underlying the exacerbation of ischemic brain damage by systemic inflammation. Our data indicate two phases of BBB disruption based on the involvement of MMP-9. The first phase is MMP-9-independent and also occurs independently of systemic inflammation suggesting a local, CNS-driven mechanism such as the cytokine/ROS-induced phosphorylation of endothelial myosin light chain. This phase of BBB disruption is reversible when there is no additional peripheral inflammatory stimulus. Systemic inflammation causes a secondary MMP-9-dependent phase of disruption that is systemically driven (likely by neutrophils) and compounds the initial disturbance and results in sustained and irreversible BBB dysfunction. The deleterious effects of systemic inflammation on ischemic brain damage highlight the important position of the BBB as a point of convergence for CNS-driven and systemically driven inflammatory processes. veh, Vehicle.
The present findings may have clinical implications beyond representing a mechanistic explanation for the poorer outcome in stroke patients presenting with preceding infection. We observed a higher incidence of HT in peripheral IL-1β-challenged mice that was prevented by inhibition of MMP-9. Although speculative, this suggests stroke patients presenting with an elevated systemic inflammatory profile (e.g., caused by preceding infection) may be at increased risk of MMP-9-mediated neurovascular proteolysis and HT, particularly when recombinant tissue plasminogen activator, an inducer/activator of MMP-9 (Sumii and Lo, 2002; Cuadrado et al., 2008), is administered for thrombolysis. Although there are currently no clinical data comparing rates of HT in recombinant tissue plasminogen activator-treated patients with and without infection, this may be an important interaction to explore. In addition to infection, stroke patients frequently present with comorbid diseases associated with chronic elevations in systemic inflammation, such as atherosclerosis and diabetes. It will be important to investigate the impact of such conditions on stroke outcome and to test the efficacy and safety of putative stroke therapies in the context of acute and/or chronic systemic inflammation in further preclinical studies.
In summary, our findings underline the critical involvement of peripheral inflammatory pathways in stroke pathophysiology and support the concept that priming of the innate immune system has a deleterious impact on stroke outcome.
Footnotes
This work was supported by the Medical Research Council, UK. We thank Drs. Sandra Campbell and Daniel Anthony (University of Oxford, Oxford, UK) for the SJC anti-neutrophil antibody, Drs. Robert Fernandez and Peter March for guidance with confocal microscopy, and Dr. Andy Vail for statistical advice.
References
- Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE. Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol. 2007;179:2576–2583. doi: 10.4049/jimmunol.179.4.2576. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Teale JM. Evidence for differential changes of junctional complex proteins in murine neurocysticercosis dependent upon CNS vasculature. Brain Res. 2007;1169:98–111. doi: 10.1016/j.brainres.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- Bonfil RD, Sabbota A, Nabha S, Bernardo MM, Dong Z, Meng H, Yamamoto H, Chinni SR, Lim IT, Chang M, Filetti LC, Mobashery S, Cher ML, Fridman R. Inhibition of human prostate cancer growth, osteolysis and angiogenesis in a bone metastasis model by a novel mechanism-based selective gelatinase inhibitor. Int J Cancer. 2006;118:2721–2726. doi: 10.1002/ijc.21645. [DOI] [PubMed] [Google Scholar]
- Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- Chamorro A, Urra X, Planas AM. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke. 2007;38:1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- Clayton TC, Thompson M, Meade TW. Recent respiratory infection and risk of cardiovascular disease: case-control study through a general practice database. Eur Heart J. 2008;29:96–103. doi: 10.1093/eurheartj/ehm516. [DOI] [PubMed] [Google Scholar]
- Cuadrado E, Ortega L, Hernandez-Guillamon M, Penalba A, Fernandez-Cadenas I, Rosell A, Montaner J. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008;84:207–214. doi: 10.1189/jlb.0907606. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. 2006;26:1966–1975. doi: 10.1161/01.ATV.0000232525.65682.a2. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Role of interleukin-1 in infectious diseases. Immunol Rev. 1992;127:119–146. doi: 10.1111/j.1600-065x.1992.tb01411.x. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol. 2008;7:341–353. doi: 10.1016/S1474-4422(08)70061-9. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Hallenbeck JM, del Zoppo GJ, Rothwell NJ, Tyrrell PJ, Hopkins SJ. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- Favrais G, Schwendimann L, Gressens P, Lelièvre V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol Dis. 2007;25:496–505. doi: 10.1016/j.nbd.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005;29:999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- Kuhlmann CR, Tamaki R, Gamerdinger M, Lessmann V, Behl C, Kempski OS, Luhmann HJ. Inhibition of the myosin light chain kinase prevents hypoxia-induced blood-brain barrier disruption. J Neurochem. 2007;102:501–507. doi: 10.1111/j.1471-4159.2007.04506.x. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007a;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Allan SM, Rothwell NJ. Systemic inflammation and stroke: aetiology, pathology and targets for therapy. Biochem Soc Trans. 2007b;35:1163–1165. doi: 10.1042/BST0351163. [DOI] [PubMed] [Google Scholar]
- Meairs S, Wahlgren N, Dirnagl U, Lindvall O, Rothwell P, Baron JC, Hossmann K, Engelhardt B, Ferro J, McCulloch J, Kaste M, Endres M, Koistinaho J, Planas A, Vivien D, Dijkhuizen R, Czlonkowska A, Hagen A, Evans A, De Libero G, et al. Stroke research priorities for the next decade–a representative view of the European scientific community. Cerebrovasc Dis. 2006;22:75–82. doi: 10.1159/000093098. [DOI] [PubMed] [Google Scholar]
- Moutsopoulos NM, Madianos PN. Low-grade inflammation in chronic infectious diseases: paradigm of periodontal infections. Ann N Y Acad Sci. 2006;1088:251–264. doi: 10.1196/annals.1366.032. [DOI] [PubMed] [Google Scholar]
- Müller-Ladner U, Pap T, Gay RE, Neidhart M, Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:102–110. doi: 10.1038/ncprheum0047. [DOI] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol. 2001;22:571–579. doi: 10.1016/s1471-4906(01)02023-3. [DOI] [PubMed] [Google Scholar]
- Palasik W, Fiszer U, Lechowicz W, Czartoryska B, Krzesiewicz M, Lugowska A. Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur Neurol. 2005;53:188–193. doi: 10.1159/000086355. [DOI] [PubMed] [Google Scholar]
- Parry-Jones AR, Liimatainen T, Kauppinen RA, Gröhn OH, Rothwell NJ. Interleukin-1 exacerbates focal cerebral ischemia and reduces ischemic brain temperature in the rat. Magn Reson Med. 2008;59:1239–1249. doi: 10.1002/mrm.21531. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Piontek J, Winkler L, Wolburg H, Müller SL, Zuleger N, Piehl C, Wiesner B, Krause G, Blasig IE. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 2008;22:146–158. doi: 10.1096/fj.07-8319com. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–2618. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Mouihate A, Pittman QJ. Peripheral inflammation exacerbates damage after global ischemia independently of temperature and acute brain inflammation. Stroke. 2007;38:1570–1577. doi: 10.1161/STROKEAHA.106.476507. [DOI] [PubMed] [Google Scholar]
- Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- Utagawa A, Truettner JS, Dietrich WD, Bramlett HM. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol. 2008;211:283–291. doi: 10.1016/j.expneurol.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9) Crit Rev Biochem Mol Biol. 2002;37:375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysolmerski RB, Lagunoff D. Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci U S A. 1990;87:16–20. doi: 10.1073/pnas.87.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Del Bigio MR. Immune pre-activation exacerbates hemorrhagic brain injury in immature mouse brain. J Neuroimmunol. 2005;165:75–82. doi: 10.1016/j.jneuroim.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Zeller JA, Lenz A, Eschenfelder CC, Zunker P, Deuschl G. Platelet-leukocyte interaction and platelet activation in acute stroke with and without preceding infection. Arterioscler Thromb Vasc Biol. 2005;25:1519–1523. doi: 10.1161/01.ATV.0000167524.69092.16. [DOI] [PubMed] [Google Scholar]
- Zhang JW, Gottschall PE. Zymographic measurement of gelatinase activity in brain tissue after detergent extraction and affinity-support purification. J Neurosci Methods. 1997;76:15–20. doi: 10.1016/s0165-0270(97)00065-4. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Tejima E, Lo EH. Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke. 2007;38:748–752. doi: 10.1161/01.STR.0000253500.32979.d1. [DOI] [PubMed] [Google Scholar]