

Abstract
Osmosensory neurons transduce osmotic signals into a neural spike code that commands behavioral and endocrine responses that mediate body fluid homeostasis. Although changes in osmoregulatory reflex gain are known to occur under physiological and pathological conditions, the basis for this modulation is unknown. Here, we show that angiotensin II amplifies osmosensory transduction by enhancing the proportional relationship between osmolality, receptor potential, and action potential firing in rat supraoptic nucleus neurons. This effect is mediated by a phospholipase C- and protein kinase C-dependent increase in cellular mechanosensitivity that is associated with a rapid increase in cortical actin filament density. Preventing this increase with cytochalasin D eliminated the enhancement of mechanosensitivity, whereas enhancing actin filament density with jasplakinolide potentiated mechanosensitivity and occluded the effects of angiotensin II. These results indicate that a receptor-mediated increase in cortical actin density can enhance osmosensitivity in acutely isolated supraoptic neurons.
Keywords: actin, angiotensin II, cytoskeleton, osmotic pressure, osmosensitivity, supraoptic nucleus, vasopressin
Introduction
Osmosensory neurons play a fundamental role in the control of extracellular fluid (ECF) osmolality. In mammals, these neurons detect small changes in ECF osmolality and transduce them into proportional changes in action potential firing rate. This neural spike code is relayed to effector neurons that modulate the intake and excretion of salt and water in a manner that promotes osmotic homeostasis (Bourque et al., 2007; Bourque, 2008). Previous studies have shown that the osmotic control of osmoregulatory responses can be modulated under various physiological conditions. For example, the slope of the relationship between plasma osmolality and vasopressin (VP) (antidiuretic hormone) secretion is potentiated by hypovolemia or hypotension and is attenuated by hypervolemia or hypertension (Dunn et al., 1973; Robertson and Athar, 1976; Robertson et al., 1976). Although this modulation of osmoregulatory gain plays a key role in the coordination of cardiovascular and hydromineral homeostasis (Verbalis, 2003), the basis for this modulation is unknown.
Previous studies have shown that neurons in the subfornical organ project axons into the supraoptic nucleus (SON) (Renaud et al., 1983; Wilkin et al., 1989), where they excite the magnocellular neurosecretory cells (MNCs) that release vasopressin from the neurohypophysis (Sgro et al., 1984). In vivo, subfornical organ neurons are activated by hypovolemia (Ishibashi et al., 1985; Potts et al., 2000) and hypotension (Nicolaidis et al., 1983), suggesting that this neural pathway contributes to the potentiation of osmotically evoked vasopressin release under these conditions. Subfornical organ neurons projecting to the supraoptic nucleus contain angiotensin II (Ang II) (Nicolaidis et al., 1983), a peptide known to mediate the excitation of MNCs during electrical stimulation of the subfornical organ (Jhamandas et al., 1989). However, in addition to these direct excitatory effects, Ang II has been shown to enhance the osmotic activation of MNCs (Akaishi et al., 1980; Sladek et al., 1982), and mice lacking the Ang II type-1 (AT1) receptor show attenuated vasopressin release during hyperosmolality compared with wild-type mice (Morris et al., 1999). Thus, Ang II may amplify osmoregulatory gain by an effect in the supraoptic nucleus.
The excitation of MNCs under hypertonic conditions is attributable in part to their intrinsic osmosensitivity (Mason, 1980; Voisin and Bourque, 2002; Bourque et al., 2007). When exposed to an increase in osmolality MNCs generate a depolarizing receptor potential that promotes an increase in the rate of action potential discharge (Oliet and Bourque, 1993). This osmoreceptor potential is mediated by the mechanical activation of Gd3+-sensitive, stretch-inhibited, cation (SIC) channels during hyperosmotic shrinking (Oliet and Bourque, 1996). Interestingly, Ang II has been shown to increase the probability of opening of SIC channels and to enhance the excitatory response of isolated MNCs to hyperosmotic stimuli (Chakfe and Bourque, 2000). Although the basis for this effect is not known, a recent study has shown that the osmotic modulation of SIC channels varies in proportion with the density of actin filaments (F-actin) (Zhang et al., 2007). We therefore hypothesized that Ang II can amplify osmosensory transduction through a receptor-mediated increase in F-actin density.
Materials and Methods
Preparation of acutely isolated MNCs.
MNCs were acutely isolated from adult (80–120 g) male Long–Evans rats (Charles River) as described previously (Chakfe and Bourque, 2000; Zhang and Bourque, 2003; Zhang et al., 2007), using a protocol approved by the Animal Care Committee of McGill University and in accordance with the guidelines of the Canadian Council on Animal Care. Briefly, unanesthetized rats were decapitated, and brains were quickly removed from the cranial vault. Coronal slices (∼0.8–1 mm thick) were cut through the hypothalamus using a razor blade. Tissue blocks (∼1 mm3) containing part of the SON were dissected out using iridectomy scissors and were incubated for 90 min at 33.5°C in 10 ml of oxygenated (100% O2) piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES)-buffered saline (in mm: 120 NaCl, 3 KCl, 1 MgCl2, 20 PIPES, 1 CaCl2, 25 glucose, pH 7.0 and 295 mosmol · kg−1) containing 0.7 g/L trypsin. Tissue blocks were then washed in trypsin-free oxygenated PIPES solution, pH 7.4 (22°C) and kept in this solution until used (0.5–5 h). Single tissue blocks in ∼0.5 ml of PIPES solution were triturated with fire polished pipettes, and the resulting suspension was plated onto four to six Petri dishes (35 mm). Cells were given at least 10 min to adhere to the plastic before the experiments, which were all performed at room temperature (20–23°C). Except where specified, cells were perfused with a standard HEPES-buffered solution comprising the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 10 HEPES, 1 CaCl2, 10 glucose, pH 7.4. These chemicals were purchased from Sigma-Aldrich.
Solutions and chemicals.
Ang II, chelerythrine (Chel), cytochalasin D (Cyt-D), phorbol 12-myristate 13-acetate (PMA), and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid tetra-potassium (BAPTA) were purchased from Sigma-Aldrich. Jasplakinolide (JSK) and Texas Red-X phalloidin were purchased from Invitrogen. Ang II and Chel were dissolved in water and stored as aliquots at −20°C. Cyt-D, PMA, and JSK were dissolved in dimethylsulfoxide (DMSO), and stored as aliquots at −20°C. These aliquots were diluted in regular HEPES or PIPES solution immediately before the experiments to yield the final concentrations indicated in the results. Except where specified, DMSO concentration was ≤0.1%. In some experiments, 10 mm BAPTA was added in the internal solution for the purpose of chelating internal Ca2+. Texas Red-X phalloidin was dissolved in PBS immediately before the experiments.
Osmometry.
Perfusion solutions contained the following (in mm): 75 Na2SO4, 3 KCl, 1 MgCl2, 10 HEPES, 1 CaCl2, 10 glucose, and mannitol (added as needed to bring solutions to the desired osmolality). Cells were first perfused with control (isotonic; 295 mosmol · kg−1) solution, and then a step change in osmolality (−60, −30, +30, or +60 mosmol · kg−1) was applied for 5 min using a fast stepper device (SF-77B; Warner Instruments), which aligned one of the adjacent barrels of a glass capillary tube assembly that delivered various solutions by gravity at a rate of 1.0∼1.2 ml min−1. Excess solution was removed via a vacuum system. Each cell was tested twice with the same osmotic stimulus, once in control solution and once in solution containing 10−7 m Ang II. Ang II was applied beginning 60 s before and during the 5 min osmotic step. The order of the trials performed on individual cells (i.e., with and without Ang II) was reversed in consecutive recordings, and in each case a 5 min recovery period was interposed between the two trials. The time course of each volume response was compared.
Measurement of cell volume.
Relative changes in cell volume [normalized volume (nV)] were determined as described previously (Zhang and Bourque, 2003). Briefly, images of the cells were captured at their maximal cross sectional area (CSA) using a phase-contrast microscope (Diaphot; Nikon) and a monochrome video camera controlled by Axon Imaging Workbench software (AIW 2.2.0.38; Molecular Devices). For each image, the CSA (in pixels) was determined by tracing the perimeter of the cell using Scion Image for Windows 4.0.2 (Scion Corporation). All values of CSA measured during the control period were averaged (CSAo) and values of nV at time t (i.e., nVt) were calculated from the CSA value at that time point (i.e., CSAt) using the following equation: nVt = [(CSAt)3/2/(CSAo)3/2]. The time course of volume change was described by fitting changes in nV as a function of time with monoexponential equations using SigmaPlot 8.0 (SPSS). The time constants and steady-state values obtained by this procedure were used to quantify the responses. Steady-state values of nV were fitted to the Boyle-van't Hoff osmotic pressure equation: (nV = (1/π) [295(1 − b)] + b), where π is the osmolality of the test solution and b is the fraction of the cell content that is osmotically inactive.
Whole-cell patch-clamp recording.
Whole-cell voltage-clamp recordings were performed using an Axopatch-1D amplifier (Molecular Devices) using pipettes (1.2 mm outer diameter glass; A-M Systems; 4–5 MΩ) pulled on a P87 Flaming-Brown puller (Sutter Instruments) and filled with internal solution containing the following (in mm): 120 K-gluconate, 1 MgCl2, 10 HEPES, 1 EGTA, 4 Na2-ATP, 1 Na-GTP, 14 phosphocreatine, pH 7.2 and 265 mosmol · kg−1. We found that isolated MNCs swell slightly when patched with pipettes containing isotonic solutions. This effect, noticeable during time-lapse imaging, may be attributable to a small positive hydrostatic pressure retained near the tip of the pipette. We have determined empirically that swelling can be eliminated when cells are patched with internal solutions whose osmolality is 20–30 mosmol/kg lower than the external medium, presumably by promoting water efflux into the more concentrated solution. Membrane current (d.c.-2 kHz) was digitized (10 kHz) via a Digidata 1200B interface coupled to a personal computer running pClamp software (version 8.2.0.235; Molecular Devices). In the current-clamp mode, the membrane potential was adjusted to a value ∼5 mV below action potential threshold and left to stabilize for at least ∼3 min before testing. Under these baseline conditions, resting MNCs fired action potentials at an average rate of 0.4 ± 0.1 Hz (n = 29). If necessary, a small amount of sustained current (less than ±10 pA) was injected to adjust the baseline membrane potential of each cell to a value ∼5 mV below action potential threshold (i.e., −50 ± 5 mV) before each osmotic stimulus. Because the depolarizing effect of Ang II reaches a maximum after ∼30 s (Chakfe and Bourque, 2000), this procedure was performed 30–40 s after introducing the peptide into the bath (i.e., 20–30 s before the osmotic stimulus). Cells that were incapable of sustained firing in response to a 1 s depolarizing current pulse were not used in these experiments. Osmotic stimuli were delivered using the Fast-Step perfusion system (see above). The pressure inside the recording pipette was monitored by a digital pressure gauge (DPM-1B Pneumatic Transducer Tester; Bio-Tek Instruments) connected to the system and was changed as required via a 20 cc syringe.
F-actin staining and confocal microscopy.
Cells were fixed by replacing the incubation medium with 4% paraformaldehyde in 0.1 m PBS for 20 min. Cells were then washed with PBS containing 1% Triton X-100 three times for 15 min and covered with PBS containing Texas Red-X phalloidin (0.165 μm; Invitrogen) for 20 min in the dark at room temperature. Cells were subsequently washed with PBS (three times for 20 min each time) and stored at 4°C until imaging was performed. Fluorescence images were acquired by an investigator blinded to the cells treatment with an upright PerkinElmer microscope equipped with a confocal scanner (UltraVIEW LCI Model CSU10-3AX; PerkinElmer Life and Analytical Sciences). Phalloidin-stained cells were illuminated by a 488 nm line of an argon–krypton laser (Melles Griot 643R; Melles Griot Laser Group) and viewed through a 40×, 0.8 numerical aperture water-immersion objective. The fluorescence emission at 525 nm was detected using a Hamamatsu Orca ER cooled CCD camera capturing 16 bit images. MetaMorph software (Universal Imaging) was used to analyze digitized images captured at the plane of the maximal CSA. All image capture parameters (laser intensity, camera gain, and exposure time) were fixed throughout the experiments. For line scan analysis of the images, three virtual lines (20 pixels wide) were positioned perpendicular to the membrane 120° apart from one another. The three fluorescence intensity profiles obtained from each cell were aligned using Microsoft Excel 2003 at the point demarcating background from cell fluorescence, and the profiles of multiple cells were averaged. Values of total cortical fluorescence intensity were obtained by averaging the intensity of a ∼2-μm-thick region traced beneath the membrane along the entire perimeter of the cell using Scion Image.
Statistical analysis.
All values in this study are reported as mean ± SEM. Linear regressions through the data were performed using SigmaPlot 8.0. Comparisons between two means were performed using Student's paired or unpaired t test, as appropriate, and data obtained from multiple groups were compared using one-way ANOVA followed by Dunn's post hoc test (SigmaStat 2.03; SPSS). Slopes of linear regressions were compared using Prism 4.0 (GraphPad Software). Differences were considered significant when p < 0.05.
Results
Ang II potentiates osmoreceptor potentials and osmotic spike coding in MNCs
To determine whether Ang II can enhance osmosensory transduction, we obtained whole-cell current-clamp recordings from isolated MNCs and compared the effects of hyperosmotic stimuli (+10 and +30 mosmol · kg−1) delivered under control conditions and 60 s after adding 10−7 m Ang II to the perfusion medium. As illustrated in Figure 1A, the peptide significantly increased the depolarizing effects of the +10 mosmol · kg−1 (control, +2.0 ± 0.3 mV, n = 7; Ang II, +3.7 ± 0.6 mV, n = 11; p = 0.033) and +30 mosmol · kg−1 stimuli (control, +2.7 ± 0.7 mV, n = 5; Ang II, +5.6 ± 0.7 mV, n = 8; p = 0.016). The slope of the relation between membrane depolarization and ECF osmolality was significantly enhanced by Ang II (control, 0.037 mV/mosmol · kg−1; Ang II, 0.098 mV/mosmol · kg−1; p < 0.01) (Fig. 1B). This peptide-induced amplification of osmoreceptor potentials was accompanied by an enhancement of osmotic spike encoding. Indeed, Ang II significantly enhanced the excitatory effects of the +10 mosmol · kg−1 (control, +0.07 ± 0.07 Hz, n = 7; Ang II, +0.80 ± 0.29 Hz, n = 11; p = 0.040) and +30 mosmol · kg−1 stimuli (control, +0.28 ± 0.06 Hz, n = 5; Ang II, +1.88 ± 0.39 Hz, n = 8; p = 0.023). Linear regression analysis (Fig. 1C) confirmed that the slope of the relation between changes in firing rate and ECF osmolality was significantly enhanced by Ang II (control, +0.011 Hz/mosmol · kg−1; Ang II, +0.057 Hz/mosmol · kg−1; p < 0.05). Thus, Ang II amplifies osmosensory transduction in isolated MNCs.
Figure 1.

Ang II increases osmosensory gain in MNCs. A, Examples of voltage recordings obtained from isolated MNCs exposed to a +10 mosmol · kg−1 hyperosmotic stimulus (black bar) in the absence (top) or presence (bottom) of 10−7 m Ang II (gray bar). B, Graph plotting the mean (±SEM) osmoreceptor potentials (depolarization) induced by osmotic stimuli applied in the absence and presence of Ang II. The solid lines are linear regressions through data points. C, Graph plotting the mean (±SEM) changes in firing rate (Δfiring) induced by osmotic stimuli applied in the absence and presence of Ang II. The lines are linear regressions through data points.
Ang II does not affect osmometry
The process of osmosensory transduction involves the effect of ECF osmolality on cell volume (i.e., osmometry) (Zhang and Bourque, 2003) and the mechanical regulation of SIC channels during changes in cell volume (i.e., mechanotransduction) (Oliet and Bourque, 1993; Zhang et al., 2007). To determine whether Ang II affects osmometry, we compared the changes in cell volume evoked by osmotic stimuli (5 min; −60, −30, +30, +60 mosmol · kg−1) applied in the absence and presence of Ang II (10−7 m, introduced 60 s before the osmotic stimulus). As illustrated in Figure 2, A and B, equivalent results were obtained under control conditions and in the presence of Ang II. Indeed, the mean steady-state values of nV observed at each osmolality were not different when examined in the absence or presence of Ang II (p > 0.05) (Fig. 2C). Moreover, as shown in Figure 2D, the time course of volume changes induced by osmotic stimuli were not affected by Ang II (p > 0.05). Thus, Ang II does not enhance osmosensory transduction through an effect on osmometry.
Figure 2.

Ang II does not affect osmometry. A, Graph plotting the mean (±SEM) values of nV recorded during osmotic stimulation (bar; amplitude indicated by the numbers at right) of MNCs under control conditions. B, Graph plotting the values of nV recorded from the same cells in the presence of Ang II. C, Graph plotting the mean (±SEM) steady-state values of nV observed under different osmotic conditions in the absence (control) and presence of Ang II. The lines are fits of the data sets using the Boyle van't Hoff equation. D, Bar graph plotting the mean (±SEM) values of the time constants describing the time course of changes in nV during osmotic stimulation in the absence (open bars) and presence (solid bars) of Ang II. Differences are not significant (p > 0.05).
Ang II increases mechanosensitivity in MNCs
To determine whether Ang II enhances the mechanical coupling between changes in cell volume and changes in membrane conductance (Zhang et al., 2007), we quantified this process by calculating the mechanosensory index (MI) of the cells during increases in membrane conductance (ΔG) induced by cell shrinking evoked by application of negative pressure (approximately −50 mmHg for 60 s) to the recording pipette. This procedure induced uniform changes in cell volume that were similar to those induced by hyperosmolality. Cells were held at −60 mV under voltage clamp and stepped to −100 mV every 10 s (500 ms pulse duration). Membrane conductance at each time point (Gt) was calculated as Gt = ΔI/0.04, where ΔI is the size of the current response to the voltage step. Values of ΔG at each time point were calculated as ΔG = Gt − G0, where G0 is the average of the Gt values collected during the control period. Because ΔG increases as a linear function of volume decrease [i.e., as a function of normalized volume decrease (nΔV); where nΔV = 1 − nV] (Zhang et al., 2007), values of MI were computed as MI = ΔG/(nΔV). As illustrated in Figure 3A, application of negative pressure caused a progressive decrease in volume and a corresponding increase in ΔG. Because values of ΔG are near zero during the baseline period, MI was only computed once a significant increase in ΔG was induced (i.e., 20 s after stimulus onset). As illustrated in Figure 3B, values of MI did not significantly vary as a function of time (p = 0.878 for 35 cells tested). Thus, for each trial, a single value of MI was calculated by averaging the MI ratios computed at all time points. The mean value of MI measured under control conditions was 3.5 ± 0.8 nS/nΔV (n = 7). As illustrated in Figure 3C, MNCs treated with Ang II displayed suction-induced decreases in cell volume that were equivalent to those observed under control conditions (nΔV in control, 0.147 ± 0.015, n = 7; Ang II, 0.140 ± 0.016, n = 7; p = 0.765). However, a significantly larger increase in ΔG was induced by suction in the presence of Ang II (+1.0 ± 0.2 nS in Ang II, n = 7; vs +0.4 ± 0.1 nS in control, n = 7; p = 0.006), resulting in a noticeably higher value of MI under those conditions (Fig. 3B,D). Indeed, the mean value of MI measured in Ang II-treated cells was 8.5 ± 0.9 nS/nΔV (n = 7), a value significantly greater than control (p < 0.05).
Figure 3.

Ang II increases mechanosensitivity. A, Effects of applying negative pressure (−50 mmHg) to the recording pipette for 60 s (bar) on a cell recorded under control conditions. The top trace shows the change in nV, the middle trace shows the membrane current recorded under voltage clamp (Vhold, −60 mV). Negative deflections are the current responses (ΔI) induced by hyperpolarizing steps (−40 mV; 500 ms). The bottom trace shows values of ΔG computed from measurements of ΔI. B, Plot of the MI values computed from the data shown in the top traces. The dashed line represents the average of all MI values computed in this experiment. C, Effects of negative pressure (−50 mmHg) on another MNC recorded in the presence of Ang II (same layout as in A). Note the larger increases in ΔI and ΔG. D, Values of MI computed from the data shown in C.
Increased mechanosensitivity amplifies osmosensory transduction
To determine whether the potentiation of osmosensory transduction caused by Ang II is attributable specifically to the effect of this peptide on mechanotransduction, we quantified the osmosensitivity of MNCs by measuring their osmosensory index (OI) (computed as for MI) during volume changes induced by hypertonic stimuli delivered in the absence (Fig. 4A) (n = 6) and presence of peptide (Fig. 4B) (n = 7). Application of a hypertonic stimulus (+60 mosmol · kg−1; 5 min) provoked a gradual decrease in nV and increase in ΔG. Although mean steady-state values of nΔV were not different in the two groups (control, 0.142 ± 0.009; Ang II, 0.140 ± 0.009; p = 0.827), values of ΔG were significantly greater in Ang II-treated cells (control, +0.53 ± 0.16 nS; Ang II, +1.2 ± 0.2 nS; p = 0.033). As for MI (Fig. 3), mean values of OI did not vary significantly at different time points during the course of the stimulus (p = 0.745). Thus, for each trial a single value of OI was calculated by averaging OI scores observed at each time point. As shown in Figure 4B, the mean value of OI in Ang II (8.44 ± 0.87 nS/nΔV) was significantly greater than that observed in control MNCs (3.27 ± 0.78 nS/nΔV; p = 0.001).
Figure 4.

Ang II potentiates osmosensory transduction by enhancing mechanosensitivity. A, Mean (±SEM) values of ΔG (top), nV (middle), and OI (bottom) observed in a group of control MNCs exposed to a +60 mosmol · kg−1 hypertonic stimulus (gray bar). B, Effect of the same hypertonic stimulus on a group of MNCs exposed to Ang II (same layout as in A). C, Bar graph plotting the mean (±SEM) values of MI and OI measured in MNCs under control conditions (open bars) and in the presence of Ang II (filled bars). *p < 0.05. n.s. indicates that the values are not significantly different. D, Plots superimposing all of the mean (±SEM) values of ΔG and corresponding nΔV observed in cells stimulated using negative pressure (circles) or osmotic pressure (squares) in the absence (open symbols) or presence (filled symbols) of Ang II. The lines show regression fits through the four data sets.
In agreement with previous observations indicating that osmosensory transduction is mediated by the mechanosensitive modulation of SIC channels (Zhang et al., 2007), values of MI and OI observed under control conditions were not significantly different from each other (p = 0.884) (Fig. 4B). Moreover, Ang II enhanced mean values of OI and MI to an equivalent extent (p = 0.8) (Fig. 4C). Indeed, when mean values of ΔG evoked by osmotic or mechanical stimuli are plotted as a function of nΔV, we observe that the data sets recorded under control conditions superimpose each other and that Ang II increases the slope of these relations by equivalent amounts (Fig. 4C). Thus, Ang II amplifies osmosensory transduction by enhancing the mechanosensitivity of MNCs.
Ang II increases mechanosensitivity via the phospholipase C–protein kinase C pathway
Previous studies have shown that AT1 receptors mediate the postsynaptic effects of Ang II on MNCs (Yang et al., 1992; Hatae et al., 2001; Moellenhoff et al., 2001) and that such receptors mediate their effects through the activation of phospholipase C (PLC) via Gq/11 (de Gasparo et al., 2000). We therefore incubated MNCs for 20 min in the presence of 1-[6-[[(17β)-3-methoxyester-1,3,5(10)-trien-17-yl]amino]-hexyl]-1H-pyrole-2,5-dione (U-73122) (2 μm), a selective inhibitor of PLC (Bleasdale et al., 1989). As in control conditions, Ang II (10−7 m) applied to vehicle (DMSO)-treated MNCs significantly increased the MI (DMSO, 2.9 ± 0.9 nS/nΔV, n = 8; DMSO plus Ang II, 7.6 ± 1.4 nS/nΔV, n = 9; p = 0.021) (Fig. 5A). However, this effect was significantly inhibited by U-73122 (control, 1.62 ± 0.47 nS/nΔV, n = 7; Ang II, 2.1 ± 0.7 nS/nΔV, n = 8; p = 0.636) (Fig. 5A).
Figure 5.
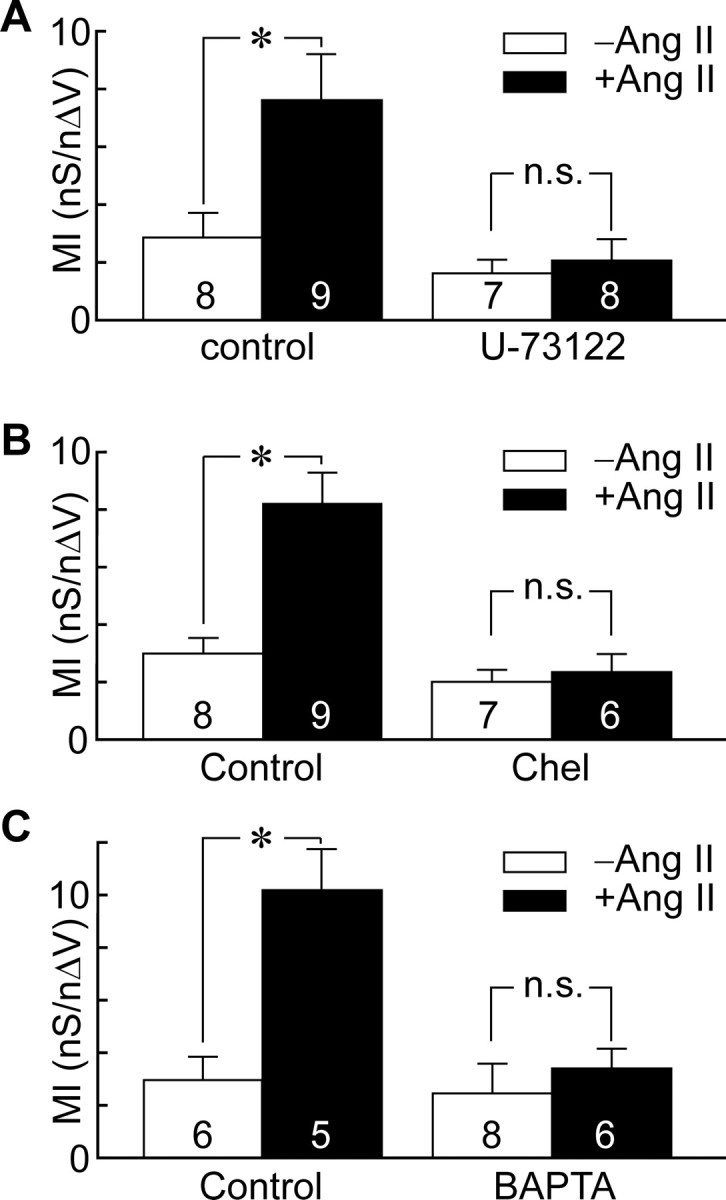
Ang II potentiates mechanosensitivity via the PLC-PKC pathway. Bar graphs show the mean (±SEM) values of MI recorded under various conditions. A, Values of MI in control (−Ang II; open bars) vs Ang II (+Ang II; filled bars) in vehicle (DMSO)-treated cells (Control) and in the presence of the PLC inhibitor U-73122. B, Values of MI observed in the absence (−Ang II; open bars) and presence (+Ang II; filled bars) of Ang II, in cells treated with vehicle (control) or Chel. C, Values of MI observed in the absence (−Ang II; open bars) and presence (+Ang II; filled bars) of Ang II, in cells patched with normal internal medium (Control) or medium containing 10 mm BAPTA. *p < 0.05. n.s., Difference not statistically significant.
Activation of PLC causes hydrolysis of PIP2 (phosphatidylinositol 4,5-bisphosphate) into inositol triphosphate and diacylglycerol. The production of diacylglycerol then leads to the activation of protein kinase C (PKC), a process which can also require release of Ca2+ from intracellular stores, depending on the PKC isoform involved (Tanaka and Nishizuka, 1994). To examine whether PKC mediates the effects of Ang II on mechanosensitivity, we first examined the effects of PMA, a potent activator of PKC (Tanaka and Nishizuka, 1994). Cells treated with 10−7 m PMA for 20 min displayed a significantly higher MI than MNCs treated with vehicle alone (8.3 ± 1.7 nS/nΔV in PMA, n = 7; vs 2.8 ± 0.9 nS/nΔV in DMSO, n = 12; p = 0.006). We next examined the effects of adding 5 μm Chel, a broad-spectrum inhibitor of PKC (Herbert et al., 1990), to the medium filling the patch pipette. After establishing the whole-cell configuration, ∼3 min was allowed for solution exchange before MI was measured. In time-matched controls, Ang II significantly enhanced the MI of MNCs (control, 3.0 ± 0.6 nS/nΔV, n = 8; vs 8.2 ± 1.1 nS/nΔV in Ang II, n = 9; p < 0.001). However, this effect was inhibited in cells loaded with Chel (2.0 ± 0.4 nS/nΔV in Chel, n = 7; vs 2.3 ± 0.6 nS/nΔV in Chel plus Ang II, n = 6; p = 0.653) (Fig. 5B).
Finally, to determine whether intracellular Ca2+ is required to mediate the effect of Ang II on mechanosensitivity, we examined the effect of including the Ca2+ chelator BAPTA (10 mm) in the recording pipette. In another group of control cells, Ang II significantly enhanced the mean MI of MNCs from 3.0 ± 0.9 nS/nΔV (n = 5) to 10.2 ± 1.6 nS/nΔV (n = 6; p = 0.004) (Fig. 5C). However, when BAPTA was included in the patch pipette, bath application of Ang II could no longer increase mean values of MI (2.5 ± 1.1 nS/nΔV in control, n = 8; vs 3.4 ± 0.8 nS/nΔV in Ang II, n = 6; p = 0.529) (Fig. 5C). Thus, Ang II increases the mechanosensitivity of MNCs via the PLC-mediated activation of a calcium-dependent form of PKC.
Ang II enhances cortical F-actin density in MNCs
Osmosensory transduction requires an intact actin cytoskeleton and transducer sensitivity appears to vary in proportion with the amount of F-actin present in the cell (Zhang et al., 2007). Ang II may therefore enhance mechanosensitivity via a PKC-dependent increase in F-actin density. As illustrated in Figure 6A, F-actin density was highest along the perimeter of MNCs, and this was visibly greater in Ang II-treated cells than controls. Quantitative line scan analysis indicated that F-actin density is greatest in the 2 μm region lying beneath the plasma membrane and that the significant increase in signal caused by Ang II is restricted to the submembrane cortex (Fig. 6B, left panel). A quantification of total cortical fluorescence confirmed that Ang II-treated cells (n = 91) show a significantly greater cortical F-actin density than controls (Ang II treated cells, 129 ± 5%, compared with controls; n = 97; p < 0.001). In contrast, Ang II failed to increase in cortical F-actin density in cells pretreated with Chel (20 μm; 15 min) (Fig. 6A,B, middle panels). The total cortical fluorescence in cells treated with Chel and Ang II was not significantly greater than in cells treated with Chel alone (Chel plus Ang II, 91.6 ± 2.7%, n = 96; vs 100.0 ± 4.2% for Chel alone, n = 103; p = 0.125). Finally, MNCs treated with 10−6 m PMA (20 min; n = 91) displayed significantly greater cortical F-actin density than vehicle (DMSO)-treated cells (128 ± 4% vs DMSO; n = 99; p < 0.001) (Fig. 6A,B, right panels). Thus, Ang II causes a PKC-dependent increase in cortical F-actin density in MNCs.
Figure 6.

Ang II induces PKC-dependent increase in cortical F-actin density. A, Confocal images showing F-actin intensity in MNCs stained with Texas Red-labeled phalloidin. The top panels show examples of cells incubated under control conditions for the treatments shown in the bottom panels. The bottom panels show examples of MNCs exposed to 1 μm Ang II for 5 min (left), Ang II and chelerythrine (Chel+Ang II; middle panel), or PMA (right). B, Line scan plots showing mean (±SEM) values of fluorescence (F-actin density; in bits) at the perimeter of cells exposed to the corresponding treatments shown in A. Note that significant differences are only observed in the ∼2 μm region that lies at the perimeter of the cells.
Increased F-actin density mediates Ang II potentiation of mechanotransduction
To examine whether the Ang II-induced increase in cortical F-actin density is necessary to mediate the effect of the peptide on mechanosensitivity, we quantified the effects of Ang II on values of MI in MNCs pretreated with Cyt-D (78 μm; 15–30 min), a drug that depolymerizes F-actin (Cooper, 1987). Ang II applied to vehicle (DMSO)-treated cells significantly increased MI (3.94 ± 0.64 nS/nΔV in controls, n = 19; vs 12.59 ± 1.92 nS/nΔV in Ang II, n = 18; p < 0.001) (Fig. 7A). However, cells treated with Cyt-D were significantly less mechanosensitive than untreated cells (MI of Cyt-D cells, 1.14 ± 0.99 nS/nΔV, n = 10; vs 3.94 ± 0.64 nS/nΔV in DMSO controls, n = 19; p = 0.021), and they failed to show a significant enhancement of their MI in response to Ang II (MI in Cyt-D plus Ang II, 1.34 ± 0.97 nS/nΔV; n = 8; p = 0.894) (Fig. 7A). Finally, if the increase in F-actin density provoked by Ang II is sufficient to mediate its effect on mechanosensitivity, then enhancing this parameter by other means should occlude the effects of the peptide. To test this hypothesis, MNCs were treated with JSK (2.5 μm; 20–30 min), a drug that promotes actin polymerization (Bubb et al., 1994). In a separate set of vehicle (DMSO)-treated controls, Ang II significantly increased MI (3.57 ± 0.88 nS/nΔV in controls, n = 6; vs 9.93 ± 2.26 nS/nΔV in Ang II, n = 6; p = 0.026). Cells treated with JSK alone were significantly more mechanosensitive than those treated with vehicle alone (MI of JSK cells, 15.89 ± 2.98 nS/nΔV, n = 9; vs 3.56 ± 0.88 nS/nΔV in DMSO controls, n = 6; p = 0.016), and they failed to show any additional enhancement of their MI in response to Ang II (MI in JSK plus Ang II, 14.67 ± 3.53 nS/nΔV; n = 8; p = 0.794) (Fig. 7B).
Figure 7.

An increase in F-actin density mediates the effects of Ang II on mechanosensitivity. Bar graphs plotting mean (±SEM) values of MI observed in the absence (−Ang II; open bars) and presence of Ang II (+Ang II; filled bars) under various conditions. A compares the effects of these treatments in the absence (control; left) or presence (right) of Cyt-D. B compares the effects of the treatments in the absence (left; control) or presence (right) of JSK. *p < 0.05. n.s., Difference not statistically significant.
Discussion
Plastic changes in osmosensory gain contribute to body fluid homeostasis
Changes in osmosensory gain play an important role in the central control of body fluid balance. For example, increases in thirst or VP release induced by ECF hyperosmolality are attenuated by concurrent hypervolemia and enhanced by concurrent hypovolemia (Dunn et al., 1973; Robertson and Athar, 1976; Robertson et al., 1976; Verbalis, 2003). These plastic changes in the osmotic modulation of behavioral and endocrine responses are important for fluid homeostasis. Indeed, the net changes in body water accretion caused by these adjustments mediate corrections in ECF volume that are beneficial under such complex physiological conditions. Although changes in osmosensory gain are known to play an important role in the neural control of body fluid balance under both physiological and pathological conditions (Verbalis, 2003), the mechanisms by which gain is regulated have remained unknown.
Ang II increases the intrinsic osmosensitivity of MNCs
A body of evidence indicates that Ang II is released into the SON during hypovolemia (Renaud et al., 1983; Sgro et al., 1984; Ishibashi et al., 1985; Jhamandas et al., 1989; Wilkin et al., 1989; Potts et al., 2000), where it can enhance the osmotic control of VP releasing MNCs (Akaishi et al., 1980; Sladek et al., 1982; Morris et al., 1999; Chakfe and Bourque, 2000). The osmotic control of electrical activity and VP release by MNCs in vivo is mediated by a variety of factors; including synaptic afferents originating from central and peripheral osmosensory neurons (Bourque et al., 2007), the osmotic control of taurine release from astrocytes (Hussy et al., 2000), and the intrinsic osmosensitivity of the MNCs (Voisin and Bourque, 2002). Changes in the sensitivity of the relationship between VP release and ECF osmolality, therefore, could be mediated by actions on any combination of these processes (Voisin and Bourque, 2002). Our results indicate that Ang II potentiates the relationship between ECF osmolality and spike coding by increasing the amplitude of the depolarizing osmoreceptor potentials generated in response to hyperosmotic stimuli (Fig. 1B). Although these results do not exclude the possibility that Ang II might modulate other factors responsible for the osmotic control of MNCs, they provide clear evidence that Ang II can enhance osmosensory gain by amplifying osmosensory transduction.
Ang II enhances osmosensory gain by amplifying mechanosensitivity
Osmosensory transduction occurs in two steps: osmometry (Zhang and Bourque, 2003) and mechanotransduction (Oliet and Bourque, 1993; Zhang et al., 2007). The modulation of this process could therefore be mediated by effects on either or both of these steps. Previous studies have shown that peptide hormones, including Ang II, can provoke changes in cell volume by affecting transmembrane ion fluxes in other types of cells (Lang et al., 1998; Fliegel and Karmazyn, 2004). It is therefore conceivable that a steepening of the slope of the relationship between cell volume and ECF osmolality could enhance osmosensory transduction in MNCs. However, our experiments indicated that Ang II does not affect osmometry in MNCs. Rather, it was found that Ang II increases the mechanosensitivity of MNCs and that this effect accounts quantitatively for the effect of the peptide on osmosensory transduction.
Ang II potentiates mechanosensitivity via a PKC-mediated increase in cortical actin density
Ang II was found to amplify mechanosensitivity through a signaling pathway involving PLC and a Ca2+-dependent form of PKC (i.e., either PKCα, PKCβ, or PKCγ) (Tanaka and Nishizuka, 1994). Furthermore, our results indicated that a PKC-dependent modulation of the actin cytoskeleton is a key step in this process. Indeed, Ang II induced a significant PKC-dependent increase in cortical F-actin density in MNCs (Fig. 6), and experiments with Cyt-D and JSK indicated that this increase in density is both necessary and sufficient to explain the effect of Ang II on mechanosensitivity in these cells (Fig. 7). How the increase in cortical actin density potentiates the mechanical activation of SIC channels remains to be determined. The basis for the regulation of SIC channels during changes in cell volume is not known, but could involve a mechanically induced actin-dependent biochemical step (e.g., modulation of enzyme activity) or a more direct actin-dependent mechanical regulation of the SIC channels (Zhang et al., 2007). Whichever is the case, our results indicate that an increase in cortical actin density somehow facilitates this process. From a mechanistic viewpoint it is likely that Ang II mediates this effect by increasing the amount of force that is transmitted to the transduction apparatus per unit change in cell volume. This hypothesis is supported by experiments showing that the stiffness of a cross-linked actin network increases with the concentration of F-actin (Gardel et al., 2004; Shin et al., 2004). Thus, if a network of actin filaments relays volume-dependent mechanical forces to the transducer element, then the amount of force applied per unit volume change could increase with F-actin density and thus enhance transducer sensitivity, as was observed in the presence of Ang II and JSK.
How does Ang II mediate an increase in cortical F actin density?
In principle, a receptor-mediated PKC-dependent increase in cortical F-actin density could be induced by the recruitment of F-actin into this subcellular compartment (Qian et al., 2002) or in response to an increase in the rate of local actin polymerization (Pilpel and Segal, 2004). However, the Ang II-mediated increase in cortical F-actin density observed in MNCs was not accompanied by a concurrent decrease in the amount of F-actin in the cytoplasm (Fig. 6B). The effects of the peptide, therefore, may involve enhanced polymerization rather than an increase in cross-linking. Indeed, studies in cultured hippocampal neurons have shown that the activation of PKC can increase F-actin density by stimulating actin polymerization via RhoA and Rac1 (Pilpel and Segal, 2004), members of the Rho family of small GTPases. Moreover, G-protein-coupled receptors can mediate the activation of Rho GTPases (Kjoller and Hall, 1999; Sah et al., 2000), and Ang II has been shown to mediate a Rho-dependent increase in F-actin density in a variety of cell types (Aoki et al., 1998; Kuwahara and Kuwahara, 2002). Interestingly, a recent study has shown that β-arrestin 1 and Gαq/11 are both required for the Ang II-dependent activation of RhoA and resultant induction of stress fiber formation in HEK293 cells transfected with the AT1 receptor (Barnes et al., 2005). Strikingly, the authors showed that maximal activation of RhoA could occur within 1 min, a time course compatible with the rapid effects of Ang II on mechanosensitivity observed in our experiments. Additional studies will be required to investigate the possible involvement of Rho family GTPases in mediating the effects of Ang II on MNCs.
Footnotes
This work was supported by Canadian Institutes of Health Research Operating Grant MOP-9939 and by a James McGill Research Chair (C.W.B.). Z.Z. was a recipient of Studentship Awards from the McGill University Health Center Research Institute and the Heart and Stroke Foundation of Canada. The Research Institute of the McGill University Health Centre receives support from Fonds de la Recherche en Santé du Québec.
References
- Akaishi T, Negoro H, Kobayasi S. Responses of paraventricular and supraoptic units to angiotensin II, sar1-ile8-angiotensin II and hypertonic NaCl administered into the cerebral ventricle. Brain Res. 1980;188:499–511. doi: 10.1016/0006-8993(80)90048-7. [DOI] [PubMed] [Google Scholar]
- Aoki H, Izumo S, Sadoshima J. Angiotensin II activates RhoA in cardiac myocytes: a critical role of RhoA in angiotensin II-induced premyofibril formation. Circ Res. 1998;82:666–676. doi: 10.1161/01.res.82.6.666. [DOI] [PubMed] [Google Scholar]
- Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 2005;280:8041–8050. doi: 10.1074/jbc.M412924200. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Bundy GL, Bunting S, Fitzpatrick FA, Huff RM, Sun FF, Pike JE. Inhibition of phospholipase C dependent processes by U-73, 122. Adv Prostaglandin Thromboxane Leukot Res. 1989;19:590–593. [PubMed] [Google Scholar]
- Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519–531. doi: 10.1038/nrn2400. [DOI] [PubMed] [Google Scholar]
- Bourque CW, Ciura S, Trudel E, Stachniak TJ, Sharif-Naeini R. Neurophysiological characterization of mammalian osmosensitive neurones. Exp Physiol. 2007;92:499–505. doi: 10.1113/expphysiol.2006.035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Chakfe Y, Bourque CW. Excitatory peptides and osmotic pressure modulate mechanosensitive cation channels in concert. Nat Neurosci. 2000;3:572–579. doi: 10.1038/75744. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- Dunn FL, Brennan TJ, Nelson AE, Robertson GL. The role of blood osmolality and volume in regulating vasopressin secretion in the rat. J Clin Invest. 1973;52:3212–3219. doi: 10.1172/JCI107521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegel L, Karmazyn M. The cardiac Na-H exchanger: a key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem Cell Biol. 2004;82:626–635. doi: 10.1139/o04-129. [DOI] [PubMed] [Google Scholar]
- Gardel ML, Shin JH, MacKintosh FC, Mahadevan L, Matsudaira P, Weitz DA. Elastic behavior of cross-linked and bundled actin networks. Science. 2004;304:1301–1305. doi: 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- Hatae T, Kawano H, Karpitskiy V, Krause JE, Masuko S. Arginine-vasopressin neurons in the rat hypothalamus produce neurokinin B and co-express the tachykinin NK-3 receptor and angiotensin II type 1 receptor. Arch Histol Cytol. 2001;64:37–44. doi: 10.1679/aohc.64.37. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Hussy N, Deleuze C, Desarménien MG, Moos FC. Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol. 2000;62:113–134. doi: 10.1016/s0301-0082(99)00071-4. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Oomura Y, Gueguen B, Nicolaidis S. Neuronal responses in subfornical organ and other regions to angiotensin II applied by various routes. Brain Res Bull. 1985;14:307–313. doi: 10.1016/0361-9230(85)90190-x. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Lind RW, Renaud LP. Angiotensin II may mediate excitatory neurotransmission from the subfornical organ to the hypothalamic supraoptic nucleus: an anatomical and electrophysiological study in the rat. Brain Res. 1989;487:52–61. doi: 10.1016/0006-8993(89)90939-6. [DOI] [PubMed] [Google Scholar]
- Kjoller L, Hall A. Signaling to Rho GTPases. Exp Cell Res. 1999;253:166–179. doi: 10.1006/excr.1999.4674. [DOI] [PubMed] [Google Scholar]
- Kuwahara M, Kuwahara M. Involvement of Rho and tyrosine kinase in angiotensin II-induced actin reorganization in mesothelial cells. Eur J Pharmacol. 2002;436:15–21. doi: 10.1016/s0014-2999(01)01591-6. [DOI] [PubMed] [Google Scholar]
- Lang F, Busch GL, Ritter M, Völkl H, Waldegger S, Gulbins E, Häussinger D. Functional significance of cell volume regulatory mechanisms. Physiol Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- Mason WT. Supraoptic neurones of rat hypothalamus are osmosensitive. Nature. 1980;287:154–157. doi: 10.1038/287154a0. [DOI] [PubMed] [Google Scholar]
- Moellenhoff E, Blume A, Culman J, Chatterjee B, Herdegen T, Lebrun CJ, Unger T. Effect of repetitive icv injections of ANG II on c-Fos and AT1-receptor expression in the rat brain. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1095–R1104. doi: 10.1152/ajpregu.2001.280.4.R1095. [DOI] [PubMed] [Google Scholar]
- Morris M, Li P, Callahan MF, Oliverio MI, Coffman TM, Bosch SM, Diz DI. Neuroendocrine effects of dehydration in mice lacking the angiotensin AT1a receptor. Hypertension. 1999;33:482–486. doi: 10.1161/01.hyp.33.1.482. [DOI] [PubMed] [Google Scholar]
- Nicolaidis S, Ishibashi S, Gueguen B, Thornton SN, de Beaurepaire R. Iontophoretic investigation of identified SFO angiotensin responsive neurons firing in relation to blood pressure changes. Brain Res Bull. 1983;10:357–363. doi: 10.1016/0361-9230(83)90104-1. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Bourque CW. Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature. 1993;364:341–343. doi: 10.1038/364341a0. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Bourque CW. Gadolinium uncouples mechanical detection and osmoreceptor potential in supraoptic neurons. Neuron. 1996;16:175–181. doi: 10.1016/s0896-6273(00)80034-3. [DOI] [PubMed] [Google Scholar]
- Pilpel Y, Segal M. Activation of PKC induces rapid morphological plasticity in dendrites of hippocampal neurons via Rac and Rho-dependent mechanisms. Eur J Neurosci. 2004;19:3151–3164. doi: 10.1111/j.0953-816X.2004.03380.x. [DOI] [PubMed] [Google Scholar]
- Potts PD, Ludbrook J, Gillman-Gaspari TA, Horiuchi J, Dampney RA. Activation of brain neurons following central hypervolaemia and hypovolaemia: contribution of baroreceptor and non-baroreceptor inputs. Neuroscience. 2000;95:499–511. doi: 10.1016/s0306-4522(99)00426-1. [DOI] [PubMed] [Google Scholar]
- Qian Y, Baisden JM, Cherezova L, Summy JM, Guappone-Koay A, Shi X, Mast T, Pustula J, Zot HG, Mazloum N, Lee MY, Flynn DC. PC phosphorylation increases the ability of AFAP-110 to cross-link actin filaments. Mol Biol Cell. 2002;13:2311–2322. doi: 10.1091/mbc.E01-12-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud LP, Rogers J, Sgro S. Terminal degeneration in supraoptic nucleus following subfornical organ lesions: ultrastructural observations in the rat. Brain Res. 1983;275:365–368. doi: 10.1016/0006-8993(83)90999-x. [DOI] [PubMed] [Google Scholar]
- Robertson GL, Athar S. The interaction of blood osmolality and blood volume in regulating plasma vasopressin in man. J Clin Endocrinol Metab. 1976;42:613–620. doi: 10.1210/jcem-42-4-613. [DOI] [PubMed] [Google Scholar]
- Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10:25–37. doi: 10.1038/ki.1976.76. [DOI] [PubMed] [Google Scholar]
- Sah VP, Seasholtz TM, Sagi SA, Brown JH. The role of Rho in G protein-coupled receptor signal transduction. Annu Rev Pharmacol Toxicol. 2000;40:459–489. doi: 10.1146/annurev.pharmtox.40.1.459. [DOI] [PubMed] [Google Scholar]
- Sgro S, Ferguson AV, Renaud LP. Subfornical organ–supraoptic nucleus connections: an electrophysiologic study in the rat. Brain Res. 1984;303:7–13. doi: 10.1016/0006-8993(84)90205-1. [DOI] [PubMed] [Google Scholar]
- Shin JH, Gardel ML, Mahadevan L, Matsudaira P, Weitz DA. Relating microstructure to rheology of a bundled and cross-linked F-actin network in vitro. Proc Natl Acad Sci U S A. 2004;101:9636–9641. doi: 10.1073/pnas.0308733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek CD, Blair ML, Ramsay DJ. Further studies on the role of angiotensin in the osmotic control of vasopressin release by the organ-cultured rat hypothalamo-neurohypophyseal system. Endocrinology. 1982;111:599–607. doi: 10.1210/endo-111-2-599. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17:471–503. doi: 10.1016/s1521-690x(03)00049-6. [DOI] [PubMed] [Google Scholar]
- Voisin DL, Bourque CW. Integration of sodium and osmosensory signals in vasopressin neurons. Trends Neurosci. 2002;25:199–205. doi: 10.1016/s0166-2236(02)02142-2. [DOI] [PubMed] [Google Scholar]
- Wilkin LD, Mitchell LD, Ganten D, Johnson AK. The supraoptic nucleus: afferents from areas involved in control of body fluid homeostasis. Neuroscience. 1989;28:573–584. doi: 10.1016/0306-4522(89)90006-7. [DOI] [PubMed] [Google Scholar]
- Yang CR, Phillips MI, Renaud LP. Angiotensin II receptor activation depolarizes rat supraoptic neurons in vitro. Am J Physiol. 1992;263:R1333–R1338. doi: 10.1152/ajpregu.1992.263.6.R1333. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Bourque CW. Osmometry in osmosensory neurons. Nat Neurosci. 2003;6:1021–1022. doi: 10.1038/nn1124. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Kindrat AN, Sharif-Naeini R, Bourque CW. Actin filaments mediate mechanical gating during osmosensory transduction in rat supraoptic nucleus neurons. J Neurosci. 2007;27:4008–4013. doi: 10.1523/JNEUROSCI.3278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]