

Abstract
Glial cell line-derived neurotrophic factor (GDNF)/Ret signaling has potent trophic effects on ventral midbrain dopaminergic, motor, sensory, and sympathetic neurons. The molecular mechanisms that restrict Ret receptor tyrosine kinase activation are not well understood. Here, we show that Lrig1, a transmembrane protein containing leucine-rich repeats and Ig-like domains in its extracellular region, acts in a negative feedback loop to regulate the activity of Ret receptor tyrosine kinase. In particular, we demonstrate that Lrig1 is capable of physically interacting with Ret and that Lrig1/Ret association inhibits GDNF binding, recruitment of Ret to lipid rafts, receptor autophosphorylation, and mitogen-activated protein kinase (MAPK) activation in response to GDNF. In neuronal cells, Lrig1 overexpression also inhibits GDNF/Ret-induced neurite outgrowth in a cell-autonomous manner. Downregulation of Lrig1 using small interference RNA knock-down experiments potentiates both neuronal differentiation and MAPK activation in response to GDNF. Together, these results provide an insight into Lrig1 function and establish a new physiological mechanism to restrict signaling and biological responses induced by GDNF and Ret in neuronal cells.
Keywords: GDNF, Ret, signaling mechanisms, Lrig1, RTK inhibitors, lipid rafts
Introduction
The development of the nervous system is tightly regulated by neurotrophic factors. The activation of receptor tyrosine kinases (RTKs) by their cognate neurotrophic factors controls a variety of cellular processes such as cell proliferation, migration, differentiation, axonal and dendritic growth, and survival (Ullrich and Schlessinger, 1990). Neurotrophins and glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) play a critical role in orchestrating development and maintenance of different populations of central and peripheral neurons. The best characterized member of the neurotrophins is nerve growth factor (NGF), which supports the survival of specific populations of sensory, sympathetic, and CNS neurons via the activation of its receptor tyrosine kinase TrkA (Huang and Reichardt, 2001, 2003).
GDNF family ligands are a relatively new family of neurotrophic factors that is composed of GDNF, neurturin, artemin, and persephin (Airaksinen and Saarma, 2002). GDNF was originally discovered by its ability to promote the survival of ventral midbrain dopaminergic neurons (Lin et al., 1993). In addition, GFLs promote the survival and control the differentiation of motor neurons (Henderson et al., 1994; Li et al., 1995; Oppenheim et al., 1995) and many peripheral neurons, including sympathetic and sensory neurons (Airaksinen et al., 1999). GFLs promote these trophic effects via the activation of the RTK Ret. A distinctive feature of the receptor complex for GDNF family ligands is the requirement of two types of receptor subunits, one specialized in ligand binding, represented by the glycosyl-phosphatidyl inositol (GPI)-anchored coreceptor GFRα (Jing et al., 1996; Treanor et al., 1996), and another involved in transmembrane signaling, represented by the receptor tyrosine kinase Ret (Durbec et al., 1996; Trupp et al., 1996). Both receptors are necessary for GDNF signaling, because GFLs are only able to bind and activate Ret when they are together with GFRα, forming a heterocomplex.
Because overactivation of RTK signaling has been implicated in the onset and progression of different human disorders and cancer, it is essential to understand how RTKs are downregulated and deactivated. To avoid signaling errors that ultimately lead to abnormal cellular behavior and disease, cellular mechanisms have evolved to ensure that appropriate signaling thresholds are achieved and maintained during the right period of time (negative signaling). Unlike positive signaling effectors, which are relatively well understood, signaling attenuation is currently under intensive study. During the last years, biochemical and genetic analyses of developmental processes have led to the identification and characterization of the mechanism of action of several RTK signaling inhibitors (Rubin et al., 2005). These studies have revealed the importance of negative feedback control of RTK function as a mechanism to ensure signaling thresholds compatible with the induction of a physiological response.
Leucine-rich repeats (LRRs) are protein–protein interaction domains found in proteins with diverse structure and function (Chen et al., 2006). In particular the leucine-rich repeat and Ig-like domain protein, Lrig1, appears as a member of an emerging family of nervous system enriched proteins containing 15 LRRs and three Ig domains in its extracellular region (Suzuki et al., 1996; Nilsson et al., 2001). Recent studies indicate that Lrig1 acts as a negative regulator of ErbB family and Met receptor tyrosine kinases by enhancing receptor ubiquitination and degradation (Suzuki et al., 2002; Gur et al., 2004; Laederich et al., 2004; Shattuck et al., 2007). These observations prompted us to investigate whether Lrig1 might act as an inhibitor of neurotrophic factor receptor tyrosine kinase signaling.
Here we show a physical interaction between Ret and Lrig1, which inhibits GDNF binding, recruitment of Ret to lipid rafts, receptor autophosphorylation, and MAPK activation in response to GDNF. In summary, our results indicate that Lrig1 is part of a negative feedback loop that restricts both Ret signaling and biological response to GFLs.
Materials and Methods
Cell lines and recombinant proteins.
MN1 is an immortalized motor neuron cell line that is responsive to GDNF (Salazar-Grueso et al., 1991; Paratcha et al., 2001). MN1-Lrig1 cells (clones L2, L15, and L20) were generated by stable transfection of the motor neuron MN1 cells with human Flag-Lrig1 construct. Experimental analysis was performed at least in two clones overexpressing Flag-Lrig1. PC12 is a rat pheochromocytoma cell line that is responsive to NGF. GDNF and GFRα1-Fc were purchased from R&D Systems (Abingdon, UK). NGF was purchased from Promega (Madison, WI).
Cell transfection, plasmids, and pharmacological treatments.
Transient transfections of COS cells were performed using the calcium phosphate method, and cells were harvested 48 h later. Primary superior cervical ganglion (SCG) neurons were transfected by electroporation. Briefly, dissociated neurons were suspended in 50 μl of Amaxa (Cologne, Germany) electroporation buffer with 3 μg of total plasmid DNA. Suspended cells were then transferred to electroporation cuvettes and electroporated with a square wave pulse generator (Harvard Apparatus, Holliston, MA) using three pulses of 100 V for 5 ms. MN1 cells were transfected using Fugene-6 (Roche, Basel, Switzerland) or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) reagents with full-length Flag-Lrig1. Plasmid cDNA encoding full-length Flag-tagged Lrig1 was kindly provided by Dr. Håkan Hedman (Umeå University, Umeå, Sweden). Plasmid cDNA encoding GFP was obtained from Clontech (Saint-Germain-en-Laye, France).
Pretreatment with epoxomicin (10 μm) and proteasome inhibitor I (PSI, 20 μm) was done for 30 min at 37°C before GDNF stimulation. The proteasome inhibitors were purchased from EMD Biosciences (San Diego, CA). Cells were treated with the protein inhibitor cycloheximide (15 μg/ml; Sigma, St. Louis, MO) in the absence or in the presence of GDNF (50 ng/ml) for 15 and 120 min.
Sympathetic neuron cultures.
SCG neurons, from embryonic day 21 (E21), Sprague Dawley rats (Scanbur, Karlslunde, Denmark) were prepared as previously described (Ledda et al., 2002). Briefly, the ganglia were dissociated with trypsin and collagenase, seeded onto poly-ornithine and laminin-coated plates, and maintained in DMEM:F12 medium containing 10% fetal bovine serum and AraC (10 μm) and supplemented with NGF (25 ng/ml) or GDNF (50 ng/ml) plus GFRα1 (150 ng/ml). To monitor the time course of Lrig1 induction after treatment with GDNF (100 ng/ml) or GDNF plus GFRα1-Fc (300 ng/ml), neurons were starved in serum-free medium without NGF for 12 h.
Real-time PCR.
The expression of Lrig1 and TATA box binding protein (Tbp) were analyzed. Total RNA was isolated from PC12 cells treated with NGF (50 ng/ml) or MN1 and rat SCG neurons stimulated with GDNF (100 ng/ml) or GDNF plus GFRα1-Fc (300 ng/ml) for different periods of time using RNA-easy columns (Qiagen, Valencia, CA). Single-stranded cDNA was synthesized using Multiscribe reverse transcriptase and random hexamers (Applied Biosystems, Foster City, CA). The cDNA was amplified using the following primer sets: Tbp: forward, 5′-GGG GAG CTG TGA TGT GAA GT-3′; reverse, 5′-CCA GGA AAT AAT TCT GGC TCA-3′. Mouse Lrig1: forward, 5′-TCT GCA GGA AGT GTA CCT CAA CAG-3′; reverse, 5′-GAG AGA CAA CTC CTA TGG AAG CAG T-3′. Rat Lrig1: forward, 5′-CTG AAG GGC CAG AGC ATT T-3′; reverse, 5′-TGA TGA TCT GTG GCT TTG GAA A-3′.
Real-time PCR was performed using a LightCycler rapid thermal cycler system (PerkinElmer, Wellesley, MA) according to the manufacturer's instructions. Reactions were performed in 25 μl volume. Nucleotides, TaqDNA polymerase, and buffer were included in the LightCycler-DNA master SYBR Green I mix (Applied Biosystems).
Preparation of lipid rafts, detergent-soluble fractions, and total cell lysates.
Cell monolayers were lysed for 60 min at 4°C in buffer containing 0.5% Triton X-100 and protease and phosphatase inhibitors. Samples were taken to 40% sucrose in an ultracentrifuge, and a discontinuous 5–25% sucrose gradient was layered on top, followed by overnight centrifugation at 175,000 × g. In some cases (see Fig. 3A), 1 ml fractions were collected from the top of the gradient for direct SDS-PAGE analysis. Rafts (fractions 2–4) were collected, washed in buffer, recentrifuged for 2 h, and resuspended in buffer with inhibitors. Protein was quantified and normalized among different raft preparations. Detergent-soluble fractions (fractions 9–12) were pooled for subsequent Ret immunoprecipitation. For total cell lysates, cells were lysed in buffer containing 1% Triton X-100 and 0.1% SDS plus inhibitors.
Figure 3.

Lrig1 restricts Ret receptor tyrosine kinase phosphorylation and MAPK activation. A, Ret phosphorylation in parental MN1 and MN1-Lrig1 cells (clon L20) treated with GDNF (50 ng/ml) as indicated. Total lysates were immunoprecipitated (IP) with anti-Ret antibodies followed by immunoblot (IB) with antibodies against phosphotyrosine (P-Tyr). Reprobing of the same blot with anti-Ret antibodies is shown. Bottom shows Flag-Lrig1 expression in total extracts. B, The histogram shows the quantification of Ret phosphorylation in three stable clones (L2, L15, and L20). Results are presented as average ± SD from three independent experiments. *p < 0.005 (Student's t test). C, MAPK activation (P-MAPK) in cell lysates of MN1 parental and MN1-Lrig1 cells (clon L20) treated with GDNF and detected by IB. Reprobing of the same blot with anti β-tubulin and anti-Flag antibodies is shown. D, The histogram shows the quantification of MAPK phosphorylation in three stable clones. Results are presented as average ± SD from three independent experiments. *p < 0.05 (Student's t test).
Immunoprecipitation and Western blotting.
Cells were lysed at 4°C in buffer containing 0.5% Triton X-100, 1% β-octylglucoside plus protease and phosphatase inhibitors. Protein lysates were clarified by centrifugation and analyzed by immunoprecipitation and Western blotting as previously described (Paratcha et al., 2001). All blots were scanned in a Storm 860 FluorImager (Molecular Dynamics, Sunnyvale, CA), and quantifications were done with ImageQuant software (Molecular Dynamics). Numbers below the lanes indicate fold of induction relative to control normalized to total levels of target protein.
The antibodies were obtained from various sources as follows: anti-phosphotyrosine (p-tyr) and anti-Ret were from Santa Cruz Biotechnology (Santa Cruz, CA); anti-P-MAPK (Thr-202/Tyr-204) was from New England Biolabs (Beverly, MA); anti-βIII-tubulin was from Promega; anti-HA was from Roche; anti-Flag antibodies were from Sigma; anti-Lrig1 was a gift from Satoshi Itami (University of Osaka, Osaka, Japan); anti-Lrig1 151 was from Agrisera (Vännäs, Sweden); anti-poly-ubiquitin (clone FK1) was from MBL (Nagoya, Japan); anti-Fyn was from EMD Biosciences; and anti-GFRα1 was from R&D Systems.
Pull-down, chemical cross-linking, and binding assays.
For pull-down assays, MN1 or COS cells, transfected with empty vector or Flag-Lrig1, were incubated with recombinant GFRα1-Fc (300 ng/ml) in the presence or absence of GDNF (100 ng/ml). After 3 h at 4°C, the cells were lysed in buffer containing 0.5% Triton X-100, 1% β-octylglucoside plus protease and phosphatase inhibitors. Samples were incubated with gentle rocking for 2 h at 4°C with protein G Sepharose beads. After washing, the beads were resuspended in loading buffer, boiled, and separated by SDS-PAGE. Bound protein was detected by Western blot using anti-Ret or anti-Flag antibodies.
125I-GDNF (GE Healthcare, Piscataway, NJ) was chemically cross-linked with ethyl-dimethyl-aminopropyl-carbodimide (EDAC) supplemented with sulfo-NHS (Pierce) as described previously (Paratcha et al., 2003). Briefly, affinity labeling to parental MN1, MN1-Lrig1, or COS cells transfected with Flag-Lrig1 construct was performed during 4 h incubation at 4°C in PBS supplemented with Ca2+ and BSA. This was followed by chemical cross-linking, EDAC neutralization with 500 mm glycine, and two washes in PBS. After washing with PBS, the cells were lysed in buffer containing 0.5% Triton X-100 and 1% β-octyl-d-glucopiranoside (Sigma) to ensure complete solubilization of membrane lipid rafts. Cell lysates were immunoprecipitated using anti-Ret or anti-Flag antibodies, separated by SDS-PAGE, blotted onto polyvinylidene difluoride membranes and exposed to phosphoscreens. The screens were developed using Storm 860 PhosphoImager and ImageQuant software (Molecular Dynamics).
Immunofluorescence and in situ hybridization.
Cryostat sections of E12.5 and newborn [postnatal day 0 (P0)] mice were blocked with 10% donkey serum and incubated with goat polyclonal anti-Ret extracellular domain (dilution, 1/200; R&D Systems) and affinity-purified rabbit polyclonal anti-Lrig1 antibodies (Lrig1–151 from Agrisera; dilution, 1/50). Secondary antibodies were from Jackson ImmunoResearch (Suffolk, UK).
Nonradioactive in situ hybridization on fixed cryostat sections (14 μm) of E12.5 mice was performed as previously described (Kele et al., 2006). Digoxigenin-labeled riboprobes (antisense or sense) were prepared by in vitro transcription using T3 and T7 polymerase from the 0.96 kb EcoRI-XhoI fragment of the mouse Lrig1 cDNA subcloned into pBluescript KS (Stratagene, La Jolla, CA). The hybridization was performed at 60°C, followed by incubation with nitroblue tetrazolium plus 5-bromo-4-chloro-3-indolyl phosphate substrates (Roche).
Photographs were obtained using a Zeiss (Thornwood, NY) Axioplan 2 microscope and the Open Lab software.
Neuronal survival and differentiation.
For MN1 and PC12 cell differentiation assays, the cells were plated on collagen- (Millipore, Billerica, MA) and poly-d-lysine- (Sigma) coated coverslips, respectively, and cultured in 1% serum-containing medium supplemented with GDNF (100 ng/ml) and soluble GFRα1-Fc (300 ng/ml) as previously described (Paratcha et al., 2001) or NGF (50 ng/ml). After 48 or 72 h, the cells were fixed with 4% paraformaldehyde (PFA) and stained with Alexa-conjugated phalloidin (Invitrogen). After staining, confocal microscopy was performed in a Zeiss confocal microscope with 25× objective lenses. The number of cells bearing neurites longer than 1.5 or 2 cell bodies, were quantified relative to the total number of neurons counted in 10 random fields of three different wells in each experiment. MN1 cell differentiation was evaluated in three independent experiments.
Neural differentiation assays were performed in dissociated cultures of E21 rat SCG neurons. Primary cultures were prepared as previously described (see above). Neurons were transfected with GFP or GFP plus Flag-Lrig1 by electroporation and cultured in the presence of GDNF (50 ng/ml) plus GFRα1 (150 ng/ml) for 36 h. Then, the cells were fixed with 4% PFA and stained with anti-Flag and anti-βIII tubulin to identify neuronal cells. A total of 62 control neurons and 85 Lrig1-transfected neurons were evaluated. Neuronal survival was evaluated using the nuclear stain 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen). GFP-positive neurons containing fragmented or condensed nuclear staining were scored as apoptotic cells and not computed in the differentiation assays. Quantification of neurite length was done with a Zeiss LSM510 confocal microscope using the Axiovision software (Zeiss) version 2.01.
siRNA transfection.
Lrig1 siRNA sequence (Dharmacon, Chicago, IL) was 5′-GAUCAUCACCCAGCCUGAG-3′ corresponding to nucleotides 1494–1512 in the mouse Lrig1 coding sequence. MN1 cells were transfected two times with Lipofectamine 2000 (Invitrogen) using a final concentration of 50 nm of the corresponding siRNA. For differentiation assays, the Lrig1 siRNA was transfected in combination with the GFP expression vector.
Results
Lrig1 interacts with the Ret receptor tyrosine kinase and is induced by GDNF signaling in neuronal cells
The molecular mechanisms that restrict Ret and TrkA RTK activation are not well understood. Previous studies showed that Lrig1 negatively regulates ErbB family and Met receptor tyrosine kinases by enhancing receptor ubiquitination and degradation (Gur et al., 2004; Laederich et al., 2004; Shattuck et al., 2007). To investigate whether Lrig1 might regulate the signaling properties of Ret and TrkA neurotrophic factor receptors, we first examined its ability to interact physically with both receptors. To investigate this possibility, we performed a coimmunoprecipitation assay in COS cells transfected with either Ret or hemagglutinin (HA)-tagged TrkA receptors in the absence or presence of Flag-tagged Lrig1. Figure 1A shows that Ret, but not TrkA, could be specifically coimmunoprecipitated with Flag-tagged Lrig1 construct. Using MN1 cell clones (MN1-Lrig1) generated by stable transfection with Flag-Lrig1, endogenous Ret could also be coimmunoprecipitated with Flag-tagged Lrig1 (Fig. 1E).
Figure 1.

Lrig1 interacts with the Ret receptor tyrosine kinase and is induced by GDNF signaling in neuronal cells. A, Coimmunoprecipitation between Flag-tagged Lrig1 (Flag-Lrig1) and Ret (left) or HA-TrkA (right) overexpressed in COS cells. Cell extracts were analyzed by immunoprecipitation (IP) with anti-Flag antibodies followed by immunoblot (IB) with antibodies against Ret or HA. Reprobing of the same blots with anti-Flag antibody is shown below. The bottom panels show Ret or HA-TrkA expression in total extracts. B, C, Quantitative analysis of Lrig1 mRNA expression by real-time PCR in MN1 cells (gray and black bars) (B) and SCG neurons (C), treated with GDNF (50 ng/ml) in the presence or absence of soluble GFRα1-Fc (150 ng/ml) during the indicated times. Histogram in B also shows Lrig1 mRNA expression in PC12 cells (white bars) treated with NGF (50 ng/ml). The levels of Lrig1 mRNA were normalized using the expression of the house-keeping gene Tbp. Shown are averages ± SD of triplicate determinations. *p < 0.01 versus control (Ctrl) group (1-way ANOVA followed by Dunnett's test). D, Western blot analysis of Lrig1 protein expression in MN1 cells treated with GDNF (100 ng/ml) or GDNF plus GFRα1-Fc (300 ng/ml). Reprobing control was done with antibodies against β-tubulin. Fold change relative to β-tubulin is indicated. E, Interaction between endogenous Ret and Flag-Lrig1 in MN1-Lrig1 cells in the absence of GDNF. Analysis was done by IP with antibodies against Flag epitope, followed by IB with anti-Ret antibodies. The bottom shows endogenous Ret expression in total MN1 cell extracts. F, Interaction between endogenous Ret and Lrig1 in parental MN1 cells treated with GDNF and GFRα1 (4 h). Analysis was done by IP with control or anti-Lrig1 antibodies, followed by IB with anti-Ret antibodies. Reprobing of the same blot with Lrig1 antibodies is shown below. The bottom shows endogenous Ret expression in total MN1 cell extracts.
Recent advances made in the understanding of the physiological mechanisms that restrict RTK signaling have revealed the importance of negative-feedback control of RTK function as a mechanism to ensure signaling thresholds compatible with the induction of a physiological response (Dikic and Giordano, 2003; Rubin et al., 2005). A common feature of these feedback loops is the ligand-dependent induction of RTK attenuators. Based on this concept, we have investigated whether Lrig1 is induced after GDNF stimulation of neuronal cells. To examine this possibility, we examined the mRNA expression of Lrig1 in MN1 cells and rat SCG neurons. Real-time PCR analysis revealed a significant induction (2- to 3.5-fold) of Lrig1 mRNA after stimulation of MN1 cells with GDNF (cis stimulation) or GDNF plus GFRα1-Fc (trans stimulation). As expected, GDNF in the presence of soluble GFRα1-Fc potentiated the upregulation of Lrig1 mRNA (Fig. 1B). Similarly, we observed an increase of Lrig1 mRNA in SCG neurons treated with GDNF and GDNF plus GFRα1-Fc (Fig. 1C). In contrast, we could not detect a substantial increase of the Lrig1 mRNA at different time points after NGF stimulation of PC12 cells, a cell line expressing both p75NTR and TrkA receptors (Fig. 1B). Lrig1 upregulation was additionally confirmed by immunoblotting of MN1 cell lysates treated with GDNF or GDNF plus GFRα1-Fc (Fig. 1D; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Association between endogenous Lrig1 and Ret receptor was also observed in lysates of MN1 cells treated with GDNF and soluble GFRα1-Fc for 4 h, revealing that the new encoded protein physically associates with Ret receptor (Fig. 1F).
To determine whether Lrig1 could play an in vivo role in Ret signaling, we examined the localization of Lrig1 in transverse sections of dorsal root ganglion (DRG) and spinal cord neurons by in situ hybridization and immunofluorescence using an anti-Lrig1 antibody raised to a nonconserved domain in other Lrig family members (Nilsson et al., 2001; Holmlund et al., 2004). At E12.5, Lrig1 expression was detected mainly in the ventricular and marginal zones of the dorsal half of the spinal cord and DRGs, revealing a protein distribution that mirrors the mRNA distribution (supplemental Fig. 2A–F, available at www.jneurosci.org as supplemental material). In particular, the early expression pattern of Lrig1 in the dorsal spinal cord suggests a role for Lrig1 in the control of Erb family receptor signaling and proliferation of embryonic spinal cord progenitor cells (Hayakawa-Yano et al., 2007). Detection of mouse Lrig1 by immunofluorescence and Western blot was additionally controlled by knock-down of endogenous Lrig1 expression in MN1 cells using siRNA (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Immunofluorescence staining of tissue sections of P0 mice revealed a striking colocalization of Lrig1 and Ret expression in DRG and spinal cord motor neurons (Fig. 2A–I). This data suggests that endogenous Lrig1/Ret interactions may play a physiological role in DRG and spinal cord motor neuron development.
Figure 2.

Colocalization of Lrig1 and Ret in DRG and spinal cord (SC) motor neurons. A, Colocalization of Lrig1 and Ret in transverse spinal cord and DRG sections from P0 mice by immunofluorescence. Yellow indicates regions of colocalization. Scale bar, 50 μm. D–I, The areas shown in D–F and G–I represent higher-magnification images of the boxes B and C, respectively. Scale bars: D–F, 30 μm; G–I, 25 μm.
Lrig1 restricts GDNF-mediated activation of Ret receptor tyrosine kinase and downstream signaling pathways
To evaluate the effect of Lrig1 interaction with Ret, we examined two biochemical events associated with Ret receptor activation, receptor autophosphorylation and stimulation of mitogen-activated protein kinases (MAPKs). In these experiments, parental MN1 cells and three clones overexpressing Lrig1 (MN1-Lrig1 cells, clones L2, L15 and L20) were serum-starved and treated with or without GDNF for 15 min. The level of Ret activation was evaluated by immunoprecipitation followed by immunoblotting with anti-phosphotyrosine antibodies. The MAPK activity was assessed by probing cell lysates with a specific antibody that recognizes the phosphorylated forms of these proteins. MN1 cell clones expressing Lrig1 showed a significant reduction in Ret tyrosine phosphorylation (Fig. 3A,B; supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) and MAPK activation (Fig. 3C,D; supplemental Fig. 3C, available at www.jneurosci.org as supplemental material) compared with parental cells. Immunoblots of Ret and GFRα1 in parental and MN1 clones overexpressing Lrig1 showed similar levels of Ret and GFRα1 expression (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), indicating that the differences observed in Ret and MAPK activation are not the consequence of a reduced expression of GDNF receptors. Altogether, these results indicate that Lrig1 interacts with Ret to inhibit receptor activation and GDNF signaling.
Lrig1 abrogates the presence and activation of Ret in lipid raft microdomains
Lipid rafts have emerged as membrane platforms specialized for signal transduction (Simons and Toomre, 2000; Paratcha and Ibanez, 2002). Previous work showed that in the absence of GDNF, Ret receptors are located outside raft compartments in nonordered membrane regions (Tansey et al., 2000; Paratcha et al., 2001). GDNF binding to lipid-anchored GFRα1 molecules induces the recruitment of Ret receptor to lipid raft compartments, a dynamic event required for effective GDNF signaling.
The ligand-independent interaction observed between Ret and Flag-tagged Lrig1 in MN1-Lrig1 cells (Fig. 1E) suggested that Lrig1 might be localized outside lipid rafts. To examine this possibility, we isolated raft and nonraft fractions from MN1-Lrig1 cells lysed with ice-cold Triton X-100 followed by centrifugation using density gradients. As expected, Lrig1 and Ret were found to be outside lipid raft compartment in basal conditions (Fig. 4A), suggesting a preferential association between Ret and Lrig1 in nonraft fractions. In agreement with previous data (Paratcha et al., 2001), the GPI-anchored GFRα1 coreceptor was exclusively detected in raft microdomains (fraction 2 of the gradient) (Fig. 4A).
Figure 4.
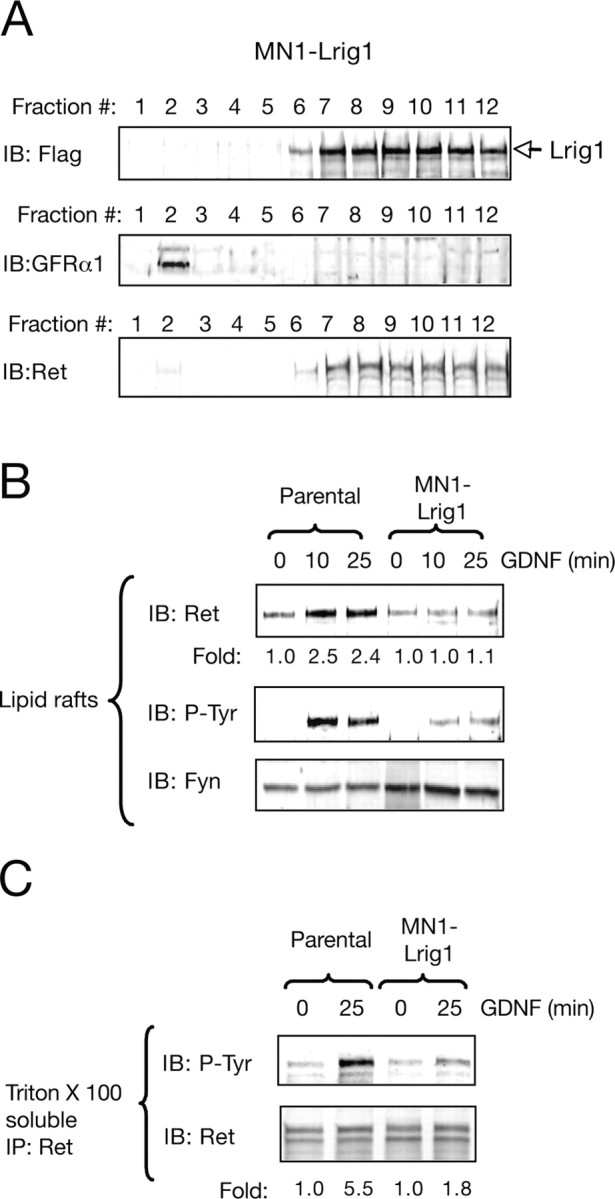
Lrig1 abrogates the presence and activation of Ret in lipid raft microdomains. A, Sucrose gradient fractions of Triton X-100 lysates prepared from MN1-Lrig1 cells analyzed by immunoblotting (IB) for Lrig1, GFRα1, and Ret. B, Raft fractions (2–4) from parental MN1 and MN1-Lrig1 cells (clon L20) treated with GDNF (50 ng/ml) as indicated and analyzed by IB with Ret and phosphotyrosine (P-Tyr) antibodies. Reprobing of the same blot with anti-Fyn antibodies is shown below. Fold change relative to control is indicated. The experiment was repeated three times with similar results. C, Detergent soluble fractions (9–12) from parental MN1 and MN1-Lrig1 cells (clon L20) treated with GDNF analyzed by IB with Ret and p-Tyr antibodies. Reprobing of the same blot with anti-Ret antibodies are indicated in the bottom. The experiment was repeated three times with similar results. Fold change normalized to the levels of Ret is indicated.
These observations prompted us to examine the role of Lrig1 in the GDNF-induced recruitment of Ret to lipid rafts. Although stimulation of parental MN1 cells with GDNF showed a robust recruitment and activation of Ret in lipid raft, the exogenous expression of Lrig1 resulted in a significant reduction of Ret tyrosine phosphorylation and localization in these microdomains (Fig. 4B; supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). These results indicate that the transmembrane protein Lrig1 might attenuate the recruitment and activation of the receptor inside the raft, sequestering it outside the raft compartment.
It has been postulated that once activated, Ret may be partitioned in a dynamic equilibrium between raft and nonraft compartments (Paratcha et al., 2001). To examine the activity of Ret outside raft domains in the presence of increased levels of Lrig1, the tyrosine phosphorylation of Ret in this compartment (Triton X-100 soluble fractions) was analyzed. A reduced activation of Ret outside raft was also observed in the MN1 cell clones expressing Lrig1 (Fig. 4C; supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). These observations are in agreement with an initial reduction in the translocation and activation of Ret receptor tyrosine kinase inside the raft compartment.
Lrig1 attenuates Ret receptor tyrosine kinase activation by reducing GDNF binding to Ret
Previous work has demonstrated that Lrig1 restricts ErbB and Met receptor signaling by enhancing receptor ubiquitination and degradation (Gur et al., 2004; Laederich et al., 2004; Shattuck et al., 2007). On the other hand, it is known that Ret receptor downregulation via ligand-induced ubiquitination and proteasomal degradation is a critical regulatory mechanism that restricts functional responses to GDNF (Scott et al., 2005; Pierchala et al., 2006). To examine whether Lrig1 inhibits Ret activation by promoting receptor ubiquitination and proteasomal degradation, a pharmacologic approach was taken. For this purpose, MN1-Lrig1 cells were treated with epoxomicin or PSI, two potent and highly specific proteasome inhibitors, before stimulation with GDNF. The phosphotyrosine levels of Ret were determined after 15 min of ligand stimulation and compared with cells that were not treated with these inhibitors. As shown in Figure 5A, proteasomal inhibition with epoxomicin did not rescue the Lrig1-mediated Ret receptor inactivation detected in MN1-Lrig1 cells that have not been exposed to epoxomicin, suggesting that the attenuation of Ret activation by Lrig1 is independent of proteasomal degradation. In agreement with this, ectopic expression of Lrig1 was not associated with a significant reduction in the level of Ret expression (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material) as has been described previously for other receptor tyrosine kinases regulated by Lrig1 (i.e., ErbB and Met). Furthermore, we examined whether Lrig1 could influence the ubiquitination and degradation of Ret in MN1 cells treated with GDNF. An increased ubiquitination of Ret was clearly associated with its activation level (Fig. 5B). Thus, ectopic expression of Lrig1 resulted in a reduced Ret activation and ubiquitination. Likewise, prolonged exposure of parental MN1 cells to GDNF in the presence of the protein synthesis inhibitor, cycloheximide (CHX), showed a high downregulation of Ret (Fig. 5C), in agreement with previous data (Scott et al., 2005; Pierchala et al., 2006). In contrast, in the presence of Lrig1, this effect was greatly attenuated and correlated with a lower degree of Ret activation (Fig. 5C). These results are in contrast with a role of Lrig1 in the promotion of Ret degradation by ubiquitination and suggest that in this case the level of ubiquitination is a consequence of the reduced activation of the receptor and not the cause. In conclusion, these data indicate that Ret inhibition by Lrig1 is not associated with receptor degradation.
Figure 5.

Lrig1 attenuates Ret receptor tyrosine kinase activation by reducing GDNF binding to Ret. A, Ret phosphorylation in parental MN1 and MN1-Lrig1 cells treated with the proteasome inhibitor epoxomicin (10 μm) and stimulated with GDNF (50 ng/ml) as indicated. Total lysates were analyzed by immunoprecipitation (IP) with anti-Ret antibodies followed by immunoblot (IB) with antibodies against phosphotyrosine (P-Tyr). Reprobing of the same blot with anti-Ret antibodies are indicated in the bottom. The experiment was repeated two times with similar results. B, Ret ubiquitination in parental MN1 and MN1-Lrig1 cells treated with the PSI (20 μm) and stimulated with GDNF (50 ng/ml) as indicated. Total lysates were analyzed by IP with anti-Ret antibodies followed by IB with antibodies against poly-ubiquitin (Poly-Ub) and phosphotyrosine (P-Tyr). Reprobing of the same blot with anti-Ret antibodies are indicated in the bottom. The experiment was repeated two times with similar results. C, Parental and MN1 clones overexpressing Lrig1 were treated with the protein synthesis inhibitor CHX (15 μg/ml) in the absence or in the presence of GDNF (50 ng/ml) for the indicated period of times. Ret proteins were analyzed by IP from cell lysates, and receptor levels were assessed by IB. Cell lysates probed with β-tubulin are shown below. Fold of change normalized to the levels of β-tubulin is indicated. D, Affinity labeling of parental and MN1-Lrig1 cells (left) and Flag-Lrig1 transfected COS cells (right) with 125I-GDNF, followed by chemical cross-linking and IP with the indicated antibodies. The membranes were reprobed (IB) with Ret or Flag antibodies. Expression of Flag-Lrig1 proteins is shown in total cell lysates. Similar results were obtained in two additional MN1-Lrig1 clones.
To further investigate the mechanistic details underlying Lrig1 abrogation of Ret activity, we analyzed the role of Lrig1 in GDNF binding to Ret. To this end, we performed affinity labeling experiments, followed by chemical cross-linking of 125I-GDNF to parental or MN1 cells overexpressing Lrig1. Immunoprecipitation of total cell lysates of affinity-labeled cells with antibodies against Ret resulted in a Lrig1-dependent decrease in the binding of radiolabeled GDNF to Ret (Fig. 5D, left). We also examined whether the inhibition of GDNF binding to Ret was attributable to ligand sequestration by direct GDNF interaction with Lrig1. For this purpose, COS cells transfected with the cDNA encoding Flag-Lrig1 were cross-linked with 125I-GDNF and immunoprecipitated with antibodies against Flag. This experiment showed that radiolabeled GDNF was unable to bind Lrig1 (Fig. 5D, right). Together, these results indicate that although Lrig1 restricts the ability of GDNF to interact with and activate Ret, Lrig1 does not physically associate with GDNF ligand.
Lrig1 inhibits neuronal differentiation of the motor neuron cell line MN1 and sympathetic neurons in response to GDNF and soluble GFRα1
A common feature of GPI-linked receptors is that they are found as both membrane-bound and soluble forms. In our previous work, we demonstrated that exogenous GFRα molecules can function in a non-cell-autonomous manner to present GDNF to Ret receptors in trans, a process that potentiates intracellular signaling and neurite outgrowth of developing sensory and sympathetic neurons (Paratcha et al., 2001; Ledda et al., 2002).
Based on this evidence, we investigated whether Lrig1 could restrict Ret tyrosine kinase activation by soluble GFRα1 supplied together with GDNF in trans. The data presented in Figure 6A indicate that Lrig1 was also able to inhibit Ret activation by GDNF supplemented together with exogenous GFRα1 in trans. In cells overexpressing Lrig1, cell surface pull-down of Ret with soluble GFRα1-Fc in the presence of GDNF resulted in a reduced precipitation of Ret receptors (Fig. 6B). This result indicates that Lrig1 transmembrane proteins can interfere with the association between Ret and GFRα1 induced by GDNF.
Figure 6.

Lrig1 inhibits neuronal differentiation of the immortalized motor neuron cell line MN1 in response to GDNF and GFRα1. A, Ret phosphorylation in parental MN1 and MN1-Lrig1 cells (clon L20) treated with GDNF (50 ng/ml) plus GFRα1-Fc (150 ng/ml) as indicated. Total lysates were immunoprecipitated (IP) with anti-Ret antibodies followed by immunoblot (IB) with antibodies against phosphotyrosine (P-Tyr). Reprobing of the same blot with anti-Ret antibodies are indicated in the bottom. The experiment was repeated two times with similar results. B, Pull down of endogenous Ret expressed in parental MN1 and MN1-Lrig1 cells with soluble GFRα1-Fc (150 ng/ml) added to living cells in the presence of GDNF (50 ng/ml). Cell lysates probed with β-tubulin are shown below. Fold change relative to β-tubulin is indicated. Similar results were obtained in two independent experiments. C, Pull down of Flag-Lrig1 expressed in COS cells with soluble GFRα1-Fc (150 ng/ml) added to living cells in the absence or presence of GDNF (50 ng/ml). Pull downs and cell lysates were probed by IB with antibodies against Flag. D, MN1 cell differentiation mediated by GDNF (100 ng/ml) and soluble GFRα1-Fc (300 ng/ml) is inhibited by Lrig1 overexpression. Photomicrographs show parental MN1 and MN1-Lrig1 (clon L20) cells stained with phalloidin, which reveals polymerized actin filaments. Scale bar, 25 μm. E, The histogram shows the quantification of the relative number of parental MN1 and MN1-Lrig1 (clon L20) cells bearing neurites longer than two cell body diameters after 72 h. of treatment with GDNF in the presence of soluble GFRα1-Fc. The results are presented as averages ± SD of a representative experiment performed in triplicate. *p < 0.01 (1-way ANOVA followed by Student–Newman–Keuls test). F, The histogram shows the quantification of the relative number of GFP-positive PC12 cells bearing neurites longer than 1.5 cell body diameter after 72 h of treatment with NGF (50 ng/ml). PC12 cells transfected with GFP in the absence (empty vector) or in the presence of Flag-tagged Lrig1 construct are indicated. The results are presented as averages ± SD of a representative experiment performed in triplicate. **p < 0.001 (1-way ANOVA followed by Student–Newman–Keuls test).
The inhibitory effect of Lrig1 on the ability of Ret to bind GFRα1-Fc in complex with GDNF suggested that Lrig1 might directly interact with the complex GFRα1/GDNF. To examine this possibility, we performed a pull-down assay in COS cells transfected with or without Lrig1. No Lrig1 could be recovered in the presence of GFRα1-Fc and GDNF, indicating that these molecules are not interacting (Fig. 6C). Together, these data suggest that Lrig1 interferes with the interaction of the heterocomplex GFRα1/GDNF with Ret.
Exogenous GFRα1 in the presence of GDNF can induce a robust neuronal differentiation of MN1 cells, characterized by increased cell spreading and development of long neurites (Fig. 6D) (Paratcha et al., 2001; Ledda et al., 2002). Interestingly, MN1 clones overexpressing Flag-Lrig1, as well as transiently transfected MN1 cells, failed to morphologically differentiate in response to GDNF and soluble GFRα1 (Fig. 6D,E; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In contrast, Lrig1 did not have an effect in PC12 neuronal differentiation induced by NGF (Fig. 6F).
We then examined the role of Lrig1 in the neurite outgrowth induced by GDNF and soluble GFRα1-Fc in SCG neurons. In agreement with the results obtained in MN1 cells, the neurite outgrowth stimulated by GDNF and GFRα1 was significantly reduced by Lrig1 overexpression, indicating that Lrig1 is able to restrict the neurite outgrowth triggered by GDNF (neurite length: control, 149.6 ± 34.3 μm; Lrig1, 87.8 ± 12.7 μm; n = 4; p < 0.05) (Fig. 7A,B, left histogram, C). Survival of GFP-positive neurons transfected with Flag-Lrig1 or control vector and treated with GDNF and GFRα1-Fc was also evaluated. No differences were observed in the percentage of neuron survival between control transfected and Lrig1-overexpressing SCG neurons (% neuron survival: control, 100 ± 2.7%, n = 3; Lrig1, 98 ± 2.1%, n = 3; p > 0.05) (Fig. 7B, right histogram). These results indicate that the partial inhibition of Ret by Lrig1 is enough to restrict neuronal differentiation but not survival.
Figure 7.
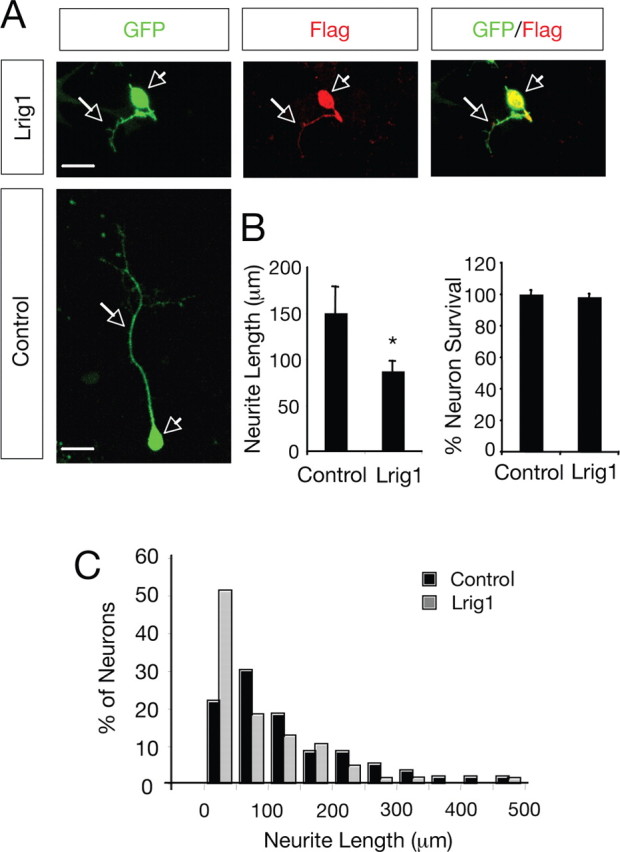
Lrig1 inhibits neurite outgrowth of SCG neurons in response to GDNF and GFRα1. A, Dissociated SCG neurons transfected with GFP in the absence (Control) or in the presence of Flag-tagged Lrig1 construct were cultured with GDNF and soluble GFRα1-Fc. After 36 h in culture, the neurons were fixed and stained with anti-Flag antibodies. Scale bar, 20 μm. Arrowheads indicates neuronal cell bodies and arrows denote neurites. B, Left, Histogram showing the inhibition of neurite outgrowth in SCG neurons by exogenous expression of Lrig1. The results are average ± SEM of four independent experiments. *p < 0.05 (Student's t test). Right, Histogram showing the neuronal survival in SCG neurons by exogenous expression of Lrig1. Neuronal survival was evaluated using the nuclear stain DAPI. GFP-positive neurons containing fragmented or condensed nuclear staining were scored as apoptotic cells. The results are average ± SD of a representative experiment performed in triplicate. C, The histogram shows the distribution of neurons carrying neurites in different length categories after transfection with GFP in the absence (Control) or in the presence of Flag-tagged Lrig1. Note the noticeable shift to the left of the distribution of neurons that received the Lrig1 construct.
Knock-down of Lrig1 by siRNA potentiate MAPK activation and neuronal differentiation of MN1 cells in response to GDNF
A siRNA duplex directed to the mouse Lrig1 mRNA sequence was used to inhibit endogenous Lrig1 mRNA levels in MN1 cells. After 4 h of GDNF treatment in the presence of soluble GFRα1, a significant reduction in the levels of the Lrig1 mRNA was detected by real-time PCR (Fig. 8A). Furthermore, a substantial reduction of endogenous Lrig1 protein was also detected by immunoblot (Fig. 8B). In agreement with a role for Lrig1 in the negative control of Ret activation, a significant potentiation of neuronal differentiation in MN1 cells was observed in cells transfected with Lrig1 siRNA and stimulated with GDNF plus GFRα1 for 48 h (Fig. 8C). Likewise, Lrig1 knock-down resulted in an increase of MAPK activity in response to GDNF (Fig. 8D).
Figure 8.

Knock-down of Lrig1 by siRNA potentiates MAPK activation and neuronal differentiation of MN1 cells in response to GDNF. A, Lrig1 mRNA levels were analyzed by real-time PCR in MN1 cells transfected with scrambled [control (Ctrl)] or Lrig1 siRNA followed by 4 h of treatment with GDNF and soluble GFRα1-Fc. Fold changes relative to control cells (nontreated cells, dotted line) are indicated. Quantitative analysis is shown as averages ± SD of triplicate determinations. The levels of Lrig1 mRNA were normalized using the expression of the house-keeping gene Tbp. *p < 0.005 (Student's t test). B, Endogenous levels of Lrig1 protein were analyzed by immunoblot (IB) in MN1 cells transfected with scrambled (Ctrl) or Lrig1 siRNA after treatment with GDNF and soluble GFRα1-Fc. Numbers below the lanes indicate fold changes relative to control cells (nontreated cells) normalized to the levels of β-tubulin. C, Morphological differentiation of MN1 cells induced by GDNF (50 ng/ml) and soluble GFRα1 (150 ng/ml) was assessed in cells transfected with scrambled (Ctrl) or Lrig1 siRNA together with a plasmid encoding GFP. The histogram shows the quantification of the relative number of GFP positive neurite-bearing cells longer than 1.5 cell diameters in the different conditions. The results are shown as average ± SEM of a representative experiment performed in quadruplicates. ***p < 0.001; **p < 0.01; *p < 0.05 (1-way ANOVA followed by Student–Newman–Keuls test). D, Lrig1 knock-down on MAPK activation was analyzed in MN1 cells treated with GDNF (25 ng/ml) for 15 min. Numbers below the lanes indicate fold changes in MAPK activation normalized to the levels of β-tubulin. The experiment was repeated three times with similar results.
Discussion
Unlike positive signals, which are relatively well understood, the molecular mediators of receptor signaling desensitization are currently under intensive study. Activated receptors initiate a complex chain of events, named negative receptor signaling, that restricts the strength and duration of positive signals and modulates the level of cellular activation. Recent studies have revealed that receptor tyrosine kinase activity is tightly regulated through the coordinated action of several protein inhibitors that function at multiple levels of the signaling cascade and at different time points after receptor engagement (Dikic and Giordano, 2003). Although neurotrophins and GFLs launch common intracellular signaling cascades, including the Ras/MAP kinase and phosphatidylinositol-3 kinase/Akt, differences in the mechanism of receptor downregulation between both TrkA and Ret receptors have been reported (Scott et al., 2005; Arevalo et al., 2006; Pierchala et al., 2006).
In this study, we establish a new mechanism to restrict signaling and biological responses induced by GDNF and its receptor tyrosine kinase, Ret, in neuronal cells. In particular, we show that Lrig1 acts in a negative feedback loop to regulate Ret, but not TrkA, activation. Our data suggest that whereas in the absence of Lrig1, GDNF binding to lipid-anchored GFRα1 molecules induces a rapid and robust recruitment of Ret to lipid raft compartment (Fig. 9A), the presence of high levels of Lrig1 inhibits the interaction between Ret and the GDNF/GFRα1 complexes, retaining Ret molecules outside the raft compartment (Fig. 9B). Thus, the physical interaction between Lrig1 and Ret results in the inhibition of Ret receptor tyrosine phosphorylation, MAPK activation, and neuronal differentiation in response to GDNF (Fig. 9B). Although our data indicate a role of Lrig1 in the control of Ret signaling, we cannot rule out that other family members (Lrig2 and Lrig3) may also play a prominent role as regulators of Ret activity.
Figure 9.
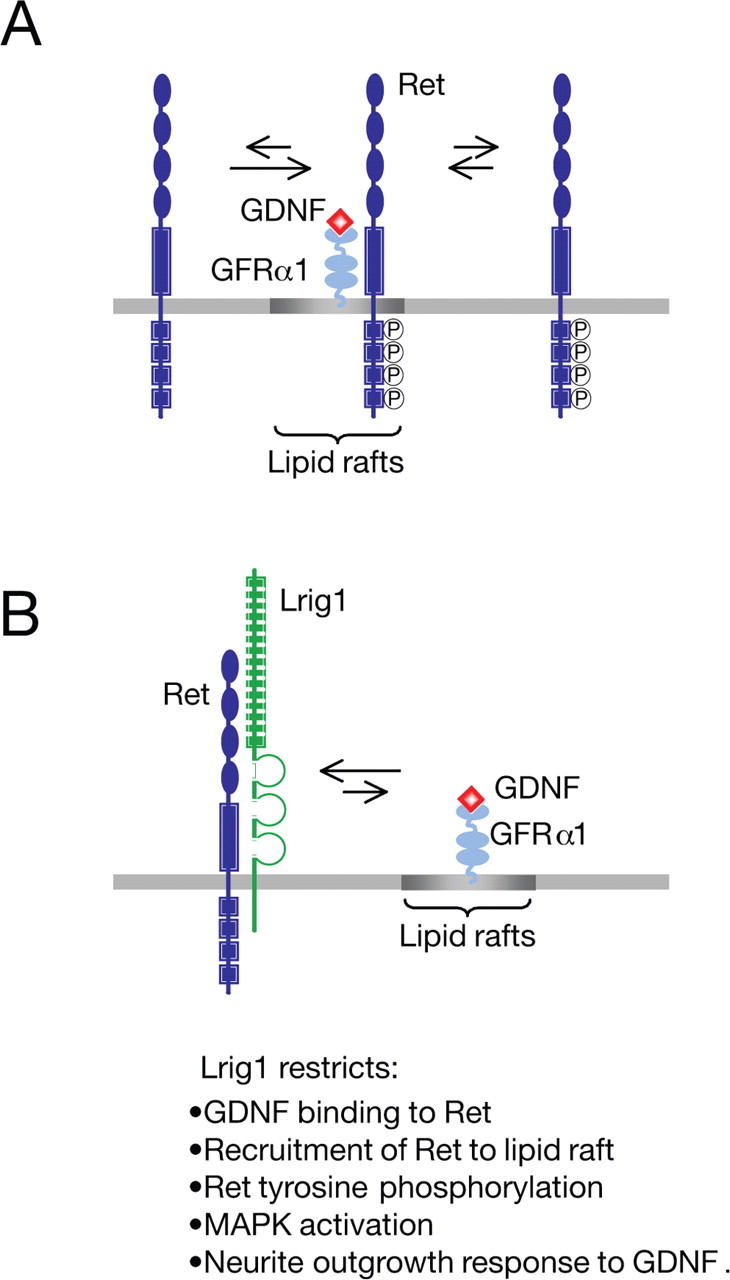
Model describing the proposed role of Lrig1 as a physiological inhibitor of Ret activation and recruitment to lipid rafts. A, During activation, GDNF binds to GPI-anchored GFRα1 coreceptors in lipid rafts, resulting in recruitment and activation of Ret in this compartment. Once activated, Ret is in equilibrium between raft and nonraft compartments. B, In the presence of Lrig1, a physical complex between Ret and Lrig1 is established. This interaction inhibits recruitment of Ret to lipid raft, ligand binding, receptor autophosphorylation, Ret-dependent downstream signaling, and neuronal differentiation in response to GDNF.
The elucidation of the mechanisms that control RTK activity is today seen as one of the major challenges in biomedical science. Even when the physiological relevance of Ret signaling inhibition by Lrig1 requires additional investigation, the data presented in this study suggest that strategies directed to knock-down Lrig1 expression in selective target neurons could enhance therapeutic activities of GDNF in the treatment of nerve injury and neurodegeneration.
Negative control of RTK signaling by Lrig1: different modes of action
RTKs coordinate a wide variety of biological processes and are therefore subjected to multiple mechanisms of control. With regard to the endpoint of the RTK signal transmission, negative regulators can be classified as either reversibles or irreversibles (Dikic and Giordano, 2003). The first ones include inhibitors that interfere with the intensity and extent of the signal in a defined period of time. Examples of reversible or transient inhibition include ligand sequestration and binding inhibition, attenuation of RTK phosphorylation, and inhibitory proteins that counteract downstream signaling pathways. In contrast, irreversible or definitive signaling inhibition includes molecules involved in RTK ubiquitination, endocytosis, and degradation (i.e., Cbl and Nedd families of ubiquitin ligases).
The identification and characterization of the mechanism of action of several RTK signaling inhibitors have established the importance of negative feedback loops as a mechanism to control and guarantee signaling thresholds compatible with the induction of a physiological response. Examples of this type of negative regulators include Sprouty (Impagnatiello et al., 2001; Sasaki et al., 2003; Mason et al., 2006), Mig6 (Pante et al., 2005; Ferby et al., 2006), and Lrig1 proteins (Gur et al., 2004; Laederich et al., 2004).
The structural similarity of Lrig1 with Kekkon1, an epidermal growth factor receptor (EGFR) inhibitor previously described in Drosophila, led to the prediction that Lrig1 could interact and attenuate EGF signaling in mammalian cells. Based on this evidence, it has been reported that Lrig1 can act as a negative feedback regulator of the mammalian ErbB receptors. Although both Lrig1 and Kekkon1 interact with the EGFR, their mechanisms of action differ substantially. The physical interaction of Kekkon1 with ErbB receptors interferes with ligand binding and receptor activation (Ghiglione et al., 1999, 2003). On the other hand, Lrig1 appears to restrict mammalian ErbB receptor signaling by enhancing Cbl-mediated receptor ubiquitination and degradation (Gur et al., 2004; Laederich et al., 2004). In addition, it has been recently demonstrated that Lrig1 could also interact with and destabilize the Met receptor in a Cbl-independent manner regardless of its activation status (Shattuck et al., 2007). Although the precise mechanism by which Lrig1 destabilizes Met receptor is still unknown, it has been proposed that Lrig1 likely acts to facilitate the association of Met with the protein degradation machinery. Based on the premise that the LRR domain is the critical interacting domain between Lrig1 and EGF/ErbB receptors, Goldoni et al. (2007) have recently demonstrated that a soluble ectodomain of Lrig1 containing only the LRR domain is sufficient to antagonize both ligand-independent and ligand-dependent EGFR activation in a non-cell-autonomous manner. In contrast to the full-length Lrig1 protein, the inhibition occurs without noticeable effects on internalization and degradation of the receptors, revealing that in mammals Lrig1 could also restricts EGFR activation by a Cbl-independent mechanism. Interestingly, the inhibition of Ret by Lrig1 described here is in concordance with both the interference of ligand binding and abrogation of EGFR activation reported for Kekkon1 in insect cells and with the negative control of EGFR function reported for the soluble ectodomain of Lrig1, which restricts receptor activation without changes in receptor stability.
It is known that the ability of cell adhesion molecules to influence developmental processes can result both from its adhesive and its signaling properties. On the basis of its structural features, it has been proposed that Lrig1 could influence neuronal development functioning as a cell type-specific adhesion molecule (Chen et al., 2006). In this regard, members of the Amigo family, a group of transmembrane proteins also containing LRRs and Ig domains, exhibit both homophilic and heterophilic binding activity that facilitate neuronal growth of hippocampal neurons (Kuja-Panula et al., 2003). Alternatively, Lrig1 might also function as a cell-surface receptor by itself, either signaling intracellularly through their cytoplasmic domain or by engagement with other transmembrane proteins in a signaling complex. In particular, the cytoplasmic portion of Lrig1 has neither a kinase domain nor binding sites for classical signaling partners of cell adhesion molecules, like Src kinases. However, the possibility that Lrig1 may associate with a yet unidentified kinase cannot be ignored.
Negative regulation of Ret signaling
The activity of the RTKs is tightly regulated, given that they play a crucial role orchestrating different developmental processes. A prevalent mechanism to control RTK activity is its downregulation by E3 ubiquitin ligase-mediated receptor degradation (Haglund et al., 2003). Monoubiquitination acts as an internalization signal, regulating endocytic trafficking and delivery of activated receptor to lysosomes to be degraded. On the other hand, polyubiquitination is a necessary event in the targeting of proteins for proteasomal degradation. In particular, ligand-dependent degradation of Ret is accomplished via ubiquitination of the receptor by E3 ligases such Cbl. Unlike EGFR, which recruits Cbl through direct interaction with the phosphorylated residues on the active receptor, Ret recruits Cbl by an indirect mechanism that involves the adaptor molecules Shc and Grb2 (Scott et al., 2005). Recently it has been reported that the proteasome is the predominant mechanism for the degradation of Ret (Pierchala et al., 2006), although the mechanisms behind transmembrane protein degradation through this pathway are still unclear. In the same study, the authors uncovered an additional role for lipid rafts in GDNF signaling describing that these membrane domains sequester phosphorylated Ret away from proteasome-dependent degradation, increasing the half-life of activated Ret and its downstream signaling. Another mechanism involved in the negative control of Ret signaling is the dephosphorylation of the activated receptor by protein tyrosine phosphatases. In this regard, recent studies have identified LAR (leukocyte common antigen-related), SHP1, and RPTPJ (receptor-type protein tyrosine phosphatase J) as phosphatases that physically interact with specific Ret oncoproteins, reducing its tyrosine phosphorylation, downstream signaling, and oncogenic activity (Hennige et al., 2001; Qiao et al., 2001; Iervolino et al., 2006).
Recent studies have demonstrated that the RTK antagonist, Sprouty, is a critical regulator of GDNF/Ret-mediated signaling and kidney development (Basson et al., 2005; Ishida et al., 2007). These studies indicate that Sprouty inhibits the responsiveness of the Wolffian duct to GDNF signaling ensuring the formation of a single ureteric bud during kidney development. Even when these genetic studies identified Sprouty as an antagonist of GDNF/Ret signaling during kidney formation, the mechanistic aspects of this inhibition as well as its relevance for neuronal development remain to be investigated.
In this work, we describe a novel and alternative mechanism to negatively regulate GDNF signaling. In particular, we demonstrate that Lrig1 physically associates with Ret and that Lrig1/Ret interaction inhibits GDNF binding, recruitment of Ret to raft domains, receptor autophosphorylation, and MAPK activation in response to GDNF.
GDNF is a known survival and differentiation factor for midbrain dopaminergic and spinal cord motor neurons, two neuronal subpopulation involved in Parkinson's disease and amyotrophic lateral sclerosis, respectively. Thus, understanding the mechanisms that control GDNF-induced Ret signaling will open new therapeutic opportunities for the treatment of these neurodegenerative disorders. Based on our results, it is logical to hypothesize that upregulation of Lrig1 in these two neuronal subpopulations may contribute to the pathogenesis of neurodegenerative diseases. Therefore, an outstanding question is to understand where this mechanism operates physiologically. In principle, the prominent and overlapping pattern of Lrig1 and Ret expression in newborn DRG and spinal cord motor neurons appears as primary locations where Ret inhibition by Lrig1 might be regulating important biological aspects of neuronal development. Our findings also provide an insight into the mode of action of Lrig1 protein and establish a new endogenous mechanism to restrict Ret activation and GDNF signaling in neuronal cells, with potential therapeutic relevance for nerve injury and neurodegeneration.
Despite the prominent expression of Lrig1 in the nervous system, morphohistological analysis of Lrig1-deficient mice revealed no apparent defects in neural tissue, suggesting that functional redundancy by other members of the Lrig family might be compensating its deficiency (Suzuki et al., 2002). Clearly, the analyses performed so far do not rule out defects in specific subpopulations of glial and neuronal cells. Therefore, a more detailed analysis of the nervous system of Lrig1 knock-out mice deserves further investigation. These additional studies will help to understand the physiological contribution of Lrig1 for nervous system function, development, and regeneration.
Footnotes
Financial support was obtained from the Swedish Medical Research Council (33SX-14773-01A and K2007-68X-20483-01-3), Karolinska Institute (2006FoBi0639), and the Royal Swedish Academy of Science. F.L. was supported by an assistant researcher position from the Swedish Medical Research Council (33P-15416-01A), and G.P. was supported by an assistant researcher position from the Swedish Medical Research Council (33PS-14809-01A) and by a tenure track position from Karolinska Institute. We thank Dr. Håkan Hedman for the Flag-tagged Lrig1 construct, Dr. Satoshi Itami for anti-Lrig1 antibodies, Eliana Sobarzo for technical assistance, Therese Sjöblom for secretarial help, and Jorge Aquino, Ernesto Restrepo, Line Lundfald, and Helena Mira for experimental assistance.
References
- Airaksinen MS, Titievsky A, Saarma M. GDNF family neurotrophic factor signalling: four masters, one servant? Mol Cell Neurosci. 1999;13:313–325. doi: 10.1006/mcne.1999.0754. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell. 2005;8:229–239. doi: 10.1016/j.devcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Chen Y, Aulia S, Li L, Tang BL. AMIGO and friends: an emerging family of brain-enriched, neuronal growth modulating, type I transmembrane proteins with leucine-rich repeats (LRR) and cell adhesion molecule motifs. Brain Res Rev. 2006;51:265–274. doi: 10.1016/j.brainresrev.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S. Negative receptor signalling. Curr Opin Cell Biol. 2003;15:128–135. doi: 10.1016/s0955-0674(03)00004-8. [DOI] [PubMed] [Google Scholar]
- Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, Sariola H, Pachnis V. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 1996;381:789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fassler R, Klein R. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12:568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- Ghiglione C, Carraway KL, III, Amundadottir LT, Boswell RE, Perrimon N, Duffy JB. The transmembrane molecule kekkon 1 acts in a feedback loop to negatively regulate the activity of the Drosophila EGF receptor during oogenesis. Cell. 1999;96:847–856. doi: 10.1016/s0092-8674(00)80594-2. [DOI] [PubMed] [Google Scholar]
- Ghiglione C, Amundadottir L, Andresdottir M, Bilder D, Diamonti JA, Noselli S, Perrimon N, Carraway IK. Mechanism of inhibition of the Drosophila and mammalian EGF receptors by the transmembrane protein Kekkon 1. Development. 2003;130:4483–4493. doi: 10.1242/dev.00617. [DOI] [PubMed] [Google Scholar]
- Goldoni S, Iozzo RA, Kay P, Campbell S, McQuillan A, Agnew C, Zhu JX, Keene DR, Reed CC, Iozzo RV. A soluble ectodomain of LRIG1 inhibits cancer cell growth by attenuating basal and ligand-dependent EGFR activity. Oncogene. 2007;26:368–381. doi: 10.1038/sj.onc.1209803. [DOI] [PubMed] [Google Scholar]
- Gur G, Rubin C, Katz M, Amit I, Citri A, Nilsson J, Amariglio N, Henriksson R, Rechavi G, Hedman H, Wides R, Yarden Y. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 2004;23:3270–3281. doi: 10.1038/sj.emboj.7600342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- Hayakawa-Yano Y, Nishida K, Fukami S, Gotoh Y, Hirano T, Nakagawa T, Shimazaki T, Okano H. Epidermal growth factor signaling mediated by Grb2 associated binder1 is required for the spatiotemporally regulated proliferation of Olig2-expressing progenitors in the embryonic spinal cord. Stem Cells. 2007;25:1410–1422. doi: 10.1634/stemcells.2006-0584. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- Hennige AM, Lammers R, Hoppner W, Arlt D, Strack V, Teichmann R, Machicao F, Ullrich A, Haring HU, Kellerer M. Inhibition of Ret oncogene activity by the protein tyrosine phosphatase SHP1. Endocrinology. 2001;142:4441–4447. doi: 10.1210/endo.142.10.8453. [DOI] [PubMed] [Google Scholar]
- Holmlund C, Nilsson J, Guo D, Starefeldt A, Golovleva I, Henriksson R, Hedman H. Characterization and tissue-specific expression of human Lrig2. Gene. 2004;332:35–43. doi: 10.1016/j.gene.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Iervolino A, Iuliano R, Trapasso F, Viglietto G, Melillo RM, Carlomagno F, Santoro M, Fusco A. The receptor-type protein tyrosine phosphatase J antagonizes the biochemical and biological effects of RET-derived oncoproteins. Cancer Res. 2006;66:6280–6287. doi: 10.1158/0008-5472.CAN-06-0228. [DOI] [PubMed] [Google Scholar]
- Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J Cell Biol. 2001;152:1087–1098. doi: 10.1083/jcb.152.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida M, Ichihara M, Mii S, Jijiwa M, Asai N, Enomoto A, Kato T, Majima A, Ping J, Murakumo Y, Takahashi M. Sprouty2 regulates growth and differentiation of human neuroblastoma cells through RET tyrosine kinase. Cancer Sci. 2007;98:815–821. doi: 10.1111/j.1349-7006.2007.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis JC, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- Kele J, Simplicio N, Ferri ALM, Mira H, Guillemot F, Arenas E, Ang S-L. Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development. 2006;133:495–505. doi: 10.1242/dev.02223. [DOI] [PubMed] [Google Scholar]
- Kuja-Panula J, Kiiltomaky M, Yamashiro T, Rouhiainen A, Raubala H. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J Cell Biol. 2003;160:963–973. doi: 10.1083/jcb.200209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laederich MB, Funes-Duran M, Yen L, Ingalla E, Wu X, Carraway KL, III, Sweeney C. The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J Biol Chem. 2004;279:47050–47056. doi: 10.1074/jbc.M409703200. [DOI] [PubMed] [Google Scholar]
- Ledda F, Paratcha G, Ibanez CF. Target-derived GFRalpha1 as an attractive guidance signal for developing sensory and sympathetic axons via activation of Cdk5. Neuron. 2002;36:387–401. doi: 10.1016/s0896-6273(02)01002-4. [DOI] [PubMed] [Google Scholar]
- Li L, Wu W, Lin LF, Lei M, Oppenheim RW, Houenou LJ. Rescue of adult mouse motoneurons from injury-induced cell death by glial cell line-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:9771–9775. doi: 10.1073/pnas.92.21.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Vallbo C, Guo D, Golovleva I, Hallberg B, Henriksson R, Hedman H. Cloning, characterization, and expression of human LIG1. Biochem Biophys Res Commun. 2001;284:1155–1161. doi: 10.1006/bbrc.2001.5092. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Johnson JE, Lin LF, Li L, Lo AC, Newsome AL, Prevette DM, Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- Pante G, Thompson J, Lamballe F, Iwata T, Ferby I, Barr FA, Davies AM, Maina F, Klein R. Mitogen-inducible gene 6 is an endogenous inhibitor of HGF/Met-induced cell migration and neurite growth. J Cell Biol. 2005;171:337–348. doi: 10.1083/jcb.200502013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha G, Ibanez CF. Lipid rafts and the control of neurotrophic factor signaling in the nervous system: variations on a theme. Curr Opin Neurobiol. 2002;12:542–549. doi: 10.1016/s0959-4388(02)00363-x. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Baars L, Coulpier M, Besset V, Anders J, Scott R, Ibanez CF. Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 2001;29:171–184. doi: 10.1016/s0896-6273(01)00188-x. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Pierchala BA, Milbrandt J, Johnson EM., Jr Glial cell line-derived neurotrophic factor-dependent recruitment of Ret into lipid rafts enhances signaling by partitioning Ret from proteasome-dependent degradation. J Neurosci. 2006;26:2777–2787. doi: 10.1523/JNEUROSCI.3420-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S, Iwashita T, Furukawa T, Yamamoto M, Sobue G, Takahashi M. Differential effects of leukocyte common antigen-related protein on biochemical and biological activities of RET-MEN2A and RET-MEN2B mutant proteins. J Biol Chem. 2001;276:9460–9467. doi: 10.1074/jbc.M008744200. [DOI] [PubMed] [Google Scholar]
- Rubin C, Gur G, Yarden Y. Negative regulation of receptor tyrosine kinases: unexpected links to c-Cbl and receptor ubiquitylation. Cell Res. 2005;15:66–71. doi: 10.1038/sj.cr.7290268. [DOI] [PubMed] [Google Scholar]
- Salazar-Grueso EF, Kim S, Kim H. Embryonic mouse spinal cord motor neuron hybrid cells. Neuroreport. 1991;2:505–508. doi: 10.1097/00001756-199109000-00002. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Taketomi T, Kato R, Saeki K, Nonami A, Sasaki M, Kuriyama M, Saito N, Shibuya M, Yoshimura A. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat Cell Biol. 2003;5:427–432. doi: 10.1038/ncb978. [DOI] [PubMed] [Google Scholar]
- Scott RP, Eketjall S, Aineskog H, Ibanez CF. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem. 2005;280:13442–13449. doi: 10.1074/jbc.M500507200. [DOI] [PubMed] [Google Scholar]
- Shattuck DL, Miller JK, Laederich M, Funes M, Petersen H, Carraway KL, III, Sweeney C. LRIG1 is a novel negative regulator of the Met receptor and opposes Met and Her2 synergy. Mol Cell Biol. 2007;27:1934–1946. doi: 10.1128/MCB.00757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sato N, Tohyama M, Wanaka A, Takagi T. cDNA cloning of a novel membrane glycoprotein that is expressed specifically in glial cells in the mouse brain. LIG-1, a protein with leucine-rich repeats and immunoglobulin-like domains. J Biol Chem. 1996;271:22522–22527. doi: 10.1074/jbc.271.37.22522. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Miura H, Tanemura A, Kobayashi K, Kondoh G, Sano S, Ozawa K, Inui S, Nakata A, Takagi T, Tohyama M, Yoshikawa K, Itami S. Targeted disruption of LIG-1 gene results in psoriasiform epidermal hyperplasia. FEBS Lett. 2002;521:67–71. doi: 10.1016/s0014-5793(02)02824-7. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Baloh RH, Milbrandt J, Johnson EM., Jr GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/s0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Treanor JJ, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumae U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]