

Abstract
Infection, ischemia, trauma, and neoplasia elicit a similar inflammatory response in the CNS characterized by activation of microglia, the resident CNS monocyte. The molecular events leading from CNS injury to the activation of innate immunity is not well understood. We show here that the intracellular chaperone heat shock protein 60 (HSP60) serves as a signal of CNS injury by activating microglia through a toll-like receptor 4 (TLR4)-dependent and myeloid differentiation factor 88 (MyD88)-dependent pathway. HSP60 is released from CNS cells undergoing necrotic or apoptotic cell death and specifically binds to microglia. HSP60-induced synthesis of neurotoxic nitric oxide by microglia is dependent on TLR4. HSP60 induces extensive axonal loss and neuronal death in CNS cultures from wild-type but not TLR4 or MyD88 loss-of-function mutant mice. This is the first evidence of an endogenous molecular pathway common to many forms of neuronal injury that bidirectionally links CNS inflammation with neurodegeneration.
Keywords: innate immunity, toll-like receptor, neurodegeneration, inflammation, heat shock protein, microglia
Introduction
Inflammation in the CNS is a double-edged sword (Wyss-Coray and Mucke, 2002). The predictable advantage of an inflammatory response in the CNS is protection from pathogens and neoplasms and clearing of nonviable cellular remains. An unanticipated drawback of inflammation is the possibility of accelerated neurodegeneration. Two special circumstances render the CNS uniquely vulnerable to injury after inflammation. First, neurons are particularly susceptible to cell death mediated by unstable oxygen and nitrogen species commonly generated during the respiratory burst of the innate immune response (Boje and Arora, 1992; Chao et al., 1992). Second, regeneration in the CNS mostly fails as the combined consequence of molecular inhibitors of axonal growth (Goldberg and Barres, 2000; Fournier and Strittmatter, 2002) and suboptimal repopulation by resident neuronal precursors (Björklund and Lindvall, 2000). The relevance of this relationship between neurodegeneration and inflammation is gradually emerging as models for neurodegenerative diseases, such as Alzheimer's disease and multiple sclerosis, suggest that neuronal cell death is not fully cell autonomous and that anti-inflammatory interventions may have clinical benefits (Perry et al., 2003).
Activation of microglia occurs in essentially all diseases of the CNS and was an early observation of histologists in the late 19th and early 20th centuries (Nissl, 1899; del Rio Hortega, 1932; Perry, 1994; Kreutzberg, 1996). Although inflammation is a near universal response to CNS injury, it is unclear how the inflammatory response is evoked after neuronal cell death and axonal degeneration. Several pieces of evidence led to our hypothesis that heat shock protein 60 (HSP60) may provide a signal to microglia, alerting the occurrence of CNS injury, through activation of the toll-like receptor 4 (TLR4)-myeloid differentiation factor 88 (MyD88) pathway. Stressed cells release proteins, including HSPs, which induce maturation and activation of innate immune cells (Chen et al., 1999; Basu et al., 2000; Somersan et al., 2001). The mammalian TLRs are conventionally considered pattern recognition receptors for conserved molecular motifs derived from pathogens (termed pathogen-associated molecular patterns) (Akira et al., 2006). However, although Toll serves an innate immune function in adult Drosophila, its critical patterning function during embryogenesis requires an endogenously produced ligand (Stein et al., 1991). Endogenous activators of mammalian TLRs have been identified, including HSPs, which represent a collection of evolutionarily conserved molecular chaperones, although debate exists regarding potential contamination of HSPs with endotoxins (Ohashi et al., 2000; Vabulas et al., 2001; Asea et al., 2002; Akira et al., 2006). We show here, by rigorously excluding the possibility of lipopolysaccharide (LPS)-related effects, that HSP60 is indeed an endogenous activator of TLR4 and as such may serve as a general danger signal for CNS injury. Our findings are consistent with the hypothesis that endogenous molecules, such as the HSPs, derived from injured or dying cells, serve as danger signals to the innate immune system (Matzinger, 2002).
Materials and Methods
Animals.
C.C3H-Tlr4Lps-d (lpsd), BALB/cJ, and C57Bl/6J mice and Sprague Dawley rats were purchased from The Jackson Laboratory (Bar Harbor, ME). C57Bl/6J MyD88−/−mice were generously provided by Dr. Shizuo Akira (Osaka University, Department of Host Defense, Osaka, Japan) and were obtained from Dr. Hidde L. Ploegh (The Whitehead Institute, Cambridge, MA). The C.C3H-Tlr4Lps-d (lpsd) mouse is the resulting strain derived by backcrossing the TLR4 mutation from the C3H/HeJ mouse with BALB/cJ mice over 20 generations. The lpsd phenotype originated from C3H/HeJ mice caused by a point mutation at codon 712 of the coding region for TLR4. Macrophages from this strain fail to induce inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and IL-6 (Poltorak et al., 1998; Hoshino et al., 1999; Qureshi et al., 1999). In this work, we use the term lpsd rather than C.C3H-Tlr4Lps-d.
Primary cultures of mixed CNS cells.
Mixed CNS cells were generated from forebrains of embryonic day 17 (E17) mice. After removal of meninges and vessels, cortices were triturated with trypsin for 20 min at 37°C. Cells were mechanically dissociated and suspended in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. Cells were then plated onto poly-d-lysine-coated glass slides (BD Biosciences, San Jose, CA) and maintained in humidified 10% CO2/90% air at 37°C.
Primary cultures of cortical neurons.
Primary cultures of enriched cortical neurons were generated from forebrains of E17 mice, as described previously (Murphy et al., 1990). Briefly, after removal of meninges and vessels, cortices were triturated and dissociated with papain in EBSS (Invitrogen) for 5 min at 37°C. After mechanical dissociation, cells were resuspended in 0.25% Trypsin inhibitor and 0.25% bovine serum albumin in EBSS and incubated at 37°C for 5 min. Cells were harvested by centrifugation at 1000 × g for 5 min. The cell concentration was adjusted to 1 × 106 cells/ml in MEM with GlutaMAX media (Invitrogen) supplemented with 10% FBS and penicillin/streptomycin. 0.5 ml of the cell-suspension was added into each well on poly-d-lysine-coated glass slides. Cultures were grown in humidified 5% CO2/95% air at 37°C. Immediately after plating, immunostaining revealed 90–95% purity for neurons.
Primary cultures of purified microglia, oligodendrocytes, and astrocytes.
Cell cultures of purified glial cells were generated from forebrains either of 2-d-old Sprague Dawley rats or of E17 mice as described previously (Vartanian et al., 1995).
HSP60 toxicity assay.
For HSP60 toxicity studies, 10 μg/ml recombinant human HSP60-Low Endotoxin (catalog number ESP-540; Stressgen, Victoria, Canada) was added to cell cultures for indicated times. The contamination with LPS of the HSP60 preparation as declared by the manufacturer was <50 EU/mg and ≤1.67 EU/mg, as determined by an independent laboratory specializing in endotoxin testing (Mikrobiologisches Labor, Münster, Germany). Control cultures were treated with PBS. For additional control experiments, HSP60 was used in combination with 10 μg/ml Polymyxin B sulfate (Sigma, Munich, Germany) or denaturated by heat (20 min at 100°C), trypsin, or proteinase K (Sigma). LPS (0111:B4) was purchased from List Biological Laboratories (Campbell, CA).
Induction of apoptosis and necrosis in CNS cells.
Apoptosis and necrosis of mixed CNS cultures were induced 24 h after plating the cells onto poly-d-lysine-coated glass sides. Apoptosis was induced by treating the cells with 2 ng/ml mouse TNF-α (Peprotech, Rocky Hill, NJ) and 35 μm cycloheximide (Sigma) for 16 h (Scaffidi et al., 2002). Necrosis was induced by treating the cells with 30 μm carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (Sigma) in combination with 5 μm ionomycin (Sigma) for 16 h (Scaffidi et al., 2002). Cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI), whereas culture supernatants were concentrated by repetitive centrifugation using microcon filter devices with a nominal molecular weight limit of 30,000 Daltons (Millipore, Billerica, MA). Recovered culture supernatants were applied to a 12% SDS-PAGE gel. HSP60 derived from CNS cells was detected by immunoblotting using a mouse monoclonal antibody against HSP60 (SPA-807; Stressgen). Protein signals were visualized by enhanced chemiluminescence detection, and intensities of the signals were quantified by LumiAnalyst software (Boehringer, Ingelheim, Germany).
Measurement of nitrite.
The content of NO in microglial cultures was analyzed by measuring the stable end product nitrite in culture supernatants. The amount of accumulated nitrite (NO2−) was determined by using the colorimetric Griess reaction (Sigma) as described previously (Chen et al., 1999). The inducible nitric oxide synthase (iNOS) inhibitor aminoguanidine (AG) was also obtained from Sigma.
Immunofluorescence microscopy.
Cells were fixed with 4% paraformaldehyde (PFA), washed with PBS, and treated with blocking solution for 20 min. Cultures were then incubated with the primary antibody [anti-neuronal-specific nuclear protein (NeuN), anti-neurofilament, microtubule-associated protein (MAP-2), and glial fibrillary acidic protein (GFAP); all purchased from Chemicon, Temecula, CA] overnight at 4°C. Subsequently, cells were incubated with the relevant secondary antibody (all purchased from Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature. Immunofluorescence images were obtained using a Nikon (Tokyo, Japan) Eclipse 660 microscope and a Spot cooled CCD. Microglia were stained with isolectin IB4 (Invitrogen, Carlsbad, CA). For O4 immunofluorescence, live cultures were incubated with a monoclonal antibody against O4 (ATCC, Teddington, UK) for 20 min and fixed with 4% PFA followed by an incubation of the cells with the relevant secondary antibody.
According to the instructions of the vendor, recombinant HSP60 was labeled with Alexa Fluor 488 using a protein labeling kit from Invitrogen. For live staining with HSP60-Alexa, cells were incubated with Alexa 488-conjugated HSP60 at 4°C or at 37°C, as indicated, for 40 min. Cells were visualized by immunofluorescence microscopy.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining of CNS cultures was conducted using the In Situ Cell Death Detection kit, TMR red, following the instruction manual (Roche, Basel, Switzerland).
Short interfering RNA transfection of human embryonic kidney 293 cells and neurotoxicity assay.
RNA interference of heat shock 60 kDa protein-1 (HSPD1), the coding gene for HSP60, was performed using two different short interfering RNA (siRNA) duplexes (A and B) from Qiagen (Hilden, Germany). The coding strands for HSPD1 siRNA were CAGGGTTTGGTGACAATAGAA (HSPD1 A) and AAGGCTTCGAGAAGATTAGCA (HSPD1 B). An Alexa Fluor 488-labeled nonsilencing siRNA duplex (Qiagen) was used as a negative control. For transfection, human embryonic kidney 293 (HEK293) cells were seeded in 6-well plates (350,000 cells/well). After 24 h, cells were transfected with siRNA (50 nmol) using Lipofectamine RNAiMax (Invitrogen) according to the recommendations of the manufacturer. After an additional 48 h, HEK293 cells were lysed by several freeze/thaw cycles at −80°C/37°C. Recovered cell lysates were applied to a 10% SDS-PAGE gel, and the efficient knock down of HSP60 was verified by immunoblotting using the mouse monoclonal antibody directed against HSP60 (Stressgen). Next, cell lysates of transfected and untransfected HEK293 cells were added to cocultures of neurons and microglia derived from wild-type mice. After 72 h, neuronal viability was assessed by immunostaining with the NeuN antibody and parallel DAPI staining, as described above.
Overexpression of HSP60 in HEK293 cells.
Human HSP60 cDNA was amplified from HEK293 cells by PCR (forward primer, 5′-TAAAGCTTATGCTTCGGTTACCCACAGTC; reverse primer, 5′-GCGGCCGCTTAGAACATGCCACCTCCCATACC), cloned into the pCR3.1-TOPO vector using a TOPA-TA cloning kit (Invitrogen) and subsequently subcloned into peGFP-N1 (Clontech, Mountain View, CA) by replacing the enhanced green fluorescent protein (eGFP) sequence with the hHSP60 cDNA, resulting in plasmid pehHSP60. The original peGFP-N1 vector was used as a negative control in transfection experiments. The correct sequence of the hHSP60 cDNA was confirmed by automated DNA sequencing. HEK293 cells were transfected with peGFP-N1 or pehHSP60 using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer instructions for 24 h. Expression levels of HSP60 were analyzed by immunoblotting. Lysis of transfected HEK293 cells was induced, and lysates were used for neurotoxicity studies as described above.
Statistical analysis.
Data are expressed as mean ± SD. Statistical differences between groups were determined using two-tailed Student's t test or one-way ANOVA with subsequent Bonferroni post hoc analysis. Differences were considered statistically significant when p < 0.05.
Results
A defect in TLR4 function protects neurons from CNS trauma
The typical isolation procedure of CNS cells from mouse brain can, in a sense, be considered a severe form of CNS trauma. While preparing mixed CNS cultures from lpsd mice, we consistently made the observation that resulting cultures contained more cells and appeared to be less vulnerable during culturing compared with cultures derived from BALB/cJ or Swiss Webster mice. Lpsd mice (see Materials and Methods) are characterized by hypo-responsiveness to LPS as a consequence of a point mutation rendering the cytosolic domain of TLR4 incapable of signal transduction (Poltorak et al., 1998; Hoshino et al., 1999; Qureshi et al., 1999). To investigate the relationship between TLR4 function and recovery of neurons from disrupted brain tissue, we generated mixed CNS cultures from forebrains of lpsd and genetically matched BALB/cJ E17 mice (Fig. 1). Embryos and forebrains of both strains had similar weights and did not differ regarding macroscopical and microscopical appearance (data not shown). Cells were isolated by mechanical disruption and trypsinization and then cultured in DMEM supplemented with FBS (see Materials and Methods). After 3 d in culture, neurons were identified by concomitant staining with anti-MAP-2 and anti-neurofilament antibodies (Fig. 1A) as well as by individual staining with the indicated antibodies (Fig. 1B,C). Immunofluorescence microscopy and statistical analysis of the ratio of DAPI- and MAP-2-positive cells/mm2 (Fig. 1B) revealed a 60% increase in total cell number (DAPI) and a 40% increase in neurons in lpsd cultures compared with wild-type cultures (p ≤ 0.001, Student's t test; for the comparison of BALB/cJ and lpsd cells). Furthermore, mixed CNS cultures derived from lpsd mice displayed enhanced neurofilament density compared with wild-type cultures (Fig. 1C).
Figure 1.
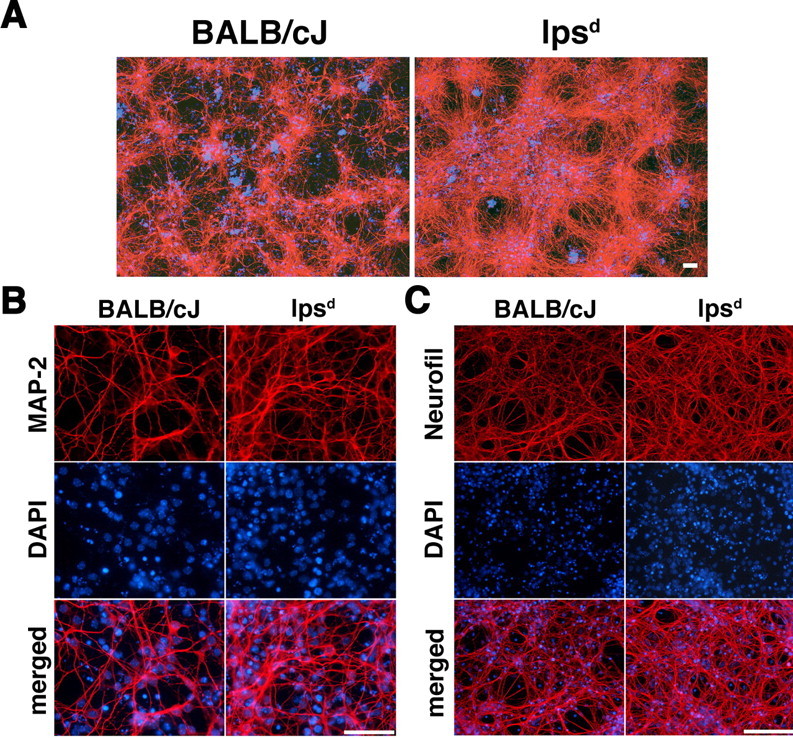
Mixed CNS cultures derived from lpsd mice are less vulnerable and contain a higher number of neurons compared with cultures derived from wild-type mice. Mixed CNS cultures from four pooled mouse forebrains derived from BALB/cJ or lpsd mice were prepared and cultured for 3 d. A–C, Subsequent parallel (A) and individual (B, C) immunostaining with antibodies against MAP-2 and neurofilament marked neurons. Parallel DAPI staining revealed the total number of cells by staining all nuclei. Scale bars, 50 μm. For quantitation of DAPI- and MAP-2-positive cells in B, six high-power fields per coverslip were analyzed. For each condition, experiments were performed in triplicates. The data shown are representative of >10 individual experiments.
HSP60 induces neuronal injury dependent on a functional TLR4 pathway
LPS is the well-described exogenous ligand for TLR4 (Poltorak et al., 1998; Hoshino et al., 1999; Qureshi et al., 1999). However, because we observed that TLR4 loss-of-function improved the quantity of viable neurons recovered from mouse forebrains, we postulated that an endogenous ligand must act on TLR4. We hypothesized that this ligand would be released from injured cells during the process of cell isolation, bind to TLR4, and lead to activation of the TLR4 pathway resulting in neuronal injury. HSP60 has been described previously as an endogenous ligand of the TLR4 complex. The proinflammatory effect of HSP60 on murine bone marrow-derived macrophages is dependent on functional TLR4 (Ohashi et al., 2000). We investigated whether HSP60 induces proinflammatory properties and neuronal injury in CNS cultures.
CNS cultures were generated from lpsd and BALB/cJ (wild-type) mice. Both cultures were treated with 10 μg/ml HSP60 or PBS (control) for 2 d. Neuronal cell bodies and dendrites as well as axons were visualized by anti-MAP-2 (Fig. 2A) and anti-neurofilament (Fig. 2C) immunostaining, respectively. Whereas treatment with HSP60 led to a major loss of axons and dendrites in mixed CNS cultures derived from BALB/cJ mice, treatment with HSP60 had no effect on the number of MAP-2- and neurofilament-positive neurons derived from lpsd mice. Statistical analysis of the total number of MAP-2-positive cells/mm2 confirmed these results (Fig. 2B). Incubation of mixed CNS cultures from BALB/cJ mice with HSP60 decreased the number of MAP-2-positive cells fivefold. In contrast, neurons in lpsd-derived cultures were not affected by HSP60 treatment. To determine the purity of the HSP60 preparation, 15 μg of the sample were analyzed by SDS-PAGE and subsequent Coomassie staining (Fig. 2D). A single band at the expected size was detected.
Figure 2.
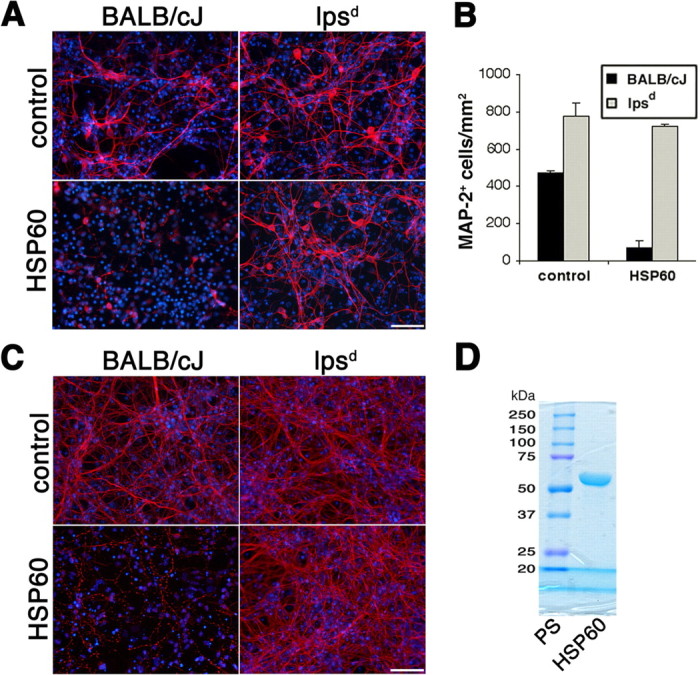
HSP60 induces neuronal cell death dependent on a functional TLR4 pathway. Mixed CNS cultures from forebrains of BALB/cJ and lpsd mice were incubated with 10 μg/ml HSP60 or PBS (control) for 2 d. A, C, Cultures were stained with antibodies against MAP-2 (A) and neurofilament (C) to mark neurons, whereas all nuclei were stained with DAPI. Scale bars, 50 μm. B, Quantitation of MAP-2-positive neurons from BALB/cJ and lpsd mice in the presence or absence of HSP60. Six high-power fields per coverslip were analyzed. For each condition, experiments were performed in triplicates. The data shown are representative of four individual experiments. The results are presented as mean ± SD (p < 0.001, Student's t test) for the comparison of BALB/cJ cells with or without HSP60. D, Fifteen micrograms of the HSP60 preparation were analyzed by SDS-PAGE and subsequent Coomassie staining. PS, Protein standards.
Together, the above results demonstrate a neurotoxic effect induced by HSP60 and imply an obligatory role for TLR4 in HSP60-mediated neuronal injury in vitro. TLR4 thus seems to mediate the innate immune response to both HSP60 and LPS.
HSP60-induced neuronal injury is not caused by LPS contamination
Although we used a low endotoxin-charge of recombinant HSP60 obtained from Stressgen (see Materials and Methods), a significant concern in establishing a link between HSP60 and functional TLR4 in signaling neurodegeneration is potential contamination of recombinant HSP60 with LPS (Chen et al., 1999; Kol et al., 1999; Gao and Tsan, 2003; Quintana and Cohen, 2005). We have shown in previous work that LPS induces neuronal cell death in neuronal cultures via TLR4 expressed in microglia (Lehnardt et al., 2003). Similarly, in this report, we demonstrate an HSP60-induced neurotoxic effect in mixed CNS cultures as well as in neuronal cultures enriched with microglia via TLR4 after 2 d of incubation (see results above) (Fig. 4). To resolve that the neurotoxic effects induced by HSP60 were attributable to HSP60 itself and not contaminating LPS, we performed a series of controls (Fig. 3). Cortical neurons prepared from BALB/cJ mice forebrains and supplemented with purified microglia from BALB/cJ mice were incubated with HSP60 or LPS (Fig. 3A). The antibiotic polymyxin B sulfate (PMBS) is a cationic cyclic polypeptide that acts primarily by binding membrane phospholipids and disrupting the cytoplasmic membane, inducing pore formation in bacterial cell walls. PMBS binds to the lipid-A portion of the LPS in the cell membrane of Gram-negative bacteria and is a specific LPS antagonist. Whereas the neurotoxic effects of HSP60 were undiminished in the presence of PMBS, LPS-induced neurotoxicity was abolished in the presence of PMBS. Furthermore, the neurotoxicity of HSP60 (10 μg/ml), but not LPS (1 μg/ml), was abrogated by each of three distinct protein denaturation regimes: heat denaturation and proteolysis by either proteinase K or trypsin. These results confirm that the observed neurotoxic effects were caused by a protein rather than by LPS.
Figure 4.

Neurons treated with HSP60 undergo apoptotic cell death dependent on a TLR4-MyD88 pathway in microglia. Cortical neurons were prepared from BALB/cJ mice forebrains. Purified neurons alone as well as neurons cocultured with purified microglia from BALB/cJ (wt) or lpsd mice were incubated with 10 μg/ml HSP60 or PBS. A, After 2 d, cultures were fixed and stained both with NeuN antibody and with IB4 to mark neurons and microglia, respectively. B, Quantitation of NeuN-positive neurons as a percentage of all DAPI-positive cells in purified cultures and cultures supplemented with BALB/cJ (WT)- or lpsd-derived microglia in the presence or absence of HSP60. Results are presented as mean ± SD (p < 0.001, Student's t test) for comparison of the group, in which wild-type microglia and HSP60 are added, with other groups. C, Cells were incubated with 10 μg/ml HSP60 or Camptothecin and analyzed by a TUNEL assay. D, Cortical neurons were prepared from C57Bl/6J mice forebrains. Purified neurons alone as well as neurons cocultured with purified microglia from C57Bl/6J (wt) or MyD88−/− mice were incubated with 10 μg/ml HSP60 or PBS. After 2 d, cultures were fixed and stained both with MAP-2 antibody and with IB4 to mark neurons and microglia, respectively. E, Quantitation of MAP-2-positive neurons as a percentage of all DAPI-positive cells in purified and C57Bl/6J (wt) or MyD88−/− microglia-enriched cultures in the presence or absence of HSP60. Results are presented as mean ± SD (p < 0.001, Student's t test) for comparison of the group, in which wild-type microglia and HSP60 are added, with other groups. For all quantitations (B, E), six high-power fields per coverslip were analyzed. For each condition, experiments were performed in triplicates. The data shown are representative of four individual experiments each. Scale bars, 50 μm.
Figure 3.
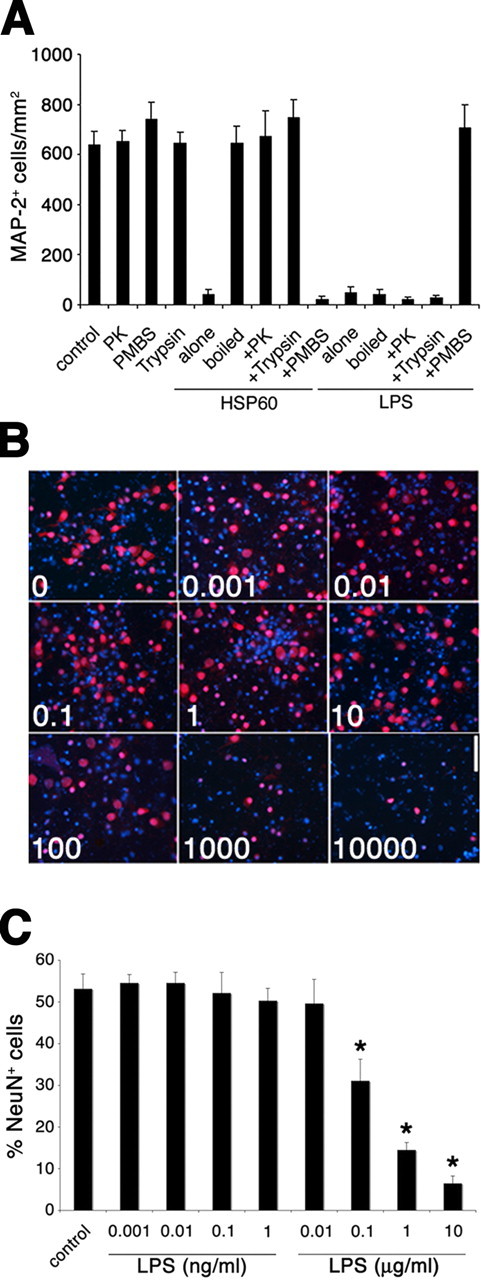
HSP60-induced neuronal cell death is not attributable to LPS contamination. Cortical neurons were prepared from BALB/cJ mice forebrains and were supplemented with purified microglia from BALB/cJ mice. A, Cells were incubated with 10 μg/ml HSP60 or 1 μg/ml LPS. Neurotoxic effects of HSP60 were abrogated by pretreatment of samples with trypsin and proteinase K (PK) or by boiling for 20 min before incubation. Neurotoxic effects of HSP60 were not abrogated by combined treatment of cultures with PMBS. In parallel, LPS and PBS (control) were also treated under the same conditions before incubation. After 2 d, cultures were fixed and stained both with MAP-2 antibody and with DAPI to mark neurons and nuclei, respectively. The results are presented as mean ± SD. Cells were incubated with various doses of LPS, as indicated, for 2 d. B, Staining with the NeuN antibody and DAPI (units are in nanograms per milliliter). C, Quantitation of NeuN-positive neurons as a percentage of all DAPI-positive cells in cultures supplemented with BALB/cJ microglia. Scale bar, 50 μm. Results are presented as mean ± SD (*p < 0.001, Student's t test) for comparison of the indicated group to the control group. All experiments were repeated three times.
To further rule out the possibility of contaminating LPS, we conducted dose–response experiments to determine the LPS concentration that would be required to induce neuronal injury in our coculture system. BALB/cJ cortical neurons supplemented with BALB/cJ microglia were incubated with various concentrations of LPS, as indicated, for 2 d. Staining with NeuN antibody and DAPI (Fig. 3B) and subsequent quantification of NeuN-positive cells (Fig. 3C) revealed that 0.1 μg/ml LPS was required to induce significant neuronal injury under these circumstances. Whereas the incubation with 0.1 μg/ml LPS led to a decrease of the ratio of NeuN-positive cells to DAPI-positive cells from 53% (absolute numbers of NeuN-positive cells, control 15.0 ± 5.7; absolute number of DAPI-positive cells, 28.1 ± 5.0) to 31% (absolute number of NeuN-positive cells, 7.4 ± 1.8; absolute number of DAPI-positive cells, 24.2 ± 5.0), 10 μg/ml LPS was required to decrease the percentage of NeuN-positive cells from control values to 7% (absolute number of NeuN-positive cells, 1.7 ± 0.8; absolute number of DAPI-positive cells, 25.4 ± 4.2). In our experiments, we used a low-endotoxin HSP60 charge, and the maximum contamination of this HSP60 preparation with LPS as declared by the manufacturer is <50 EU/mg, which is equal to <5 ng of LPS per milligram of recombinant protein. The same HSP60 batch was tested by an independent commercial laboratory specializing in endotoxin quantification (Mikrobiologisches Labor, Münster, Germany). Using the Chromogenix Coamatic Chromo-Limulus amoebocyte lysate assay, an LPS content of ≤1.67 EU/mg protein was determined. In our experimental set up (at a dose of 10 μg/ml HSP60), the theoretical amount of LPS in the assay was <0.05 ng/ml (estimated by Stressgen) or ≤0.00167 ng/ml (as determined by Mikrobiologisches Labor), which is far below the concentration threshold required to induce measurable neuronal cell loss under our assay conditions. At least 10 μg/ml LPS was required to induce a similar decrease in the ratio of NeuN-positive cells to DAPI-positive cells as we observed in cocultures treated with HSP60 for 2 d (Fig. 4A,B,D,E). This dose exceeds the theoretical content of LPS in our HSP60 preparation, as determined by the Mikrobiologisches Labor, by almost seven orders of magnitude. Thus, the observed neurotoxic effects after incubation with HSP60 are not caused by LPS contaminating the HSP60 preparation.
Together, these results are consistent with the immunostimulatory effects of HSP60 previously described by several laboratories (Chen et al., 1999; Kol et al., 1999; Ohashi et al., 2000; Quintana and Cohen, 2005) and argue against the possibility of LPS contamination as the causative factor of neurotoxicity in our experiments.
HSP60-induced neuronal injury displays characteristics of apoptosis and is dependent on a functional TLR4-MyD88 pathway in microglia
To determine whether the effects of HSP60 on neuronal survival were cell autonomous or extrinsic, we compared the effect of HSP60 on purified neurons to cocultures of neurons and microglia (Fig. 4). Cultures were treated with HSP60 for 2 d and then stained for neurons (NeuN) and microglia (IB4) to allow microscopic analysis (Fig. 4A). HSP60 had no effect on neuronal survival in purified neuronal cultures. In contrast, when purified microglia from wild-type mice were added to enriched neuronal cultures, we observed severe loss of neurons in the presence of HSP60. As a control, the addition of purified microglia from lpsd mice to wild-type neurons had no effect on neuronal survival after HSP60 treatment, confirming the requirement for intact TLR4 signaling. The statistical analysis of the ratio of NeuN-positive cells to DAPI-positive cells confirmed these results (Fig. 4B). Absolute numbers of NeuN-positive cells were: neurons alone, 17.3 ± 5.4; neurons plus wild-type microglia, 20.2 ± 5.4; neurons plus lpsd microglia, 19.1 ± 4.2; neurons plus HSP60, 18.2 ± 3.2; neurons plus wild-type microglia and HSP60, 2.5 ± 1.4; neurons plus lpsd microglia and HSP60, 18.8 ± 4.3.
To investigate whether the observed HSP60-induced neuronal cell death was of apoptotic nature, we conducted a TUNEL assay after incubation with HSP60 for 1 d under the experimental conditions described above (Fig. 4C). Distinct TUNEL staining was observed only in enriched neuronal cultures supplemented with wild-type microglia and treated with HSP60. Neuronal cultures supplemented with lpsd-derived microglia were almost completely negative for TUNEL staining, confirming the finding that HSP60-induced neuronal injury is dependent on a functional TLR4 pathway in microglia. In parallel, neuronal cultures cocultured with wild-type microglia were incubated with 0.1 m Camptothecin as a positive control for TUNEL staining.
Activation of TLR4 recruits the adapter protein MyD88 through interacting TLR domains. MyD88 is critical in the TLR-mediated signaling pathway (Medzhitov et al., 1998; Horng and Medzhitov, 2001). MyD88-deficient mice do not show inflammatory response to LPS, including production of inflammatory mediators by macrophages, proliferation of B cells, and endotoxin shock (Kawai et al., 1999). Several reports have demonstrated that MyD88 is essential for cellular responses mediated by most members of the TLR family (Takeuchi et al., 1999; Hacker et al., 2000; Schnare et al., 2000; Hayashi et al., 2001). To determine whether HSP60-induced neurotoxicity requires functional MyD88, we purified microglia from MyD88−/− mice and added these cells to highly enriched neuronal cultures derived from wild-type mice. Cultures were then incubated with HSP60. Immunostaining with MAP-2 revealed that addition of HSP60 to cultures supplemented with microglia derived from MyD88−/− mice had no effect on neuronal survival, whereas neurons in cultures supplemented with microglia from mice of the wild-type strain C57Bl/6J suffered cell death (Fig. 4D). Statistical analysis of the ratio of MAP-2-positive neurons to DAPI-positive cells confirmed this result (Fig. 4E). Absolute numbers of MAP-2-positive cells were: neurons alone, 8.2 ± 2.9; neurons plus wild-type microglia, 9.3 ± 2.2; neurons plus MyD88−/− microglia, 9.9 ± 2.3; neurons plus HSP60, 8.9 ± 3.2; neurons plus wild-type microglia and HSP60, 0.9 ± 0.9; neurons plus MyD88−/− microglia and HSP60, 9.5 ± 4.5. Neuronal cultures generated from wild-type mice cocultured with microglia derived from MyD88−/− mice were incubated with 0.1 m Camptothecin, and subsequent MAP-2 and DAPI staining revealed neuronal cell death, thus demonstrating that these neurons are indeed susceptible to injury (Fig. 4D). Together, neuronal death induced by HSP60 requires a functional TLR4-MyD88 pathway in microglia.
Microglia are the only cell type in the CNS that binds HSP60-Alexa
Microglia express TLR4, which is required for the effects of LPS on circulating monocytes and microglia (Medzhitov and Janeway, 2000; Bsibsi et al., 2002; Lehnardt et al., 2002). We also determined in previous work that astrocytes, oligodendrocytes, and neurons do not express TLR4 (Lehnardt et al., 2003). The above results suggest that the neurotoxic effect of HSP60 requires the presence of microglia. Thus, we investigated whether microglia or other cell types in the CNS specifically bind HSP60. Recombinant HSP60 was labeled with Alexa dye. Primary cultures of microglia, neurons, oligodendrocytes, and astrocytes were then incubated with HSP60-Alexa at a concentration of 1 μg/ml for 40 min on ice, or at 37°C. Live cells were then visualized by fluorescence microscopy (Fig. 5). Microglia bound HSP60-Alexa, whereas neurons, oligodendrocytes, and astrocytes did not show any labeling with HSP60-Alexa. Whereas HSP60 labeling at 4°C revealed only membrane staining of microglia, incubation at 37°C led to the detection of HSP60-Alexa at the microglial cell membrane as well as in intracellular vesicles, suggesting internalization.
Figure 5.

Microglia, but not neurons, oligodendrocytes, or astrocytes, bind HSP60-Alexa. Primary cultures of rat microglia, neurons, oligodendrocytes, and astrocytes were incubated with 1 μg/ml HSP60-Alexa at 4°C or 37°C and analyzed by immunofluorescence. Parallel cultures were immunostained for microglia (IB4), neurons (MAP-2), oligodendrocytes (O4), and astrocytes (GFAP). Scale bar, 50 μm.
Apoptotic and necrotic CNS cells release HSP60 that causes neuronal injury in vitro
Based on the above data, we postulated that CNS injury results in the release of HSP60, which causes activation of microglia in a TLR4-dependent manner. In turn, activated microglia cause additional neuronal injury by releasing neurotoxic molecules. If this hypothesis was true, then one must assume that CNS cells undergoing cell death release HSP60.
To investigate whether injured cells release HSP60 (Fig. 6), we prepared mixed CNS cultures from the forebrains of wild-type mice and induced apoptosis or necrosis by treatment with 5 ng/ml mouse TNF-α and 35 μm cycloheximide or 5 μm ionomycin and 20 μm CCCP, respectively (Scaffidi et al., 2002). DAPI staining of the drug-treated cells revealed a typical nuclear morphology of apoptotic and necrotic cells (Fig. 6A). Culture supernatants were freed of cells and organelles by centrifugation and analyzed by SDS-PAGE followed by immunoblotting with a mouse antibody against HSP60 (Fig. 6B). Supernatants from cells under control conditions did not contain HSP60. HSP60 was clearly detected in supernatants from both apoptotic and necrotic cells. We observed a greater amount of HSP60 in supernatants from necrotic cells compared with supernatants from apoptotic cells. These data demonstrate that HSP60 is released by apoptotic and necrotic CNS cells and is therefore in principle capable of activating cell surface receptors.
Figure 6.
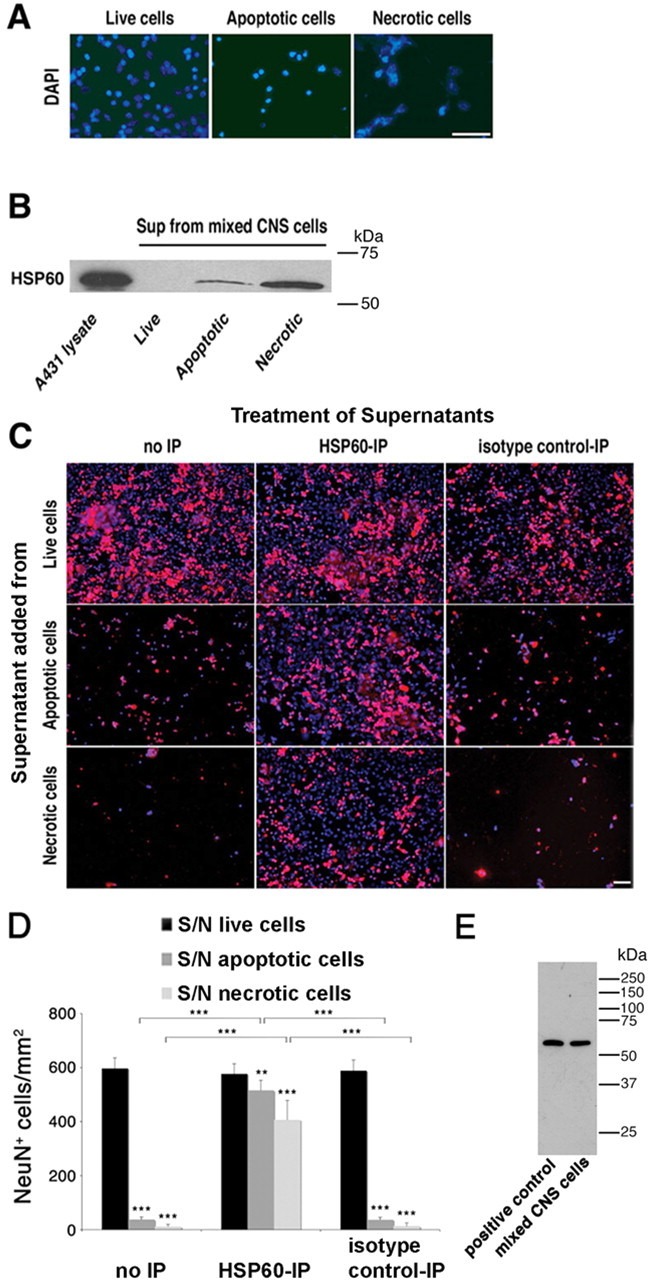
HSP60 is released by apoptotic and necrotic CNS cells and causes neuronal injury. Mixed CNS cultures derived from wild-type mice were forced to undergo apoptosis or necrosis. A, Untreated and drug-treated cells were stained with DAPI. B, Culture supernatants (Sup) of treated cells were analyzed by SDS-PAGE and subsequent immunoblotting with an HSP60 antibody. Cell lysate derived from A431 cells served as a positive control. C, Culture supernatants of treated cells were immunoprecipitated with an HSP60 antibody or normal mouse IgG. Neuronal-enriched mixed CNS cultures derived from wild-type mice were treated with either plain or immunoprecipitated (with an antibody against HSP60 or with normal mouse IgG) culture supernatants of live, apoptotic, and necrotic cells. Cells were then immunostained with NeuN to mark neurons and with DAPI. D, Quantitation of NeuN-positive neurons in CNS cultures treated with supernatants (S/N) from live, apoptotic, or necrotic CNS cells with or without immunoprecipitation (IP) against HSP60. Six high-power fields per coverslip were analyzed. For each condition, experiments were performed in triplicates. The data shown are representative of three individual experiments. The results are presented as mean ± SD (**p < 0.01, ***p < 0.001; Student's t test) for the comparison of S/N from apoptotic or necrotic cells versus S/N from live cells and for the comparison of S/N treated with anti-HSP60 antibody versus isotype control antibody or no treatment. E, Immunoblot of wild-type mouse-derived mixed CNS cells and heat-shocked HeLa cells (positive control) probed with the SPA-807 antibody. Scale bars, 50 μm.
As described above, neuronal injury induced by recombinant HSP60 is mediated by a TLR4-dependent pathway in microglia. We therefore addressed the question of whether endogenous HSP60 released by apoptotic and necrotic cells injures neurons similar to recombinant HSP60, and if so, whether we could block this neurotoxic effect by specific depletion with HSP60 antibodies. To this end, we induced apoptosis or necrosis in mixed CNS cultures as described above and harvested the supernatants from these cultures. The cytotoxic agents were removed by repetitive filtration on size-exclusion filter devices (Millipore, Bedford, MA), and supernatants were added to neuronal enriched mixed CNS cultures. In parallel, HSP60 was depleted from the supernatants of apoptotic and necrotic cells by repetitive immunoprecipitation with a mouse monoclonal antibody directed against HSP60 (SPA-807; Stressgen) or normal mouse IgG as an isotype control. Cultures were stained with the NeuN antibody and DAPI (Fig. 6C). Nondepleted supernatants of both apoptotic and necrotic cells induced neuronal injury and loss, although the supernatant derived from apoptotic cells showed neurotoxic effects to a lesser extent than the supernatant from necrotic cells. The supernatant from untreated cells served as control, and its addition to mixed CNS cultures did not affect the survival of neurons compared with untreated cultures (data not shown). In contrast, supernatants from apoptotic and necrotic cells from which HSP60 was depleted by immunoprecipitation before addition to neuronal cultures showed much reduced toxicity compared with nondepleted supernatants or supernatants treated with isotype control antibodies. Statistical analysis of the total number of NeuN-positive cells/mm2 confirmed these results (Fig. 6D). To ensure that the antibody against HSP60 is monospecific, a complete immunoblot of mixed CNS cells derived from mouse was probed with this antibody (Fig. 6E). Only a single band was detected in both heat-shocked HeLa cell lysate (positive control) and mixed CNS cells.
In summary, these results demonstrate that endogenous HSP60 released from dying CNS cells is capable of causing neuronal injury and loss.
Silencing of HSP60 protects neurons from cell death induced by cell lysates from HEK293 cells
To confirm the specificity of HSP60-induced neurotoxicity observed above, we used two different siRNA duplexes targeting the mRNA for HSP60. HEK293 cells were transfected with the siRNA duplexes, and subsequent immunoblotting of lysed cells with the antibody against HSP60 revealed the efficient knock down of HSP60 by both siRNA duplexes (Fig. 7A). Untransfected cells (control) and cells transfected with a nonsilencing siRNA (neg. siRNA) served as controls. Specificity of the knock down was proven by the lack of effect on levels of the unrelated protein β-actin (Fig. 7A). Densitometric analysis of the HSP60 expression levels confirmed the effectiveness of the knock down (Fig. 7B). Next, cocultures of neurons and microglia were incubated with cell lysates from transfected or untransfected HEK293 cells for 72 h. Immunostaining with the NeuN antibody revealed that cell lysates from untransfected cells and from cells transfected with the nonsilencing control siRNA induced neuronal cell loss. In contrast, silencing of HSP60 by either siRNA A or siRNA B significantly reduced neurotoxic effects induced by the respective cell lysates, although neuronal toxicity was not completely abrogated (Fig. 7C). Quantification of NeuN-positive cells verified these results, revealing a significant reduction in neuronal cell loss as a consequence of HSP60 knock down (Fig. 7D). The absolute numbers of NeuN-positive cells were as follows: control, 18.5 ± 3.5; untransfected, 1.9 ± 1.1; negative siRNA, 1.5 ± 1.1; siRNA HSPD1 A, 5.2 ± 3.0; siRNA HSPD1 B, 6.8 ± 2.1. These results confirm the specificity of HSP60 as the cause of neurotoxicity in our experiments.
Figure 7.
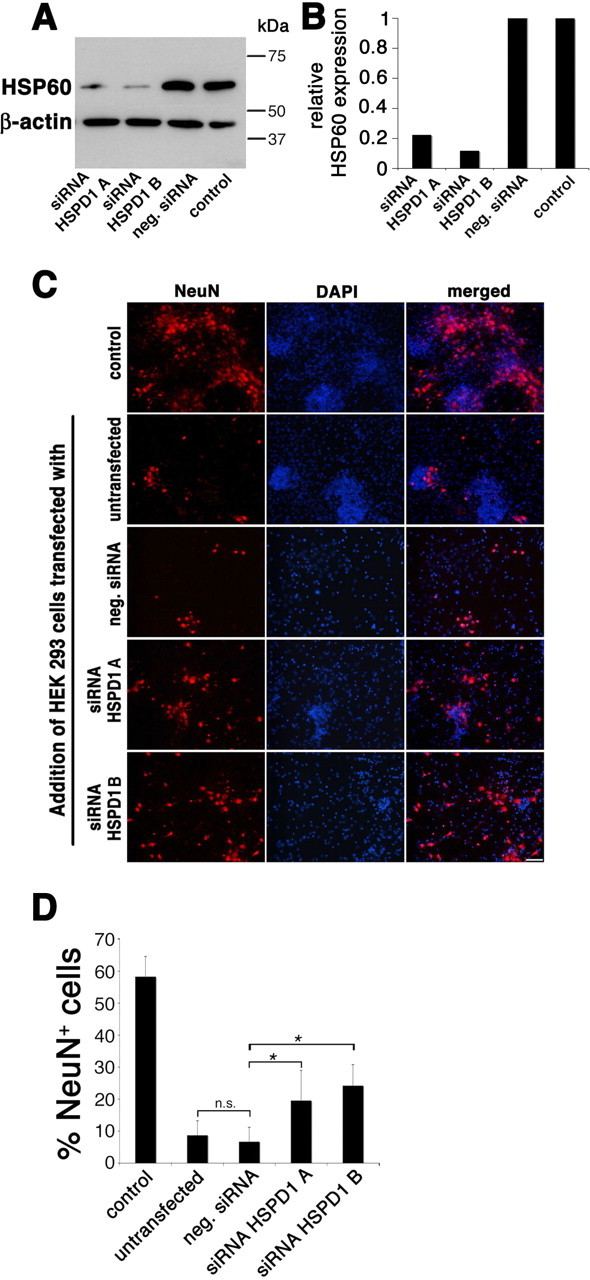
Silencing of HSP60 reduces neurotoxicity of HEK293 cell lysates added to cocultures of neurons and microglia. HEK293 cells were transfected with siRNA A and B targeting HSP60. Untransfected cells (control) and cells transfected with nonsilencing siRNA (neg. siRNA) served as controls. A, Immunoblot of lysed cells probed with the SPA-807 antibody directed against HSP60. β-Actin served as a loading control. B, Densitometric analysis of the immunoblot signals after detection by chemiluminescence. C, Cocultures of neurons and microglia derived from wild-type mice were treated with cell lysates of transfected and untransfected HEK293 cells and were subsequently immunostained with NeuN to mark neurons, and with DAPI. Scale bar, 50 μm. D, Quantitation of NeuN-positive neurons as a percentage of all DAPI-positive cells in cocultures treated with HEK293 cell lysates. Six high-power fields per coverslip were analyzed. For each condition, experiments were performed in duplicates. The data shown are representative of four individual experiments. The results are presented as mean ± SD (*p < 0.001) for the comparison of neg. siRNA with siRNA HSPD1A and for the comparison of neg. siRNA with siRNA HSPD1B (one-way ANOVA and post hoc Bonferroni's test).
Overexpression of HSP60 increases neurotoxicity induced by cell lysates from HEK293 cells
HEK293 cells were transfected with plasmids encoding for hHSP60 or GFP (control). Immunoblotting of cell lysates with an antibody against HSP60 displayed overexpression in hHSP60-transfected cells (Fig. 8A). Densitometric analysis of HSP60 expression levels confirmed overexpression of HSP60 by ∼2.6-fold in hHSP60-transfected cells compared with control-transfected cells. Cocultures of neurons and microglia were incubated with cell lysates from hHSP60- or control-transfected HEK293 cells for 72 h. Because the baseline level of HSP60 expression in HEK293 cells is high, lysates from hHSP60- and control-transfected HEK293 cells were diluted equally to such an extent that lysates from control-transfected cells induced no neuronal damage compared with untreated cells. Serial dilutions (1:2, 1:3, 1:5, 1:10, 1:15) were performed. Lysates from control-transfected cells showed strong neurotoxicity in the 1:2 through 1:5 dilutions (absolute numbers of NeuN-positive cells: untreated, 22.0 ± 3.6; control-transfected 1:2 dilution, 0.3 ± 0.4; control-transfected 1:3 dilution, 0.5 ± 0.5; control-transfected 1:5 dilution, 0.5 ± 0.5). Toxicity was somewhat reduced in the 1:10 dilution (absolute number of NeuN-positive cells, 6 ± 3.6) but remained significantly higher than in the untreated group (p = 0.025). In the 1:15 dilution, cell viability showed no significant difference between lysates from control-transfected cells (absolute number, 17.5 ± 2.3) and untransfected controls anymore (p = 0.923). Therefore, lysates from control-transfected and HSP60-transfected cells were used in the 1:15 dilution in subsequent experiments to visualize differences in neuronal toxicity induced by overexpression of HSP60. Staining with the NeuN antibody and DAPI and subsequent quantification of positive cells revealed that lysates from cells overexpressing HSP60 induced a significantly higher degree of neuronal loss than lysates from control-transfected cells (percentage NeuN-positive of DAPI-positive cells: 24.2 vs 59.6%; p < 0.001) (Fig. 8B). Absolute numbers of NeuN-positive cells were as follows: untreated, 18.7 ± 4.7; control-transfected, 17.2 ± 4.9; HSP60-transfected, 5.9 ± 4.3. These results substantiate the specificity of HSP60 as the cause of neuronal damage in our experiments.
Figure 8.

Overexpression of HSP60 increases neurotoxicity of HEK293 cell lysates added to cocultures of neurons and microglia. HEK293 cells were transfected with plasmids encoding hHSP60 or GFP (control). A, Immunoblot of lysed cells probed with the SPA-807 antibody directed against HSP60. Left lane, Control-transfected; right lane, HSP60-transfected. β-Actin served as a loading control. B, Quantitation of NeuN-positive neurons as a percentage of all DAPI-positive cells in cocultures of neurons and microglia derived from wild-type mice treated with cell lysates of hHSP60- or control-transfected HEK293 cells. Cell lysates were diluted 1:15. Quantitation of NeuN-positive neurons was performed by analyzing four high-power fields per coverslip. For each condition, experiments were performed in duplicates. The data shown are pooled from three individual experiments with three different preparations of transfected cells. The results are presented as mean ± SD (*p < 0.001) for the comparison of control-transfected cells with hHSP60-transfected cells (one-way ANOVA and post hoc Bonferroni's test).
HSP60 leads to TLR4-dependent release of neurotoxic NO from microglia
Ohashi et al. (2000) demonstrated that induction of NO release from bone marrow-derived macrophages by HSP60 is dependent on TLR4. In the brain, reactive oxygen species contribute to neuronal death (Chao et al., 1992; Brosnan et al., 1994). We have shown recently that activation of TLR2 leads to the secretion of neurotoxic NO from microglia (Lehnardt et al., 2006). First, we investigated whether the supernatants of dying CNS cells are capable of inducing NO secretion by microglia. To this end, purified wild-type microglia were incubated with supernatants of apoptotic and necrotic CNS cells for 24, 48, and 72 h (Fig. 9A). Subsequently, culture media were analyzed for nitrite content by colorimetric Griess reaction. Both supernatants of apoptotic and necrotic CNS cells significantly induced NO production in microglia. LPS served as a positive control. To determine the contribution of NO to HSP60-induced neurotoxicity, wild-type cortical neurons cocultured with microglia derived from wild-type mice were treated with either 10 μg/ml HSP60 alone or in combination with the iNOS inhibitor aminoguanidine, starting 1.5 h before stimulation with HSP60, for 72 h (Fig. 9B). Cells were then stained for NeuN, and DAPI and were quantified. Absolute numbers of NeuN-positive cells were as follows: control, 17.6 ± 6.1; HSP60, 1.8 ± 1.6; AG, 17.3 ± 3.1; HSP60 plus AG, 8.6 ± 3.5. Thus, whereas HSP60 induced a major reduction in the number of neurons compared with control conditions, HSP60-induced neurotoxicity was significantly less pronounced in cultures pretreated with aminoguanidine.
Figure 9.

HSP60 induces release of neurotoxic NO from microglia through a TLR4-dependent pathway. A, Purified microglia derived from BALB/cJ mice were incubated with supernatants of apoptotic and necrotic mixed CNS cells for the indicated duration. The amount of nitrite in the culture media was determined by the Griess reaction. B, Quantitation of NeuN-positive neurons as a percentage of all DAPI-positive cells in cocultures of neurons and microglia incubated with either 10 μg/ml HSP60 alone or in combination with the iNOS inhibitor aminoguanidine (200 μm). Cultures were fixed after 72 h and immunostained with NeuN and DAPI, and neurons were quantified. Results are presented as mean ± SD (*p < 0.001, Student's t test) for the comparison of the control group with HSP60 alone or with HSP60 plus AG and for the comparison of HSP60 alone with HSP60 and AG. C, D, Purified microglia derived from BALB/cJ and lpsd mice were incubated for 48 h with increasing concentrations of HSP60 (C) or were treated with 10 μg/ml HSP60 for various incubation times (D). The amount of nitrite in the culture supernatant was determined by the Griess reaction. For each condition, experiments were performed in duplicates. The data shown are representative of three individual experiments. Results are presented as mean ± SD.
To test whether production of NO in microglia caused by HSP60 treatment is dependent on TLR4, we incubated purified microglia of BALB/cJ and lpsd mice with HSP60 at various concentrations (Fig. 9C) and for various durations (Fig. 9D). The culture supernatants were saved and analyzed for NO by Griess reaction. Whereas microglia from BALB/cJ mice responded to the challenge by HSP60 in a dose- and time-dependent manner, NO was not released from microglia isolated from lpsd mice in response to HSP60 treatment. Thus, stimulation of microglial NO production by HSP60 is dependent on TLR4.
Discussion
Stressed, injured, and dying cells release signals into their local environment that activate resident innate immune cells. These signals function as the first warning to the organism, indicating that normal cellular function has been disrupted. Several molecules have been identified, which, when released from injured or dying eukaryotic cells, activate innate immunity. These endogenous activators of innate immunity are evolutionarily conserved proteins or nucleotides with the common feature of being “foreign” to the extracellular environment in a normal tissue (Basu et al., 2000; Sauter et al., 2000; Schnurr et al., 2000; Termeer et al., 2000; Somersan et al., 2001). The novelty of our data in this context is that such endogenous activators may also play a role in CNS injury. Collectively, these observations support the “Danger Model” of innate immune activation proposed by Matzinger, in which dendritic cells can be activated by endogenous molecules as well as by pathogens (Matzinger, 2002). The danger model considers the possibility that pattern recognition receptors, such as TLRs, originally evolved as receptors for injury-related signals (Matzinger, 2002). However, the original function of TLRs may well be a developmental one, because in lower phylogenetic species such as Drosophila, Toll is required for dorsoventral patterning of the fly embryo (Anderson et al., 1985).
Considerable data exist showing that microglial activation occurs in the setting of neuronal injury models (Streit and Kreutzberg, 1988; Kreutzberg, 1996). Such activation may be triggered by endogenous activation of TLRs in the CNS, as shown for TLR2 (Babcock et al., 2006). However, a possible endogenous mediator of this effect remains unidentified.
We demonstrate here that HSP60 triggers neuronal injury by activating microglia in a TLR4- and MyD88-dependent manner. Recent work had concluded that the effects of HSP60 on macrophages were likely to be caused by LPS, the well-established ligand of TLR4, contaminating recombinant HSP60 preparations (Gao and Tsan, 2003). In contrast, Cohen and colleagues reported that pure HSP60 as well as HSP70 indeed activate primary macrophages by using human HSP60 produced in an eukaryotic cell-free system, which is free of bacterial contaminants (Quintana and Cohen, 2005). In accordance with this finding, several lines of evidence in our study demonstrate that HSP60 causes neurotoxic effects, and these effects were not caused by LPS, as we confirmed by several approaches. First, we used a low-endotoxin HSP60 charge in our study, and this preparation was used after dilution to such an extent that the theoretical content of LPS (as tested by an independent laboratory) was equal or below 0.00167 ng/ml. We show here that this concentration of endotoxin is by far not sufficient to induce neuronal cell death in our experiments. Gao and Tsan (2003) previously reported that this specific HSP60 preparation (ESP-540; Stressgen) does not, in contrast to other preparations, cause an increase in TNF-α release by murine macrophages, taking this for confirmation of the absence of relevant amounts of contaminating LPS. However, it is unclear why HSP60 did not cause TNF-α release from macrophages in their experiments, whereas we observe such release from microglia (data not shown). One can speculate that the different cell types or different experimental conditions account for this difference. Second, we rigorously exclude the possibility that the TLR4-dependent effect of HSP60 in CNS cultures is caused by contaminating LPS by its insensitivity to the addition of the LPS inhibitor polymyxin B sulfate, and its elimination by denaturation of HSP60 using heat, trypsin, or proteinase K. Instead, we identified HSP60 as the causative neurotoxic factor derived from injured CNS cells by demonstrating a highly similar effect induced by recombinant HSP60 and endogenous HSP60 derived from mammalian CNS cells. Finally, we found that neurotoxicity of HEK293 cell lysates was reduced after silencing of HSP60 using two different siRNA duplexes. Conversely, we showed that neuronal cell loss was increased when cell lysates from HSP60-overexpressing cells were used for neurotoxicity assays.
Although near complete abrogation of neuronal injury was observed after immunodepletion of supernatants from necrotic or apoptotic cells with HSP60-specific antibodies, some microglia-dependent injury still occurred. Likewise, silencing of HSP60 in HEK293 cells only led to a partial protection of neurons. Although the most likely explanation for these findings is that some HSP60 remained after immunoprecipitation and that gene silencing in HEK293 cells was not complete, it is possible that other endogenous ligands are released from injured CNS and HEK293 cells, which activate innate immunity. Additional endogenous ligands of TLR4, such as HSP70 and β-Defensin, have been identified (Ohashi et al., 2000; Vabulas et al., 2001; Biragyn et al., 2002). Although it has been reported that HSPs are released after necrotic but not apoptotic cell death (Basu et al., 2000), our data suggest that the difference is quantitative rather than qualitative. Nevertheless, we cannot completely exclude the possibility that CNS cultures treated with cycloheximide and TNF-α to induce apoptosis contain an undetected amount of necrotic cells that lead to release of HSPs.
During CNS development, a vast number of cells undergo apoptosis in competition for targets and growth factors without activating innate immunity (Sastry and Rao, 2000). The question of why an inflammatory response is initiated by apoptosis in the adult but not during development is not entirely clear. Because the resident innate immune system does not establish residency until the perinatal period, a local inflammatory response might not be possible until after this developmental event. Alternatively, it is possible that cells stressed because of various genetic and environmental insults (e.g., Huntington's disease, MPTP) that ultimately lead to apoptosis (Przedborski and Jackson-Lewis, 1998; Mattson, 2000) release factors that are not released during apoptosis in normal development.
In this report, we firmly establish a molecular mechanism by which CNS injury leads to release of HSP60, which in turn activates innate immunity in a TLR4- and MyD88-dependent manner. We thus propose a model, in which CNS injury activates innate immunity through the release of HSP60 and possibly other endogenous molecules. The consequences of an engaged innate immune response lead to additional neuronal injury and death. The implication of this model is that inflammation and neurodegeneration are bidirectionally linked through conserved macromolecules such as the HSPs and innate immune receptors such as the toll-like receptors.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB-TRR43/A1 (S.L.) and a grant from the Rahel Hirsch Program, Charité-Universitaetsmedizin Berlin (S.L.). We thank Drs. Hidde L. Ploegh and Olaf Ninnemann for their advice on the experimental design. We also thank Lena Smirnova for helpful discussions. MyD88-deficient mice were generated by Dr. S. Akira and were obtained through Drs. Douglas Golenbock and Hidde L. Ploegh.
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell. 1985;42:791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Wirenfeldt M, Holm T, Nielsen HH, Dissing-Olesen L, Toft-Hansen H, Millward JM, Landmann R, Rivest S, Finsen B, Owens T. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J Neurosci. 2006;26:12826–12837. doi: 10.1523/JNEUROSCI.4937-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- Björklund A, Lindvall O. Self-repair in the brain. Nature. 2000;405:892–893:895. doi: 10.1038/35016175. [DOI] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Chen W, Syldath U, Bellmann K, Burkart V, Kolb H. Human 60-kDa heat-shock protein: a danger signal to the innate immune system. J Immunol. 1999;162:3212–3219. [PubMed] [Google Scholar]
- del Rio Hortega P. Microglia. In: Penfield W, editor. Cytology and cellular pathology of the nervous system. Vol 2. New York: Hoeber; 1932. pp. 481–534. [Google Scholar]
- Fournier AE, Strittmatter SM. Regenerating nerves follow the road more traveled. Nat Neurosci. 2002;5:821–822. doi: 10.1038/nn0902-821. [DOI] [PubMed] [Google Scholar]
- Gao B, Tsan MF. Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem. 2003;278:22523–22529. doi: 10.1074/jbc.M303161200. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Barres BA. The relationship between neuronal survival and regeneration. Annu Rev Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- Hacker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med. 2000;192:595–600. doi: 10.1084/jem.192.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Horng T, Medzhitov R. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc Natl Acad Sci USA. 2001;98:12654–12658. doi: 10.1073/pnas.231471798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kol A, Bourcier T, Lichtman AH, Libby P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest. 1999;103:571–577. doi: 10.1172/JCI5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Nissl F. Über einige Beziehungen zwischen Nervenzellerkrankungen und gliösen Erscheinungen bei verschiedenen Psychosen. Arch Psychiatr. 1899;32:1–21. [Google Scholar]
- Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Perry V. Austin, TX: RG Landes; 1994. Macrophages and the nervous system. [Google Scholar]
- Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13:35–38. [PubMed] [Google Scholar]
- Quintana FJ, Cohen IR. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol. 2005;175:2777–2782. doi: 10.4049/jimmunol.175.5.2777. [DOI] [PubMed] [Google Scholar]
- Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry PS, Rao KS. Apoptosis and the nervous system. J Neurochem. 2000;74:1–20. doi: 10.1046/j.1471-4159.2000.0740001.x. [DOI] [PubMed] [Google Scholar]
- Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000;10:1139–1142. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- Schnurr M, Then F, Galambos P, Scholz C, Siegmund B, Endres S, Eigler A. Extracellular ATP and TNF-alpha synergize in the activation and maturation of human dendritic cells. J Immunol. 2000;165:4704–4709. doi: 10.4049/jimmunol.165.8.4704. [DOI] [PubMed] [Google Scholar]
- Somersan S, Larsson M, Fonteneau JF, Basu S, Srivastava P, Bhardwaj N. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol. 2001;167:4844–4852. doi: 10.4049/jimmunol.167.9.4844. [DOI] [PubMed] [Google Scholar]
- Stein D, Roth S, Vogelsang E, Nusslein-Volhard C. The polarity of the dorsoventral axis in the Drosophila embryo is defined by an extracellular signal. Cell. 1991;65:725–735. doi: 10.1016/0092-8674(91)90381-8. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Kreutzberg GW. Response of endogenous glial cells to motor neuron degeneration induced by toxic ricin. J Comp Neurol. 1988;268:248–263. doi: 10.1002/cne.902680209. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Termeer CC, Hennies J, Voith U, Ahrens T, Weiss JM, Prehm P, Simon JC. Oligosaccharides of hyaluronan are potent activators of dendritic cells. J Immunol. 2000;165:1863–1870. doi: 10.4049/jimmunol.165.4.1863. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–31339. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- Vartanian T, Li Y, Zhao M, Stefansson K. Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med. 1995;1:732–743. [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]