

Abstract
α-Synuclein (α-syn) has been implicated in the pathogenesis of many neurodegenerative disorders, including Parkinson's disease. These disorders are characterized by various neurological and psychiatric symptoms based on progressive neuropathological alterations. Whether the neurodegenerative process might be halted or even reversed is presently unknown. Therefore, conditional mouse models are powerful tools to analyze the relationship between transgene expression and progression of the disease. To explore whether α-syn solely originates and further incites these alterations, we generated conditional mouse models by using the tet-regulatable system. Mice expressing high levels of human wild-type α-syn in midbrain and forebrain regions developed nigral and hippocampal neuropathology, including reduced neurogenesis and neurodegeneration in absence of fibrillary inclusions, leading to cognitive impairment and progressive motor decline. Turning off transgene expression in symptomatic mice halted progression but did not reverse the symptoms. Thus, our data suggest that approaches targeting α-syn-induced pathological pathways might be of benefit rather in early disease stages. Furthermore, α-syn-associated cytotoxicity is independent of filamentous inclusion body formation in our conditional mouse model.
Keywords: conditional, α-synuclein, neurodegeneration, Parkinson's disease, mouse model, dark cells
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease, affecting ∼1% of the elderly population with a higher prevalence in men (Dluzen and McDermott, 2000). Clinically, PD is characterized by severe and progressing motor symptoms (bradykinesia, resting tremor, rigidity, and postural instability), which are mainly linked to the degeneration of dopaminergic neurons in the substantia nigra pars compacta, coupled with a depletion of dopamine (DA) and metabolites in the nigrostriatal projections (Bernheimer et al., 1973). PD-related motor symptoms are frequently predated by olfactory dysfunction (Doty et al., 1988) and in later stages accompanied by cognitive decline (Riekkinen et al., 1998; Bruck et al., 2004), suggesting an involvement of different brain areas in disease progression. Microscopically, proteinacious inclusions, termed “Lewy bodies” (LBs) are frequently detected in remaining nigral neurons of postmortem PD brains. The presence of these cytoplasmatic inclusions might be cytotoxic (Hurtig et al., 2000; Giasson et al., 2002; Braak et al., 2004), but there is increasing evidence that, in contrast, they might be cytoprotective by sequestering toxic protein structures (Gispert et al., 2003; Tanaka et al., 2004; Inden et al., 2005; Bodner et al., 2006). α-Synuclein (α-syn) was identified as the major filamentous component of LBs (Spillantini et al., 1998). The identification of three point mutations (A53T, A30P, and E46K) in the α-synuclein gene linked to autosomal dominant (AD) PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004) support a central role for α-syn in the pathogenesis of PD. Most importantly, duplications and triplications of wild-type (wt) α-syn have been identified as a cause of ADPD (Singleton et al., 2003; Chartier-Harlin et al., 2004) indicating that an overload of α-syn protein is sufficient to result in nigral cell loss. However, the pathomechanism underlying wt or mutated α-syn-induced cytotoxicity to the nigrostriatal dopaminergic system remains unclear so far. To elucidate the role of α-syn in the pathogenesis of PD, transgenic (tg) models were created overexpressing human α-syn in the brain. Some of theses models represented LB-like formations (Masliah et al., 2000; Kahle et al., 2001; Tofaris et al., 2006). However, α-syn tg mice failed to model neurodegeneration of the nigrostriatal system as an important precondition to use these models in preclinical trials. Furthermore, expression of all previously established models was permanent and did not answer the questions of whether α-syn is the solely elicitor and enhancer of dopaminergic cell death and whether disease progression can be halted or even reversed after onset of neuropathology by cessation of transgene expression.
Thus, we generated tet-off conditional tg mice expressing human wt α-syn under control of neuronal specific promoters. We demonstrate here that mice expressing human wt α-syn develop degeneration of neurons in the substantia nigra and hippocampus as well as reduced hippocampal neurogenesis, leading to progressive motor decline and cognitive impairment. These phenotypic alterations were independent of the formation of α-syn-positive fibrillary aggregates. More importantly, progression of the phenotype could be halted in aged symptomatic mice when α-syn expression was switched off.
Materials and Methods
Constructs.
Wt human α-syn cDNA (nucleotides 46–520; GenBank accession no. NM_000345) was cloned downstream of the tTA responsive promoter of the pUHD 10-3 expression vector (Gossen et al., 1995) into the XbaI restriction site. Mice expressing the tTA construct under control of calcium/calmodulin-dependent protein kinase IIα (CaMKIIα) promoter (Mayford et al., 1996) (designated CaM promoter in this study) or the hamster prion protein promoter (PrnP) have been described previously (Tremblay et al., 1998). The PrnP promoter is designated PrP promoter in this study.
Transgenic mice.
DNA fragments were injected into fertilized eggs at a concentration of ≈2 ng/μl. Tg mice were generated and founder mice were identified by Southern blot analyses according to standard procedures. α-Syn tg responder lines (α-syn and α-syn1) were established by crossbreeding with C57BL/6 mice. Offspring were analyzed by PCR analysis of tail DNA, using primers specific for the tTA sequence (for CaM- or PrP-expressing mice: tTA-F, 5′-GAC GAG CTC CAC TTA GAC GG-3′; tTA-R, 5′-TAC TCG TCA ATT CCA AGG GC-3′) or the tet-responsive element (for α-syn tg mice: pTRE-F, 5′-GAG ACG CCA TCC ACG CTG T-3′; pTRE-R, 5′-CAG TCT AGT TGT GGT TTG TCC A-3′), respectively. The relative number of DNA copies was estimated via quantitative real-time PCR (qPCR) on a LightCycler 2.0 (Roche, Mannheim, Germany) using a LightCycler FastStart DNA MasterPlus SYBR Green I kit (Roche) and mouse tail genomic DNA. Reactions were performed in a 20 μl mixture containing 10 pmol of each primer, 5 μl of 1:50 diluted DNA and 1× SYBR Green Mix (Roche). qPCR was performed in duplicate and normalized to a reference gene (β-actin; β-actin-F, 5′-CGTACCACAGGCATTGT-3′; β-actin-R, 5′-GATGTCACGCACGATTTC-3′). Primer sequences to detect copy number of the α-syn transgene were located in the pTRE sequence (qTRE-F, 5′-CGGGTCGAGTAGGCGTGTA-3′; qTRE-R, 5′-ACAGCGTGGATGGCGTCT-3′). The PCR amplification efficiency was determined for each gene using serial dilutions of genomic DNA. The relative amount of DNA copy number in two independent responder lines was calculated according to Pfaffl (2001).
To cease expression of the transgene (designated “gene off”), mice were treated with 2 mg/ml doxycycline (dox) (Sigma, Munich, Germany) in a 5% sucrose solution in the drinking water. For long-term treatment (>2 months), sucrose was weekly reduced in 0.1% steps until a final concentration of 0.2% sucrose was reached. All mice were kept in normal light/dark cycle (12 h light/dark) and had ad libitum access to food and water.
Light and electron microscopical immunohistochemistry.
Mice were anesthetized deeply with CO2 and perfused intracardially with 4% paraformaldehyde (PFA) in 0.1 m PBS. Brains were removed from the skull and postfixed overnight in the fixative, embedded in paraffin, and sagittally cut into 7 μm sagittal sections. After dewaxing in xylene and rehydrating in decreasing stages of ethanol, slides were microwaved for 15 min in 10 mm sodium citrate, pH 6.0, and washed with PBS. Endogenous peroxidase was blocked using 0.3% hydrogen peroxide. After blocking with 5% normal rabbit serum (NRS) in PBS and 0.3% Triton X-100, sections were washed with PBS, and the primary 15G7-α-syn antibody (1:2; diluted in PBS and 1% NRS) (Kahle et al., 2000) was added and incubated at room temperature (RT) for 2 h. The secondary antibody was coupled with biotin (Vector Laboratories, Burlingame, CA), diluted 1:200 in PBS and 1% NRS, and added after washing the slides with PBS. After incubation for 1 h and washing with PBS, an avidin–biotin enhancer complex coupled with peroxidase (ABC Elite; Vector) was added and incubated at RT for 2 h. After washing with PBS, 3,3′-diaminobenzidine (DAB) (Sigma) was added, and the reaction was stopped in double-distilled water after 8 min. Slides were lightly counterstained with hematoxylin (Merck, Darmstadt, Germany), dehydrated, and mounted using CV mount medium (Leica, Bensheim, Germany).
For detailed light and electron microscopical analysis, mice were deeply anesthetized with pentobarbital (40 mg of Nembutal, i.p.) and transcardially perfused by a 4% PFA. Fixed brains were removed from the skull, coronally cut into 50 μm vibratome sections, and immunostained as recently described (Petrasch-Parwez et al., 2007). Briefly, after reduction and blocking, sections were incubated for 72 h at 4°C with anti-calbindin D-28k (1:2000) (CB-38a; Swant, Bellinzona, Switzerland), anti-tyrosine hydroxylase (TH) (1:1000) (AB 152; Chemicon, Hofheim, Germany), or 15G7-α-syn (1:10) (Kahle et al., 2000) in 10% normal goat serum or 10% NRS. After rinsing in PBS, sections were placed in biotinylated goat anti-rabbit (1:1000) (Vector) or rabbit anti-rat (1:500) (Vector) in 2% bovine serum albumin for 24 h at 4°C. Finally, sections were incubated in ABC (1:1000 in PBS) and visualized with DAB. For light microscopical analysis, sections were viewed using an Axioplan 2 imaging microscope (Carl Zeiss, Oberkochen, Germany) equipped with an AxioCam HR color digital camera (Zeiss) and the AxioVision 4.3 software package (Zeiss).
Sections for electron microscopical analysis were immunostained with omission of Triton X-100 and incubation of 15G7-α-syn antibody (1:2), postfixed with 2% osmium tetroxide, dehydrated, and flat-embedded in Araldite (Serva, Heidelberg, Germany). Semithin (0.75 μm) and ultrathin sections (100 nm) were cut with a Leica Ultracut UCT microtome and stained with 1% toluidine blue, pH 9.0. Ultrathin sections were contrasted with 5% aqueous uranyl acetate and lead citrate, pH 12. Semithin sections were documented by a DP71 camera (Olympus, Münster, Germany) and mounted on an Olympus light microscope photodocumented and measured by the analysis soft imaging system.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed on 50 μm sections of 110-week-old CaM_α-syn and corresponding control mice according to the manufacturer's instructions (Promega, Madison, WI). Staining efficiency was controlled using thymus sections as controls, which yielded the expected results (data not shown).
Cell counting.
Number of TH-positive cells were estimated with systematic random sampling of about every second section to yield 9–11 sections per mouse per substantia nigra (SN). TH-reactive neurons of four control and four CaM_α-syn mice (aged ∼110 weeks) were counted. An optical fractionator was used to estimate total cell number (West, 1993). Region definitions were based on the Paxinos and Franklin (2001) mouse brain atlas, and stereological analysis was done as described previously (Maetzler et al., 2007). Cells undergoing dark cell degeneration can be identified either in toluidine blue-stained semithin sections or electron microscopically in ultrathin sections. The areas inspected were measured by the analysis soft imaging system and comprised the substantia nigra pars compacta and pars reticulata of two CaM_α-syn mice (9 and 20 months of age), and two corresponding 9-month-old control brains. Dark and healthy neurons (see Fig. 5 H) were quantified on 10 semithin sections of the respective areas, all at bregma level, ∼5.4–5.6 mm, according to Paxinos and Franklin (2001). All counted sections were at least 7.5 μm apart from each other, and only healthy lightly stained neurons with a distinct nucleus and darkly toluidine-blue stained cells were counted.
Figure 5.
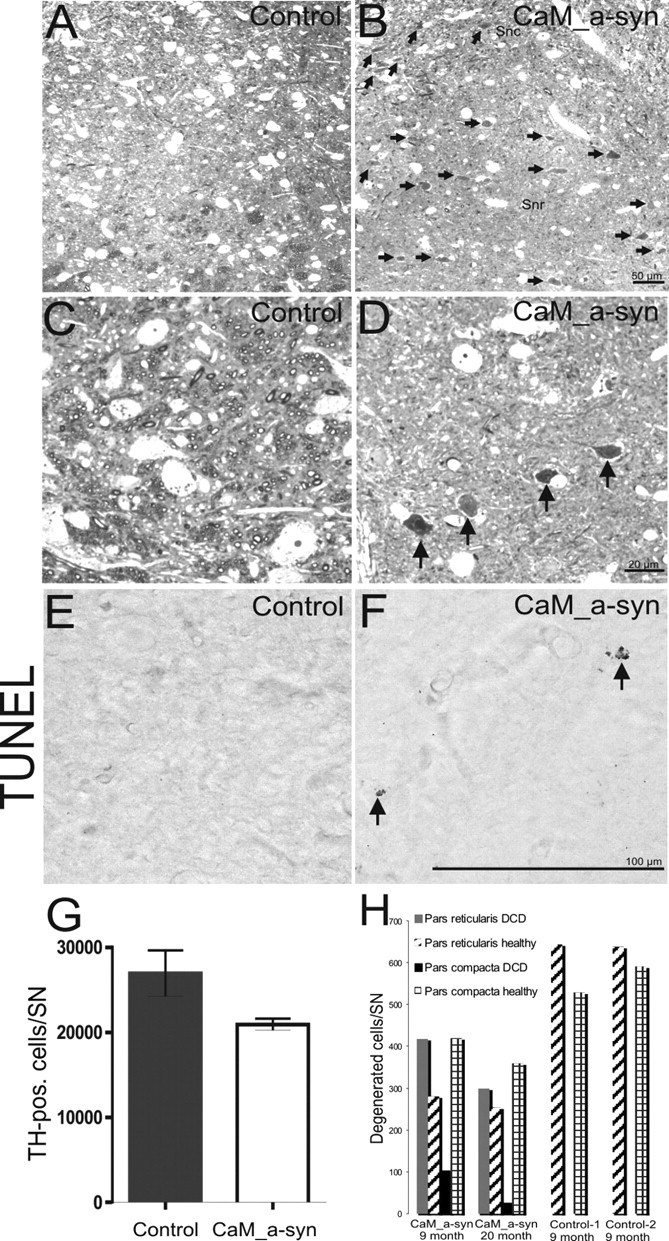
Nigral cell death in CaM_α-syn mice. A, B, Toluidine blue-stained sections of a 9-month-old control mouse (A) and a CaM_α-syn littermate (B) showed numerous dark cells (arrows) in the pars reticulata (SNr) in the tg α-syn mouse that were absent in the control. Various dark cells were also distributed among healthy neurons in the pars compacta (SNc). C, D, Higher enlarged pictures of the pars reticulata of the control showed the large lightly stained healthy neurons (C) and the shrunken dark degenerated cells (arrows) in the respective area of the CaM α-syn mouse (D). F, Single apoptotic cells (arrows) were detected in CaM_α-syn mice, were absent in control mice (E). G, Graph showing stereological counting of TH-positive SN neurons revealed a strong tendency of reduction, bordering significant level (p = 0.07). Error bars indicate SEM. H, Counting of dark cells in two CaM α-syn mice revealed a high amount of dark cells (DCD) in the substantia nigra pars reticulata and a minor amount of dark cells in the pars compacta. In both controls, dark cells were missing.
Double-labeling immunofluorescence.
After washing with PBS and blocking in 10% normal donkey serum (NDS) containing 0.3% Triton X-100 in PBS for 1 h, 20-μm-thick free-floating cryosections were incubated with 15G7-α-syn (1:5) for 24 h, and then washed with PBS, and subsequently incubated with anti-TH (1:500) for 24 h at 4°C. After subsequent washes, sections were labeled using Cy-2-coupled anti-rabbit antibody (1:200; 2 h; RT), washed again, and incubated in Cy-3-coupled anti-rat antibody (1:200; 2 h; RT) secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA), each diluted in PBS and 3% NDS. After washing with PBS, sections were mounted in Vectashield DAPI (4′,6′-diamidino-2-phenylindole) mounting medium (Vector). Sections were analyzed using a Nikon light microscope (Eclipse 80i; Nikon, Tokyo, Japan), Nikon objectives (Plan Apo, VC 2×, NA, 1.0; Plan Apo, VC 40×, NA, 1.1), motorized specimen stage for automatic sampling (Märzhäuser, Wetzlar, Germany), electronic microcator (Heidenhain, Traunreut, Germany), a dedicated Nikon HiSN fluorescence system, a Nikon cooled DS-5Mc camera, and imaging software (Stereo Investigator; MicroBrightField, Williston, VT).
Sequential extraction of α-syn.
Subcellular fractionation of human cerebral cortex samples and whole mice brains was performed as described previously (Tofaris et al., 2006). Briefly, human tissue was homogenized in 10 vol and mouse tissue in 5 vol of TBS+ (Tris-buffered saline plus Complete protease inhibitor; Roche Diagnostics, Mannheim, Germany) and spun for 30 min at 120,000 × g. The resulting supernatants represented the TBS+ soluble fraction. The pellet was then extracted sequentially with 1 ml of TBS+ containing 1% of Triton X-100, and TBS+, 1 m sucrose, after extraction with RIPA buffer (50 mm Tris-HCl, pH 7.4, 175 mm NaCl, 5 mm EDTA, 1% NP-40, and 0.5% sodium deoxycholate) and 0.1% SDS. The detergent-insoluble pellet was solubilized in 8 m urea/5% SDS.
Study of neurogenesis.
CaM_α-syn mice were compared with littermate controls and dox-treated gene-off CaM_α-syn mice (16 weeks on/8 weeks off) and respective treated controls. For details of experimental design, see Table 1. All tested animals were 16 weeks old at the beginning of the experiment. To determine the impact on neurogenesis, these mice received daily intraperitoneal injections of bromodeoxyuridine (BrdU) (50 mg/kg; Sigma) for 5 consecutive days starting at 20 weeks of age and were killed 4 weeks after the BrdU injections. Immunohistochemistry and double-labeling immunofluorescence with antibodies against BrdU, proliferating cell nuclear antigen (PCNA), doublecortin (DCX), and NeuN, and quantification of immunopositive cells was performed as previously described (Winner et al., 2004). Statistical analysis was performed using the one-way ANOVA and post hoc Bonferroni's multiple-comparison test (Prism; Graph Pad Software, San Diego, CA). The data are expressed as mean values ± SEM. A value of p < 0.05 was considered significant.
Table 1.
Experimental design

Top, Injection paradigm and doxycycline treatment (gene off) of conditional CaM_α-syn mice and respective controls. Bottom, Cell number and fate of newborn cells in the hippocampal dentate gyrus. Data are presented as mean ± SEM. BrdU, Bromodeoxyuridine. *p < 0.05 compared with control mice; + p < 0.05 compared with CaM_α-syn mice.
Quantification of DA, 3,4-dihydroxyphenylacetic acid, homovanillic acid, serotonin, and 5′-hydroxyindoleacetic acid.
Untreated CaM_α-syn mice (gene on), treated CaM_α-syn mice (gene off), and corresponding controls were analyzed for postmortem tissue content of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), serotonin, and 5′-hydroxyindoleacetic acid (5-HIAA). After cervical dislocation, the brain was rapidly removed and placed on an ice-cooled plate for selective dissection of the main olfactory bulb, striatum, and substantia nigra. Brain samples were sonicated in 20-fold 0.1 m perchloric acid and centrifuged at 23,000 rcf for 30 min at 4°C. A total of 20 μl of supernatant was injected onto a C18 column (Dionex, Germering, Germany). The mobile phase, pH 4.3, consisted of 90% 50 mm sodium acetate, 35 mm citric acid, 105 mg/L octane sulfonic acid, 48 mg/L sodium EDTA solution, and 10% methanol. Flow rate was 1 ml/min. Peaks were detected by an ED50 electrochemical detector (Dionex), and the detector potential was set at 700 mV. Data were collected and processed using the Chromeleon computer system (Dionex). Data are reported as mean ± SEM. Statistical analysis was done using Student's t test for pairwise comparisons and ANOVA with post hoc Newman–Keuls test. A value of p < 0.05 was considered significant.
MicroPET studies.
Five untreated CaM_α-syn mice, three respective treated CaM_α-syn, and seven control mice were used for positron emission tomography (PET) imaging studies. Mice were anesthetized with ether while placing a 30 ga catheter in the tail vein, which had a dead volume of <100 μl. To avoid blood coagulation and for flushing, it was filled with a 2% heparine/NaCl mixture. During imaging, mice were anesthetized with 1.5% isoflurane (Vetland Anesthesia System, Louisville, KY) at a flow rate of 0.8 L/min oxygen. PET imaging studies were performed with a microPET FOCUS 120 scanner (Siemens Preclinical Solutions, Knoxville, TN) yielding a spatial resolution of ∼1.3 mm in the reconstructed image. Data were acquired in list mode over 3600 s and histogrammed into 64 time frames. Mice were injected with [11C]d-threo-methylphenidate (11C-MP), which was synthesized by alkylation of the free acid of N-protected d-threo-methylphenidate using [11C]methyliodide (Ding et al., 1994a). High specific activity [11C]CH3I was prepared in an automated module (PETtrace MeI Microlab; GE Healthcare, Uppsala, Sweden). The radiolabeling was performed in a PET tracer synthesizer for 11C methylations from GE Healthcare. The total synthesis time was 60 min, and specific activities of 75–140 GBq/μmol at end of synthesis were obtained. The mean injected activity of 11C-MP was 22 ± 18 MBq, which corresponds to a mean carrier of 959 ± 1276 pmol, whereas the maximum was <5445 pmol to avoid saturation effects. The injected tracer volume was between 25 and 100 μl. Data were acquired in fully three-dimensional (3D) mode, and images were reconstructed using filtered back projection with a matrix size of 128 × 128, and a zoom of 3. A standard 3D region of interest (ROI) for the striatum was applied to all mice. This standard ROI was created manually (AsiPro software; Siemens) with the aid of isocontours selected to yield a diameter of 2 mm for the striatum in accordance with the mouse stereotaxic atlas (Paxinos and Franklin, 2001). 3D-ROIs of the striatum and the cerebellum were used to generate time–activity curves (TACs) for quantitative analysis. Kinetic receptor modeling was applied using Logan-Plots (Logan, 2000) to determine the modified binding potential for dopamine transporter (DAT). Statistical analysis was performed using the unpaired two-tailed Student's t test. For power analysis, an α value p = 0.05 and 80% power was used (JMP 5.1 software). The data are expressed as mean values ± SEM. A value of p < 0.05 was considered significant.
Western blot analysis.
Mice were killed by CO2 and tissues were frozen at −80°C. Total protein extracts were obtained by homogenization in TES buffer (50 mm Tris, pH 7.5, 2 mm EDTA, 100 mm NaCl) supplemented with a mixture of protease inhibitors (Complete; Roche Applied Science, Mannheim, Germany). After the addition of Nonidet NP-40 (Sigma) to a final concentration of 1%, and incubation at 4°C for 30 min, homogenates were centrifuged at 20,000 rcf for 20 min at 4°C. The cleared supernatant was supplemented with glycerol and stored at −80°C. Protein extracts (20 μg each) were fractionated on 17% SDS-PAGE and blotted on a nitrocellulose membrane (Whatman, Dassel, Germany). Immunoblots were blocked in 5% dry milk in TBST buffer (10 mm Tris, pH 7.5, 0.15 m NaCl, 0.1% Tween 20) and subsequently probed with 15G7-α-syn antibody (1:5) or 7544-anti-α-syn antibody (1:200) (Kahle et al., 2000) or Mc42 (1:3000) (Transduction Laboratories, Lexington, KY). Anti-β-actin (1:10,000) (A4700; Sigma) was used as internal control. Bound antibodies were visualized with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (ECL or ECLplus; GE Healthcare) and exposure to hyperfilm (GE Healthcare). ImageQuant software 5.0 (GE Healthcare) was used to determine the optical density of protein bands, and all data were normalized to the expression of β-actin.
Rotating rod and motor skill analysis.
Motor training was performed at the beginning of the dark phase of the daily cycle. Body weight was recorded every 6 weeks at the beginning and the end of the session. Eight CaM_α-syn mice and eight corresponding α-syn single-tg controls were tested on an accelerating rotarod (TSE, Bad Homburg, Germany). The speed of the rod was set to increase from 4 to 40 rpm over a 5 min period. The latency to fall from the rod was measured in two trials per day over a 5 d period, after a training session over 5 successive days (two trials per day). Each test trial lasted for a maximum of 6 min. At least 30 min recovery time was allowed between trials. The data for each group of animals was averaged. Assessment of motor ability was first measured in 18-week-old untreated mice and was repeated every 6 weeks over a 30 week period. Then, all tested mice were given 2 mg/ml dox through drinking water and tested again over a 16 week period.
Statistical data analysis was performed using JMP 5.1 (SAS Institute, Cary, NC). Data are presented as mean ± SEM. The sex ratio within the four groups was compared by a contingency analysis. Effects of the factor “group” and the factor “sex” and their interaction on the overall mean motor performance were assessed by a multifactorial ANOVA. A linear regression analysis was used to analyze the time course of motor performance between sessions in both the tg group and the control group. Linear regression was calculated using either the formula y = b + m 1 t for t < T and y = b + m 1 T + m 2(t − T) (for animals having a differential slope between testing sessions: t > T) or y = b + mt (for animals having no differential slope between testing sessions). A t test for paired observations was used to compare the slopes within the groups before and after treatment. A two-sample t test was chosen to test for differences in the time course of performance between the two groups. Linear regression was used for comparison of weight increase between the tested groups. The slopes and the intercepts of the tg and the control mice were compared with a two-sample t test. To test for differences between motor skills learning curves of tg mice and controls, a paired t test was used. A value of p < 0.05 was considered significant.
Morris water maze experiments.
Four female 52-week-old CaM_α-syn mice and respective control littermates were tested for their ability of spatial learning and long-term memory by performing Morris water maze (MWM) behavioral experiments as described by Winkler et al. (2000); for details of experimental design, see Figure 9 C. Briefly, for data analysis, the pool (diameter, 112.5 cm) was divided into four equal quadrants (southwest, southeast, northwest, northeast). The escape platform (10 × 10 cm2) was positioned in the center of quadrant 3 and submerged 1 cm under the water surface. For cued training, platform position was marked with a small colored flag, extending 7 cm above the water surface. For learning, mice were trained to find the hidden and unmarked platform (conventional reference memory MWM training) (Janus, 2004) for 5 d (two trials per day) and were allowed to remain on the platform for 30 s after each trial. The intertrial intervals were 240 s. For assessment of spatial memory, a probe trial was performed 24 h after the final trial of the learning session; the platform was removed from the pool and the path that the mice swam in a period of 60 s was recorded. The retention was performed on day 13 (two trials, each 60 s). After the hidden platform version of the MWM, mice were tested in the cued reference memory MWM test (cued maze) for 2 d (two trials per day) to exclude vision deficits. To assess working memory, the hidden escape platform was changed on day 19 from southwest to northeast (platform switch) and mice were tested in four trials, each lasting 60 s. To prevent the usage of nonspatial searching strategies, the release points for the mice were counterbalanced in the pool for each trail. The data are expressed as mean values ± SEM. Statistical analysis was performed using two-way ANOVA repeated measurement (Prism; Graph Pad Software). The significance level was set at p < 0.05.
Figure 9.

Motor dysfunction affected motor skill learning and cognitive impairment in CaM_α-syn mice. A, To test motor coordination, the time of CaM_α-syn (n = 8) and respective age- and sex-matched α-syn single-tg control mice (n = 8) to stay on an accelerated rotarod was measured in sessions, consisting of two trials for 5 d, every 6 weeks. Accelerated rotarod documented impaired motor performance in CaM_α-syn mice (open circles) compared with littermate controls (filled circles). Progressive motor impairment started at the age of 30 weeks documented by linear regression, which revealed decreasing time endurance on the rotarod of −13.4 s of the following four sessions (mean slope), which was significantly worse compared with the performance of the control group. Doxycycline treatment to cease expression of the human α-syn is indicated by arrow (gene off). Linear regression analysis revealed halting of progressive motor impairment after cessation of the transgene in symptomatic 58-week-old tg mice, indicated by amelioration of calculated slope to −4.2 s per session. No significant difference in body weight was observed between the CaM_α-syn mice and control group. B, To assess motor skill learning, the difference in time endurance of balancing on the accelerated rotarod between trial 1 and trial 2 of the first tested day in session 1 of 18-week-old CaM_α-syn and control mice was analyzed. Significant improvement was documented only in control mice, revealing impaired motor skill learning in CaM_α-syn mice. C, MWM paradigm: CaM_α-syn tg mice (n = 4) and control mice (n = 4) first received 5 d of cued training (2 trials per day), in which they learned locating a hidden platform to escape from the water. Then, spatial learning was assessed in one probe trial in which the platform is not present. Retention was tested 7 d after the initial learning period (2 trials). To exclude vision impairment, a cued water maze paradigm was performed for 2 d. Working memory was assessed by switching the platform location. D, No significant difference in learning session was found between the two groups. E, After a 7 d retention, CaM_α-syn tg mice demonstrated impaired function of long-term memory by spending significantly more time in the water to find the hidden platform compared with the control group. Error bars indicate SEM. *p < 0.05.
Results
Generation of conditional models of PD
We generated conditional mouse models of PD using the tetracycline-regulated tet-off system (Gossen and Bujard, 1992). As multiplication of the wt α-syn locus is sufficient to cause PD in human individuals (PARK4) (Singleton et al., 2003; Chartier-Harlin et al., 2004), tg mouse lines were generated overexpressing the human wt α-syn gene. Two responder lines (designated α-syn and α-syn1) were chosen to establish stable lines by crossbreeding with C57BL/6 mice. By using quantitative real-time PCR of tail genomic DNA, we did not find differences in copy number of the integrated transgene between these two responder lines.
For inducible expression of human α-syn in tg mouse brain, an additional mouse line was required expressing the tetracycline-controlled transactivator (tTA) tg under the control of a neuron-specific promoter. Subsequently α-syn tg mice were crossbred with either a prion-protein promoter line (designated PrP) (Tremblay et al., 1998) or a calcium/calmodulin-dependent protein kinase IIα promoter line (designated CaM) (Mayford et al., 1996) (Fig. 1 A). Heterozygote crossing of the CaM line or the PrP line with mice of either the syn or the syn1 line, yielded among single-tg mice also double-tg mice (designated CaM_α-syn or CaM_α-syn1, PrP_α-syn or PrP_α-syn1). To abolish expression of the human α-syn gene, double-tg mice were treated for a minimum of 30 d with 2 mg/ml dox administered to the drinking water.
Figure 1.

Conditional mouse design and expression pattern of human α-syn in brains of tg mice. A, tTA expression was either driven by the PrP or the CaM promoter. In its active form, tTA binds to the tetO7 sequence of the minimal promoter and initiates expression of the downstream localized human α-syn gene. Expression can be inhibited by the presence of dox, because it binds to tTA, making it inactive. B, Subregional brain dissection for Western blot analysis. The reconstructed atlas template is according to the stereotaxic atlas (Paxinos and Franklin, 2001). C, Western blot analyses (20 μg per lane) of human α-syn in dissected tg mouse brains using the 15G7-α-syn antibody and β-actin as internal loading control. After administration of dox for 3 weeks (gene off), reduced protein levels in adult double-tg mice of each line were observed. Human brain was used as positive control. D, Band intensities were quantified using ImageQuant software and normalized to the β-actin staining in the same line to correct for variations in loading. After normalization, the amount of human α-syn was calculated in percentage relative to expression intensity of the human brain signal. +, Unspecific background staining of membrane. Ob, Olfactory bulb; Cx, cortex; Bg, basal ganglia; Ce, cerebellum; Bs, brainstem; Hu, human brain.
Expression pattern of tg α-syn in conditional mice
To assess tg α-syn expression, brains of double-tg mice were dissected in five subregions (Fig. 1 B). α-Syn expression level was calculated by quantifying α-syn signals on Western blots in relation to expression signals of human α-syn obtained from human prefrontal cortex (positive control) (Fig. 1 D). β-Actin was used as control for equivalent loading of protein extracts. In PrP_α-syn1 mice, expression was limited to the olfactory bulb, whereas PrP_α-syn mice showed weak signals in the olfactory bulb, cortex, basal ganglia, and cerebellum (Fig. 1 C). Expression was particularly enhanced by using the CaM promoter line. CaM_α-syn mice showed elevated levels of human α-syn in the cortex and basal ganglia (Fig. 1 D). However, expression of human α-syn was absent in the cerebellum of these mice (Fig. 1 C). Compared with PrP_α-syn1 mice, CaM_α-syn1 mice presented enhanced expression in olfactory bulb, cortex, and basal ganglia (Fig. 1 D). The signal was specific to the human α-syn protein because brain tissues of non-tg controls and of α-syn knock-out mice (Abeliovich et al., 2000) did not display any signal in the expected regions (data not shown). In the olfactory bulb, the relative levels of overexpression of human α-syn, compared with the mouse endogenous α-syn levels were 0.6-fold for PrP_α-syn, 1.5-fold for PrP_α-syn1, 2.0-fold for CaM_α-syn, and 3.3-fold for CaM_α-syn1 mice (data not shown). We then determined whether expression of the human α-syn gene could be abolished by administration of dox in the drinking water. Western blot analyses revealed that dox decreased tg protein levels up to 94% depending on both the specific brain region and the expression intensity detected in protein extracts of untreated mice. Dox treatment ceased expression of human α-syn best in the olfactory bulb (≈94%) and slightly less efficient in the basal ganglia, cortex, and cerebellum (≈70%) and in the brainstem (≈50%) (Fig. 1 D). Although we detected some residual human α-syn in treated CaM_α-syn mice (Fig. 1 C), long-term treatment (∼60 d) was able to fully downregulate expression in mice older than 12 months (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
We focused our analyses to PrP_α-syn and especially on CaM_α-syn mice, because these mice showed similar regulation pattern by dox treatment in comparison with the respective PrP_α-syn1 or CaM_α-syn1 mice but showed much higher tg expression.
In these lines, α-syn expression was immunohistochemically substantiated by regional α-syn immunostaining (Fig. 2). CaM_α-syn mice revealed strong immunoreactivity in the olfactory bulb, olfactory tubercle, cortex, caudate–putamen, globus pallidus, hippocampus, thalamus, and substantia nigra (Fig. 2 A–C). PrP_α-syn mice showed a similar, but much weaker α-syn expression. Additional tg α-syn staining was observed in the molecular and granular layers of the cerebellum (Fig. 2 D–F). In the olfactory bulb, PrP_α-syn mice showed expression limited to the glomerular layer (Fig. 2 D), whereas CaM_α-syn mice revealed more prominent staining of the granular layer (Fig. 2 A). The more sensitive immunohistochemical staining of double-tg mice of both lines treated with dox for 30 d showed that turning off transgene expression in adult mouse brain (aged >3 months) was less effective than dox administration from birth (Fig. 2, compare G–L for treated adult mice with M–R for dox application from birth on). In treated adult CaM_α-syn mice, human α-syn expresssion was abolished in neuronal cell bodies of the hippocampal pyramidal cell layer and the cortex. However, residual staining was still detectable in neuropil of the cortex, the molecular layer of the dentate gyrus (Fig. 2 H), and the substantia nigra (Fig. 2 I). Similarly, in treated adult PrP_α-syn mice, residual expression was observed in the cerebellar molecular layer (Fig. 2 K), the thalamus (Fig. 2 J), and the substantia nigra (Fig. 2 L). This residual expression was not attributable to leaky expression because mice single-tg for the human α-syn showed no transgene expression (data not shown). Additionally, when mice were born and raised with dox, tg α-syn expression was turned off in all brain regions of CaM_α-syn (Fig. 2 M–O) and PrP_α-syn mice (Fig. 2 P–R), respectively.
Figure 2.

Distribution of human α-syn expression in treated and untreated conditional tg mice. Sagittal sections showed regional immunostaining for human α-syn in the brain of adult untreated CaM_α-syn mice (A–C) and PrP_α-syn mice respectively (D–F). A, Strong immunoreactivity was detected in the olfactory bulb (Ob), olfactory tubercle (Tu), cortex (Cx), caudate–putamen (CPu), globus pallidus (GP), substantia nigra (SN), thalamus (Th), and hippocampus (Hc) in CaM_α-syn mice. B, In the CA1 area of the Hc, α-syn immunoreactivity was observed in a subpopulation of the pyramidal cells (PY) (arrows), various dendrites in the stratum radiatum (RAD) (arrowhead), and in the neuropil of the molecular layer (mol). C, Intense α-syn staining was observed in both the substantia nigra pars compacta (pc) and pars reticulata (pr). D, PrP_α-syn mice displayed minor expression in the Ob, the CPu, the GP, the Hc, and the pr in comparison with CaM_α-syn mice. Additional staining was observed in the cerebellum (Ce). E, Expression of α-syn was detected in the molecular layer (MOL) and in the granular layer (GL), without staining of the Purkinje cell layer (P). F, Staining was prominent in the pc but less expressed in the pr. Doxycycline treatment over a 3 week period of adult CaM_α-syn mice (G–I) and adult PrP_α-syn mice (J–L) partly ceased transgene expression. H, I, In CaM_α-syn mice, treatment resulted in complete loss of immunoreactivity in the stratum oriens (OR), PY, and RAD of Hc, whereas the mol, the overlying Cx (H), and neuronal processes of the SN (I) were still stained, although less prominent. J–L, In treated PrP_α-syn mice, residual α-syn expression was detected in the Th (J) but abolished in the GL (not in the MOL) of the Ce (K) and reduced in the pc and pr of the SN (L). α-Syn staining was missing in CaM_α-syn mice (M–O) and PrP_α-syn mice (P–R), when mice were born and raised (over a minimum of 8 weeks) with dox. Scale bar: B, C, E, H, I, K, N, O, Q, R, 20 μm; A, D, G, J, M, P, 200 μm; F, L, 5 μm.
CaM_α-syn mice revealed expression of human α-syn in the nigrostriatal system at levels comparable with endogenous α-syn of the midbrain-region of control mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To investigate the tg α-syn expression in dopaminergic neurons of these mice, the olfactory bulb and the substantia nigra were further studied by double-fluorescence immunohistochemistry. Punctate α-syn staining was detected in the glomerular layer of the olfactory bulb (Fig. 3 A) (for higher magnification, see Fig. 3 E), an area that comprises TH-positive neurons (Fig. 3 B) (for higher magnification, see Fig. 3 F). Ultrastructural analysis of the glomerula revealed tg α-syn in unmyelinated axons (Fig. 3 D) and in synaptic terminals (Fig. 3 H), which was in agreement with the positive spots shown by fluorescence staining (Fig. 3 E). Consistently, we did not find somatic colocalization with TH (Fig. 3 C) (for higher magnification, see Fig. 3 G). The strong α-syn fluorescence labeling in the substantia nigra (Fig. 3 I) (for higher magnification, see Fig. 3 M) corresponded to a high amount of α-syn-positive dendrites, myelinated and unmyelinated axons as observed in semithin (Fig. 3 P) and ultrathin sections (Fig. 3 L) of the respective area. Additionally, α-syn was colocalized with TH (Fig. 3 J) (for higher magnification, see Fig. 3 N) in nigral dopaminergic neurons (merged: Fig. 3 K,O) presenting with immunoreactive spots of varying size, also observed in semithin sections (Fig. 3 P). Electron microscopical analysis detected α-syn-positive patches in the cytoplasm often associated with lysosomes or mitochondria (data not shown). However, immunopositive patches as investigated ultrastructurally were not composed of filaments. They therefore closely resembled microaggregates and not LB-like structures, in particular because α-syn-positive patches did not stain with anti-ubiquitin antibodies (data not shown).
Figure 3.
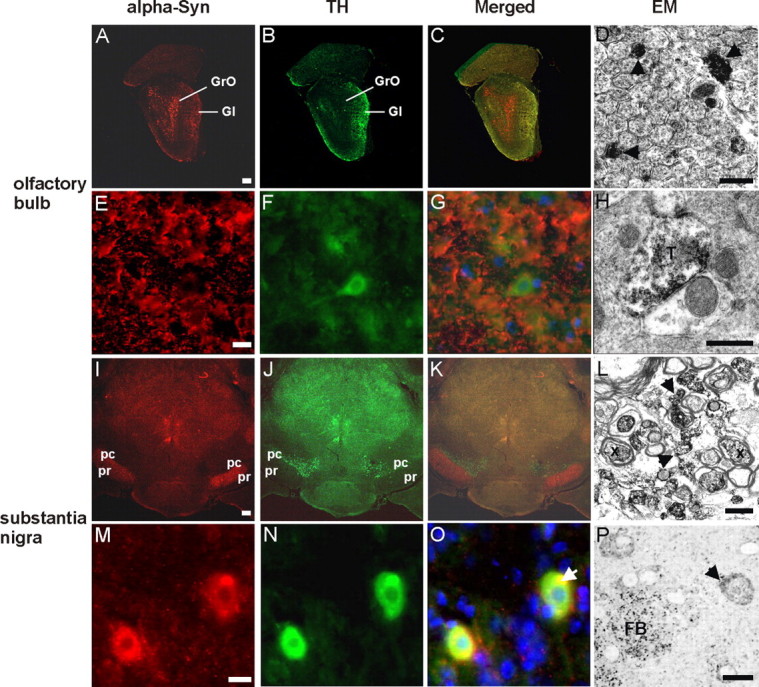
Confocal images of α-syn and TH double-staining immunofluorescence and electron microscopy of α-syn expression in the olfactory bulb and the substantia nigra. Frontal brain sections of 20-month-old CaM_α-syn mice were double-labeled against α-syn (red) and TH (green) to detect transgene expression in dopaminergic structures of the olfactory bulb (A–H) and substantia nigra (I–P). A, α-syn staining was detected in the granular (GrO) and, to a minor extent, in the glomerular layer (Gl) of the olfactory bulb; the latter contains also TH-positive structures (B). C, Merged image shows a partial overlap of α-syn expression and TH in the Gl. E, F, Higher magnification of the Gl revealed prominent punctate α-syn immunostaining of the neuropil (arrows) (E) and TH immunoreaction also in neuronal somata (F). G, Merged image shows no colocalization of α-syn and TH-positive neurons, but a moderate colocalization in the neuropil. D, H, Electron microscopy of the Gl confirmed α-syn accumulation in the neuropil localized in unmyelinated axons (D, arrows) and synaptic terminals (T) (H). I, In the SN, α-syn was most prominent in the pars reticulata (pr) and less expressed in the pars compacta (pc), which is comprised of TH-positive cells (J). K, Merged image illustrated overlapping of α-syn and TH staining in the intersectional area between pc and pr. M, Higher magnification of this area detected α-syn in neuronal somata with accumulations varying in size, in some cases being colocalized with TH (N) as clearly identified in the merged image (O, arrow). L, Electron microscopy of the intersectional area revealed α-syn in myelinated (X) and unmyelinated (arrows) axons. P, Semithin section confirmed different accumulation sizes of α-syn in neuronal somata (arrow) and in fiber bundles (FB). Scale bar: A–C, I–K, 1 mm; E–G, M–O, 100 μm; D, H, L, 500 nm; P, 1 μm.
Consistently, sequential extraction of human α-syn from whole brain of a 12-month-old and an 18-month-old tg mouse revealed tg signals in cytoplasmic (TBS) and membrane (T/T) fractions. However, urea-soluble extracts were devoid of tg α-syn (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Electron microscopy of neuropathology in CaM_α-syn mice
In CaM_α-syn mouse brains, strong tg α-syn immunoreactivity was prominent in cell processes of the substantia nigra pars compacta and pars reticulata (Fig. 4 B). The subregions were differentiated by adjacent TH-stained sections (Fig. 4 A). Electron microscopical analysis of this region detected degenerated neurons with darkened nuclear and cytoplasmic appearance, slightly collapsed nuclear envelope, and increased accumulation of lysosomes (Fig. 4 C). Large lipid-like droplets were identified between nerve fibers of the pars reticulata often associated with α-syn-positive axons (Fig. 4 D). Several dystrophic myelinated (Fig. 4 E) and unmyelinated nerve fibers (Fig. 4 F) were filled with electron-dense organelles and dark condensed mitochondria. α-syn-positive patches dispersed within these dystrophic axons suggest a close association of degenerating structures with the transgene (Fig. 4 F).
Figure 4.
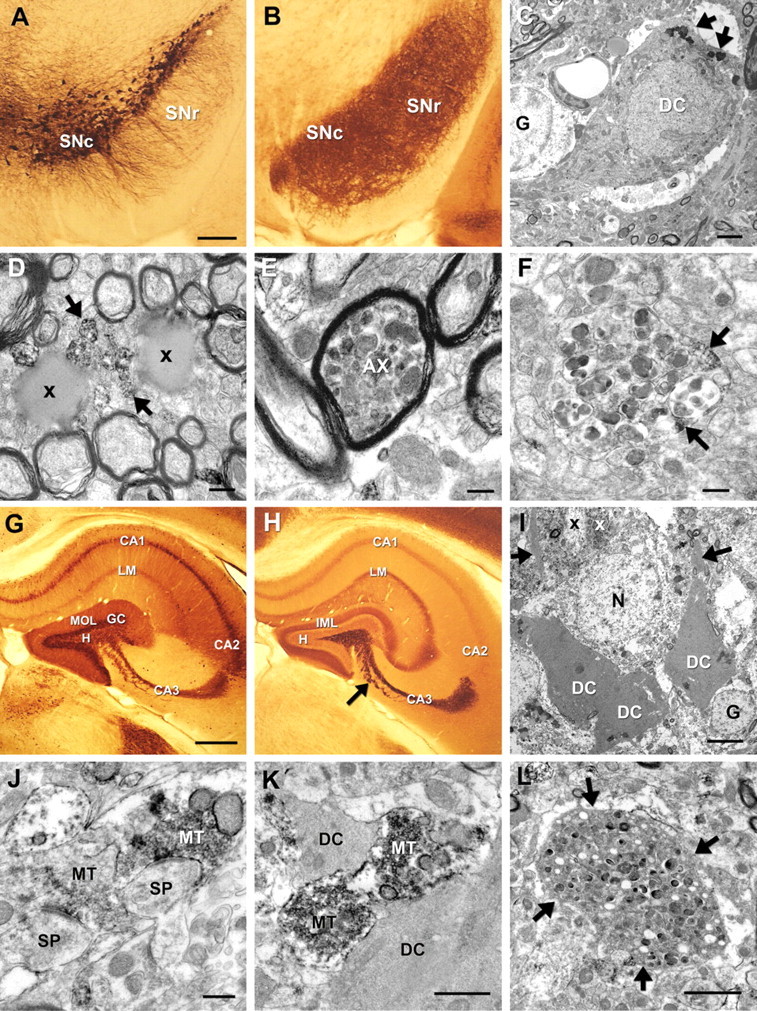
Immunoelectron microscopy of neurodegeneration and associated α-syn in 20-month-old CaM_α-syn mice in the substantia nigra and hippocampus. A, TH-stained vibratome section showed dopaminergic neurons in the substantia nigra pars compacta (SNc) and their cell processes extending toward the pars reticulata (SNr). B, Adjacent section displayed strong 15G7-α-syn immunoreactivity in the SNc and SNr. C, The nigral neuron (DC) neighbored to a glial cell (G) localized at the border of the SNc to the SNr showed signs of dark cell degeneration. The nucleus appeared slightly collapsed and the cytoplasm darkened with pronounced accumulation of lysosomes (arrows). D, Large lipid droplets (x) were dispersed among the fiber bundles of the SNr closely associated with immunopositive unmyelinated nerve fibers (arrows). E, Several myelinated axons (AX) contained dark condensed organelles, also observed in unmyelinated structures (F) associated with α-syn immunopositive patches (arrows). G, Calbindin immunostaining showed the different subareas of the hippocampus. H, The adjacent α-syn-stained section exhibited labeling of the pyramidal cell layer in the CA1, the stratum lacunosum moleculare (LM), and the inner molecular layer (IML) of the dentate gyrus. Prominent α-syn immunoreactivity was obvious in the curved stripe of mossy fibers and terminals in the CA3 area sharply contrasting to the lightly stained hilus (H) of the dentate gyrus. I, Dark degenerated neurons (DC) (for localization, see arrow in H) were dispersed among normal neurons (N). The dark mossy fiber bundles (white “x”), frequently positive for α-syn immunoreactivity, were localized between the proximal dendrites (x), some of which exhibited signs of degeneration (arrows). J, K, Many α-syn-positive mossy fiber terminals (MT) formed asymmetric contacts with immunonegative dendritic spines (SP) (J) or were associated with DC (K). L, Dystrophic structures (arrows) with condensed organelles were also identified in the dentate gyrus. Scale bar: A, B, 100 μm; C, 1 μm; D–F, 200 nm; G, H, 100 μm; I, 4 μm; J, K, 500 nm; L, 2 μm.
In the hippocampus, α-syn immunoreactivity revealed a specific distribution pattern (Fig. 4 H) [for subregional differentiation, see adjacent calbindin-stained sections (Fig. 4 G)]. Tg α-syn was moderately expressed in the pyramidal cell layer of CA1, the stratum lacunosum moleculare and the medial molecular layer of the dentate gyrus, whereas prominent expression was found in the CA3 area (Fig. 4 H). Electron microscopical analysis of this area further detected groups of dark degenerated pyramidal cells scattered between unaffected neurons (Fig. 4 I). α-syn accumulated mosaic-like in mossy fibers, which are localized between the proximal dendrites of the CA3 neurons (Fig. 4 I), some of which also showed signs of degeneration. Dark cell degeneration was also apparent on CA1 pyramidal cells as well as on interneurons and granule cells of the dentate gyrus, all showing various stages of neurodegeneration (data not shown). Many α-syn-positive mossy fiber terminals, which may show swollen vesicles and enlarged mitochondria, formed asymmetric contacts with immunonegative dendritic spines (Fig. 4 J). Several immunopositive terminals were found to be associated with dark degenerated cell parts (Fig. 4 K). Structures with condensed organelles with varying degree of degeneration (Fig. 4 L) closely resembling the axonal pathology as already shown in Figure 4, E and F, were also identified in the CA3 area and in the hilus of the dentate gyrus.
To characterize the extension of the cell death in the substantia nigra, mice were analyzed with regard to their numbers of dopaminergic neurons in the substantia nigra pars compacta. Counting of dopaminergic cells revealed a strong tendency of reduction bordering significant level (p = 0.07) (Fig. 5 G). In two CaM_α-syn mice (9 and 20 months of age), counting of dark cells revealed a high amount of dark cells in the substantia nigra compared with the remaining healthy cells (Fig. 5 B,D). Degenerated cells were most prominent in the pars reticulata and to a minor extent also seen in the pars compacta (Fig. 5 H). In contrast, dark cells were missing in both 9-month-old control mice (for representative images, see Fig. 5 A,C; and for cell numbers, see Fig. 5 H, graph).
Although dark cell degeneration was obviously the prominent cell death in CaM_α-syn mice, TUNEL staining of adjacent midbrain sections revealed single apoptotic cells (Fig. 5 F), which were not seen in aged-matched controls (Fig. 5 E). Therefore, apoptosis may also contribute to cell death at a lower rate.
Neurochemical alterations
The impact of α-syn expression on both DA and serotonin synthesis and turnover in dopaminergic brain areas was assessed in the olfactory bulb, striatum, and midbrain (including the substantia nigra) of aged CaM_α-syn mice (aged 102 ± 8 weeks) in comparison with controls (aged 106 ± 9 weeks). A statistically significant reduction of DA level was detected in the olfactory bulb of CaM_α-syn mice (Fig. 6 A). In contrast, CaM_α-syn mice in which α-syn expression was ceased for 37 weeks beginning at 58 weeks of age demonstrated DA content comparable with the level of the equally treated and age-matched control group (Fig. 6 A). DA was also reduced in the striatum without reaching statistical significance (Fig. 6 B). There were some effects on the serotonergic system in the olfactory bulb and the midbrain area, but this alteration was only detected in dox-treated animals and was therefore likely nonrelated to α-syn overexpression (Fig. 6 A,C). Thus, expression of tg α-syn affected synthesis of DA in the olfactory bulb of CaM_α-syn mice. In all other brain regions, synthesis and metabolism of DA and serotonin were preserved.
Figure 6.
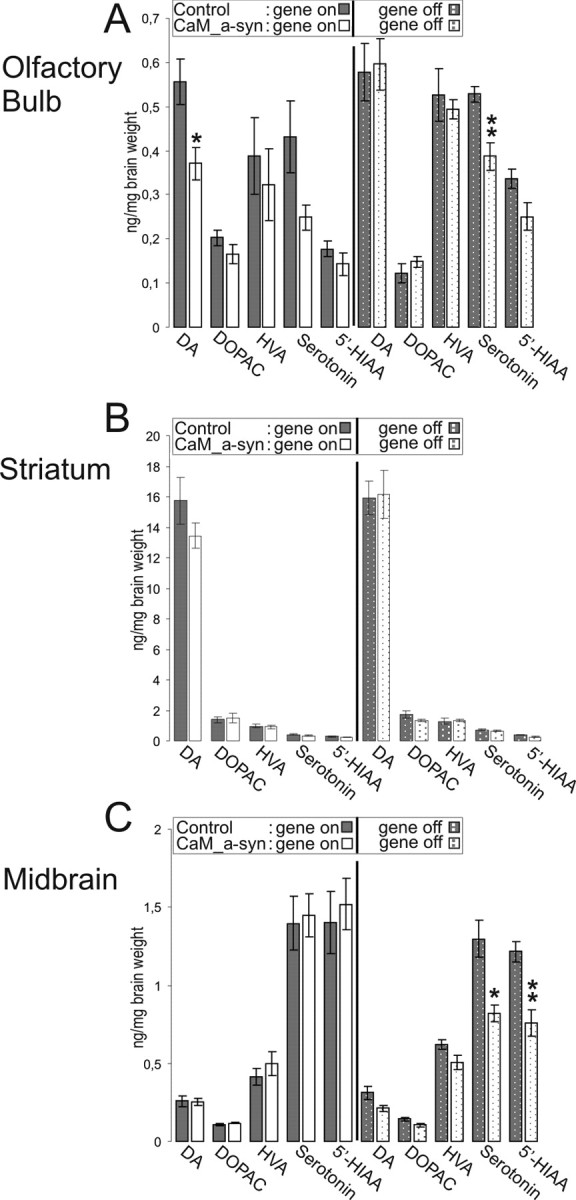
Effect of α-syn expression on neurotransmitter content in brain of old-aged untreated (gene on) and treated (gene off) conditional mice. A–C, Content of neurotransmitter was measured in the olfactory bulb (A), striatum (B), and the midbrain region (including the substantia nigra; C). Untreated (gene on) CaM_α-syn mice (males, n = 1; females, n = 5) and controls (males, n = 2; females, n = 6), treated (gene off) CaM_α-syn mice (males, n = 2; females, n = 2) and equally treated controls (males, n = 3; females, n = 2) were used for HPLC analysis. CaM_α-syn mice showing significant reduction of DA in the olfactory bulb, which was reserved by treatment with dox. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, compared with corresponding group.
Imaging of DAT activity by [11C]d-threo-methylphenidate microPET
Because reduction of nigral dopaminergic neurons is correlated with reduction of DAT binding in the striatum of human PD (Antonini et al., 2001) and in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxin PD models (Brownell et al., 2003; Honer et al., 2006) microPET analysis of the DAT binding potential using 11C-MP was performed. The microPET images showed a clearcut visualization of the striatum and revealed qualitatively the reduced tracer uptake of the CaM_α-syn mouse (Fig. 7 A) compared with the control mouse (Fig. 7 B). Subsequent calculation of striatal and cerebellar time–activity curves (exemplarily shown for the CaM_α-syn mouse in Fig. 7 C) yielded a decrease of 20% in striatal radioactivity levels of CaM_α-syn mice (n = 5; aged 88 ± 5 weeks) compared with control littermates (n = 7; aged 89 ± 4 weeks), indicating a reduction of DAT binding sites at presynaptic terminals. In contrast, in CaM_α-syn mice, in which transgene expression was ceased for 24 weeks, starting at the age of 58 weeks (n = 3; aged 82 weeks) striatal 11C-MP binding potential was only reduced by 9% when compared with littermate controls. However, the differences among the three groups (control, CaM_α-syn, gene-off CaM_α-syn) did not reach statistical significance (p = 0.437; one-way ANOVA) (Fig. 7 D).
Figure 7.

Striatal DAT binding potential quantification in conditional mice. A, B, Representative transverse color-coded microPET images (of all frames over 60 min acquisition time) of striata were conducted with [11C]d-threo-methylphenidate of a CaM_α-syn (A) and the respective control mouse (B). C, Time–activity curve of striatum and cerebellum of a CaM_α-syn mouse were normalized to the injected dose. TACs from striatum and cerebellum were well separated, indicating that the injected tracer did not cause saturation effects or unspecific binding. D, Quantitative analysis of DAT binding potential in the striatum of CaM_α-syn mice (n = 5), respective controls (n = 5), and dox-treated CaM_α-syn mice (n = 3; gene off) revealed reduced DAT binding without reaching statistical significance in CaM_α-syn mice. Error bars indicate SEM.
Analysis of neurogenesis
In the dentate gyrus of the hippocampal formation of the mammalian brain, the capacity for neurogenesis is preserved into adulthood. Because strong α-syn immunoreactivity was detected in mossy fibers and terminals of the dentate gyrus, its impact on mitotic activity, migration, differentiation, and survival of newly generated neurons was analyzed in conditional mice (Table 1). There was no significant difference of PCNA-positive cells suggesting that the same amount of new cells were generated in treated or untreated CaM_α-syn mice, when compared with littermate controls. However, the number of BrdU-positive cells was significantly reduced in CaM_α-syn mice, indicating decreased survival of newly generated cells. Because the ratio of neuronal (NeuN-positive) profiles among BrdU-positive cells was unaltered, the total number of newly generated neurons (measured as the number of BrdU-positive cells multiplied by the percentage of BrdU/NeuN profiles; exemplary double-labeling shown in Fig. 8 G–I) was significantly reduced by 49% in CaM_α-syn mice compared with respective control littermates (Fig. 8, compare D, E). Dox treatment of control mice revealed a decreased neurogenesis, most likely because of the missing interacting partner (tTA) in these mice. Importantly, CaM_α-syn mice in which transgene expression was ceased for 8 weeks starting at 16 weeks age did not show significantly reduced neurogenesis, demonstrating a direct influence of transgene α-syn expression on hippocampal neurogenesis in CaM_α-syn mice (Fig. 8 F). To specify undue influence of α-syn expression on phases of maturation of adult proliferating progenitor cells, young neuroblasts were counted by immunostaining against DCX. Concordant with less neurogenesis, the total number of neuroblasts was significantly reduced in CaM_α-syn mice. This effect was not present in the dox-treated CaM_α-syn mice (Fig. 8, compare A–C). Therefore, overexpression of transgene α-syn affected survival of neuronal committed progenitors and young neurons at the stage of integration into the hippocampal dentate gyrus and downregulation of transgene expression resulted in recovery.
Figure 8.

Reversed hippocampal decrease in adult neurogenesis of conditional CaM_α-syn mice. For studies of neurogenesis, 24-week-old CaM_α-syn mice, littermate controls, and treated CaM_α-syn mice were analyzed. Newly generated DCX-labeled neuroblasts were quantified in the hippocampal dentate gyrus (A–C). A significant decrease in DCX-expressing profiles was present in the CaM_α-syn group (B) compared with the control group (A), and this effect was not detectable in mice with ceased transgene expression (C). A reduction of BrdU-labeled, and therefore newly generated, cells (arrows) was present in CaM_α-syn mice (E) in relation to the control group (D) and was not observed in treated (gene off) CaM_α-syn mice (F). Using colocalization of BrdU and NeuN, there was no significant difference in the percentage of neuronal differentiation of BrdU-positive cells among controls, CaM_α-syn mice, and treated (gene off) CaM_α-syn mice, exemplarily shown in G–I: Separate confocal analysis of the fluophores green (488 nm; BrdU; G), red (568 nm; NeuN; H), and merged in the identical focal plane (I). BrdU, Bromodeoxyuridine. Scale bars: A–C, 50 μm; D–F, 400 μm; G–I, 20 μm.
Behavioral phenotype
Because PD is a neurodegenerative disorder with typical motor symptoms, we assessed motor performance and motor skill learning using an accelerating rotarod apparatus.
Repeated testing of adult CaM_α-syn mice over a 30 week period revealed progressive impairments in the motor performance compared with age- and sex-matched controls (Fig. 9 A, gene on). There were some gender effects, because both female CaM_α-syn mice and female control mice performed better than the respective male mice; however, their proportion and therefore contribution to test results was equal in both groups (conducted by a contingency analysis) (data not shown). Mice were tested on the rotarod for the first time at the age of 18 weeks and already showed significant impairment. However, progressive decline did not start before the age of 30 weeks, indicating that these mice could cope with impact of human α-syn expression on motor brain nuclei over a certain period. After the age of 30 weeks, CaM_α-syn mice revealed a deterioration of motor performance of −13.4 s (mean slope) every following test session. In contrast, controls displayed a decrease of time endurance of only −3.3 s (mean slope), which was likely age related (Fig. 9 A) and significantly better compared with the CaM_α-syn mice (p = 0.0272). To determine whether mice were able to recover at a late stage of disease, 58-week-old mice were treated with dox and tested again over a 19 week period (Fig. 9 A, gene off). The control group was treated equally to exclude influences on performance caused by administration of the antibiotic. CaM_α-syn mice did not recover from this loss of performance but showed an ameliorated performance, because the rate of decreasing time endurance on the rotarod was ameliorated significantly from −13.4 to −4.2 s per session after dox treatment (p = 0.038), which is not significantly different than the mean slope of control mice (p = 0.85). Dox treatment did not change the mean slope of motor performance in control mice. No significant differences in weight were observed between both groups over the tested period (Fig. 9 A).
Because rotarod measurement can also be used as paradigm for learning of complex movements (Buitrago et al., 2004), we analyzed accelerated rotarod results with regard to performance improvement within trial 1 and trial 2 of the first tested session. A two-tailed unpaired t test revealed no significant difference in trial 1 between controls and CaM_α-syn mice (p = 0.5430). However, short-term improvement between trial 1 and trial 2 was only observed for control mice (p = 0.0016) but not for CaM_α-syn mice (p = 0.4393), indicating impaired motor skill learning in CaM_α-syn mice (Fig. 9 B).
Because α-syn pathology was detected in the hippocampus of CaM_α-syn mice (Fig. 4 I–L), animals were tested in a MWM to analyze the impact on spatial learning and memory (for paradigm, see Fig. 9 C). For this, 52-week-old animals were trained to escape to a submerged platform in a tank filled with opaque water. No significant difference in swimming speed and acquisition was detected during reference memory training and all mice significantly reduced their escape latency during the training period (F (9,70) = 4.2, p < 0.001 for the trial) (Fig. 9 D). A probe trial without the platform, performed 24 h after the final learning trial, did not show any significant difference in time spent in the platform target area between controls and CaM_α-syn mice (p = 0.84). Seven days after this probe trial, mice were tested for retention. During retention, animals were allowed to search for a period of 60 s. CaM_α-syn mice showed a significant impairment in retention compared with their control littermates (F (1/14) = 5.1; p < 0.05) (Fig. 9 E). Assessment of working memory by a platform switch from southwest to northeast bordered significance level (F (1/28) = 3.9; p = 0.06) (data not shown).
Discussion
By using the tetracycline-regulated system, we generated the first conditional mouse models of PD that express human wt α-syn protein in different brain regions using the PrP and the CaMKIIα promoter, respectively. When compared with PrP_α-syn mice, CaM_α-syn mice showed a stronger immunoreactivity in forebrain and midbrain areas, including the nigrostriatal system. In contrast, CaM_α-syn1 mice and PrP_α-syn1 mice showed weaker tg signals, which were limited to the forebrain. This phenomenon corresponds to line-to-line variabilities, caused by copy number and integration site of the tg constructs, which also has been described for permanent (Maskri et al., 2004) and conditional (Mayford et al., 1996) models previously. In both of our responder lines, α-syn and α-syn1, copy numbers were identical, indicating that position effects attributable to integration of the transgene are responsible for differences in the expression.
Although Western blot analyses showed a tight transcriptional control by dox in adult mice, the more sensitive immunohistochemical staining revealed residual immunoproduct in the neuropil of several brain areas. However, by expanding treatment endurance, expression was completely suppressed, excluding leaky expression even in old-aged CaM_α-syn mice.
We focused additional analyses on CaM_α-syn mice, which showed prominent alteration of the nigrostriatal system. Consistent with findings in previously published α-syn tg mice (Masliah et al., 2000; Matsuoka et al., 2001; Richfield et al., 2002; Rockenstein et al., 2002; Gispert et al., 2003; Tofaris et al., 2006), human α-syn protein was also localized in the cytoplasm of nigral dopaminergic neurons. Electron microscopical analyses revealed prominent accumulation of tg α-syn in nigral axons and association of human α-syn with lysosomes. The latter may indicate activation of autophagy, which is suggested to accompany aggregation and neurodegeneration (Wang et al., 2006) and which has also been found in cellular systems overexpressing α-syn (Cuervo et al., 2004; Sarkar et al., 2007). Ultrastructural analyses also detected dark cells in the substantia nigra and axonal pathology with condensed mitochondria and lipid droplets. This might be attributable to a disruption of the intracellular transport of lipids and organelles, which has been described for overexpression of human α-syn in cell culture (Gosavi et al., 2002) and MPTP-treated tg mice (Song et al., 2004). However, tg α-syn was mainly patchlike distributed in the cytoplasm of nigral cells without forming fibrils, and sequential extraction also did not reveal human α-syn in the insoluble fraction. Decrease of dopaminergic neurons and a considerable amount of dark degenerated cells gave evidence that this region may display a similar vulnerability to α-syn overexpression as observed in PD affected humans. Because degeneration occurred independently of fibrillary structures, it is possible that these structures may not contribute to cytotoxicity or may even protect cells from neurodegeneration.
In our CaM_α-syn mice, cell death was observed in the hippocampus and the substantia nigra, resembling dark-cell degeneration pattern previously described in other neurodegenerative diseases as Huntington's disease (Turmaine et al., 2000) and spinocerebellar ataxia 7 (Custer et al., 2006) but has not been shown in PD so far. However, cellular and axonal pathology detected in the hippocampus closely reflected α-syn pathology seen in tissues of patients with PD or dementia with Lewy bodies by Galvin et al. (1999). Additionally, tg α-syn accumulated in the stratum lacunosum moleculare and in mossy fibers and their terminals, which may impair synaptic transmission in hippocampal perforant pathway projections critical for consolidation and retention of long-term memory (Steffenach et al., 2002; Remondes and Schuman, 2004). Using MWM analysis, 12-month-old CaM_α-syn mice displayed confirmatively impaired long-term memory, when tested 7 d later for their platform preference (retention) (Fig. 8 E). Thus, overexpression of tg α-syn might cause an impairment of the synaptic output or neuronal plasticity.
In CaM_α-syn mice, progressive motor decline started after the age of 30 weeks. Nonprogressive motor deficits as detected in younger mice might originate from difficulties in learning fine motor skills, which is based on an intact circuit, including sensorimotor cortex, cerebellum, and basal ganglia (Hikosaka et al., 2002). Consistently, CaM_α-syn mice did not show intrasession improvement on the first day tested, although performance of the first trial did not differ significantly compared with those of littermate controls. Therefore, progression of motor impairment resembled features of PD in humans, as motor symptoms also starting mild with impairment of fine motor skills and getting progressively severe.
Importantly, progressive motor decline could not be reversed but halted in symptomatic mice by turning off tg α-syn expression, substantiated by the calculated negative slope of CaM_α-syn mice, which was ameliorated after treatment with dox. This further indicates that tg α-syn directly induced progressive motor decline and, additionally, that disease progression depends on continuous presence of transgene expression. However, the nonreversibility of symptoms in treated CaM_α-syn mice suggested that neurodegeneration and nigral axon pathology was not ameliorated by a compensatory effect as increased neurogenesis, which might be a frequent response to injury in rodents (Arvidsson et al., 2002; Nakatomi et al., 2002; Zhao et al., 2003; Winner et al., 2006) and human brain (Huisman et al., 2004). Analysis of neurogenesis of 6-month-old CaM_α-syn mice consistently revealed a negative impact on progenitor cells in the granular cell layer of the dentate gyrus. In comparison with mice permanently expressing human wt α-syn under control of the PDGF-β promoter (Winner et al., 2004), CaM_α-syn mice showed a similar reduction of ∼50% of newly differentiated neurons in the granular cell layer of the dentate gyrus, paralleled by a significant reduction of DCX-positive neuroblasts. By using our conditional system, we could show for the first time that impaired neurogenesis was primarily attributable to human α-syn expression because it was not seen in dox-treated CaM_α-syn mice and could be reversed after cessation of the transgene in young-aged mice.
To assess the integrity of the dopaminergic system, HPLC- and microPET analyses were performed. The effect of transgene expression on neurotransmitter level in aged CaM_α-syn mice showed significant alteration of DA synthesis in the olfactory bulb and this effect was reversed in age-matched mice treated with dox. However, although degeneration of nigral cells was evident by cell counting and electron microscopy, only a tendency toward reduced DA levels was observed in the striatum. With respect to other published α-syn-expressing mouse lines, only those carrying doubly mutated or truncated human α-syn showed significant reduced DA levels in the striatum, however, without displaying an overt loss of dopaminergic neurons (Richfield et al., 2002; Tofaris et al., 2006). Furthermore, only neurotoxin models (MPTP; 6-OHDA) showed significantly reduced striatal DA content (for review, see Bové et al., 2005), and in these mice cell death occurs subacutely and not slowly progressing over time. It is suggested that the loss of dopaminergic synapses is partially compensated by increased DA metabolism in the surviving terminals in PD patients (for review, see Zigmond et al., 1990; Bezard and Gross, 1998). Thus, in CaM_α-syn mice, a compensatory mechanism to nigral cell degeneration by decreased reuptake and/or increased DA synthesis to maintain sufficient DA levels or even by compensatory sprouting of remaining dopaminergic TH-positive fibers (Song and Haber 2000) is likely.
Detailed analyses of integrity of striatal dopaminergic nerve terminals were assessed by microPET studies in vivo. 11C-MP binding to DAT reflects density of striatal dopaminergic nerve terminals (Ding et al., 1994b), suggested to be reduced as a result from degenerated neurons and axons seen on ultrastructural level in the substantia nigra of aged CaM_α-syn mice. However, binding of 11C-MP was only tendentiously decreased in CaM_α-syn mice and with a large variation in all tested groups. A conducted power analysis of data of control and CaM_α-syn mice revealed that at least 51 mice had to be investigated to obtain significant results among controls, CaM_α-syn mice, and treated CaM_α-syn mice. As with HPLC, significantly reduced DAT binding by using microPET analysis has only been published in animals with subacute and selective destruction of dopaminergic neurons (Nguyen et al., 2000; Xing et al., 2000; Brownell et al., 2003; Takamatsu et al., 2004; Inaji et al., 2005; Lauwers et al., 2007). Thus, our data suggest that evaluation of integrity of dopaminergic system as a single system affected by transgene expression might be more precarious than assessment of behavioral neuropathological impairment, reflecting tg influence on the whole brain network.
In conclusion, we have generated and characterized the first conditional mouse model of PD, fulfilling many of the main criteria of the human condition, including cognitive impairment and degeneration of nigral cells, accompanied by a clear progressive motor deficiency. Degeneration of nigral neurons in this model might be the most important feature and has not been reported for other tg or knock-out PD models yet. By turning the transgene off at later disease stages, we were able to slow down the progression of the disease. We therefore consider this model ideal to evaluate novel therapeutical strategies as neuronal cell loss and impaired neurogenesis contribute to a progressive neurological phenotype, delivering an easy neuropathological read-out. The rate of treatment efficacy in future preclinical trials might be directly compared in vivo with the same mouse lines in which the transgene is turned off.
Footnotes
This work was supported by German Research Foundation Grant DFG RI 682/6-1/3 (O.R.), Intramural Research Funding Program Forum F468 (E.P.-P.), Medical Faculty Bochum, and Bavarian State Ministry of Sciences, Research, and the Arts ForNeuroCell Grant (B.W., J.W.). We thank our colleagues for fruitful discussion, in particular Dr. Berg, Dr. Kahle, and Dr. Machulla. We thank Dr. Jucker, Dr. Calhoun, and C. Schaefer for help with stereology. We thank S. Leitner, S. Kuhlmann, H. W. Habbes, and M. Löbbecke-Schumacher for excellent technical assistance; K. Mahesch for help with Morris water maze analysis; F. Marxreiter for evaluation of hippocampal neurogenesis; and Dr. Dietz for detailed statistical analysis of rotarod performance.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Antonini A, Moresco RM, Gobbo C, De Notaris R, Panzacchi A, Barone P, Calzetti S, Negrotti A, Pezzoli G, Fazio F. The status of dopamine nerve terminals in Parkinson's disease and essential tremor: a PET study with the tracer [11-C]FE-CIT. Neurol Sci. 2001;22:47–48. doi: 10.1007/s100720170040. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE. Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog Neurobiol. 1998;55:93–116. doi: 10.1016/s0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci USA. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bové J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson's disease. NeuroRx. 2005;2:484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Brownell AL, Canales K, Chen YI, Jenkins BG, Owen C, Livni E, Yu M, Cicchetti F, Sanchez-Pernaute R, Isacson O. Mapping of brain function after MPTP-induced neurotoxicity in a primate Parkinson's disease model. NeuroImage. 2003;20:1064–1075. doi: 10.1016/S1053-8119(03)00348-3. [DOI] [PubMed] [Google Scholar]
- Bruck A, Kurki T, Kaasinen V, Vahlberg T, Rinne JO. Hippocampal and prefrontal atrophy in patients with early non-demented Parkinson's disease is related to cognitive impairment. J Neurol Neurosurg Psychiatry. 2004;75:1467–1469. doi: 10.1136/jnnp.2003.031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago MM, Schulz JB, Dichgans J, Luft AR. Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol Learn Mem. 2004;81:211–216. doi: 10.1016/j.nlm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Ding YS, Sugano Y, Fowler JS, Salata C. Synthesis of the racemate and individual enantiomeres of [11C]methylphenidate for studying presynaptic dopaminergic neuron with positron emission tomography. J Labelled Comp Radiopharm. 1994a;34:989–997. [Google Scholar]
- Ding YS, Fowler JS, Volkow ND, Gatley SJ, Logan J, Dewey SL, Alexoff D, Fazzini E, Wolf AP. Pharmacokinetics and in vivo specificity of [11C]dl-threo-methylphenidate for the presynaptic dopaminergic neuron. Synapse. 1994b;18:152–160. doi: 10.1002/syn.890180207. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, McDermott JL. Gender differences in neurotoxicity of the nigrostriatal dopaminergic system: implications for Parkinson's disease. J Gend Specif Med. 2000;3:36–42. [PubMed] [Google Scholar]
- Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology. 1988;38:1237–1244. doi: 10.1212/wnl.38.8.1237. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci USA. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Nakamura K, Sakai K, Nakahara H. Central mechanisms of motor skill learning. Curr Opin Neurobiol. 2002;12:217–222. doi: 10.1016/s0959-4388(02)00307-0. [DOI] [PubMed] [Google Scholar]
- Honer M, Hengerer B, Blagoev M, Hintermann S, Waldmeier P, Schubiger PA, Ametamey SM. Comparison of [18F]FDOPA, [18F]FMT and [18F]FECNT for imaging dopaminergic neurotransmission in mice. Nucl Med Biol. 2006;33:607–614. doi: 10.1016/j.nucmedbio.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Huisman E, Uylings HB, Hoogland PV. A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson's disease. Mov Disord. 2004;19:687–692. doi: 10.1002/mds.10713. [DOI] [PubMed] [Google Scholar]
- Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- Inaji M, Okauchi T, Ando K, Maeda J, Nagai Y, Yoshizaki T, Okano H, Nariai T, Ohno K, Obayashi S, Higuchi M, Suhara T. Correlation between quantitative imaging and behavior in unilaterally 6-OHDA-lesioned rats. Brain Res. 2005;1064:136–145. doi: 10.1016/j.brainres.2005.09.055. [DOI] [PubMed] [Google Scholar]
- Inden M, Kondo J, Kitamura Y, Takata K, Nishimura K, Taniguchi T, Sawada H, Shimohama S. Proteasome inhibitors protect against degeneration of nigral dopaminergic neurons in hemiparkinsonian rats. J Pharmacol Sci. 2005;97:203–211. doi: 10.1254/jphs.fp0040525. [DOI] [PubMed] [Google Scholar]
- Janus C. Search strategies used by APP transgenic mice during navigation in the Morris water maze. Learn Mem. 2004;11:337–346. doi: 10.1101/lm.70104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson's disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–2225. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lauwers E, Beque D, Van Laere K, Nuyts J, Bormans G, Mortelmans L, Casteels C, Vercammen L, Bockstael O, Nuttin B, Debyser Z, Baekelandt V. Non-invasive imaging of neuropathology in a rat model of alpha-synuclein overexpression. Neurobiol Aging. 2007;28:248–257. doi: 10.1016/j.neurobiolaging.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Logan J. Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl Med Biol. 2000;27:661–670. doi: 10.1016/s0969-8051(00)00137-2. [DOI] [PubMed] [Google Scholar]
- Maetzler W, Berg D, Schalamberidze N, Melms A, Schott K, Mueller JC, Liaw L, Gasser T, Nitsch C. Osteopontin is elevated in Parkinson's disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiol Dis. 2007;25:473–482. doi: 10.1016/j.nbd.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Maskri L, Zhu X, Fritzen S, Kuhn K, Ullmer C, Engels P, Andriske M, Stichel CC, Lubbert H. Influence of different promoters on the expression pattern of mutated human alpha-synuclein in transgenic mice. Neurodegener Dis. 2004;1:255–265. doi: 10.1159/000085064. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, Brownell AL, Iris Chen YC, Livni E, Coyle JT, Rosen BR, Cavagna F, Jenkins BG. Detection of the effects of dopamine receptor supersensitivity using pharmacological MRI and correlations with PET. Synapse. 2000;36:57–65. doi: 10.1002/(SICI)1098-2396(200004)36:1<57::AID-SYN6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Petrasch-Parwez E, Nguyen HP, Löbbecke-Schumacher M, Habbes HW, Wieczorek S, Riess O, Andres KH, Dermietzel R, von Hörsten S. Cellular and subcellular localization of huntingtin aggregates in the brain of a rat transgenic for Huntington disease. J Comp Neurol. 2007;501:716–730. doi: 10.1002/cne.21272. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Remondes M, Schuman EM. Molecular mechanisms contributing to long-lasting synaptic plasticity at the temporoammonic-CA1 synapse. Learn Mem. 2003;10:247–252. doi: 10.1101/lm.59103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- Riekkinen P, Jr, Kejonen K, Laakso MP, Soininen H, Partanen K, Riekkinen M. Hippocampal atrophy is related to impaired memory, but not frontal functions in non-demented Parkinson's disease patients. NeuroReport. 1998;9:1507–1511. doi: 10.1097/00001756-199805110-00048. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Song DD, Haber SN. Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J Neurosci. 2000;20:5102–5114. doi: 10.1523/JNEUROSCI.20-13-05102.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenach HA, Sloviter RS, Moser EI, Moser MB. Impaired retention of spatial memory after transection of longitudinally oriented axons of hippocampal CA3 pyramidal cells. Proc Natl Acad Sci USA. 2002;99:3194–3198. doi: 10.1073/pnas.042700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu H, Kakiuchi T, Noda A, Uchida H, Nishiyama S, Ichise R, Iwashita A, Mihara K, Yamazaki S, Matsuoka N, Tsukada H, Nishimura S. An application of a new planar positron imaging system (PPIS) in a small animal: MPTP-induced parkinsonism in mouse. Ann Nucl Med. 2004;18:427–431. doi: 10.1007/BF02984486. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay P, Meiner Z, Galou M, Heinrich C, Petromilli C, Lisse T, Cayetano J, Torchia M, Mobley W, Bujard H, DeArmond SJ, Prusiner SB. Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc Natl Acad Sci USA. 1998;95:12580–12585. doi: 10.1073/pnas.95.21.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- Winkler J, Ramirez GA, Thal LJ, Waite JJ. Nerve growth factor (NGF) augments cortical and hippocampal cholinergic functioning after p75NGF receptor-mediated deafferentation but impairs inhibitory avoidance and induces fear-related behaviors. J Neurosci. 2000;20:834–844. doi: 10.1523/JNEUROSCI.20-02-00834.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Lie DC, Rockenstein E, Aigner R, Aigner L, Masliah E, Kuhn HG, Winkler J. Human wild-type alpha-synuclein impairs neurogenesis. J Neuropathol Exp Neurol. 2004;63:1155–1166. doi: 10.1093/jnen/63.11.1155. [DOI] [PubMed] [Google Scholar]
- Winner B, Geyer M, Couillard-Despres S, Aigner R, Bogdahn U, Aigner L, Kuhn G, Winkler J. Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp Neurol. 2006;197:113–121. doi: 10.1016/j.expneurol.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Xing D, Chen P, Keil R, Kilts CD, Shi B, Camp VM, Malveaux G, Ely T, Owens MJ, Votaw J, Davis M, Hoffman JM, BaKay RA, Subramanian T, Watts RL, Goodman MM. Synthesis, biodistribution, and primate imaging of fluorine-18 labeled 2beta-carbo-1′-fluoro-2-propoxy-3beta-(4-chlorophenyl)tr opanes. Ligands for the imaging of dopamine transporters by positron emission tomography. J Med Chem. 2000;43:639–648. doi: 10.1021/jm9902234. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci USA. 2003;100:7925–7930. doi: 10.1073/pnas.1131955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]