

Abstract
Cyclin-dependent kinase 5 (cdk5) and glycogen synthase kinase 3β (GSK3β) have been implicated in pathogenic processes associated with Alzheimer's disease because both kinases regulate tau hyperphosphorylation and enhance amyloid precursor protein (APP) processing leading to an increase in amyloid β (Aβ) production. Here we show that young p25 overexpressing mice have enhanced cdk5 activity but reduced GSK3β activity attributable to phosphorylation at the inhibitory GSK3β–serine 9 (GSK3β–S9) site. Phosphorylation at this site was mediated by enhanced activity of the neuregulin receptor complex, ErbB, and activation of the downstream phosphatidylinositol 3 kinase/Akt pathway. Young p25 mice had elevated Aβ peptide levels, but phospho-tau levels were decreased overall. Thus, cdk5 appears to play a dominant role in the regulation of amyloidogenic APP processing, whereas GSK3β plays a dominant role in overall tau phosphorylation. In older mice, GSK3β inhibitory phosphorylation at S9 was reduced relative to young mice. Aβ peptide levels were still elevated but phospho-tau levels were either unchanged or showed a trend to increase, suggesting that GSK3β activity increases with aging. Inhibition of cdk5 by a specific inhibitor reduced cdk5 activity in p25 mice, leading to reduced Aβ production in both young and old mice. However, in young mice, cdk5 inhibition reversed GSK3β inhibition, leading to an increase in overall tau phosphorylation. When cdk5 inhibitor was administered to very old, nontransgenic mice, inhibition of cdk5 reduced Aβ levels, and phospho-tau levels showed a trend to increase. Thus, cdk5 inhibitors may not be effective in targeting tau phosphorylation in the elderly.
Keywords: Alzheimer's disease, tau, cdk5, GSK3β, neuregulin receptors, APP, Aβ
Introduction
Alzheimer's disease (AD) is characterized by the intracellular accumulation of hyperphosphorylated tau into tangles and the extracellular accumulation of amyloid β (Aβ) peptide into amyloid plaques. Tau is a microtubule binding protein that stabilizes and promotes microtubule polymerization (Pelech, 1995; Michaelis et al., 2002). The phosphorylation state of tau changes during development and adulthood, and the appearance of aberrantly phosphorylated sites correlates with disease status. Phosphorylation status is therefore carefully balanced by the activity of multiple protein kinases and phosphatases. Cyclin dependent kinase 5 (cdk5) and glycogen synthase kinase 3β (GSK3β) have been identified as prime candidates for pathogenesis (Planel et al., 2002; Drewes, 2004) because they both generate disease-associated phospho-epitopes on tau, and they colocalize with filamentous tau aggregates in the brains of patients (Imahori and Uchida, 1997; Shelton and Johnson, 2004) and in a transgenic mouse model of tauopathy (Imahori and Uchida, 1997; Ishizawa et al., 2003). cdk5 and GSK3 also regulate Aβ production in vivo (Phiel et al., 2003; Cruz et al., 2006). There is a high degree of redundancy between the two kinases, and several of the phosphorylation sites on targets overlap, thus the relative activity of each in terms of amyloid precursor protein (APP) processing and tau phosphorylation is still unclear.
Independent studies suggest a strong correlation between cdk5 activity and neuregulin receptor pathways (Fu et al., 2001; Li et al., 2003). Neuregulin-1 (NRG-1) is part of a family of growth factors that include glial growth factor, heregulin, and neu differentiation factor (Carraway and Burden, 1995). NRG receptors (ErbB) are required for the myelination process (Chen et al., 2006; Lemke, 2006), and both the level of NRG-1 and the activity of its receptor are very high during myelination in the first neonatal week (Willem et al., 2006). It is then dramatically downregulated in the adult nervous system (Kerber et al., 2003). Cdk5 activity upregulates the neuregulin/ErbB pathway by phosphorylating T871 in ErbB2 and/or S1120 in ErbB3 (Li et al., 2003). Phosphorylation of ErbB2/3 appears to serve as a priming event required for additional activation of the receptor by its corresponding ligands, and it thereby regulates downstream pathways such as phosphatidylinositol 3-kinase (PI3K) activity and acetylcholine receptor expression (Fu et al., 2001).
Our studies show that cdk5 can negatively regulate GSK3β activity through neuregulin/ErbB signaling. Aβ generation correlates with enhanced cdk5 activity, whereas tau phosphorylation status overall correlates with GSK3β activity. Pharmacological inhibition of cdk5 decreased Aβ production, but, in young mice, it led to release of GSKβ inhibition and increased phosphorylation of tau. In old mice, Aβ was still reduced, but phospho-tau levels were not significantly changed. Thus, in the elderly, cdk5 inhibitors may not be as effective as GSK3β inhibitors for targeting both pathways, and this may have implications for AD patients.
Materials and Methods
Animals.
p25 mice (Ahlijanian et al., 2000) were generated from hemizygous, cryopreserved stock mice supplied by The Jackson Laboratory (Bar Harbor, ME), bred to homozygosity. Mice were used at 4 or 5 d [birthdate set as postnatal day 0 (P0)], 3 months, or 12 months of age. Controls were age- and strain-matched (FVB) animals. Nontransgenic (Ntg) mice (24–28 months of age) used for the inhibitor study were Swiss-Webster. All animals were maintained and killed according to National Institutes of Health and Institutional Animal Care and Use Committee guidelines.
Immunoblot analysis.
All mice were killed by cervical dislocation, and brains were immediately removed, dissected, and kept on dry ice. Hemibrains were weighed and homogenized without thawing in 5× radioimmunoprecipitation (RIPA) buffer (50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, and 10 μl/ml protease inhibitor mix). Samples were either centrifuged for 10 min at 20,000 × g at 4°C, supernatant was quickly diluted with loading buffer, or quickly frozen on dry ice for storage. The supernatant or direct homogenate were diluted 1:10 in O+ buffer [62.5 mm Tris-HCl, pH 6.8, 10% (w/v) glycerol, 5% 2-mercaptoethanol, 2.3% SDS, 1 mm EGTA, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, and 10 μl/ml protease inhibitor mix] and incubate at 85°C for 10 min. Depending on the antibody used, 5–20 μg of protein were analyzed with SDS-PAGE. Extraction and immunoblotting was performed as described previously (Noble et al., 2003), with appropriate primary and secondary antibody dilutions. Data shown represent two mice from each group.
Kinase activity assay.
GSK3β, cdk5, and extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) activity assays were performed as described (Planel et al., 2001). Briefly, dissected cortices were homogenized in 5× w/v of modified RIPA buffer [50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, and 10 μl/ml protease inhibitor cocktail P8340 (Sigma, St. Louis, MO)] and centrifuged at 20,000 × g for 30 min at 4°C. Kinases from brain extracts were immunoprecipitated for 1 h at 4°C with complexes of Dynabeads (M 280 sheep anti-rabbit or anti-mouse IgG; Invitrogen, Carlsbad, CA) and GSK3β (610201; BD Transduction Laboratories, San Jose, CA), ERK/MAPK (9102, p44/42 MAPK; Cell Signaling Technology, Danvers, MA), or cdk5 antibodies (C-8; Santa Cruz Biotechnology, Santa Cruz, CA) prepared according to the instructions of the manufacturer. Assays with immunoprecipitated kinases were conducted in the presence of 200 μm ATP, with 100 μg/ml recombinant tau as the substrate (P2717; Invitrogen). Kinase activities were assessed by Western blotting and quantification of tau phosphorylation with phospho-tau-specific antibodies [Tau (pS199) or (pS404); Invitrogen].
CP-681301 and CI-1033 treatment of mice.
CP-681301 (Pfizer, Groton, CT) was administered by subcutaneous injection at 5.8 mg/kg. Older animals were injected on 2 consecutive days, 24 h apart. Neonatal p25 mice were administered a single dose and killed 6 h later. Administration of the drug in older animals was noted to lead to slight hypothermia, which has been shown to result in hyperphosphorylation of tau (Planel et al., 2007). All mice were therefore kept at 37°C for 30 min before they were killed, and rectal temperature was monitored at the time of death. All mice were 37 ± 1.5°C. Animals were killed 3 h after last injection and dissected as described previously. CI-1033 (Pfizer) was administered by subcutaneous injection at 40 mg/kg. Neonatal mice were administered a single dose and killed 4 h later. All mice were kept at 37°C for 30 min before death.
Tissue culture and CI-1033 treatment.
SH-SY5Y cells were grown in DMEM supplemented with 10% fetal bovine serum and appropriate antibiotics (Invitrogen) in a 5% CO2 humidified incubator at 37°C. Medium was replenished every 3 d. Cells were placed in serum-free medium 12 h before treatment. CI-1033 at the indicated concentration was added to the culture 2 h before epidermal growth factor (EGF) treatment. EGF at 100 ng/ml or vehicle was added 10 min before collecting the cells.
Aβ ELISA.
Soluble murine Aβ1–40 and Aβ1–42 from a region including cortex and hippocampus was assessed using antibodies Aβx-40 (JRF/cAβ40) and Aβx-42 (JRF/cAβ42/26) for capture and JRF/AβN-25 (human) JRF/rAβ1–15/2 for detection (Dr. Sonia Jung, Centocor R & D Inc., Radnor, PA), as described previously (Schmidt et al., 2005a,b). Several sets of mice (n = 6–12 per group) were assessed for each experiment.
Statistical analysis.
Statistical analysis was performed with a two-tailed t test or correlation test (see Fig. 1). All data presented are means ± SEM. All arbitrary units were converted to the percentage value of the control or vehicle-treated value. Asterisks indicate statistically significant differences: *p < 0.05 and **p < 0.01.
Figure 1.

p25/cdk5 activation inhibits GSK3β through the NRG receptor pathway. A, Representative immunoblots of cdk5, p35/p25, pGSK3β–S9, GSK3β, β-catenin, and pβ-catenin on brain extracts from Ntg (n = 7) and p25 (n = 7) mice at postnatal day 4. pGSK3β–S9 levels normalized to GSK3β total protein were significantly increased in p25 mice compared with Ntg mice (p < 0.01). Four different sets of animals were examined with similar results; the most representative set is shown. p25 mice showed significantly increased levels of β-catenin and significantly decreased levels of the GSK3β target pβ-catenin. B, Representative immunoblots showing levels of neuregulin signaling components, pAkt-S473, Akt, pPI3K–Y199 (p55 subunit), and pErbB2–Y877. β-Tubulin was used for loading control; there was a significant increase of pAkt–S473 (p < 0.05), pPI3K–Y199 (p < 0.05), and pErbB2–Y877(p < 0.05) in p25 mice compared with Ntg mice. C, Correlation analysis of pErbB2 and pGSK3β–S9 in Ntg and p25 mice showed a significant correlation between pErbB2–Y877 and pGSK3β–S9 (p < 0.01).
Antibodies.
Anti-cdk5 (C-8), anti-p35/25 (C-19), anti-pErbB (Tyr 1056-R; sc-33040-R), anti-ErbB4 (sc-71070), anti-ErbB2 (3B5; sc-33684), and anti-Neuregulin-1 (sc-28916) were purchased from Santa Cruz Biotechnology. Anti-human Aβ1–17 (6E10), anti-Aβ17–24 (4G8), and anti-soluble APPβ (sAPPβ) were from Signet Laboratories (Dedham, MA). Anti-β-actin (AC-74) and anti-β-tubulin were purchased from Sigma. The monoclonal antibody against β-site APP-cleaving enzyme 1 (BACE1) (3D5) was a kind gift from Dr. R. Vassar (Northwestern University, Chicago, IL). Monoclonal antibodies C/1.61 (full length and C83/C99) and M3.2 (sAPPα) were a kind gift from Dr. P. Mathews (Nathan Kline Institute, Orangeburg, NY). Anti-GSK3β (Ser9), anti-ErbB2 (Tyr877), anti-Akt, anti-Akt (Ser473), anti-Akt (Thr308), anti-ERK 1/2, anti-ERK (Thr202/Tyr204), and anti-PI3Kp55 (Tyr199) were purchased from Cell Signaling Technology. Anti-GSK3β monoclonal antibody was from BD Transduction Laboratories. Tau antibodies PHF-1 (pS396/pS404), CP13 (pS202), and MC6 (pS235) were kind gifts from Dr. Peter Davis (Albert Einstein University, Bronx, NY). Tau-1 (dephosphorylated tau) was from Millipore (Billerica, MA). Polyclonal anti-total tau antibody was purchased from DakoCytomation (Glostrup, Denmark). Anti-Tau pS422, Tau pS199, Tau pT231, Tau pS404, and Tau pS422 were from Invitrogen. Monoclonal anti-tau AT8 (pS202/pT205) and AT270 (pT181) were purchased from Pierce (Rockford, IL).
Results
cdk5 activation by p25 overexpression inhibits GSK3β in young mice
Overexpression of p25 in the transgenic mice was confirmed by immunoblotting (Fig. 1A). Enhanced activity of cdk5 attributable to elevation of p25 in the transgenic mice was assessed after immunoprecipitation of the kinase from cortical extracts (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Interestingly, a significant reduction in cdk5 protein level (p < 0.05) was observed in p25 mice attributable to autophosphorylation of p35, which signals ubiquitination/degradation (Patrick et al., 1998; Hisanaga and Saito, 2003) of the kinase, thus shortening the turnover time. Autophosphorylation of p35 was observed as a shift in the mobility of p35 in p25 mice.
GSK3 activity is regulated by phosphorylation at serine 9 (S9) of GSK3β (S21 of GSK3α) or Tyr 216 (Wang et al., 1994). Phosphorylation at S9 inactivates GSK3β, and activation of GSK3β requires dephosphorylation at S9 and autophosphorylation at Y216 (Cole et al., 2004). The effect of p25-mediated cdk5 activation on GSK3β phosphorylation status was assessed in vivo. Mouse cortical extracts from Ntg mice and p25 overexpressing (p25) mice were analyzed shortly after birth on postnatal day 4 or 5. Brain extracts were probed for GSK3β total protein and GSK3β phosphorylated at S9 in Ntg (n = 7) and p25 (n = 7) mice (Fig. 1A), The level of GSK3β–S9 was quantified and normalized to total GSK3β levels. There was a significant increase in S9 phosphorylation in p25 mice (142.2 ± 16.2) compared with Ntg mice (100.0 ± 10.4; p < 0.05) (Fig. 1A). To further assess the activation status of GSK3β in vitro and in vivo, we examined GSK3β kinase activity in vitro after immunoprecipitation of the kinase from Ntg and p25 cortical extracts. A significant reduction in GSK3β activity was observed in p25 mice (36.5 ± 11.8) compared with Ntg mice (100.0 ± 17.7; p < 0.05). To confirm that GSK3β activity is inhibited in vivo, we examined the level of one of its substrates, β-catenin, and its phosphorylation status. β-Catenin is a key downstream effector of the Wnt signaling pathway (Cadigan and Nusse, 1997), and GSK3β destabilizes β-catenin by phosphorylating sites S33, S37, and T41 (Yost et al., 1996), thus promoting its degradation. The level of β-catenin was significantly enhanced in p25 mice (129.3 ± 8.5) compared with Ntg (100.0 ± 1.8) mice (p < 0.01), whereas the phosphorylation at S33/37/T41 relative to total β-catenin levels was significantly reduced in p25 mice (52.8 ± 7.4) compared with Ntg mice (100.0 ± 17.9; p < 0.05), supporting our observations that GSK3β activity is reduced in these mice.
p25 overexpression activates PI3K/Akt pathway in vivo
There is no evidence suggesting that GSK3β is a direct substrate for cdk5. The best known GSK3 inhibiting kinase is Akt, which phosphorylates GSK3β at S9 (Krasilnikov, 2000). Akt is activated by phosphorylation at S473 or T308 by 3-phosphoinositide-dependent kinase-1 (PDK1). Furthermore, PDK1 activity is dependent on the level of phosphatidylinositol (3,4,5)-trisphosphate, which is the key product of PI3K and phosphatidylinositol (3,4,5)-bisphosphate (Alessi et al., 1997). PI3K/Akt pathway activation was monitored in p25 mice (Fig. 1B). The levels of Akt, active Akt, and active PI3K were determined by immunoblot analysis with specific antibodies against total Akt, phospho-Akt (S473), and active PI3K (p55 subunit pY199). The level of Akt protein was comparable in Ntg and p25 mice, whereas the active forms of Akt (Ntg, 100 ± 15.0 vs p25, 151.0 ± 12.6; n = 7 and n = 6, respectively) and PI3K (Ntg, 100 ± 7.4 vs p25, 147.1 ± 20.1; n = 7 and n = 6, respectively) were significantly increased in p25 mice (p < 0.01) (Fig. 1B)
Enhanced GSK3β–S9 phosphorylation correlates with increased ErbB2 activation in p25 mice
PI3K plays a pivotal role in many cellular signaling systems, and several extracellular signals have been reported to activate the PI3K pathway (Franke et al., 1997; Garcia et al., 2006). In previous reports, cdk5 was shown to phosphorylate the neuregulin receptor ErbB at ErbB2–T871 or ErbB3–S1120 and activate the downstream PI3K pathway (Li et al., 2003). Phosphorylation at these sites facilitates autophosphorylation of the receptor at multiple sites including Y877, which was the site used to monitor activation status. The activation status of ErbB2 was examined, and a significant increase in pErbB2 was observed in p25 (147.1 ± 20.1; n = 6) compared with Ntg mice (100.0 ± 7.4; n = 7; p < 0.01) (Fig. 1B). To examine the correlation between GSK3β–S9 phosphorylation and ErbB2 activation, results for GSK3β–S9 versus pErbB2–Y877 were plotted (Fig. 1C). A high degree of correlation was observed between pGSK3β–S9 and pErbB2–Y877 (p < 0.01). The robust phosphorylation of neuregulin signaling components at their activation sites and the significant correlation with GSK3β–S9 levels indicates that neuregulin receptor activation is a key intermediate step that connects enhanced cdk5 activity with GSK3β inhibition.
Inhibition of ErbB signaling attenuates PI3K/Akt pathway and GSK3β inhibition in vitro and in vivo
To confirm that ErbB receptor signaling mediates the inhibitory phosphorylation of GSK3β, we examined the effects of a potent and clinically relative pan-ErbB irreversible inhibitor, CI-1033 (Smaill et al., 2000; Flaherty and Brose, 2006). To enhance ErbB-mediated signaling in SH-SY5Y cells, the neuregulin family agonist EGF was applied. CI-1033 was then added at doses ranging from 10 to 1000 nm. As expected, there was a significant increase in Akt phosphorylation as well as pGSK3β−S9 in the presence of EGF but with no or low dose (10 nm) CI-1033 (Fig. 2A). CI-1033 was, however, very effective at abolishing the effects of EGF at 100 nm and higher concentration (Fig. 2A). We then examined the effects of CI-1033 in the neonatal mice. In Ntg mice, subdermal injection of CI-1033 (40 mg/kg) for 4 h induced significant reduction of pAkt–S473 (vehicle, n = 5, 100.0 ± 6.3 vs CI-1033, n = 4, 80.2 ± 2.8; p < 0.05) and reduced phosphorylation of GSK3β-S9 (vehicle, n = 5, 100.0 ± 3.4 vs CI-1033, n = 4, 78.7 ± 5.3; p < 0.01) (Fig. 2B). Furthermore, CI-1033 treatment blocked Akt phosphorylation and GSK3β inhibitory phosphorylation in p25 mice (Fig. 2C). These data confirm that neuregulin/ErbB signaling regulates Akt pathway activation and GSK3β inhibition both in vitro and in vivo.
Figure 2.

Inhibition of ErbB receptor activation reverses GSK3β inhibition in vitro and in vivo. A, Representative blots showing the dose–response effects of the ErbB inhibitor CI-1033 in blocking EGF-induced activity of ErbB in SH-SY5Y cells. EGF-induced phosphorylation of the inhibitory S9 site on GSK3β and the activation site S473 on Akt. Administration of CI-1033 reversed the effects of EGF at doses above 10 nm. B, Representative blots showing the effect of CI-1033 (I) compared with vehicle (V) in nontransgenic mice. C, Representative blots showing that p25 overexpression increased the level of pGSK3β–S9 and pAkt–S473 levels in 5-d-old mice (Ntg/Veh vs p25/Veh) and the ErbB inhibitor CI-1033 reversed the effects. Quantitative analysis of densitometry data are shown on the right.
Reduced NRG receptor activation and PI3K signaling pathway in cdk5-deficient mice
To further confirm the role of cdk5 in GSK3β inhibition, components of the signaling pathway were examined in cdk5-deficient (KO) mice. GSK3β and Akt levels were similar between Ntg and cdk5−/− KO mice. However, reduction of the Akt/PI3K signaling pathway was shown by reduced levels of pGSK3β–S9, pAkt–S473, and pPI3K–Y199 (Fig. 3). This is consistent with previous reports for mice with cdk5 or p35 deficiency (Hallows et al., 2003; Li et al., 2003). Furthermore, we found a reduction in ErbB2 receptor phosphorylation (pY877) at the activation site in cdk5−/− KO mice compared with Ntg mice.
Figure 3.
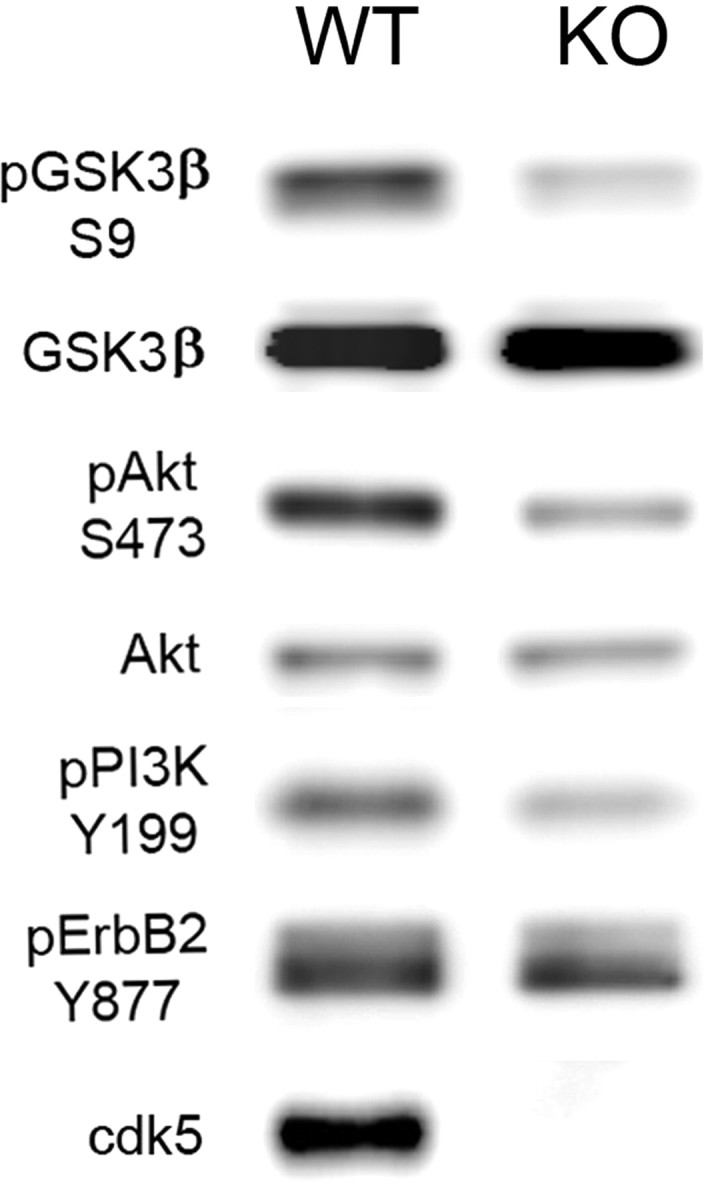
Analysis of ErbB receptor and PI3K/Akt pathway in cdk5-deficient mice. Representative immunoblots showing GSK3β, pGSKβ3–S9, Akt, pAkt–Ser473, pPI3K–Y199 (p55 subunit), and pErbB2–Y877 in brain extracts from fetal WT and cdk5-deficient (KO) mice. pGSKβ3–S9, pAkt–Ser473, pPI3K–Y199, and pErbB2–Y877 levels were all reduced in the cdk5 KO mice.
Phosphorylation status of tau in p25 mice
One mutual substrate for GSK3β and cdk5 is tau protein. Because the young p25 transgenic mice have enhanced cdk5 activity but reduced GSK3β activity, they are an ideal system in which to analyze the differential effect of cdk5 and GSK3β on tau phosphorylation in vivo. The site-specific phosphorylation of tau was assessed with eight different antibodies that cover most of the sites recognized by cdk5 or GSK3β from in vitro studies (Ishiguro et al., 1991; Illenberger et al., 1998; Reynolds et al., 2000). The data were assigned to three groups (Fig. 4). In the first group, pS231, pT181 (AT270), pS202 (CP13), and pS396/pS404 (PHF-1) showed no significant difference between Ntg and p25 mice. These sites are recognized by both cdk5 and GSK3β, and the activation of cdk5 appears to be compensated for by the inhibition of GSK3β, leading to no net change in phosphorylation status (Fig. 4A). In the second group, pS235 (MC6) (Ntg, n = 7, 100.0 ± 4.6 vs p25, n = 6, 128.2 ± 4.0; p < 0.01) and pS202/pT205 (AT8) (Ntg, n = 7, 100.0 ± 6.1 vs p25, n = 6, 131.3 ± 6.9; p < 0.01) showed significantly increased levels of phosphorylation (Fig. 4B). These sites respond positively to cdk5 activity, although it could not be determined whether cdk5 acted directly on these sites or indirectly through modulation of other phosphorylating agents. In the third group, phosphorylation at pS199 was significantly reduced (Ntg, n = 7, 100.0 ± 6.1 vs p25, n = 6, 131.3 ± 6.9; p < 0.01) (Fig. 4C). This site appears to be targeted primarily by GSK3β.
Figure 4.

Phosphorylation of tau in p25 mice. A, Representative immunoblots for total tau and tau pS231, pT181 (AT270), pS202 (CP13), and pS396/S404 (PHF-1). The level of phosphorylation at these sites did not change significantly between Ntg and p25 mice as they are targeted by both cdk5 and GSK3β. B, Representative immunoblots for pS235 (MC6) (p < 0.01) and pS202/pT205 (AT8) (p < 0.01). Phosphorylation at these sites was significantly increased, and they are thus considered primarily cdk5 responsive. C, Representative immunoblots for pS199 and Tau1 (dephosphorylated tau). Phosphorylation at the pS199 site was significantly decreased (p < 0.01), and it is thus considered GSK3β responsive. Results with Tau1 showed that, overall, tau in p25 mice was dephosphorylated compared with tau in Ntg mice.
Tau1 recognizes tau that is dephosphorylated at specific sites. We found significantly increased Tau1 epitope in the p25 mice, consistent with a general reduction in tau phosphorylation (Ntg, n = 7, 100.0 ± 5.4 vs p25, n = 6, 118.5 ± 3.4; p < 0.05). The total tau antibody recognizes tau independent of phosphorylation status at the N terminus. It recognized two distinct bands, a 56 kDa tau band and a 60 kDa hyperphosphorylated tau band. Showing a mobility shift in tau using a phosphorylation-independent total tau antibody is a strong indicator of tau hyperphosphorylation (Morishima-Kawashima et al., 1995). In p25 mice, the prominent tau band at 56 kDa did not change significantly, but the high mobility phospho-tau band was significantly reduced (Ntg, n = 7, 100.0 ± 7.9 vs p25, n = 6, 71.2 ± 2.0; p < 0.01) compared with Ntg mice (Fig. 4A) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). On overexposed blots, the reduced intensity of the hyperphosphorylated band was also observed with several other phosphorylation-dependent antibodies with increased exposure time (data not shown). These results were consistent with the Tau1 signal that demonstrates that p25 mice have significantly lower levels of phosphorylated tau than Ntg mice. GSK3β inhibition therefore appears to play a dominant role in reducing the overall phosphorylation level of tau, at least in neonatal p25 mice. These results are consistent with a previous publication that reported negative effects of cdk5 activation on tau phosphorylation in different p25 mice at a young age (Plattner et al., 2006).
Enhanced β-site APP processing and Aβ production in p25 mice
GSK3β (Rockenstein et al., 2007) and GSK3α (Phiel et al., 2003) have both been shown to play a role in the amyloidogenic pathway, and cdk5 has been shown to increase Aβ production in an inducible p25 mouse (Cruz et al., 2006). APP processing and Aβ production were therefore examined in the young p25 mice. APP is cleaved by β-secretase (BACE) to produce C99 (also known as β-CTF) from the carboxy terminal fragment (CTF), and a secreted fragment, sAPPβ, is released into the extracellular space. APP can also be cleaved by α-secretase to produce C83 (also known as α-CTF) and sAPPα, which precludes β-processing. Three different sets of animals were examined (n > 6 in each experimental group) with similar results. There was a significantly greater level of C99 and sAPPβ (Ntg, n = 6, 100.0 ± 13.5 vs p25, 215.7 ± 30.8; p < 0.01), as well as increased BACE1 levels (Ntg, n = 6, 100.0 ± 18.2 vs p25, n = 5, 200.5 ± 31.6; p < 0.05) in the p25 mice compared with Ntg mice (Fig. 5A). In three different sets of mice, we noticed a trend for increased sAPPα (data not shown) and C83, but the increase was not statistically significant for either fragment in any set of mice. Consistent with enhanced β processing in p25 mice, there was a significant increase in murine Aβ production in p25 mice compared with Ntg mice (Fig. 5B). Aβ1–42 was significantly higher in all sets of p25 mice (p < 0.05). Aβ1–40 was more variable at this developmental stage in the three sets of animals. However, the mean level of Aβ1–40 was higher than in Ntg mice for all three groups (p < 0.1), but only the group with the largest number of animals (n = 12 for Ntg mice and n = 10 for p25 mice) showed a statistically significant difference (p < 0.05) (Fig. 5B).
Figure 5.

Effect of p25/cdk5 activation on APP processing and Aβ production in vivo. A, APP processing and Aβ production were examined in Ntg and p25 mice. In p25 mice, there was a significant increase in BACE levels (p < 0.05), accompanied by significantly increased β processing indicated by enhanced sAPPβ (p < 0.01) and increased level of APP C99 (β-CTF). FL, Full length. B, ELISA analysis of soluble Aβ levels in Ntg and p25 mice. There was a significant increase in Aβ40 and Aβ42 in p25 mice compared with Ntg mice.
Effect of p25 overexpression in adult mice
To assess whether the interplay between GSK3β and cdk5 continued beyond the neonatal period, the status of S9 phosphorylation, the activation status of Akt/PI3K, and phospho-tau levels were examined in p25 and wild-type (WT) mice at 3 months of age (Fig. 6A,B) and at 12 months of age (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). At 3 months of age, the level of pGSK3β–S9 was diminished compared with the neonatal mice (Fig. 6C), but it was still elevated (Ntg, n = 6, 100.0 ± 17.4 vs p25, n = 5, 160 ± 17.8; p < 0.05) compared with the Ntg controls, suggesting that GSK3β was still inhibited at this age. pErbB2–Y877, pAkt–S473, and pPI3K–Y199 were all elevated in p25 mice compared with Ntg mice, but the data did not reach statistical significance, suggesting that prolonged expression of p25, or aging, diminished the effect of cdk5 on ErbB-mediated signaling. Phosphorylated tau epitopes including S396/404, S199, and Tau1 (overall phospho-tau) were not significantly different between p25 and Ntg mice (Fig. 6B), suggesting that tau phosphorylation was balanced by the activity of GSK3β, cdk5, and possibly other kinases or phosphatases at this age. Phosphorylation at the S199 site showed a trend to decrease but did not reach statistical significance. Although cdk5 is still overactive in the p25 mice at 3 and 12 months of age (Ahlijanian et al., 2000 and data not shown), the cdk5-responsive site S235 was undetectable at 3 and 12 months of age. At 12 months of age, pGSK3β–S9 and pAkt–S473 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and phospho-tau levels (data not shown) showed no significant change between Ntg and p25 mice.
Figure 6.

Effect of p25/cdk5 activation on GSK3β regulation and tau phosphorylation in adult mice. A, Representative blots and statistical analysis showing GSK3β, GSK3β–S9, and ErbB signaling components including pAktS473, total Akt, pPI3KY199, pErbB2Y877, and p35/p25 in Ntg (n = 5) and p25 (n = 6) mice at 3 months of age. B, Representative blots showing phospho-tau at sites pS396/pS404 (PHF-1), pS199, dephosphorylated tau (Tau1), total tau, and actin levels (loading control). Although there was a trend to decrease for S199, overall levels of tau phosphorylation level indicated by Tau1 showed no change at this age. C, Comparison between the GSK3β–S9 levels for 5-d-old and 3-month-old mice (3 mice each shown). Inhibitory phosphorylation at the S9 site was significantly reduced in 3-month-old mice.
Pharmacological inhibition of cdk5 prevents GSK3β–S9 phosphorylation, increases phospho-tau, but reduces Aβ production in p25 mice
To test whether the effect of p25-mediated enhanced cdk5 activity on the GSK3β and PI3K pathways could be reversed by cdk5 inhibition, we administered a specific cdk5 inhibitor, CP-681301 (Pfizer). Three groups of neonatal mice were examined: Ntg mice treated with vehicle (n = 7), p25 mice treated with vehicle (n = 7), and p25 mice treated with CP-681301 (n = 6). As expected, CP-681301 treatment reduced the level of tau phosphorylated at the cdk5-responsive site S235 (recognized by MC6) (Fig. 7A). Interestingly, it also reversed the inhibition of GSK3β activity as shown by reduced phosphorylation at the S9 site (Fig. 7A,B). This release of inhibition correlated with an increase in phospho-tau pS199 in drug-treated mice relative to vehicle-treated controls. Overall, CP-681301 administration correlated with increased tau phosphorylation as indicated by the increased signal of the high-molecular-weight tau band in the total tau immunoblots (Fig. 7A,B) and decreased levels of dephosphorylated tau (Tau1). As expected, p25 overexpression led to an increase in Aβ production and inhibition of cdk5 reversed these effects (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 7.

Effect of cdk5 inhibitor CP-681301 on GSK3β regulation and tau phosphorylation in neonatal p25 mice. A, Representative blots showing the effect of CP-681301 on GSK3β, GSK3β–S9, and tau phosphorylated at sites pS199 and pS235, relative to total tau, and dephosphorylated tau (Tau1) in neonatal Ntg mice treated with vehicle (Ntg/Veh; n = 7), p25 mice treated with vehicle (p25/Veh; n = 7), and p25 mice treated with CP-681301 (p25/CP; n = 6). Actin was used as a loading control for this study. B, Statistical analysis of GSK3β–S9 phosphorylation, pS199, and total dephosphorylated tau (Tau1) in Ntg mice treated with vehicle (Ntg/Veh), p25 mice treated with vehicle (p25/Veh), and p25 mice treated with cdk5 inhibitor CP-681301 (p25/CP). The cdk5 inhibitor reversed the effects of overactive cdk5 in the p25 mice, leading to reduced inhibitory phosphorylation of GSK3β and increased phosphorylation of tau at relevant sites.
Pharmacological inhibition of cdk5 in aged, nontransgenic mice reduced Aβ production but not phosphorylated tau
Elderly patients may be candidates for kinase inhibitor therapeutics to target amyloid and/or tangle biogenic pathways. We therefore examined the effect of the cdk5 inhibitor on phospho-tau levels and APP processing in aged, Ntg mice. Plattner et al. (2006) reported that, in contrast to young mice, very old p25 had significantly increased GSK3β activity, leading to tau hyperphosphorylation at GSK3β relevant sites that could be reduced through administration of the GSK3 inhibitor lithium. However, administration of lithium to aged Ntg mice had little effect on phospho-tau levels (S396/S404 or S202/205 sites). To assess whether inhibition of cdk5 would reduce phospho-tau levels in aged (24–28 months) Ntg mice, CP-681301 was administered for 2 d, and the level of tau phosphorylated at the S199, S396/404, and Tau1 sites was monitored (Fig. 8). Mice administered with the cdk5 inhibitor showed either unchanged phosphorylation status of tau (three of six mice) or slightly increased phosphorylation of tau (three of six mice, especially pS199). Interestingly, the level of pGSK3β–S9 was significantly decreased in CP-treated mice relative to vehicle-treated controls, similar to that seen in younger mice, suggesting that reduced cdk5 activity results in increased GSK3 activity at this age (Fig. 8A). Similar to mice at younger ages, inhibition of cdk5 led to a highly significant reduction of Aβ peptides (Fig. 8B). These data suggest that, although inhibition of cdk5 is very effective at reducing Aβ levels, cdk5 does not target tau effectively, and inhibition may even be detrimental as it enhances GSK3β activity over and above what has been reported in Ntg aged mice (Plattner et al., 2006).
Figure 8.

Effect of cdk5 inhibitor CP-681301 on GSK3β regulation, tau phosphorylation, and Aβ production in vivo in aged mice. A, Representative blots showing the effect of CP-681301 treatment on GSK3β, GSK3β–S9, dephosphorylated tau (Tau1), phosphorylated tau at sites pS199 and pS235, and actin in Ntg mice treated with vehicle (Veh; n = 6) or with CP-681301 (CP; n = 6). B, Analysis of soluble Aβ1–40 and Aβ1–42 levels in Ntg mice treated with vehicle or with cdk5 inhibitor CP-681301. Mice used in this study were between 24 and 28 months of age.
Discussion
We have shown that p25-mediated enhanced activity of cdk5 inhibits GSK3β in neonatal mice by phosphorylation at the S9 site. Our data suggests that one way that inhibitory phosphorylation of GSK3β can be mediated is by phosphorylation of the neuregulin receptor ErbB at its activation site, leading to consequent activation of the PI3K/Akt pathway. Because young p25 mice have both overactive cdk5 and inhibited GSK3β, we were able to show that, of the two, cdk5 is the predominant kinase that regulates APP processing, whereas GSK3β is the predominant kinase regulating tau phosphorylation. Inhibition of cdk5 had a significant effect on Aβ production in old, nontransgenic mice, but it had little effect on tau phosphorylation status, suggesting that cdk5 inhibitors may not effectively target both pathways in AD.
Despite having significantly enhanced cdk5 activity, GSK3β was inhibited in young p25 mice. Assessment of the phosphorylation status of tau in neonatal mice identified sites that were hyperphosphorylated, unchanged, or hypophosphorylated, but, importantly, the net result of the interplay between phosphorylation modulators was that tau was dephosphorylated overall (indicated by increased Tau1 signal and reduced levels of high mobility, hyperphosphorylated tau). Thus, it is important to examine several sites when considering the impact of phosphorylation modulators. We also examined p25 mice at 3 and 12 months of age. By 3 months of age, pGSK3β–S9 levels were significantly reduced compared with neonatal levels, but the levels were still higher than those in Ntg animals at a similar age, suggesting some level of GSK3β inhibition remained. The reduction in inhibitory phosphorylation most likely reflects the age-associated increase in GSK3β activity that has been reported previously in aged mice (Plattner et al., 2006). At the ages studied, no significant difference in tau phosphorylation status was detected between p25 and Ntg mice at the sites of interest.
In the young mice, the potent cdk5 inhibitor CP-681301 attenuated pathways that phosphorylate GSK3β at the S9 site and thereby increased its activity, leading to increased total tau phosphorylation overall, as indicated by reduced dephosphorylated tau (Tau1) levels and enhanced higher-molecular-weight tau. Aged, nontransgenic mice administered the cdk5 inhibitor also showed a decrease in S9 inhibition and, for some epitopes such as S199, showed a trend to increase, although overall there no significant change in phospho-tau levels.
A previous study (Noble et al., 2003) has shown that, consistent with our data, tau was not hyperphosphorylated in ∼12-month-old p25 mice compared with Ntg littermates, but it was hyperphosphorylated in littermates coexpressing mutant, human tau (P301L) and p25 compared with tau-only littermates. In the bigenic p25/tau mice, the formation of aggregated tau was accelerated by the p25-mediated activation of cdk5. Because the interplay between kinases and phosphatases may be altered in the diseased brain and because their activity toward tau may be different when it is in a pathological state, it will be interesting to determine whether the effect of a cdk5 inhibitor will be different when administered to mice with plaque and/or tangle pathology compared with normal, aged mice.
Data from an inducible p25 mouse show that increased cdk5 activity correlated with enhanced BACE protein expression, increased β-site APP processing, and increased Aβ production in adult mice (Cruz et al., 2006). We have shown that p25/cdk5 activation leads to increased APP processing and production of Aβ peptides in our mouse model and, importantly, that these effects occur in mice in which GSK3β is inhibited and tau is hypophosphorylated. The effect of cdk5 on APP processing was very robust because Aβ production was significantly increased in neonatal p25 mice as well as in adult p25 mice. It was also fully reversible through administration of a cdk5 inhibitor to both p25 mice and aged nontransgenic mice.
In the p25 transgenic mice, induction of cdk5 activity by p25 overexpression was accompanied by phosphorylation of the ErbB receptor and PI3K/Akt at their activation sites, which is consistent with activation of the neuregulin signaling pathway. Activation of Akt was consistent with increased GSK3β–S9 phosphorylation and reduced GSK3β activity. Conversely, inhibition of cdk5 enhanced GSK3β activity by facilitating the dephosphorylation of S9. Our results are consistent with previous studies showing that cdk5 is involved in the neuregulin-mediated regulation of neuromuscular junction development (Li et al., 2003) and that cdk5 can phosphorylate ErbB2/3 to activate the PI3K/Akt pathway (Fu et al., 2001). Neuregulin levels and the activity of the receptor peak in neonatal mice, but they greatly reduced after maturation of the nervous system (Carraway and Burden, 1995). This is consistent with our data showing that, relative to Ntg mice at the same age, neonatal p25 mice had the highest level of GSK3β–S9 phosphorylation, 3-month-old p25 mice had diminished GSK3β–S9 phosphorylation, and aged mice (12 months old) showed no significant difference between p25 and Ntg mice.
The effect of cdk5 activity on components of the neuregulin signaling pathway was confirmed in cdk5-deficient mouse embryos, in which a reduced level of regulating phosphorylation for ErbB/PI3K/Akt and GSK3β was observed compared with WT controls. Our findings confirm and extend data from similar, low-level expressing young p25 mice (Plattner et al., 2006) that cdk5 negatively regulates GSK3β activity in vivo. However, neuregulin signaling is unlikely to be the only pathway involved, because published data has shown that GSK3β coimmunoprecipitated with cdk5, the catalytic protein phosphatase 2A (PP2A) subunit PP2Ac, and the scaffolding/regulatory PP2A subunit PR65/A in similar p25 mice (Plattner et al., 2006), implying a functional association of these molecules and implicating PP2A as a mediator of the signal between cdk5 and GSK3β. Other data also implicated phosphatases in the cdk5–GSK3β interaction (Morfini et al., 2004). Although cdk5 clearly initiates the effects seen on APP processing and tau phosphorylation in the p25 mice, it is possible that kinases other than GSK3β are also involved, either directly or indirectly, in mediating the outcome. One kinase that is also regulated by PI3K/Akt/GSK3β signaling is ERK. Phosphorylated ERK–T202/Y204 levels were increased in the neonatal p25 mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), but, although there was trend to increase, significantly increased ERK activity was not observed. Regulation is also likely to change with aging because the negative impact of p25 on GSK3β activity was significantly reduced in both sets of aged p25 mice.
Our observations in neonatal mice support observations from fetal mice that cdk5 has a protective role to play in development, because it inhibits apoptosis by activation of the PI3K/Akt pathway and promotes cell survival pathways in vivo (Li et al., 2003). Other studies suggest that short-term exposure to elevated p25 can be beneficial as transient p25 expression has also been shown to improve synaptic dendritic spine formation (Fischer et al., 2005). However, prolonged p25 expression in the same inducible mouse causes neurodegeneration and cognitive deficit (Fischer et al., 2005). Regulation of ErbB/neuregulin signaling by p25/cdk5 has implications other than in degeneration, tau phosphorylation, or APP processing. Several studies have implicated aberrant neuregulin signaling in schizophrenia. NRG-1 has been identified as a strong susceptibility candidate gene linked to schizophrenia (Lachman et al., 2006; Norton et al., 2006), and the NRG-1 receptor (ErbB4) and the substrate Akt have been identified as weak susceptibility candidate genes (Straub and Weinberger, 2006). Biochemical studies suggest that neuregulin signaling is enhanced in postmortem schizophrenia patients, leading to suppressed NMDA receptor function (Hahn et al., 2006; Norton et al., 2006). Given that the cdk5 inhibitor CP-681301 leads to reduced ErbB/neuregulin signaling in our mice, pharmacological inhibition of cdk5 may have therapeutic implications for other diseases in which the neuregulin pathway has been implicated, such as schizophrenia.
In general, the p25 mouse serves as a model system in which we can distinguish the effect of GSK3β and cdk5 activity on two pathways of potential significance in AD pathogenesis, aberrant tau phosphorylation, and APP processing (Fig. 9). Overall, our results suggest that a complex interplay between phosphorylating agents occurs in vivo, and the dynamics can change at different stages of development or maturity. The effect on important, disease-implicated substrates such as tau and APP can therefore be unpredictable and care should be taken when assessing the utility of inhibitor treatments for neurodegenerative diseases. The identification of ErbB/neuregulin signaling as a mediator of the interplay between cdk5 and GSK3β is of relevance to development as well as to pathogenic processes leading to neurodegenerative disease and possibly neuropsychiatric disorders.
Figure 9.

Schematic illustration of cdk5 and GSK3β interaction and its effects on AD-related pathways including tau phosphorylation and Aβ production.
Footnotes
This work was supported by National Institute on Aging Grant AG172116, National Institute of Neurological Disorders and Stroke Grant NS48447, and Alzheimer's Association Zenith Award (K.E.D.). We are grateful to Dr. H. Pant (National Institutes of Health) for extracts from cdk5 null mice and to Dr. S. Jung (Centocor R & D Inc., Radnor, PA) for anti-Aβ 40/42 antibodies. We thank Dr. P. Davies for the gift of tau antibodies and Dr. P. Mathews for the gift of APP antibodies.
Y.W., E.P., M.H., H.Y.F., L.W., L.L., W.H.Y., and K.E.D. declare no competing financial interests. L.-F.L. works for Pfizer and supplied the CP-681301 and CI-1033 drugs.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Carraway KL, III, Burden SJ. Neuregulins and their receptors. Curr Opin Neurobiol. 1995;5:606–612. doi: 10.1016/0959-4388(95)80065-4. [DOI] [PubMed] [Google Scholar]
- Chen S, Velardez MO, Warot X, Yu ZX, Miller SJ, Cros D, Corfas G. Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J Neurosci. 2006;26:3079–3086. doi: 10.1523/JNEUROSCI.3785-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004;377:249–255. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid β in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci. 2004;29:548–555. doi: 10.1016/j.tibs.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–838. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Brose MS. Her-2 targeted therapy: beyond breast cancer and trastuzumab. Curr Oncol Rep. 2006;8:90–95. doi: 10.1007/s11912-006-0042-4. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Fu AK, Fu WY, Cheung J, Tsim KW, Ip FC, Wang JH, Ip NY. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nat Neurosci. 2001;4:374–381. doi: 10.1038/86019. [DOI] [PubMed] [Google Scholar]
- Garcia Z, Kumar A, Marques M, Cortes I, Carrera AC. Phosphoinositide 3-kinase controls early and late events in mammalian cell division. EMBO J. 2006;25:655–661. doi: 10.1038/sj.emboj.7600967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Hallows JL, Chen K, DePinho RA, Vincent I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci. 2003;23:10633–10644. doi: 10.1523/JNEUROSCI.23-33-10633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisanaga S, Saito T. The regulation of cyclin-dependent kinase 5 activity through the metabolism of p35 or p39 Cdk5 activator. Neurosignals. 2003;12:221–229. doi: 10.1159/000074624. [DOI] [PubMed] [Google Scholar]
- Illenberger S, Zheng-Fischhofer Q, Preuss U, Stamer K, Baumann K, Trinczek B, Biernat J, Godemann R, Mandelkow EM, Mandelkow E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol Biol Cell. 1998;9:1495–1512. doi: 10.1091/mbc.9.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer's disease. J Biochem (Tokyo) 1997;121:179–188. [PubMed] [Google Scholar]
- Ishiguro K, Omori A, Sato K, Tomizawa K, Imahori K, Uchida T. A serine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci Lett. 1991;128:195–198. doi: 10.1016/0304-3940(91)90259-v. [DOI] [PubMed] [Google Scholar]
- Ishizawa T, Sahara N, Ishiguro K, Kersh J, McGowan E, Lewis J, Hutton M, Dickson DW, Yen SH. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am J Pathol. 2003;163:1057–1067. doi: 10.1016/S0002-9440(10)63465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber G, Streif R, Schwaiger FW, Kreutzberg GW, Hager G. Neuregulin-1 isoforms are differentially expressed in the intact and regenerating adult rat nervous system. J Mol Neurosci. 2003;21:149–165. doi: 10.1385/JMN:21:2:149. [DOI] [PubMed] [Google Scholar]
- Krasilnikov MA. Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation. Biochemistry (Mosc) 2000;65:59–67. [PubMed] [Google Scholar]
- Lachman HM, Pedrosa E, Nolan KA, Glass M, Ye K, Saito T. Analysis of polymorphisms in AT-rich domains of neuregulin 1 gene in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2006;141:102–109. doi: 10.1002/ajmg.b.30242. [DOI] [PubMed] [Google Scholar]
- Lemke G. Neuregulin-1 and myelination. Sci STKE. 2006;2006 doi: 10.1126/stke.3252006pe11. pe11. [DOI] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278:35702–35709. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Dobrowsky RT, Li G. Tau neurofibrillary pathology and microtubule stability. J Mol Neurosci. 2002;19:289–293. doi: 10.1385/JMN:19:3:289. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. discussion 371–380. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Norton N, Moskvina V, Morris DW, Bray NJ, Zammit S, Williams NM, Williams HJ, Preece AC, Dwyer S, Wilkinson JC, Spurlock G, Kirov G, Buckland P, Waddington JL, Gill M, Corvin AP, Owen MJ, O'Donovan MC. Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2006;141:96–101. doi: 10.1002/ajmg.b.30236. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–24064. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- Pelech SL. Networking with proline-directed protein kinases implicated in tau phosphorylation. Neurobiol Aging. 1995;16:247–256. doi: 10.1016/0197-4580(94)00187-6. discussion 257–261. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides Tau protein kinase I / glycogen synthase kinase 3beta and Cyclin-dependant kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Planel E, Sun X, Takashima A. Role of GSK-3 beta in Alzheimer's disease pathology. Drug Dev Res. 2002;56:491–510. [Google Scholar]
- Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantitation. Methods Mol Biol. 2005a;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Methods Mol Biol. 2005b;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Johnson GV. Cyclin-dependent kinase-5 in neurodegeneration. J Neurochem. 2004;88:1313–1326. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- Smaill JB, Rewcastle GW, Loo JA, Greis KD, Chan OH, Reyner EL, Lipka E, Showalter HD, Vincent PW, Elliott WL, Denny WA. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J Med Chem. 2000;43:1380–1397. doi: 10.1021/jm990482t. [DOI] [PubMed] [Google Scholar]
- Straub RE, Weinberger DR. Schizophrenia genes—famine to feast. Biol Psychiatry. 2006;60:81–83. doi: 10.1016/j.biopsych.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]