

Abstract
Tau-tubulin kinase-1 (TTBK1) is involved in phosphorylation of tau protein at specific Serine/Threonine residues found in paired helical filaments, suggesting its role in tauopathy pathogenesis. We found that TTBK1 levels were upregulated in brains of human Alzheimer' disease (AD) patients compared with age-matched non-AD controls. To understand the effects of TTBK1 activation in vivo, we developed transgenic mice harboring human full-length TTBK1 genomic DNA (TTBK1-Tg). Transgenic TTBK1 is highly expressed in subiculum and cortical pyramidal layers, and induces phosphorylated neurofilament aggregation. TTBK1-Tg mice show significant age-dependent memory impairment as determined by radial arm water maze test, which is associated with enhancement of tau and neurofilament phosphorylation, increased levels of p25 and p35, both activators of cyclin-dependent protein kinase 5 (CDK5), enhanced calpain I activity, and reduced levels of hippocampal NMDA receptor types 2B (NR2B) and D. Enhanced CDK5/p35 complex formation is strongly correlated with dissociation of F-actin from p35, suggesting the inhibitory mechanism of CDK5/p35 complex formation by F-actin. Expression of recombinant TTBK1 in primary mouse cortical neurons significantly downregulated NR2B in a CDK5- and calpain-dependent manner. These data suggest that TTBK1 in AD brain may be one of the underlying mechanisms inducing CDK5 and calpain activation, NR2B downregulation, and subsequent memory dysfunction.
Keywords: transgenic, protein kinase, Alzheimer's disease, NMDA receptor, memory, neuropathology, phosphorylation
Introduction
Tau-tubulin kinase (TTBK) family consists of TTBK1 (Sato et al., 2002) and TTBK2 (Houlden et al., 2007), and belongs to the casein kinase 1 superfamily (Manning et al., 2002). TTBK1 and TTBK2 are 60% identical and 73% similar in the first 554 aa of their sequences. TTBK2 is ubiquitously expressed in multiple tissues and was recently linked to the development of spinocerebellar ataxia type 11 (SCAT11) (Houlden et al., 2007). In SCAT11, neurofibrillary tangles (NFT), hallmarks of Alzheimer's disease, are common; neuropil threads and sparse tau-positive grains appear in the CA1 hippocampal subregion and may be involved in NFT formation in the hippocampus. TTBK1 is specifically expressed in neurons and can directly phosphorylate tau proteins both in vitro and in tissue culture cells at multiple Ser/Thr residues that are found on PHF-tau in AD brains, and is expressed in tangle-bearing neurons in the cortical region of AD brains (Sato et al., 2002). These results strongly indicate that TTBK1 is involved in tau phosphorylation in vivo and especially in AD pathogenesis. However, neither TTBK1 nor TTBK2 has been extensively characterized in AD brains or transgenic animal models.
A number of studies have been performed on the structure and assembly of tau and its phosphorylation by various kinases, including, but not limited to, cyclin-dependent protein kinase 5 (CDK5), glycogen synthase kinase-3 β (GSK3β), and c-Jun N-terminal kinase (JNK) (Ishiguro et al., 1992; Arioka et al., 1993; Drewes et al., 1997; Imahori and Uchida, 1997; Patrick et al., 1999; Sato et al., 2002). Association of CDK5 with its neuron-specific regulatory subunit p35 activates its catalytic activity, whereas cleavage of p35 to p25 by specific proteases, such as calpain–1, and formation of CDK5/p25 complexes result in its aberrant activation (Tsai et al., 1994; Patrick et al., 1999; Lee et al., 2000). CDK5 activation increases phosphorylation of axonal and synaptic molecules such as tau and NMDA receptors (Wang et al., 2003; Hawasli et al., 2007; Zhang et al., 2008). A significant increase in p25 levels and CDK5 activity was reported in human AD brains compared with non-AD brains (Lee et al., 1999; Patrick et al., 1999; Tseng et al., 2002), although others could not replicate the p25 formation in AD brains (Yoo and Lubec, 2001; Tandon et al., 2003) (for recent review, see (Giese et al., 2005)). Transgenic expression of p25 induces hyperphosphorylation of tau (Ahlijanian et al., 2000; Bian et al., 2002), with some models also showing induced progressive neurodegeneration and neurofibrillary tangle pathology (Cruz et al., 2003), adding further support to its role in AD pathogenesis.
Because there is currently no available transgenic mouse model of TTBK1 or TTBK2 to study their roles in brain pathologies, we have created transgenic mice (TTBK1-Tg) harboring 57kb human TTBK1 genomic DNA derived from P1-derived artificial chromosome (PAC) clone to reconstitute physiological expression pattern of human TTBK1 gene in mice. TTBK1 mice show CNS-specific expression of human TTBK1 transgene, which is strikingly similar to the expression pattern of endogenous mouse TTBK1 gene. TTBK1 mice develop age-dependent pathology in brain, including intraneuronal accumulation of phosphorylated neurofilament (p-NF), tau phosphorylation, cortical and hippocampal astro/microgliosis, calpain I upregulation, enhanced CDK5/p35 complex formation, NMDA receptor downregulation, and memory impairment, all recapitulating the CDK5 activation-induced neuronal dysfunction in vivo.
Materials and Methods
Transgenic animals and human specimens.
All the animal related procedures were reviewed and approved by Institutional Animal Care and Use Committee and University of Nebraska Medical Center. We have generated 4 lines of TTBK1-Tg mice (line 141, 144, 150, and 168). Human TTBK1 genomic DNA (57 kb) was isolated from RP3–330M21 PAC DNA after digestion with PmeI, PvuI, and PacI enzymes, and subjected to pulsed-field gel electrophoresis (PFGE, Bio-Rad Laboratories). The resultant 57 kb fragment was purified and its identity confirmed by Southern hybridization with a 5′ TTBK1-PCR derived probe (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). This purified fragment was injected into B6/SJL F1 mouse blastocysts at University of Michigan Transgenic Core Facility, and 4 founders were confirmed to have TTBK1 mRNA expression in the brain by Northern blotting (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). The founders were backcrossed to B6/129 F1 strain (Jackson Laboratories) for at least 5 generations before the study, and nontransgenic (non-Tg) littermates were used for the control group to minimize the effect of background difference.
Midfrontal cortex tissue samples from neurologically unimpaired subjects (n = 9) and from subjects with AD (n = 11) were obtained from the Rapid Autopsy Program at Center for Neurovirology and Neurodegenerative Disorders Brain Bank, University of Nebraska Medical Center, and the specimens were chosen based on the shortest postmortem interval (PMI) (<7 h) of age-matched specimens available. Diagnosis of AD was confirmed by pathological and clinical criteria. Tissue was frozen at autopsy and stored at –80°C until use. The demography of the specimens is listed in supplemental Table S1 and S2 for TTBK1 and p35 immunoblotting separately. Neither age nor PMI was statistically different between groups in either table (p values of Student's t test between AD and control groups: S1 age 0.054, S1 P.M.I 0.49, S2 0.168, S2 P.M.I 0.944).
Northern blotting.
Total RNA was isolated from neonate brains using Trizol (Invitrogen) according to the manufacturer's procedure. The Northern blot was hybridized with 32P-labeled cDNA probe derived from the 3′-noncoding region of human TTBK1 cDNA (1015bp), which is specific to human TTBK1 without cross-reactivity to TTBK2 gene, using DNA labeling beads (Amersham Biosciences) according to the manufacturer's instructions. The membranes were exposed to Phosphoscreen, and the images were scanned by Typhoon (GE Healthcare).
Immunoblot analysis and antibodies.
Proteins were separated by SDS-PAGE, transferred to Immobilon-P membrane (Millipore), and blocked by blocking buffer (SuperBlock with 5% skim milk, Pierce). The following antibodies were used for immunoblotting: β-actin mAb (1:10,000 dilution, Sigma-Aldrich); β-tubulin mAb (1:10,000, Abcam); T46 mAb (specific to tau 404–441, 1:1000, Invitrogen); Tau-5 mAb (human tau 218–225, 1:1000, BD Biosciences) (Porzig et al., 2007); AT8 mAb (phospho-tau Ser199, Ser202, and Thr205, 1:500, Innogenetics); PHF-1 mAb (phospho-tau Ser396 and Ser404, 1:250, gift from P. Davies); 12E8 mAb (phospho-tau Ser262 and Ser356, 1:1000, gift from P. Seubert); NMDA receptors 2A, 2B and 2D goat pAbs (C terminus, 1:1000, Santa Cruz Biotechnology); calpain 1 mAb (1:200, Santa Cruz Biotechnology); p35/p25 CDK5 activator rabbit or goat pAb (C terminus or internal region, 1:100, Santa Cruz Biotechnology); CDK5 mAb and phospho-CDK5 rabbit pAbs (C terminus and phospho-Y15, 1:200, Santa Cruz Biotechnology), phospho-GSK3α/β pAb (Ser21/9, 1:1000, Cell Signaling Technology), phospho-GSK3α/β mAb (phospho-Tyr279/216, 1:1000, Upstate Biotechnology/Millipore), GSK3β pAb (C terminus, 1:2000, Santa Cruz Biotechnology); phospho-APP pAb (phospho-Thr668, 1:1000, Cell Signaling Technology); phospho-neurofilament mAb (pNF, 1:200, Dako); TTBK1 pAb (Lot 7589, 1.67 μg/ml) (Sato et al., 2002); TTBK1 mAb (developed by immunization of BALB/c 3T3 mice with denatured purified catalytic domain of TTBK11–320, Clone F287–1.1–1E9, 13.55 μg/ml); HA mAb (1:10,000, Sigma). TTBK1 pAb and mAb cross-react with human and mouse TTBK1 but not TTBK2.
For analysis of p35/p25 expression in mouse cortex and human frontal neocortex, brain tissues were dissected from frozen specimens and homogenized in lysis buffer A [50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, 2 mm EDTA, and a proteinase inhibitor mixture (Sigma, 5 μl per 100 mg tissue)] and centrifuged at 14,000 × g at 4°C for 20 min. After determination of the protein concentration by a BCA kit (Pierce), 50 μg (for mouse brain extracts) or 100 μg (for human brain extracts) of the samples were subjected to 10% SDS-PAGE, electroblotted to Immobilon-FL membrane (Millipore), and blocked with 5% skim milk in TBST washing buffer (20 mm Tris-Cl, pH 7.4, 150 mm NaCl, 0.05% Tween 20) followed by immunoblotting using rabbit anti-p35 pAb, anti-β-tubulin mAb, or anti-β-actin mAb at the indicated dilution. After washing with TBST and incubation with horseradish peroxidase-conjugated anti-rabbit or mouse secondary antibody (Santa Cruz Biotechnology, 10,000 dilution in 5% skim milk, 1 h), the immunoreactive bands were visualized by chemiluminescence (ECL Plus, GE Healthcare) and the intensity was measured using Typhoon imaging system (GE Healthcare). The data were presented as ratiometric analysis of p35, p25, β-tubulin, and β-actin.
Immunohistochemistry.
Immunohistochemistry was performed as described previously (Yamamoto et al., 2005, 2007). Mice were killed by brief exposure to isoflurane and perfused transcardially with 25 ml of ice-cold PBS. The brains were rapidly removed, immersed in freshly depolymerized 4% paraformaldehyde for 48 h, and cryoprotected by successive 24-h immersions in 10%, 20%, and 30% sucrose in 1× PBS immediately before sectioning. Fixed, cryoprotected brains were frozen and sectioned in the sagittal plane at 30 μm using a Cryostat (Leica), with sections collected serially. Immunohistochemistry was performed using specific antibodies as listed in Table 1. Immunostained sections were visualized using Envision Plus (Dako) and DAB medium (DAB Substrate Kit, 3,3′-diaminobenzidine, SK-4100, Vector Laboratories).
Table 1.
Summary of antibodies used for immunohistochemistry
| Antibody | Epitopes | Type | Dilution | Resource | Reference |
|---|---|---|---|---|---|
| TTBK1 | TTBK1 kinase domain | M | 1:100 | Ikezu laboratory | This manuscript |
| GFAP | Glial fibrillary acidic protein | P | 1:2000 | Dako | (Yamamoto et al., 2005, 2007) |
| IBA1 | C-terminal protein | P | 1:1000 | Dr. S. Kohsaka | (Ito et al., 1998) |
| Synaptophysin | Synaptosome preparation | M | 1:2000 | Sigma | (Murphy et al., 2000) |
| Synaptotagmin | Brain synaptic junctional protein complexes | M | 1:2000 | Abcam | (Rastaldi et al., 2006) |
| Phospho-NF | Phosphorylated neurofilament | M | 1:200 | Dako | (Gorantla et al., 2007) |
| Phospho-APP | Thr668 | P | 1:250 | Cell Signaling Technology | (Kins et al., 2003) |
| Phospho-GSK3α/β | Tyr279/216 | M | 1:200 | Upstate Biotechnology | (Allen et al., 2002) |
| Phospho-GSK3α/β | Ser21/9 | P | 1:200 | Cell Signaling Technology | (Allen et al., 2002) |
| GSK3β | C-terminal protein | P | 1:200 | Santa Cruz Biotechnology | (Allen et al., 2002) |
| PHF-1 | P-tau Ser396 and Ser404 | M | 1:100 | Dr. P. Davies | (Greenberg et al., 1992) |
| AT8 | P-tau Ser199, Ser202, and Thr205 | M | 1:200 | Innogenetics | (Biernat et al., 1992) |
| 12E8 | P-tau Ser262 and/or Ser356 | M | 1:2000 | Elan Pharmaceuticals | (Seubert et al., 1995) |
| pS422 | P-tau Ser422 | P | 1:100 | Dr. A. Delacourte | (Bussière et al., 1999) |
| AT100 | P-tau Ser212 and Thr214 | M | 1:100 | Pierce | (Zheng-Fischhöfer et al., 1998) |
M, Monoclonal; P, polyclonal.
Calpain I activity assay.
Calpain I activity was measured using specific substrates and inhibitors with minor modification of the published method (Mittoo et al., 2003). Briefly, mouse hemibrains were homogenized in ice-cold lysis buffer A and centrifuged at 14,000 × g at 4°C for 20 min. After determination of the protein concentration of the extract, 100, 200, and 500 μg protein samples were incubated with or without calpain inhibitors (4 μm LLNL or 1 μm calpeptin, Calbiochem) in a 150 μl reaction buffer (50 mm Tris–HCl, pH 7.5, 50 mm NaCl, 1 mm EDTA, 1 mm EGTA, 5 mm β-mercaptoethanol, and 0.5 mm phenylmethylsulfonyl fluoride) for 10 min. Next, 50 μl of Calpain I-specific substrate II (final 20 μm, Calbiochem) and CaCl2 (final 5 mm) were added and incubated for 30 min at room temperature in darkness with gentle agitation. After the reaction, fluorescent intensity was measured at Ex. 320 nm and Em. 480 ± 20 nm using fluorometer.
CDK5 kinase assay.
The CDK5 kinase assay in the p35-immunoprecipitated complex from mouse brain was performed as described (Wang et al., 2003) with modification for the immunoprecipitation antibody. A hemibrain was homogenized in four volume of ice-cold lysis buffer containing 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, 2 mm EDTA, 1 mm sodium orthovanadate and proteinase inhibitor mixture (Sigma, 5 μl per 100 mg tissue) and centrifuged at 14,000 × g at 4°C for 20 min. The protein extracts (500 μg) were incubated with polyclonal rabbit anti-p35 (C-19, 2 μg, Santa Cruz Biotechnology) overnight at 4°C with gentle rotation, followed by the addition of 40 μl of Protein A/G-Sepharose (Santa Cruz Biotechnology) for 3 h at 4°C. The immunoprecipitates were washed four times with the lysis buffer, once with kinase buffer (without 32P-ATP and substrate), and the beads were incubated in 20 μl of kinase buffer containing 20 mm Tris-HCl, pH 7.6, 20 mm MgCl2, 2 mm MnCl2, 1 mm EDTA, 1 mm EGTA, 0.1 mm dithiothreitol, 10 μCi of [γ-32P] ATP and histone-H1 (5 μg, Fisher Scientific) at 30°C for 30 min. The reaction was stopped by the addition of Laemmli buffer and boiling for 5 min. The samples were subjected to SDS-PAGE (10% gel), and the gel was fixed and stained with R-250 Coomassie Brilliant Blue. The radioactivity signal exposed to a phosphoscreen was visualized by Typhoon system (Molecular Dynamics)
Immunoprecipitation of p35 and immunoblotting of coprecipitated protein complex.
Protein extracts from the mouse hemibrain (2 mg) were incubated with anti-p35 (C-19, 4 μg) or normal rabbit serum and Protein A/G-Sepharose as described. After washing the immunoprecipitated beads with lysis buffer three times and twice with ddH2O, the immunocomplex was eluted by of 0.1 m glycine-HCl, pH 2.5, followed by centrifugation. After neutralization of pH to 7.5 by Tris buffer, the samples were subjected to immunoblotting using goat anti-NR2B pAb, anti-calpain 1 mAb, anti-CDK5 mAb, anti-β-actin mAb, or goat anti-p35 pAb, which recognize both p35 and p25.
For separation of F-actin and G-actin in the immunocomplex, the eluted and neutralized samples were subjected to ultracentrifugation (100,000 × g for 30 min) using TSA100 rotor (Beckman) according to the published method with modifications (Barany et al., 1964). The supernatant (containing G-actin) and pellet fractions (containing F-actin) were subjected to immunoblotting using anti-β-actin mAb.
Protein mass spectrometry.
Protein identification by mass spectrometry was performed as previously described (Rozek et al., 2007). Briefly, gel pieces excised from one-dimensional electrophoresis gel were washed for 1 h at room temperature with 200 μl of 50% acetonitrile (ACN)/50 mm NH4HCO3, dried, then incubated at 37°C overnight in 0.1 μg of trypsin (Promega) plus 50 mm NH4HCO3. Resulting peptides were extracted with 60% ACN/0.1% trifluoroacetic acid (TFA), dried, and re-suspended in 0.5% TFA. Next, samples were purified using C18 Zip Tips (Millipore) according to manufacturer's procedure and re-suspended in 0.1% formic acid in water before mass spectrometric analysis. Samples were analyzed using nano-ESI-LC-MS/MS system (ProteomeX System with LCQDecaPlus, ThermoElectron) with a microcapillary RP-C18 column (New Objectives). Acquired spectra were searched against NCBI Protein database (NCBI.fasta from ftp.ncbi.nih.gov) using Sequest algorithm (BioWorks 3.2 software (ThermoElectron). Threshold for Dta generation parameters was 10,000; precursor ion mass tolerance was set at 1.4, peptide tolerance at 2.00 and fragment ions tolerance at 1.00 with two missed cleavage sites
Tissue culture, siRNA transfection, and recombinant adenovirus infection.
Primary culture of mouse cortical neurons was prepared from non-tg E16–E17 embryo and plated (1.0 × 106 cells/well in poly-d-lysine-coated 6-well plates) in Neurobasal media with 1× B27 supplement and 1 mm sodium pyruvate (all from Invitrogen) as described (Lesuisse and Martin, 2002; Yamamoto et al., 2007). After seven d differentiation and on determining that glial contamination was <15% by standard immunocytochemistry (GFAP staining for contaminated astrocytes, microtubules associated protein-2 for differentiated neurons, and Hoechst 33342 for nuclear staining), neurons were infected with HA-TTBK1 or GFP adenovirus as described (Sato et al., 2006), and one-d after the infection cells were treated with or without a calpain inhibitor (5 μm LLNL, Calbiochem) for 16 h. For the siRNA study, seven-d differentiated neurons were transfected with siRNA against mouse CDK5 (Flexitube predesigned siRNA, ID SI00947254 and SI00947247, final 40 nm concentration) or control siRNA using GeneSilencer siRNA transfection reagent (Genlatis) according to the manufacturer's instructions. Five days after the siRNA transfection, neurons were infected with HA-TTBK1 adenovirus, and one-d after the infection cells were treated with or without 5 μm LLNL for 16 h. The protein lysates were extracted from cells for immunoblotting of NR2B, HA-TTBK1, CDK5, and β-actin.
Ten day RAWM test.
Mice at 9–10 months of age (TTBK1 and non-tg littermates in B6/129 background) were introduced into the perimeter of a circular water-filled tank 91 cm in diameter and 110 cm in height with triangular inserts placed in the tank to produce six swim paths radiating out from a central area as described (Arendash et al., 2001a,b, 2004; Gordon et al., 2001; Todd Roach et al., 2004). Spatial cues for mice orientation were present on the walls of the tank. At the end of one arm, a 10 cm circular Plexiglas platform is placed, submerged 1 cm deep–hidden from the mice. The platform is located in the same arm for 4 consecutive acquisition trials (T1 through T4), and in a different arm across days. For T1-T4, the mouse starts the task from a different randomly chosen arm, excluding the arm with the platform. Each trial lasts 1 min and an error is scored each time the body of the mouse, excluding tail, enters the wrong arm, enters the arm with the platform but does not climb on it, or does not make a choice for 15 s. After T1 through T4 the mouse is returned to its cage for 30 min, and then administered a retention trial (T5). For the retention trial the start arm is the same as in T4. Each trial ends when the mouse climbs onto and remains on the hidden platform. The time taken by the mouse to reach the platform is recorded as its latency. If the mouse does not reach the platform, 60 s is recorded as its latency and the mouse is gently guided to the submerged platform. The mouse is given 20 s to rest on the platform between each trial from T1 through T4. Testing was considered complete when WT mice reached asymptotic performance (one error on trials four and five; 10 training days). The errors for the last 3 d of testing of each mouse were averaged and used for statistical analysis. The swimming speed was measured by means of the automatic video-tracking system Ethovison (version 1.92; Noldus) for 1 min.
For the balance beam task, each animal was placed at the center of a suspended beam that is segmented at 10 cm intervals (100 cm in length) and released as described (Arendash et al., 2001a). Whether the mice fell off the platform and the number of segments crossed for each trial (3 min) were recorded.
Statistical analysis.
All data were normally distributed. In case of multiple mean comparisons, data were analyzed by ANOVA, followed by Newman–Keuls multiple comparison tests using statistics software (Prism 4.0, GraphPad Software). In case of single mean comparison, data were analyzed by Student's t test. In case of RAWM, data were analyzed by two-way repeated measure ANOVA, followed by Bonferroni post test. p values < 0.05 were regarded as significant.
Results
Upregulation of TTBK1 in AD brains
We have developed both polyclonal and monoclonal antibodies against the catalytic domain of human TTBK1. Using the monoclonal antibody (Clone 1E9), we detected both endogenous full-length and processed TTBK1 around 200 and 80–100 kDa, respectively, in human brains (Fig. 1A). TTBK1 undergoes chloroquine-sensitive endoproteolysis to generate processed form, presumably because of lysosomal processing (Sato et al., 2006). The immunoreactive band pattern is consistent with the transgenic expression of human TTBK1 in TTBK1-Tg mice, and both forms of TTBK1 have kinase activities similar to those in our previous study (Sato et al., 2006). The band intensities of both full-length and processed form were significantly increased in the frontal cortical gray matter of AD cases compared with the age-matched controls (Fig. 1B), suggesting that TTBK1 expression is upregulated in AD brains.
Figure 1.

Upregulation of TTBK1 in frontal cortex of AD brains. Protein lysate was extracted from the gray matter of the frontal cortex of AD and age-matched control brains. Protein samples (100 μg/lane) were subjected to SDS-PAGE and immunoblotting using anti-TTBK1 and anti-β-actin mAbs. A, Immunoblotting of full-length (180–200 kDa) and processed TTBK1 (80–105 kDa) (top), and β-actin (bottom). B, Densitometric quantification of TTBK1/β-actin band intensity ratio for both full-length and processed TTBK1 protein (n = 9 for AD cases and 10 for control cases). *p < 0.05 as determined by Student's t test.
Human TTBK1 (hTTBK1) transgene is specific to CNS
To understand the role of TTBK1 upregulation in brain, a TTBK1-Tg mouse model was developed by microinjection of B6/SJL F1 mouse blastocyst embryo with entire hTTBK1 genomic DNA (57 kb) isolated from RP3–330M21 PAC DNA after digestion with PmeI, PvuI, and PacI enzymes, and PFGE (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). The resultant 57 kb genomic fragment was purified and its identity confirmed by Southern hybridization with a 5′ TTBK1-PCR derived probe (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Human TTBK1 gene structure consists of CpG island in the 5′ non-coding region, which is a potential promoter region for epigenetic and functional regulation (Bock et al., 2007), 13 introns, and 14 exons, spanning 57 kb in chromosome 6p21.1 (Fig. 2A). This purified fragment was injected into the mouse blastocysts, and we generated 4 lines of hTTBK1 transgenic mice (lines 141, 144, 150, and 168) as confirmed by hTTBK1 mRNA expression in the brain by Northern blotting (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Line 141 had the highest expression (20-fold higher than endogenous TTBK1) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material), and was used for the following studies (designated as TTBK1-Tg mice). Transgenic human TTBK1 mRNA is expressed as 8 kb full-length mRNA in cerebellum, spinal cord, cortex, and its 4.5 kb variant is expressed in kidney (Fig. 2B). Immunoblotting analysis of human TTBK1 protein shows similar expression pattern in TTBK1-Tg mice, which migrates around 200 (full-length) and 80–100 kDa (processed form, Fig. 1C, top). Endogenous mouse TTBK1 expression is restricted to cerebellum and cortex (bottom). There is no TTBK1 protein expression in kidney, suggesting that the 4.5 kb variant found in the Northern blotting does not contain all the exons 1–8 encoding the kinase domain of TTBK1. Overall, both endogenous and transgenic TTBK1 expression is restricted to CNS, consistent with our previous report (Sato et al., 2006).
Figure 2.

Generation of human TTBK1 transgenic mice. A, Schematic presentation of the 57 kb TTBK1 genome consisted with 5′-flanking region (containing repetitive sequences and CpG island), 14 exons (numbered boxes) and 13 introns (containing two Alu repeats in introns 1 and 12), and additional CpG island in exon 14. B, Northern blotting of mouse endogenous TTBK1 mRNA in multiple tissues. C, Immunoblotting of transgenic human TTBK1 (TTBK1-Tg, top) and endogenous mouse TTBK1 (non-Tg, bottom) in multiple tissues using anti-TTBK1 mAb.
TTBK1 and pathologic marker expression in TTBK1-Tg mice
TTBK1-Tg mice were backcrossed multiple times to develop B6/129 background strain and aged for 12–13 months for neuropathological studies. TTBK1 staining of sagittal sections of mouse brain revealed ubiquitous neuronal expression of transgenic TTBK1, as expected, using Northern and immunoblotting analyses. Especially high levels of TTBK1 were seen in subiculum (Fig. 3E,F) and external pyramidal layer of visual cortex (Fig. 3G,H) compared with the same regions in non-Tg mice (Fig. 3A–D). We also detected neurotoxicity as determined by cytoplasmic staining of phospho-NF (Neurofilament) in entorhinal cortex (EC) (Fig. 3M), subiculum (Fig. 3N), and visual cortex (Fig. 3O,P) compared with the same regions of non-Tg mice (Fig. 3I–L), suggesting neuronal dysfunction in these regions of TTBK1-Tg brain. Neurofilaments contain KSP (Lys, Ser, Pro) repeats that are consensus motifs for the proline-directed kinases. CDK5 is a proline-directed kinase that is considered to be the major kinase for the NF phosphorylation (Kesavapany et al., 2003).
Figure 3.
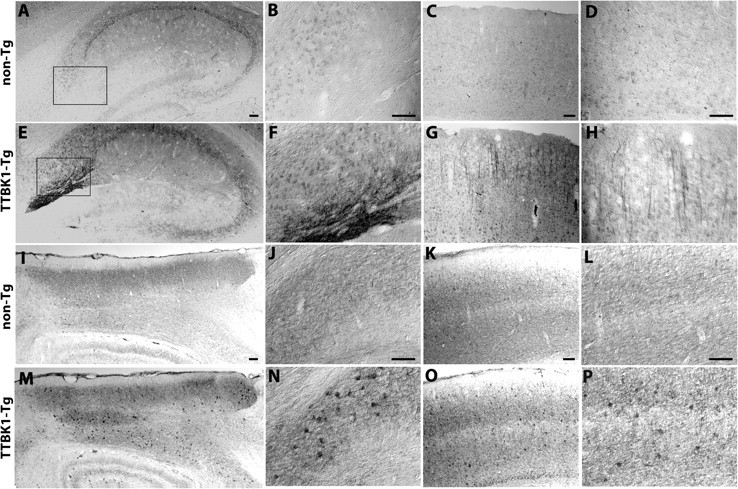
Expression of TTBK1 and phospho-NF in TTBK1-Tg mouse brain. A–H, TTBK1 staining of sagittal brain sections of aged non-Tg (A–D) or TTBK1-Tg mice (E–H) at 12–13 month-old. A, E, hippocampus; B, F, subiculum; C, G, visual cortex; D, H, external pyramidal layer of visual cortex. I–P, phospho-NF staining of non-Tg (I–L) and TTBK1-Tg mice (M–P) in the same age. I, M, entorhinal cortex; J, N, subiculum; K, O, visual cortex; L, P, the internal and external pyramidal layers of visual cortex. Original magnifications: A, C, E, G, I, K, M, O, 100×; B, D, F, H, J, L, N, P, 200×. Scale bars: (A–D, I–L), 100 μm.
TTBK1-Tg mice also show enhanced microgliosis in hippocampus and cortex (Fig. 4A–F), and astrogliosis in cortex (Fig. 4G,H) as determined by ionized calcium-binding adaptor molecule 1 (IBA1, mononuclear phagocyte marker) and glial fibrillary acidic protein (GFAP, astrocyte marker) staining. Enhanced gliosis, due to chronic TTBK1 expression, is reminiscent of neuropathology in AD brain.
Figure 4.
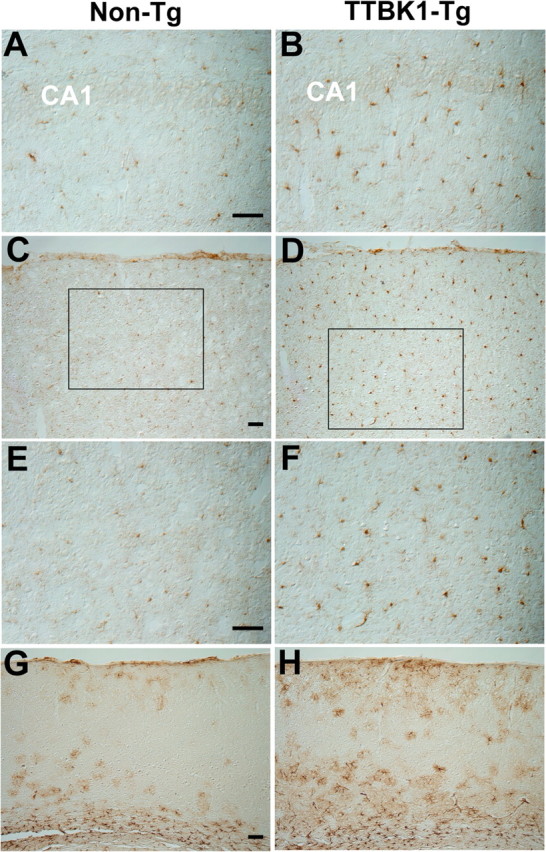
Microgliosis and astrogliosis in hippocampus and cortex of TTBK1-Tg mice. A–F, IBA1 staining of sagittal brain sections of aged non-Tg (A, C, E) or TTBK1-Tg mice (B, D, F) at 12–13 months old. A, B, hippocampus; C–F, visual cortex; E, F, high-power magnification of inset in C and D, respectively. G, H, GFAP staining of cortical region in non-Tg (G) and TTBK1-Tg mice (H). Original magnifications: (A, B, E, F), 200×; (C, D, G, H), 100×. Scale bars: (A, C, E, G), 100 μm.
Because TTBK1 is known to phosphorylate tau protein, we examined the phosphorylation status of endogenous tau protein using multiple anti-phospho-tauspecific antibodies (Fig. 5A,B). The phosphorylation at 12E8 mAb recognition site (Ser262 and Ser356) and NF (p-NF) was significantly enhanced in TTBK1-Tg mice (Fig. 5B), consistent with the enhanced p-NF staining in the entorhinal and visual cortex and subiculum. However, we could not detect Sarkosyl-insoluble form of aggregated tau protein by immunoblotting or accumulation of phospho-tau in hippocampal or cortical regions by immunohistochemistry in TTBK1-Tg mice (data not shown), suggesting that endogenous tau is not aggregated by the phosphorylation.
Figure 5.

Enhanced 12E8 phospho-tau and phospho-NF levels in TTBK1-Tg mice. A, Protein samples from cortical region from aged non-Tg and TTBK1 mice (12–13 months of age) were subjected to immunoblotting using AT8 (Ser199/202 and Thr205), PHF-1 (Ser396/404), 12E8 (Ser262/356), Tau-5 (total tau), p-APP (Thr668), p-NF, and β-tubulin antibodies, and representative blotting images were shown. B, Densitometric quantification of immunoreactive band intensity normalized by total tau protein or β-tubulin level (for p-APP and p-NF). Data are shown as percentage ratio of the total tau or β-tubulin signal. *statistical significance at p < 0.05 as determined by Student's t test (n = 4–5 for each group).
Age-dependent spatial learning impairment in TTBK-Tg mice
We examined the effect of TTBK1 expression on spatial learning function using 10-d 6-arm radial arm water maze (RAWM) test. This learning paradigm evaluates both escape latency and arm selection errors to reach the hidden platform placed in one arm in 5 trials per d for 10 d. All the mice were tested at 10 months of age. Control group (non-Tg) show clear memory acquisition based on the short and stable escape latency (Fig. 6A) and errors (Fig. 6B) by trial 4, and recall at trial 5, which is tested at 30 min after the trial 4 to evaluate their short-term memory recall ability. In contrast, TTBK1-Tg mice failed to show memory acquisition (trial 4) or recall (trial 5) based on two parameters (Fig. 6A,B), demonstrating significant memory impairment. We observed an 11% reduction in swimming speed of TTBK1-Tg mice (20.6 ± 0.83 cm/s) compared with non-Tg littermates (23.1 ± 0.81 cm/s). Although TTBK1-Tg mice swimming speed is statistically slower than non-Tg mice, this difference does not explain the greater difference in latency (22.6 and 24.1 s in trial 4 and 5 in TTBK1-Tg mice vs 6.7 and 6.8 s in trial 4 and 5 in non-tg mice) or error number (3.9 and 4.4 in trial 4 and 5 in TTBK1-Tg mice vs 1.1 and 1.2 in trial 4 and 5 in non-tg mice). The difference in swimming speed may be due to the difference in body weight (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material), showing lighter body weight of TTBK1-Tg mice compared with non-Tg mice; lighter body weight may correspond to inferior muscle development and subsequently slower swimming speeds. Other behavioral tests (such as balance beam, rotarod and forelimb grip test) did not show significant differences between the two groups (supplemental Fig. S2B,C, available at www.jneurosci.org as supplemental material), ruling out differences in sensorimotor function in the two groups. TTBK1-Tg mice (Line 141) show learning impairment as early as 7 months of age but not in Line 144 (low transgene copy line) of the same age, suggesting the earlier onset of spatial memory impairment and transgene dose-dependency (data not shown).
Figure 6.

Memory impairment in TTBK1-Tg mice as determined by radial arm water maze (RAWM). A, B, Mice at 9–10 months of age (non-Tg and TTBK1-Tg) were tested by 10-d RAWM test consisting of 5 trials (T1–T5) of hidden platform tests per day for 10 d. After T1 through T4 the mouse is returned to its cage for 30 min and then administered a retention trial (T5). The time and errors taken by the mouse to reach the platform are recorded as escape latency (A) or errors (B). *p < 0.05, **p < 0.01, or ***p < 0.001 as determined by two-way repeated-measure ANOVA with Bonferroni posttest (n = 9 for non-Tg and 10 for TTBK1-Tg group).
TTBK1 upregulates CDK5/p35 and CDK5/p25 complex formation, and calpain I activity
These neuropathological and behavioral phenotypes observed in TTBK1-Tg mice are strikingly similar to those found in p25 over-expressing mouse models. Thus, we investigated whether other tau kinases (CDK5 and GSK3β) are activated by TTBK1 expression in brain. TTBK1-Tg mice show enhanced levels of CDK5 activator p35, p25, and p25/p35 ratio (Fig. 7A,B) as determined by immunoblotting, indicative of CDK5 activation in brain. We also tested whether this mimics the CDK5 activation in AD brains using the brain samples of postmortem subjects. Both p25 and p35 levels are upregulated in the cortical gray matter of AD brains, whereas p25/p35 ratio was unchanged (Fig. 7C,D).
Figure 7.
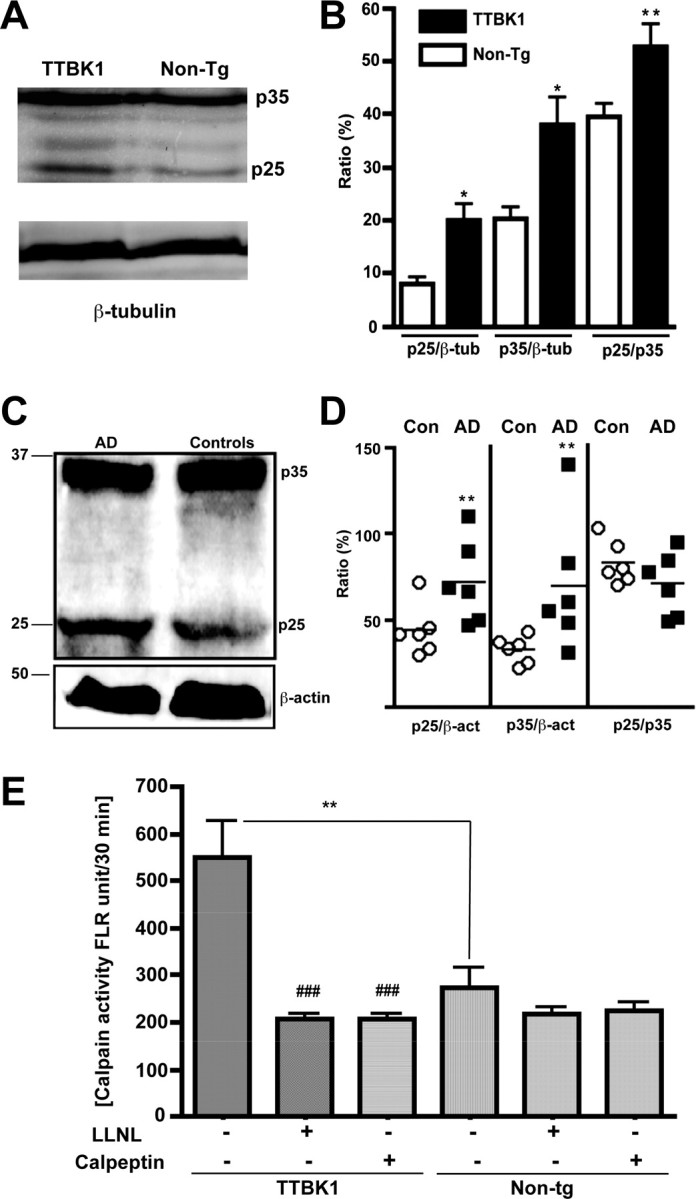
Elevated p25 and p35 subunits of CDK5 complex in TTBK1-Tg mice and AD brain, and calpain I activity in TTKB1-Tg mice. A, C, Immunoblotting of protein extracts from aged mouse cortex (A, 50 μg per lane) or human frontal cortical gray matter (C, 100 μg per lane) using anti-p35 pAb (A, C), anti-β-tubulin mAb (A), or anti-β-actin mAb (C). B, D, Chemiluminescence quantification and ratiometric presentation of p25/β-tubulin, p35/β-tubulin, or p25/p35% ratio (B) or p25/β-actin, p35/β-actin, or p25/p35% ratio (D). *p < 0.05, **p < 0.01 as determined by Student's t test (n = 5 for mouse non-Tg and TTBK1-Tg groups, and n = 6 for human AD and age-matched control groups). E, calpain I activity in the hemibrain of aged TTBK1-Tg and non-Tg mice in the presence or absence of calpain inhibitors (LLNL and calpeptin). **p < 0.01 ###0.001 vs TTBK1-Tg group without inhibitors as determined by ANOVA and Newman–Keuls post hoc.
We also examined the calpain I activity using specific substrates and inhibitors and found that calpain I activity was significantly increased in TTBK1-Tg mouse brains compared with age-matched non-Tg littermates (Fig. 7E). This enhancement may be responsible for the increased level of p25 in TTBK1-Tg mice. We have further tested whether enhanced p25 and p35 levels correspond to increased CDK5 activity in TTBK1-Tg mice. For that purpose, p35 or p25 was immunoprecipitated from a mouse brain, and the CDK5 activity of the coprecipitated CDK5/p35 or CDK5/p25 complex was measured using histone H1, a known substrate of CDK5 (Fig. 8A). We observed approximately a threefold increase in CDK5 activity in TTBK1-Tg mice (7.1 ± 0.9 × 108 cpm) compared with non-Tg mice (2.7 ± 0.5 × 108 cpm, p < 0.005), demonstrating the enhanced CDK5 activity. We also unexpectedly found that p35 or p25 coprecipitated p40 as shown in the Coomassie Brilliant Blue stained gel (Fig. 8A, top). Interestingly, the p40 band intensity is 70% reduced in TTBK1-Tg mouse group (5.44 ± 2.94 vs 12.24 ± 4.02, p < 0.05) and holds a close negative correlation with the coprecipitated CDK5 activity (Fig. 8B, p < 0.0001). This suggests that p40 competitively binds to p35 or p25, and binding of p40 to p35 or p25 inhibits the complex formation of CDK5 with p35 or p25. We subjected the stained gel to mass-spec sequencing for protein identification and determined that p40 is mouse β-actin (Table 2). Because the amount of β-actin present on the gel is not stoichiometric to the coprecipitated p35 or p25, we assume that this is polymerized F-actin. To confirm this, we eluted anti-p35 antibody-coprecipitated protein complex from protein A/G-agarose beads by 0.1M glycine, pH 2.5,, and after neutralization of the eluted samples to pH 7.5 by Tris buffer, F-actin and G-actin was separated by ultracentrifugation. The majority of the coprecipitated actin was F-actin (Fig. 8C). We also immunoprecipitated CDK5 from mouse brain using anti-CDK5 antibody, and observed no difference in it activity between TTKB1-Tg mice and non-Tg littermates, and there was no coprecipitation of p40 (data not shown). Our results suggest that although some fraction of Cdk5 formed complex and coprecipitated with p35/25, which would have higher activity than Cdk5 alone, it would be the minor faction of immunoprecipitated Cdk5. Thus, although the activity of Cdk5/p35 complex is enhanced in TTBK1-Tg mice, it did not reflect on the difference in total Cdk5 activity in brain. Together, this data suggests that TTBK1 enhances the complex formation of CDK5/p35 or CDK5/p25 via upregulation of p35 and p25 as well as dissociation of F-actin from p35 or p25 as a potential mechanism.
Figure 8.

Elevated CDK5/p35 complex activity in TTBK1-Tg mice. A, Coomassie Brilliant Blue R-250 staining of p35-immunoprecipirated samples from non-tg and TTBK1-Tg mouse brain with histone H1 (HH1) substrate (top) and autoradiogram of HH1 (32PO4-HH1, bottom). B, Scattered plot of p40 intensity and CDK5 activity from both non-tg and TTBK1-Tg mice. C, Immunoblotting of β-actin after ultracentrifugation of immunocomplex eluted from anti-p35 antibody immunoprecipitated agarose beads. T, total extract before ultracentrifugation; S, supernatant fraction (G-actin); P, pellet fraction (F-actin). D, Immunoblotting of NR2B, CDK5, calpain 1, β-actin, p35, and p25 after immunoprecipitation with anti-p35 rabbit polyclonal antibody (lane αp35) or normal rabbit IgG (lane IgG).
Table 2.
Protein coverage of p40 (mouse β-actin)
| Sequence | Mass (H+) | % Mass | aa | % aa |
|---|---|---|---|---|
| TTGIVMDSGDGVTHTVPIYEGYALPHAILR | 3185.60 | 8.13 | 122–151 | 8.60 |
| DLYANTVLSGGTTMYPGIADR | 2216.46 | 5.66 | 266–286 | 6.02 |
| EITALAPSTMK | 1162.38 | 2.97 | 290–300 | 3.15 |
| QEYDESGPSIVHR | 1517.58 | 3.87 | 334–346 | 3.72 |
| QEYDESGPSIVHRK | 1645.75 | 4.20 | 334–347 | 4.01 |
| Totals | 8153.13 | 20.81 | 76 | 21.78 |
Number of amino acids: 349 (aa 27–375), Average MW, 391,86.7; pI, 6.00.
TTBK1 downregulates NR2B in CDK5 and calpain-dependent manner
CDK5 and calpain closely regulate the postsynaptic levels of NMDA receptor (NR) subunits, especially NR2B, and affect hippocampal long-term potentiation and spatial learning (Hawasli et al., 2007; Zhang et al., 2008). Calpain also directly cleaves NR2B and reduces its cell surface expression (Simpkins et al., 2003; Wu et al., 2005, 2007). Thus, it is possible that increased activity of CDK5 and calpain I in TTBK1-Tg mouse brain lead to hippocampal neuronal dysfunction through downregulation of NR subunits. We also found that p35, calpain 1, β-actin, CDK5, and NR2B form a complex as determined by immunoblotting of the complex after immunoprecipitation of p35 (Fig. 8D). Indeed, the expression of both NR2B and NR2D were downregulated in the hippocampus of TTBK1-Tg mice compared with the age-matched non-Tg mice, whereas the expression of NR2A was unchanged (Fig. 9A,B). We also confirmed that transient expression of TTBK1 downregulated NR2B expression in primary cultured mouse cortical neurons, which are sensitive to calpain inhibitor (Fig. 10A) or silencing of endogenous CDK5 by siRNA transfection (Fig. 10B). We did not observe an additive effect from blocking both CDK5 and calpain in TTBK1-overexpresing neurons (Fig. 10B), suggesting that CDK5 is downstream of calpain with regards to the downregulation of NR2B. These findings suggest that TTBK1 induces downregulation of NR2B via upregulation of CDK5/p35 or CDK5/p25 complex formation and calpain I activity, which may account for the spatial learning impairment.
Figure 9.

Reduced NMDA receptor (NR) 2B and 2D subunits in TTBK1-Tg mouse hippocampus. A, Protein samples (50 μg per lane) from aged mouse hippocampal region were subjected to SDS-PAGE and immunoblotting using anti-NR2A, NR2B, NR2D, and β-actin antibodies. B, Densitometoric quantification of immunoreactive band normalized by the β-actin band intensity. *p < 0.05 or **p < 0.01 as determined by Student's t test (n = 4–5 in each group).
Figure 10.

Suppression of NR2B levels in primary cultured neurons after TTBK1 expression. A, Primary cultured mouse cortical neurons were infected with adenovirus expressing GFP (Ad-GFP) or HA-tagged TTBK1 (Ad-TTBK1) and treated with calpain inhibitor (5 μm LLNL). The cell extracts were subjected for immunoblotting of TTBK1 (using HA-tag antibody), NR2B, and β-actin. B, Primary neurons were transfected with siRNA for CDK5 mRNA (CDK5 siRNA) or nonspecific RNA (Con siRNA), and infected with Ad-TTBK1, followed by calpain inhibitor (LLNL) treatment. Cell extracts were subjected to immunoblotting for TTBK1, NR2B, CDK5, and β-actin.
Discussion
TTBK1 belongs to the casein kinase (CK1) group of the human kinome (Manning et al., 2002), and has previously been characterized as a tau kinase, which directly phosphorylates tau and enhances its aggregation (Sato et al., 2006). Indeed, TTBK1-Tg mice show enhanced phosphorylation of endogenous tau protein at the 12E8 recognition site (Ser262), which is extensively phosphorylated in paired helical filament (PHF) tau in AD brain but not in autopsied normal adult brain (Hasegawa et al., 1992; Morishima-Kawashima et al., 1995). This suggests that TTBK1 is involved in Ser262 phosphorylation in AD brains. Although the Ser262 site is not directly phosphorylated by the catalytic domain of TTBK1 in vitro, as determined by phosphopeptide mapping (Sato et al., 2006), we observed Ser262 phosphorylation by transient expression of full-length TTBK1 in human embryonic kidney cells (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), suggesting the TTBK1-induced activation of other tau kinases involved in Ser262 phosphorylation, such as microtubule affinity regulating kinase (MARK), cAMP-dependent protein kinase (PKA), and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Litersky et al., 1996; Drewes et al., 1997; Jenkins and Johnson, 1997). Alternatively, priming of tau phosphorylation by TTBK1 at AT8, PHF-1, and AP422 sites (Ser199, Ser202, Ser396, and Ser422) may allow MARK, PKA, or CaMKII to phosphorylate Ser262.
In addition to the effect of tau kinases on tau modifications, a significant number of studies report their tau-independent effects on neuronal dysfunction and neurodegeneration in multiple transgenic animal models. Over-expression of active GSK3β results in an AD-like phenotype (Brownlees et al., 1997), inhibition of long-term potentiation (LTP) (Hooper et al., 2007), and learning deficit in transgenic mice (Engel et al., 2006), which can be reversed by turning off GSK3β expression under the regulatory control of a conditional promoter (Lucas et al., 2001; Engel et al., 2006). However, TTBK1-Tg mice show activation of CDK5, not GSK3β, as determined by the phosphorylation of Ser9 (inhibitory phosphorylation) and Tyr216 (activating phosphorylation) (supplemental Fig. S3A,B). TTBK1-Tg mice show enhanced activity of calpain I, which may explain the enhanced p25 levels.
In our study, we observed upregulation of p25 and p35 in the frontal neocortical region of AD brain specimens of short PMI (average 3.45 h). Although there is some controversy of p25 upregulation in AD brains, especially that calpain-mediated protein degradation can be activated in postmortem brains (Taniguchi et al., 2001), our data are supportive of the upregulation of p25 in at least frontal neocortex of AD brains. However, it is not specific to p25, because both p25 and p35 were upregulated in this region.
Chronic over-expression of p25 induces significant neurodegeneration (Cruz et al., 2003) and impaired LTP (Fischer et al., 2005). However, low-level of p25 may initially act as a compensatory mechanism to increase neuroplasticity, thereby counteracting disease mechanisms that impair neuronal function (Fischer et al., 2005; Ris et al., 2005). In TTBK1-Tg mouse brain, we observed strong upregulation of CDK5/p35 or CDK5/p25 complex activity, which may be related to the novel interaction of p35 or p25 with F-actin. Interestingly another CDK5 activator, p39, can bind to actin cytoskeleton and its depolymerization by cytochalasin D leads to strong increase of p39-associated CDK5 activity (Humbert et al., 2000), suggesting similar mechanism of F-actin-regulated association of p39 with CDK5.
Several studies demonstrated that CDK5 regulates dendritic plasticity via regulation of F-actin formation through phosphorylation of multiple actin-regulatory molecules, such as WAVE1, p27kip1, Neurabin-I, and ephexin1 (Kawauchi et al., 2006; Kim et al., 2006; Causeret et al., 2007; Fu et al., 2007; Sung et al., 2008). Our data suggests that F-actin may block the formation of CDK5/p35 complex via its competitive binding to p35 as a negative feedback mechanism of CDK5 activity. However, it is not clear whether this is a direct binding of F-actin to p35 or indirect binding via known actin-binding proteins, or whether F-actin has higher affinity to p35 than CDK5. This point needs further molecular and biochemical studies with purified proteins. Perikaryal accumulation of p-NF in neurons has been shown by over-expression of CDK5 (Shea et al., 2004) and is indicative of the inhibition of NF axonal transport, frequently seen under pathological conditions. The pathologic accumulation of p-NF in the cortical region of TTBK1-Tg mice suggests chronic activation of CDK5 in vivo.
The mechanism of TTKB1-induced p25 and p35 upregulation is unknown. However, ERK activation leads to upregulation of p35 through induction of a transcriptional factor Erg1 (Harada et al., 2001), and interferon-γ also upregulates p35 through extracellular signal-regulated kinase (ERK) activation (Song et al., 2005). Because we observed astro/microgliosis in the TTBK1-Tg mouse brain, it is possible that activated microglia secrete proinflammatory cytokines for ERK activation in neurons, or TTBK1 may activate ERK through the classical ERK activation cascade, leading to p35 upregulation. Alternatively, oxidative stress can induce p35 upregulation (Strocchi et al., 2003). Both oxidative stress and ERK activity are upregulated in AD brain through amyloid plaque formation, neuroinflammation, and neuronal insults (Zhu et al., 2002; Ikezu, 2008), which may be attributed to p25 and p35 upregulation in brain.
CDK5 activity closely regulates the postsynaptic NR2B levels and LTP through CDK5 and calpain-dependent NR2B degradation (Hawasli et al., 2007). This occurs potentially through inhibition of the Src-like tyrosine kinase-mediated tyrosine phosphorylation of NR2B, which is critical to its synaptic localization (Zhang et al., 2008). In addition to CDK5, calpain also directly cleaves NR2B and downregulates its cell surface expression level. CDK5 activity is independent of calpain-mediated NR2B cleavage and instead plays a role as a scaffolding molecule for the complex formation of NR2B, p35, and calpain (Hawasli et al., 2007). TTBK1 may be facilitating this process via enhancing the dissociation of F-actin from p35, and the formation of the macromolecule complex (p35, CDK5, calpain, and NR2B). We also detected upregulation of calpain activity in TTBK1-Tg mice. Interestingly, μ-calpain levels are upregulated in AD brains, suggesting its association with TTBK1 upregulation in this report (Taniguchi et al., 2001). Our in vitro study demonstrates that TTBK1 over-expression downregulates NR2B in a CDK5- and calpain-dependent manner, further supporting the role of CDK5 and calpain for NR2B degradation in neurons.
This is the first time that a TTBK family gene has been shown to cause neuronal and cognitive dysfunction in mammalian models in vivo. The lack of tau aggregation or intraneuronal accumulation in TTBK1-Tg mice suggests this is not due to tau modifications, but more related to CDK5/p35 complex formation and calpain I activation. The effect of TTBK1 expression is in contrast to the potential role of TTBK2, which might prevent tau toxicity based on the enhanced tau pathology seen in individuals with TTBK2 mutations and a C. elegans silencing study (Kraemer et al., 2006; Houlden et al., 2007). It will be interesting to investigate single nucleotide polymorphisms or mutations in TTBK1, and TTBK2, to see how they are linked to Alzheimer's disease and other neurodegenerative disorders.
Footnotes
*S.S., J.X., and S.O. contributed equally in this study.
This work was supported by University of Nebraska Medical Center Faculty Retention Fund (T.I.), John Douglas French Alzheimer's Foundation (S.S.), and Vada Oldfield Alzheimer's Research Awards (SS and TI). US patent pending. We thank David Morgan for the consultation in spatial learning tests, Peter Shubert for 12E8 antibody, Peter Davies for PHF-1 antibody, Andre Delacourte for pS422 antibodies, Shinichi Kohsaka for IBA1 antibody, Dan Monaghan for NR2 subunit antibodies and suggestions for the NR2 data interpretation, John Davis for GSK3β antibodies, Kiyotaka Tokuraku for his expertise in actin biology, James Buescher for the maintenance and genotyping of transgenic mouse colony, and Meg Marquardt for manuscript editing.
References
- AhlijanianMK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 97:2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AllenB, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ArendashGW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM (2001a) Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res 891:42–53. [DOI] [PubMed] [Google Scholar]
- ArendashGW, Gordon MN, Diamond DM, Austin LA, Hatcher JM, Jantzen P, DiCarlo G, Wilcock D, Morgan D (2001b) Behavioral assessment of Alzheimer's transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol 20:737–744. [DOI] [PubMed] [Google Scholar]
- ArendashGW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H (2004) Environmental enrichment improves cognition in aged Alzheimer's transgenic mice despite stable beta-amyloid deposition. Neuroreport 15:1751–1754. [DOI] [PubMed] [Google Scholar]
- AriokaM, Tsukamoto M, Ishiguro K, Kato R, Sato K, Imahori K, Uchida T (1993) Tau protein kinase II is involved in the regulation of the normal phosphorylation state of tau protein. J Neurochem 60:461–468. [DOI] [PubMed] [Google Scholar]
- BaranyM, Koshland DE Jr, Springhorn SS, Finkelman F, Therattil Antony T (1964) Adenosine Triphosphate Cleavage During the G-Actin to F-Actin Transformation and the Binding of Adenosine Diphosphate to F-Actin. J Biol Chem 239:1917–1919. [PubMed] [Google Scholar]
- BianF, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KK, Walker LC (2002) Axonopathy, tau abnormalities, and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol 446:257–266. [DOI] [PubMed] [Google Scholar]
- BiernatJ, Mandelkow EM, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E (1992) The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J 11:1593–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BockC, Walter J, Paulsen M, Lengauer T (2007) CpG island mapping by epigenome prediction. PLoS Comput Biol 3:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BrownleesJ, Irving NG, Brion JP, Gibb BJ, Wagner U, Woodgett J, Miller CC (1997) Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3beta transgenes. Neuroreport 8:3251–3255. [DOI] [PubMed] [Google Scholar]
- BussièreT, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Bueé L, Delacourte A (1999) Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol 97:221–230. [DOI] [PubMed] [Google Scholar]
- CauseretF, Jacobs T, Terao M, Heath O, Hoshino M, Nikolic M (2007) Neurabin-I is phosphorylated by Cdk5: implications for neuronal morphogenesis and cortical migration. Mol Biol Cell 18:4327–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CruzJC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40:471–483. [DOI] [PubMed] [Google Scholar]
- DrewesG, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89:297–308. [DOI] [PubMed] [Google Scholar]
- EngelT, Hernández F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26:5083–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FischerA, Sananbenesi F, Pang PT, Lu B, Tsai LH (2005) Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 48:825–838. [DOI] [PubMed] [Google Scholar]
- FuWY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH, Fu AK, Greenberg ME, Ip NY (2007) Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci 10:67–76. [DOI] [PubMed] [Google Scholar]
- GieseKP, Ris L, Plattner F (2005) Is there a role of the cyclin-dependent kinase 5 activator p25 in Alzheimer's disease? Neuroreport 16:1725–1730. [DOI] [PubMed] [Google Scholar]
- GorantlaS, Liu J, Sneller H, Dou H, Holguin A, Smith L, Ikezu T, Volsky DJ, Poluektova L, Gendelman HE (2007) Copolymer-1 induces adaptive immune anti-inflammatory glial and neuroprotective responses in a murine model of HIV-1 encephalitis. J Immunol 179:4345–4356. [DOI] [PubMed] [Google Scholar]
- GordonMN, King DL, Diamond DM, Jantzen PT, Boyett KV, Hope CE, Hatcher JM, DiCarlo G, Gottschall WP, Morgan D, Arendash GW (2001) Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol Aging 22:377–385. [DOI] [PubMed] [Google Scholar]
- GreenbergSG, Davies P, Schein JD, Binder LI (1992) Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 267:564–569. [PubMed] [Google Scholar]
- HaradaT, Morooka T, Ogawa S, Nishida E (2001) ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol 3:453–459. [DOI] [PubMed] [Google Scholar]
- HasegawaM, Morishima-Kawashima M, Takio K, Suzuki M, Titani K, Ihara Y (1992) Protein sequence and mass spectrometric analyses of tau in the Alzheimer's disease brain. J Biol Chem 267:17047–17054. [PubMed] [Google Scholar]
- HawasliAH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA (2007) Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci 10:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HooperC, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S (2007) Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci 25:81–86. [DOI] [PubMed] [Google Scholar]
- HouldenH, Johnson J, Gardner-Thorpe C, Lashley T, Hernandez D, Worth P, Singleton AB, Hilton DA, Holton J, Revesz T, Davis MB, Giunti P, Giunti P, Wood NW (2007) Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet 39:1434–1436. [DOI] [PubMed] [Google Scholar]
- HumbertS, Dhavan R, Tsai L (2000) p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci 113:975–983. [DOI] [PubMed] [Google Scholar]
- IkezuT (2008) Alzheimer's disease. In: Neuroimmune pharmacology (Ikezu T, Gendelman HE, eds), pp 343–353. New York: Springer.
- ImahoriK, Uchida T (1997) Physiology and pathology of tau protein kinases in relation to Alzheimer's disease. J Biochem 121:179–188. [PubMed] [Google Scholar]
- IshiguroK, Omori A, Takamatsu M, Sato K, Arioka M, Uchida T, Imahori K (1992) Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci Lett 148:202–206. [DOI] [PubMed] [Google Scholar]
- ItoD, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57:1–9. [DOI] [PubMed] [Google Scholar]
- JenkinsSM, Johnson GV (1997) Phosphorylation of microtubule-associated protein tau on Ser 262 by an embryonic 100 kDa protein kinase. Brain Res 767:305–313. [DOI] [PubMed] [Google Scholar]
- KawauchiT, Chihama K, Nabeshima Y, Hoshino M (2006) Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol 8:17–26. [DOI] [PubMed] [Google Scholar]
- KesavapanyS, Li BS, Pant HC (2003) Cyclin-dependent kinase 5 in neurofilament function and regulation. Neurosignals 12:252–264. [DOI] [PubMed] [Google Scholar]
- KimY, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P (2006) Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442:814–817. [DOI] [PubMed] [Google Scholar]
- KinsS, Kurosinski P, Nitsch RM, Götz J (2003) Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol 163:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KraemerBC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD (2006) Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet 15:1483–1496. [DOI] [PubMed] [Google Scholar]
- LeeKY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN (1999) Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res 34:21–29. [DOI] [PubMed] [Google Scholar]
- LeeMS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405:360–364. [DOI] [PubMed] [Google Scholar]
- LesuisseC, Martin LJ (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 51:9–23. [DOI] [PubMed] [Google Scholar]
- LiterskyJM, Johnson GV, Jakes R, Goedert M, Lee M, Seubert P (1996) Tau protein is phosphorylated by cAMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J 316:655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LucasJJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ManningG, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- MittooS, Sundstrom LE, Bradley M (2003) Synthesis and evaluation of fluorescent probes for the detection of calpain activity. Anal Biochem 319:234–238. [DOI] [PubMed] [Google Scholar]
- Morishima-KawashimaM, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y (1995) Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem 270:823–829. [DOI] [PubMed] [Google Scholar]
- MurphyDD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20:3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PatrickGN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402:615–622. [DOI] [PubMed] [Google Scholar]
- PorzigR, Singer D, Hoffmann R (2007) Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem Biophys Res Commun 358:644–649. [DOI] [PubMed] [Google Scholar]
- RastaldiMP, Armelloni S, Berra S, Calvaresi N, Corbelli A, Giardino LA, Li M, Wang GQ, Fornasieri A, Villa A, Heikkila E, Soliymani R, Boucherot A, Cohen CD, Kretzler M, Nitsche A, Ripamonti M, Malgaroli A, Pesaresi M, Forloni GL, Schlöndorff D, Holthofer H, D'Amico G (2006) Glomerular podocytes contain neuron-like functional synaptic vesicles. FASEB J 20:976–978. [DOI] [PubMed] [Google Scholar]
- RisL, Angelo M, Plattner F, Capron B, Errington ML, Bliss TV, Godaux E, Giese KP (2005) Sexual dimorphisms in the effect of low-level p25 expression on synaptic plasticity and memory. Eur J Neurosci 21:3023–3033. [DOI] [PubMed] [Google Scholar]
- RozekW, Ricardo-Dukelow M, Holloway S, Gendelman HE, Wojna V, Melendez LM, Ciborowski P (2007) Cerebrospinal fluid proteomic profiling of HIV-1-infected patients with cognitive impairment. J Proteome Res 6:4189–4199. [DOI] [PubMed] [Google Scholar]
- SatoS, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A (2002) Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem 277:42060–42065. [DOI] [PubMed] [Google Scholar]
- SatoS, Cerny RL, Buescher JL, Ikezu T (2006) Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J Neurochem 98:1573–1584. [DOI] [PubMed] [Google Scholar]
- SeubertP, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, et al (1995) Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270:18917–18922. [DOI] [PubMed] [Google Scholar]
- SheaTB, Yabe JT, Ortiz D, Pimenta A, Loomis P, Goldman RD, Amin N, Pant HC (2004) Cdk5 regulates axonal transport and phosphorylation of neurofilaments in cultured neurons. J Cell Sci 117:933–941. [DOI] [PubMed] [Google Scholar]
- SimpkinsKL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR (2003) Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci 23:11322–11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SongJH, Wang CX, Song DK, Wang P, Shuaib A, Hao C (2005) Interferon gamma induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J Biol Chem 280:12896–12901. [DOI] [PubMed] [Google Scholar]
- StrocchiP, Pession A, Dozza B (2003) Up-regulation of cDK5/p35 by oxidative stress in human neuroblastoma IMR-32 cells. J Cell Biochem 88:758–765. [DOI] [PubMed] [Google Scholar]
- SungJY, Engmann O, Teylan MA, Nairn AC, Greengard P, Kim Y (2008) WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc Natl Acad Sci U S A 105:3112–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TandonA, Yu H, Wang L, Rogaeva E, Sato C, Chishti MA, Kawarai T, Hasegawa H, Chen F, Davies P, Fraser PE, Westaway D, St George-Hyslop PH (2003) Brain levels of CDK5 activator p25 are not increased in Alzheimer's or other neurodegenerative diseases with neurofibrillary tangles. J Neurochem 86:572–581. [DOI] [PubMed] [Google Scholar]
- TaniguchiS, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M (2001) Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett 489:46–50. [DOI] [PubMed] [Google Scholar]
- Todd RoachJ, Volmar CH, Dwivedi S, Town T, Crescentini R, Crawford F, Tan J, Mullan M (2004) Behavioral effects of CD40-CD40L pathway disruption in aged PSAPP mice. Brain Res 1015:161–168. [DOI] [PubMed] [Google Scholar]
- TsaiLH, Delalle I, Caviness VS Jr, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371:419–423. [DOI] [PubMed] [Google Scholar]
- TsengHC, Zhou Y, Shen Y, Tsai LH (2002) A survey of Cdk5 activator p35 and p25 levels in Alzheimer's disease brains. FEBS Lett 523:58–62. [DOI] [PubMed] [Google Scholar]
- WangJ, Liu S, Fu Y, Wang JH, Lu Y (2003) Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci 6:1039–1047. [DOI] [PubMed] [Google Scholar]
- WuHY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K (2005) Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J Biol Chem 280:21588–21593. [DOI] [PubMed] [Google Scholar]
- WuHY, Hsu FC, Gleichman AJ, Baconguis I, Coulter DA, Lynch DR (2007) Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem 282:20075–20087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YamamotoM, Horiba M, Buescher JL, Huang D, Gendelman HE, Ransohoff RM, Ikezu T (2005) Overexpression of monocyte chemotactic protein-1/CCL2 in beta-amyloid precursor protein transgenic mice show accelerated diffuse beta-amyloid deposition. Am J Pathol 166:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YamamotoM, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T (2007) Interferon-{gamma} and tumor necrosis factor-{alpha} regulate amyloid-{beta} plaque deposition and {beta}-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol 170:680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YooBC, Lubec G (2001) p25 protein in neurodegeneration. Nature 411:763–764; discussion 764–765. [DOI] [PubMed] [Google Scholar]
- ZhangS, Edelmann L, Liu J, Crandall JE, Morabito MA (2008) Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J Neurosci 28:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-FischhöferQ, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E (1998) Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 252:542–552. [DOI] [PubMed] [Google Scholar]
- ZhuX, Lee HG, Raina AK, Perry G, Smith MA (2002) The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals 11:270–281. [DOI] [PubMed] [Google Scholar]