

Abstract
Proteins containing extended polyglutamine repeats cause at least nine neurodegenerative disorders, but the mechanisms of disease-related neuronal death remain uncertain. We show that sympathetic neurons containing cytoplasmic inclusions formed by 97 glutamines expressed within human huntingtin exon1–enhanced green fluorescent protein (Q97) undergo a protracted form of nonapoptotic death that is insensitive to Bax deletion or caspase inhibition but is characterized by mitochondrial dysfunction. By treating the neurons with combined cytosine arabinoside and NGF withdrawal, we demonstrate that Q97 confers a powerful resistance to apoptosis at multiple levels: despite normal proapoptotic signaling (elevation of P-ser15–p53 and BimEL), there is no increase of Puma mRNA or Bax activation, both necessary for apoptosis. Even restoration of Bax translocation with overexpressed Puma does not activate apoptosis. We demonstrate that this robust inhibition of apoptosis is caused by Q97-mediated accumulation of Hsp70, which occurs through inhibition of proteasomal activity. Thus, apoptosis is reinstated by short hairpin RNA-mediated knockdown of Hsp70. These findings explain the rarity of apoptotic death in Q97-expressing neurons. Given the proteasomal blockade, we test whether enhancing lysosomal-mediated degradation with rapamycin reduces Q97 accumulation. Rapamycin reduces the amount of nonpathological Q25 by 70% over 3 d, but Q97 accumulation is unaffected. Interestingly, Q47 inclusions form more slowly as a result of constitutive lysosomal degradation, but faster-forming Q97 inclusions escape lysosomal control. Thus, cytoplasmic Q97 inclusions are refractory to clearance by proteasomal and lysosomal systems, leading to a toxicity that dominates over neuroprotective Hsp70. Our findings may explain the rarity of apoptosis but the inevitable cell death associated with polyQ inclusion diseases.
Keywords: Bim, cell death, Hsp70, lysosome, proteasome, Puma
Introduction
Huntington's disease (HD) is an autosomal dominant disorder caused by an abnormal polyglutamine (polyQ) expansion in the N-terminal region of the Huntingtin protein (Htt). This mutation causes the protein to misfold and aggregate, leading to selective neuronal dysfunction and death (Orr and Zoghbi, 2007). Currently, little is understood regarding the mechanisms of neuronal cell death in HD. Whereas some studies indicate the importance of apoptosis (Hackam et al., 1998; Martindale et al., 1998; Saudou et al., 1998; Sanchez et al., 1999; Jana et al., 2001; Tang et al., 2005), others have questioned this conclusion (Jackson et al., 1998; Vonsattel and DiFiglia, 1998; Moulder et al., 1999; Turmaine et al., 2000). Although death may proceed nonapoptotically, components of apoptosis, such as p53-activation, cytochrome c (cyt c) release, and even caspases, may still contribute to HD pathology without full-blown expression of apoptosis being evident (Ona et al., 1999; Chen et al., 2000; Jana et al., 2001; Bae et al., 2005; Wang et al., 2008).
The mechanisms of apoptosis and their mode of suppression by survival factors are especially well understood in sympathetic superior cervical ganglion (SCG) neurons (Pierchala et al., 2004; Putcha and Johnson, 2004; Ham et al., 2005; Wyttenbach and Tolkovsky, 2006; Pierchala et al., 2007), thus providing a good system for testing how mutant polyQ protein and the ensuing polyQ aggregation process may interact with the apoptotic pathway. Kim et al. (2004) observed that hemagglutinin (HA)-tagged polyQ127 formed nuclear aggregates whose prevalence was reduced by exogenous Hsp70 or by stabilizing endogenous Hsp70 using proteasome inhibitors. polyQ127–HA caused somal and dendrite atrophy but no death up to 7 d. Suzuki and Koike (2005) applied penetratin-linked cell-permeable peptides containing 10 or 22 polyQ repeats fused to a nuclear localization signal sequence, observing that Q22, which is usually nontoxic, formed nuclear aggregates and suppressed Trk signaling but caused necrosis-like death rather than the anticipated apoptotic death. Why apoptotic death failed despite inhibition of TrkA-mediated survival signaling was not addressed.
Cytosolic aggregates of mutant polyQ-expanded Htt are also prevalent in HD and can perturb several vital pathways, including survival signaling downstream of Trk (Song et al., 2002). Whether trophic signaling is disrupted in SCG neurons expressing cytosolic polyQ aggregates has not been tested. PolyQ toxicity crucially depends on the balance between aggregate formation and degradation rates (Colby et al., 2006; Williams et al., 2006; King et al., 2008). The factors that determine the steady-state level of polyQ aggregates in SCG neurons are not known. We have modeled mutant polyQ toxicity in SCG neurons using polyQ expressed in the native context of exon1 of human Htt (Kazantsev et al., 1999; Sathasivam et al., 1999). We report that, through its inhibition of proteasomal activity, Q97 causes Hsp70 accumulation, which in turn prevents apoptotic mechanisms at several crucial control points. Apoptosis is thereby subverted into necrosis-like death, which is not ameliorated by Hsp70 or by enhancement of the lysosomal degradation pathway using rapamycin. The dominant mode of death described here coupled to proteasome and lysosome insufficiencies may explain why the neurodegeneration observed in HD is so insidious.
Materials and Methods
Chemicals.
The following chemicals were used: clasto-lactacystin β-lactone (β-lactone), lactacystin, MG132, and proteasome substrate I (z-LLL-AMC; Calbiochem), Boc-(O-methyl)aspartyl-fluoromethylketone (BAF; MP Biomedicals), bafilomycin A1 (BA1) and cytosine arabinoside (AraC; Sigma), and Lipofectamine 2000, Lysotracker Red, Superscript II RNase H–Reverse Transcriptase, RNAaseOUT, and Taq polymerase (Invitrogen).
Antibodies.
The following antibodies were used: neurofilament (2H3; (Developmental Studies Hybridoma Bank), proteasome 20S (PW8155; Biomol), LAMP-1 (Lgp120; gift from J. Paul Luzio, University of Cambridge, Cambridge, UK), ubiquitin (SPA-200; Stressgen), LC3 (gift from Y. Uchiyama, Osaka University Graduate School of Medicine, Suita, Osaka, Japan); Hsp70 (SPA-810; Stressgen), actin (Sigma), P-ser473–Akt, Akt, P-ser63–c-Jun, P-Thr202/Tyr204–ERK1/2, P-Thr183/Tyr185–JNK, P-ser15–p53, and P-ser235/236–S6 (Cell Signaling Technology), active Bax (1D1; Neomarkers), BimEL (Millipore Bioscience Research Reagents), cyt c (BD Biosciences), CoxIV (Abcam), and green fluorescent protein (GFP) (polyclonal 8637-1, monoclonal 8371-2; BD Biosciences).
SCG and PC12 culture.
Sympathetic neurons were isolated from superior cervical ganglia of postnatal day 0–1 Wistar rat pups or mouse pups and cultured as described previously (Hawrot and Patterson, 1979; Buckmaster et al., 1991; Nobes and Tolkovsky, 1995). Use of animals followed national guidelines and was approved by the ethical committees of the institutions. Bax-null founder mice were obtained from the late Dr. Stanley Korsmeyer (Dana Farber Institute, Boston, MA) and were used after extensive crossing into the CD1 background. Animals were genotyped as described previously (Wyttenbach and Tolkovsky, 2006). Neuronal cells were enriched by two successive preplates on collagen-coated dishes and seeded on collagen-coated wells or coverslips in L15–CO2 medium containing 20 μm uridine/fluorodeoxyuridine, 3% rat serum or FBS, and 100 ng/ml 2.5S NGF. Cultures were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C. Collagen from the tail tendons of obsolete adult Wistar rats was solubilized in 0.018% acetic acid, and the concentration was adjusted to an absorbance of 0.15 at 280 nm. PC12 cells were grown on collagen-coated wells as described previously (Greene and Tischler, 1976).
Neuronal survival and apoptosis.
The undersides of four-well plates were divided into nine small squares using a needle. SCG neurons were seeded at relatively low density (1500–6000 neurons), and the number of phase bright cells was counted under phase microscopy. At least 250 neurons were counted per sample. Live cultures were stained with Hoechst 33342 (5 μg/ml) to visualize nuclear morphology, and propidium iodide (5 μg/ml) or trypan blue (0.08%) were used to determine plasma membrane integrity. To induce apoptosis, neurons were deprived of NGF by washing three times (10 min each) in L15–CO2 medium containing 3% FBS but lacking NGF. The last medium change also contained 0.5% anti-NGF antiserum raised in the laboratory and, when indicated, the dominant proapoptotic drug AraC (1 mm, neutralized before addition) (Anderson and Tolkovsky, 1999).
Adenovirus preparation and infection of neurons.
E1/E3-deficient recombinant adenoviruses (Ad) (serotype 5, subgroup C), containing full-length human Htt exon1 with polyQ fused to enhanced GFP (EGFP) at the C-terminal (abbreviated Q25, Q47, and Q97) and EGFP constructs were generated using the AdEasy system (He et al., 1998). The number of polyQ repeats was verified by sequencing. After amplification in 293 cells, virus was purified by two rounds of ultracentrifugation on CsCl gradients, desalted on a PD-10 column, and stored at −80°C. Adenoviruses were empirically titrated for equal infectivity in SCG neurons by quantifying the number of EGFP-fluorescing cells in the presence of proteasome and autophagy inhibitors and confirmed by analyzing expression by immunoblotting (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To determine polyQ inclusion body load (IB), the percentage of neurons expressing GFP containing GFP-positive (GFP+) aggregates was determined. Between 300 and 500 neurons were counted per sample.
Hsp70 short hairpin RNA cloning and transfection.
Hsp70 targeting sequences (Magrané et al., 2004) were cloned into pSilencer (Ambion) using the insert design tool provided by Ambion: Hsp70 #1, 5′-CCAAGGTGCAGGTGAACTATTCAAGAGATAGTTCACCTGCACCTTGGGCTTTTTT-3′; 5′-AATTAAAAAAGCCCAAGGTGCAGGTGAACTATCTCTTGAATAGTTCACCTGCACCTTGGGGCC-3′; Hsp70 #2, 5′- GCGAGAACCGGTCGTTCTATTCAAGAGATAGAACGACCGGTTCTCGCCCTTTTTT-3′;5′-AATTAAAAAAGGGCGAGAACCGGTCGTTCTATCTCTTGAATAG-AACGACCGGTTCTCGCGGCC-3′; and Scr, 5′-TTATTCTATTCT-CGTCAAC TTCAAGAGA GTTGACGAGAATAGAATAACCTTTTTT-3′; 5′-AATTAAAAAAGGTTATTCTATTCTCGTCAACTCTCTTGAAGTT-GACGAGAATAGAATAAGGCC-3′.
The two oligonucleotides for each targeting sequence were annealed by heating to 100°C and slow cooling to room temperature. The annealed primers were ligated into the pSilencer 1.0-U6 vector (Ambion), which was doubly cut with ApaI and EcoRI restriction enzymes (New England Biolabs). Correct insertion of the short hairpin RNA (shRNA) targeting sequences was verified by sequencing (DNA Sequencing Facility, Department of Biochemistry, Cambridge University, Cambridge, UK). Transfection of SCG neurons and PC12 cells was performed using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer, using 1 μg of each shRNA plasmid DNA mixed with 0.5 μg of H2B–GFP plasmid DNA.
Quantitative reverse transcription-PCR.
RNA was extracted from neurons seeded at 12,000 cells per well using the RNeasy minikit (Qiagen). cDNA was prepared from 0.5 μg of RNA as described previously (Wyttenbach and Tolkovsky, 2006) and amplified using Syber Green PCR master mix (Invitrogen) and an ABI Prism PCR machine/detector (GeneTool). Mean threshold values of triplicate repeats per experiment were used to normalize the relative amount of transcript to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as control. The amount of transcript was calculated using the formula (0.5xRNA/0.5cRNA) * 100, where xRNA is mean test cDNA cycle, and cRNA is mean GAPDH cDNA cycles. The following primers were used (5′ to 3′): BimEL/L/S forward, GGCCAAGCAACCTTCTGATG; BimEL/L/S reverse, GCCTTCTCCATACCAGACGG; Puma forward, TCCTCAGCCCTCCCTGTCAC; Puma reverse, CCATTTCTGGGGCTCCAGGA; and GAPDH forward, ATTGTCAGCAATGCATCCTG; GAPDH reverse, TCAGCTCTGCGATGACCTTGCC.
Immunocytochemistry and microscopy.
Dissociated SCG neurons (1500–3000 per well) were seeded on collagen-coated glass coverslips. At the end of each treatment, cultures were fixed in 3% paraformaldehyde for 20 min, followed by three washes in PBS. Some cultures were stained with propidium iodide before fixation to mark cells that had lost plasma membrane integrity. Some cultures were postfixed in methanol at −20°C for 5 min. Cells were subsequently permeabilized (0.3% Triton X-100 in PBS with 10% goat serum) for 1 h before addition of the primary antibody. Primary antibodies were diluted in the same buffer and incubated for 1 h at room temperature or overnight at 4°C, washed four times, and incubated with the appropriate secondary antibody. Hoechst 33342 was used to visualize nuclei. Coverslips were mounted in Vectashield H-1000 (Vector Laboratories) or Fluoromount-G (Southern Biotechnology Associates). Immunostaining was analyzed by fluorescence or spinning disc confocal laser microscopy using an Olympus IX70 microscope. Images were recorded using a digital CCD camera (AstraCam) connected to an UltraView LCI confocal imaging system (PerkinElmer Life and Analytical Sciences).
Immunoblotting.
Neurons (12,000–15,000 per well) were harvested by gentle pipetting, washed in PBS, and lysed in ice-cold lysis buffer (Virdee et al., 1999) containing phosphatase inhibitors and protease inhibitors (Complete; Roche Diagnostics). The supernatant remaining after centrifugation at 9000 × g for 10 min was assayed for protein (BCA kit; Sigma), the appropriate amount was denatured in SDS-PAGE sample buffer, and proteins were resolved by electrophoresis on SDS–polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane (0.2 μm pore size) using a semidry electroblotter. Membranes stained with Ponceau Red were photographed, blocked with 5% milk in TBST (50 mm Tris-Cl, pH 7.5, 0.15 m NaCl, and 0.1% Tween 20), and probed with antibodies diluted in TBST. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL) (GE Healthcare) and exposed to Kodak X-OMAT or Fuji/Konica film. For reprobing, membranes were stripped in a solution of 0.1 m glycine, pH 3, containing 1% SDS for 30 min at room temperature, followed by extensive washing in TBST. Alternatively, 0.1% NaN3 was used to irreversibly inhibit the HRP signal of the secondary antibody if a primary antibody from a different species was used subsequently. Films were scanned (Hp Scanjet 5470c), and the intensity of bands was analyzed by optical densitometry using NIH Image J analysis software (http://rsbweb.nih.gov/ij/).
Resolubilization of insoluble lysate fraction with formic acid.
Insoluble pellets that were harvested alongside proteins extracted for immunoblotting (described above), were washed twice in PBS, and resuspended in 100% formic acid for 1–2 h at 37°C (Hazeki et al., 2000). Formic acid was removed under vacuum centrifugation, and the solubilized material was resuspended in 20 μl of sample buffer containing 0.25 m Tris base to restore alkaline pH. Resuspended proteins were separated by SDS-PAGE and immunoblotted as described above.
Proteasome assay.
Dissociated SCG neurons were seeded at 60,000 cells per reaction. After 4–6 d, neurons were infected with either Ad-Q25 or Ad-Q97 and cultured for 2 d. Cells were washed three times in PBS, and nondenaturing cytosolic extracts were prepared according to the method described previously (Kisselev and Goldberg, 2005) in 30 μl of buffer containing 50 mm Tris-HCl, pH 7.5, 250 mm sucrose, 5 mm MgCl2, 2 mm ATP, 1 mm DTT, 0.5 mm EDTA, and 0.025% digitonin. After 20 min on ice, lysates were centrifuged for 10 min at 17,500 × g, protein concentration was determined by BCA analysis, and 15 μg equivalents were diluted into 50 μl of 26S proteasome assay buffer (50 mm Tris-HCl, pH 7.5, 40 mm KCl, 5 mm MgCl2, 0.5 mm ATP, 1 mm DTT, and 0.5 mg/ml BSA) in a 96-well plate. Reaction was started by adding 50 μl of 200 μm z-LLL-AMC (Calbiochem). AMC release was quantified using a fluorescent plate reader (FLUOstar OPTIMA; BMG Labtech) at 37°C from 1 to 30 h at 6 h intervals. Nonspecific hydrolysis was measured by adding 10 nm expoxomicin for 30 min before mixing with proteasome substrate. Linearity of measurement was determined using free AMC.
Statistics.
The mean value of replicates within an experiment (<10% difference) was taken as a single value when calculating SDs from multiple experiments. Usually, duplicates or triplicate wells were seeded per experiment. Multiple comparisons were made using ANOVA followed by post hoc testing (SigmaStat; Systat Software). Pairwise comparisons were conducted using two-tailed Student's t test. Either 95% confidence intervals or one-sample t test were used for calculating the significance of ratiometric values,
Results
Q97 forms insoluble cytoplasmic inclusions
SCG neurons were cultured for 4–6 d before infection with adenovirus constructs encoding human htt exon1–EGFP fusion proteins containing polyQ stretches of 97 or 25 glutamines (abbreviated to Q97 and Q25, respectively) (Fig. 1). GFP-positive neurons appeared within 24 h after infection with Ad–Q97, at first displaying mostly diffuse Q97 distribution throughout the cell but then giving rise to intensely fluorescent IBs in the cytoplasm within the next 20 h (Fig. 1A). After 2 d, the number of IB per cell, which ranged from 1 to 12, increased only slightly (Fig. 1B), but the diameter of aggregates grew considerably, accompanied by diminished diffuse cytoplasmic Q97 fluorescence (Fig. 1A). By day 3, virtually all Q97-expressing neurons contained aggregates (Fig. 1B). In contrast, Q25 expression remained cytoplasmic and diffuse up to 6 d (Fig. 1C,D), consistent with previous studies in cell lines (Wyttenbach et al., 2000). Formation of Q97 IB provoked notable changes in the organization of neurofilaments, proteasomes, and lysosomes. Neurofilaments were rearranged in both cell bodies and neurites from a filamentous form that was uniformly distributed to one encaging individual IB in 85% of the cells by day 4 (data not shown). Proteasomes were recruited to Q97 IB in 40% of the neurons after 4 d, detected with an antibody raised against 20S core subunits (Fig. 1C). Lysosomes (detected with anti-LAMP-1) also reorganized, forming what appear as “rosettes” surrounding each IB in 37% of neurons by day 4 (Fig. 1D).
Figure 1.

Q97 forms multiple cytoplasmic IBs that recruit proteasomes and lysosomes. SCG neurons infected with Ad-Q25 or Ad-Q97 were analyzed for kinetics of IB formation and expression of proteasome (anti-20S subunits) or lysosomes (anti-LAMP-1). A, Collapsed confocal Z stack of a typical neuron (whose perimeter is encircled by the dotted line) showing the nucleation of multiple, Q97-positive IBs over 20 h (from 24 to 44 h). B, The red line depicts the percentage of neurons with IBs (inclusion load). Between 2 and 4 d, neurons were further examined to analyze whether new inclusions were being formed continuously. Approximately stable proportions of neurons were found to contain 1–3, 4–6, or 7–12 IBs, with a minor fraction containing >12 IBs, as shown under the line. Note that, by 3 d, virtually all the neurons expressed cytoplasmic IBs. C, Immunostaining with anti-20S proteasome (red) of Q25- and Q97-expressing neurons (green) after 4 d; nuclei are shown in blue (Hoechst 33342). Note recruitment of proteasomes from a diffuse pattern (in Q25-positive cells) to the IB (in Q97-positive cells). D, Immunostaining with anti-LAPMP-1 (red) after 4 d shows a similar pattern of redistribution to that of the proteasomes. Scale bars, 10 μm.
Formation of Q97 IB also caused changes in the expression and organization of nonstructural proteins. Hsp70 expression was significantly elevated (Fig. 2A,C) in 85% of IB-positive neurons by day 4 (Fig. 2A, middle column). In ∼35% of the neurons, Hsp70 was localized to IB, similar to the pattern of proteasomal staining (Fig. 2A, rightmost column). An increase in ubiquitin expression also occurred, characterized mainly by IB localization (Fig. 2B, middle column), although formation of cytoplasmic bodies that did not colocalize with IBs was also found in ∼15% of cells (Fig. 2B, rightmost column). However, massive ubiquitylation of Q97 was apparent at day 2 after infection with Ad-Q97, detected when the pellet of the NP-40 lysate, which contained insoluble Q97 IBs, was resolubilized in formic acid and analyzed by immunoblotting (Fig. 2C). The resolubilized Q97 IB fraction also contained most of the Hsp70 immunoreactivity, which was also elevated from day 2 onward but not at 1 d after infection (Fig. 2C), thus lagging behind IB formation by ∼1 d. In contrast, Q25 expression neither provoked accumulation of Hsp70, nor was there any Q25 or ubiquitinylated Q25 in the pellet of the soluble fraction (Fig. 2A–C). It should be noted that the apparently more rapid accumulation of soluble Q97 compared with Q25 on day 1 (Fig. 2C) was not attributable to unequal infectivity of the two viruses but attributable to ongoing, selective degradation of Q25 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Hence, SCG neurons respond to Q97 expression and IB formation by rearranging proteasome and lysosomal components, by ubiquitylation of Q97, and by upregulating Hsp70, the latter two processes appearing coincidentally with increased growth of Q97 IB (from day 2 onwards).
Figure 2.

Q97 expression leads to the accumulation of Hsp70. SCG neurons were infected with Ad-Q25 or Ad-Q97 for 4 d, after which cells were fixed and immunostained for Hsp70 (A, red) or ubiquitin (B, red); fluorescent images show nuclei (blue) and Q25 or Q97 (green). Right column in A shows Hsp70 redistributed to IB, which occurred in ∼35% of the neurons. Right column in B shows “hotspots” of ubiquitin staining distributed independently of IB. Note selective increase of Hsp70 and ubiquitin in Q97- but not Q25-expressing neurons. Scale bars, 10 μm. C, SCG protein extracts were lysed in NP-40 buffer at days 1–4 (D1–D4) after infection. The soluble fraction was removed and the insoluble protein pellet was solubilized in formic acid as described in Materials and Methods. Both fractions were analyzed by immunoblotting with anti-GFP, to analyze the distribution of Q25 and Q97 between soluble and insoluble fractions. Arrowheads, Specific bands; asterisks, nonspecific bands. Blots from the insoluble fraction were also probed with anti-Hsp70 or anti-ubiquitin. Note specific upregulation of Hsp70 and ubiquitylated Q97, beginning from day 2 in the insoluble fraction of Q97-expressing neurons.
Q97 promotes nonapoptotic cell death
We next examined the kinetics of Q97-induced toxicity (Fig. 3). There was no difference in the rate of cell death between Q25- and Q97-expressing neurons in the first 3 d after infection with Ad-Q25 or Ad-Q97 (Fig. 3A). By day 4, however, the number of neurons expressing Q25 had stabilized, whereas the number of Q97-expressing neurons continued to decline, ∼80% of the neurons dying by day 6. Cell death of neurons expressing Q25 was suppressed in rat SCG neurons by addition of the pan-caspase-inhibitor BAF (Fig. 3B) or by deletion of Bax in mouse Bax−/− neurons (Fig. 3C), which are resistant to several types of proapoptotic insults (Deckwerth et al., 1996; Gillardon et al., 1996; Putcha et al., 1999; Kirkland and Franklin, 2003; Wyttenbach and Tolkovsky, 2006; Smith and Deshmukh, 2007). These data indicate that the early toxicity that occurred after infection with Ad-Q25 was apoptotic. However, there was no rescue of rat Q97-expressing neurons by the caspase inhibitor BAF or of Q97-expressing neurons from Bax−/− mice. Hence, Q97-expressing SCG neurons died nonapoptotically, in a Bax- and caspase-independent manner.
Figure 3.

SCG neurons die nonapoptotically in response to Q97 expression. Neurons infected with equivalent amounts of Q25 and Q97 were scored for cell death at the indicated time points by counting the number of phase-bright cells in nine selected regions. A, Mean ± SD; n = 3; **p < 0.01, Student's t test. B, A cohort of cultures was treated with 50 μm BAF. Dashed lines are from results in A. Mean ± SD; n = 3; *p < 0.05, Student's t test. C, Neurons from Bax heterozygote or knock-out mice were infected with Ad-Q25 or Ad-Q97 and scored for cell death as above. Mean ± range; n = 2, each experiment conducted in three independently infected wells. Comparisons between triplicates in each experiment were tested using Student's t test, *p < 0.05. D, SCG neurons were costained for cyt c and CoxIV and examined by confocal microscopy on day 4. Representative images of cells expressing Q25 (top row) and Q97 (bottom row) are shown. Arrow indicates cell lacking cyt c but with clustering and condensation of mitochondria, indicated by staining with anti-CoxIV. Scale bars, 10 μm. E, Quantification of the number of cells lacking mitochondrial cyt c from days 1 to 4 (mean ± SD; n = 2 for day 3; n = 4 for days 3 and 4; comparisons by t test, ***p < 0.001). F, Neurons were treated with 10 μm CsA from day 2 and analyzed for cyt c retention in mitochondria at day 4 (mean ± SD; n = 3; comparison of Q97 ± CsA by t test, **p < 0.01).
To better define the course of events leading to Q97-induced nonapoptotic death, neuronal cultures were stained for cyt c a key step in death commitment. Within 4 d from the onset of Q97 expression, up to 80% of the neurons lost immunostaining for cyt c compared with 20% of Q25-expessing neurons (Fig. 3D,E). Figure 3D shows that mitochondria in Q97-expressing neurons that had lost cyt c staining were condensed and clustered (as monitored by CoxIV staining) rather than absent as would occur in the case of mitophagy (Xue et al., 2001). Coincidental with cyt c loss, neurons containing Q97 IBs did not load the membrane potential-sensitive dye mitotracker orange, suggesting an association between dissipation of mitochondrial membrane potential and cyt c loss (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Consistent with loss of mitochondrial membrane potential, neurons containing Q97 IB showed intense fluorescence of ethidium cation 2 d after loading cells with dihydroethidium (Budd et al., 1997). No ethidium accumulated in Q25-expressing neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Cyclosporine A (CsA) has been used to rescue SCG neurons from nonapoptotic death after loss of mitochondrial membrane potential and cyt c release (Chang and Johnson, 2002). Addition of CsA 2 d after infection prevented mitochondrial dysfunction in approximately half of Q97-expressing neurons at day 4 (from 80 to 45%), assessed by the loss of mitochondrial cyt c (p < 0.01) (Fig. 3F), whereas untreated neurons were unaffected. These data are consistent with involvement of the permeability transition pore in the nonapoptotic death, although the marked toxicity of CsA to Q25-expressing cells precluded additional examination of this question. Thus, delayed nonapoptotic death is correlated with mitochondrial dysfunction.
Trk-mediated signaling is not reduced by Q97, but death-associated signals are elevated
SCG neurons depend on NGF signaling via Akt and extracellular signal-regulated kinase (ERK) for trophic and survival support (Tsui-Pierchala et al., 2000). In SCG neurons expressing a Q22–NLS peptide (Suzuki and Koike, 2005), NGF-induced Trk signaling was reduced before toxicity. We therefore first examined whether expression of Q97 caused a deficiency in NGF signaling, which may explain why neurons die. However, Figure 4A shows that there was no change in Akt or ERK phosphorylation. Moreover, phosphorylation of Akt and ERK1/2 was reduced after NGF deprivation, demonstrating that NGF signaling was constitutively active in Q97-expressing neurons (Fig. 4B). Hence, it is unlikely that Q97-expressing neurons die as a result of lack of trophic support by NGF. There was also no difference in the extent of c-Jun N-terminal protein kinase (JNK) phosphorylation between Q25- and Q97-expressing neurons, P-JNK being reduced between days 2 and 4, consistent with the presence of NGF-mediated signaling (Virdee et al., 1997). However, a notable difference between the signals present in Q25- and Q97-expressing neurons was the selective elevation of between 6- and 10-fold in P-ser63–c-Jun and BimEL expression in Q97-expressing neurons beginning at day 2 (Fig. 4A). Both proteins have been implicated in SCG neuron death after NGF deprivation (Ham et al., 2005) and are signals that are normally suppressed by NGF (Virdee et al., 1997; Biswas and Greene, 2002). However, both proteins accumulate after proteasomal inhibition regardless of receptor signaling (Jariel-Encontre et al., 1995; Biswas and Greene, 2002). In preliminary experiments, proteasome inhibition with lactacystin also induced P-ser63–c-Jun (M. A. King, unpublished data). Thus, Q97 causes a dominant toxic signal that overrides the survival signals induced by NGF.
Figure 4.

Expression of Q97 does not inhibit NGF-mediated signaling. SCG neurons expressing Q25 or Q97 were lysed at days 1–4 (D1–D4), and soluble extracts were analyzed by immunoblotting for expression/phosphorylation of various signaling proteins. A, Analysis of P-ser473–Akt (P-Akt), P-ERK1/2 (P-ERK), total ERK (tERK) (as loading control), Hsp70, P-JNK, P-ser63–c-Jun (P-c-jun), and BimEL. Note increased P-c-Jun and BimEL expression in Q97-expressing neurons beginning at day 2 but similar amounts of P-JNK (that decline with time) in both sets of neurons. B, NGF (+N, −N) was removed (as described in Materials and Methods) from cohort Q25- and Q97-expressing cultures for 24 h between days 2 and 3 and analyzed for expression of P-Akt and P-ERK, with tERK as loading control. Note similar reduction in phosphokinase staining in NGF-deprived neurons of both sets.
Q97-expressing neurons are resistant to death induced by simultaneous NGF deprivation and AraC addition
The finding that Q97 induced mitochondrial cyt c loss but no apoptosis prompted us to test whether the neurons would die by apoptosis when exposed to a potent proapoptotic signal, achieved by simultaneous withdrawal of NGF and cotreatment with AraC (abbreviated to A/-N) (Anderson and Tolkovsky, 1999). Figure 5 shows that 75% of Q25-expressing neurons treated with A/-N died apoptotically within 1 d (Fig. 5A,B), displaying typical apoptotic nuclear fragmentation also observed in uninfected neurons treated with A/-N (Fig. 5A). However, <10% of nuclei of Q97-expressing neurons showed any nuclear aberrations during exposure to A/-N (Fig. 5A,B). Additionally, Q97-expressing neurons failed to be labeled by terminal deoxynucleotidyl transferase-mediated nick end labeling, whereas apoptotic nuclei were strongly labeled (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). There was no defect in proapoptotic signaling induced by A/-N, because treatment with A/-N induced equivalent increases in P-ser15–p53 and BimEL (Wyttenbach and Tolkovsky, 2006) in uninfected, Q25- and Q97-expressing neurons (Fig. 5C). However, there was little activation and recruitment of Bax to mitochondria in Q97-expressing cells treated with A/-N, evidenced by staining with 1D1 antibody, which reveals exposure of the N terminus in active Bax (cells displaying mitochondrial 1D1-positive Bax rising from 10 ± 5 to 14 ± 5%, mean ± range; n = 2) (Fig. 5D), whereas most uninfected neurons showed intense Bax immunostaining at the mitochondria (86 ± 6%, mean ± range; n = 2). Interestingly, ∼66% of the neurons expressing Q47, which did not form insoluble IBs during this period, did display Bax activation (Fig. 6D). Hence, the deficit in activation of Bax translocation to the mitochondria appears to depend on IB formation.
Figure 5.
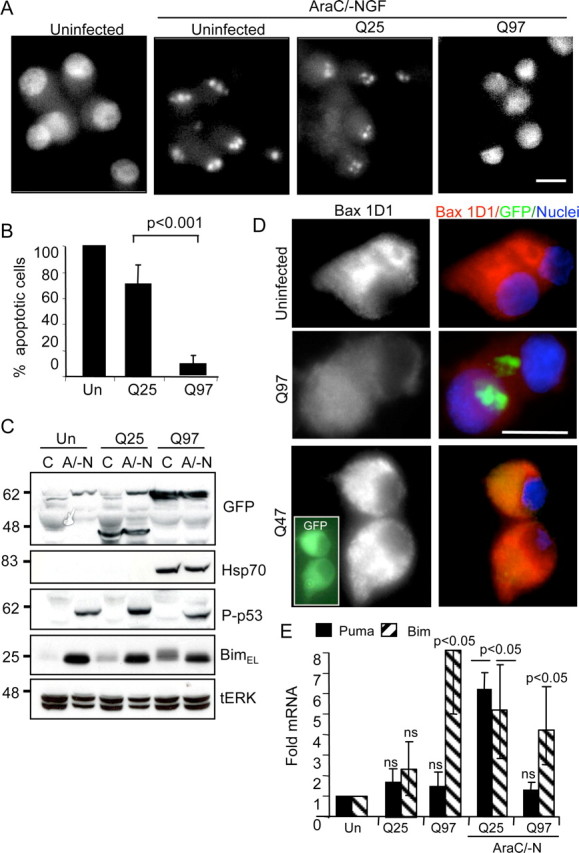
Intense proapoptotic stimuli do not kill Q97-expressing neurons by apoptosis. SCG neurons were infected with Ad-Q25 or Ad-Q97 or left uninfected. After 2 d, each type of culture was deprived of NGF and treated with 1 mm AraC (AraC/-N or A/-N) for 24 h. A, Apoptotic morphology: cells were fixed and stained with Hoechst 33342 to delineate nuclear morphology. Scale bar, 10 μm. B, Quantification of apoptosis (mean ± SD; n = 4; p < 0.001, Student's t test). C, Uninfected (Un), Q25-expressing, and Q97-expressing neurons express apoptotic signals in response to A/-N. Neurons were left untreated or treated from days 2 to 3 with 1 mm AraC in the absence of NGF (A/-N). BAF (50 μm) was added to all cultures to prevent loss of neurons undergoing apoptosis. Extracts were probed for P-ser15–p53, BimEL, and Hsp70, using tERK as loading control. D, A/-N induces Bax activation only in neurons devoid of IB. Neurons were infected with Ad-Q25, Ad-Q47, or Ad-Q97, and Bax activation was detected by immunostaining for active Bax with anti-Bax 1D1 (red); nuclei stained with Hoechst are in blue, and expression of Q25, Q47 (inset), and Q97 is indicated in green. Note normal Bax activation in Q47-expressing neurons. E, RT-PCR detection of Puma and Bim mRNA. Uninfected neurons or neurons expressing Q25 or Q97 for 2 d were left untreated or treated with 1 mm AraC in the absence of NGF but in the presence of 50 μm BAF for 12 h. The amount of mRNA was normalized to control GAPDH mRNA for each extract, setting the values from uninfected controls to 1 to calculate fold change. Mean ± SD; n = 3; p < 0.05, ANOVA; p < 0.05 versus control Tukey's honestly significant difference test except Q97/Puma, which was not significantly changed.
Figure 6.
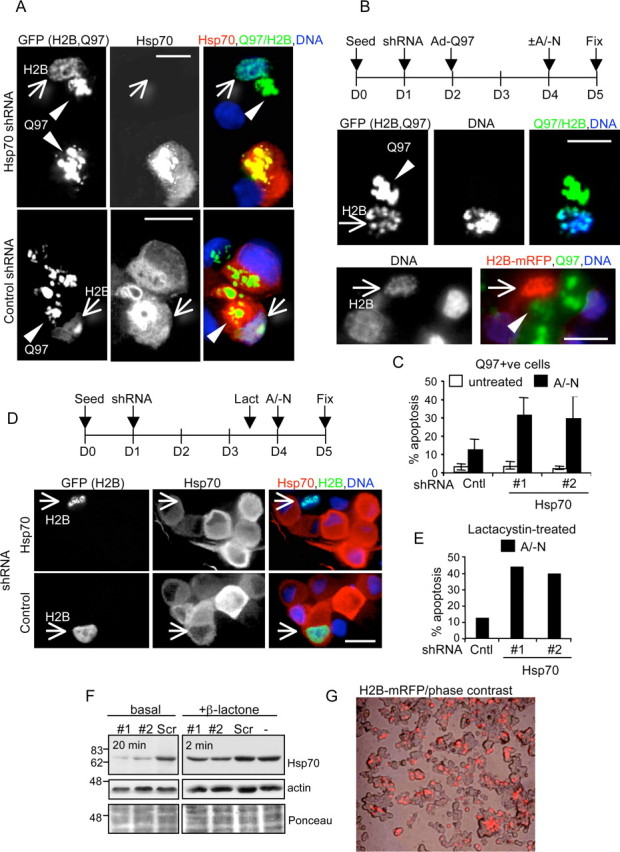
Hsp70 shRNA reduces Q97-induced Hsp70 expression and enables apoptosis. SCG neurons were cotransfected with respective shRNA constructs and either H2B–GFP or H2B–mRFP using Lipofectamine 2000 at 1 d after seeding. On day 2, neurons were either infected with Ad-Q97 and treated on day 4 with A/-N as indicated or treated with 4 μm lactacystin on day 3.5 and then treated with A/-N at day 4. Neurons were then fixed and then immunostained for Hsp70 and/or scored for apoptotic nuclei. A, Example of Q97+ neurons expressing H2B–GFP that were cotransfected with Hsp70 shRNA (top row) or control shRNA (bottom row); arrow indicates H2B–GFP-positive nucleus undergoing apoptosis in which Hsp70 is downregulated by the Hsp70 shRNA but is not maintained attributable to transfection with control shRNA. Arrowhead indicates Q97 IBs. B, Schematic of treatment regimen. Examples of Q97+ neurons expressing H2B–GFP or H2B–mRFP that were cotransfected with Hsp70 shRNA, treated with A/-N, immunostained for Hsp70, and scored for apoptosis (indicated by arrows). C, Quantitation of apoptosis in untreated or A/-N-treated Q97+ neurons expressing control (Cntl), Hsp70 #1, or Hsp70 #2 shRNA. Mean ± SD; A/-N-treated, n = 5 (nonspecific shRNA) or n = 6 (Hsp70 #1, Hsp70 #2) independent infections/transfections; untreated, n = 3 independent infections/transfections. Student's two-tailed t test: p < 0.0004 Scr control versus Hsp70 #1; p < 0.02 versus Hsp70 #2. D, Example of neurons treated with lactacystin (Lact) 12 h before treatment with A/-N for 24 h. Arrow indicates an apoptotic neuron expressing H2B–GFP cotransfected with Hsp70 shRNA and a healthy neuron expressing H2B–GFP cotransfected with nonspecific shRNA. E, Graph shows quantification of apoptosis in one experiment conducted as in D. F, PC12 cells were transfected with the indicated Hsp70 or control shRNAs in combination with H2B–mRFP or with empty vector and H2B–mRFP (−) at a ratio of 2:1 plasmid DNA were cultured for 48 h, after which one cohort was treated with 2 μm β-lactone for 14 h. Extracts were immunoblotted for Hsp70. Scr (scrambled) is the loading control. Ponceau stainings are shown as loading controls. G, Example of PC12 cells expressing Hsp70 #1 shRNA and H2B–mRFP, showing ∼50% prevalence in the population. Scale bars, 10 μm.
BH3-only proteins regulate Bax translocation to the mitochondria (Adams and Cory, 2007). We have shown previously that Puma is the crucial BH3-only protein that mediates AraC-induced (p53-dependent) apoptosis as well as >60% of apoptosis induced by NGF deprivation, the remainder being Bim dependent (Wyttenbach and Tolkovsky, 2006). Puma-induced apoptosis requires its transcriptional activation. An analysis of Puma mRNA by quantitative reverse transcription (RT)-PCR 3 d after infection showed that treatment with A/-N for 12 h was sufficient to cause a sixfold increase in Puma mRNA in Q25-expressing neurons, whereas there was no significant increase in Puma mRNA expression in Q97-expressing neurons (Fig. 5E). Q97 expression did not inhibit transcription per se, because Bim mRNA was still elevated fourfold to eightfold in Q97-expressing neurons in the absence or presence of A/-N treatment, similar to the fivefold increase in Bim mRNA induced by A/-N in Q25-expressing neurons (Fig. 5E). To examine whether the lack of Puma was limiting the amount of Bax recruited to the mitochondria and thus the reason for lack of apoptosis in response to A/-N, neurons were infected with Ad-Puma and, after 24 h, were inspected for Bax recruitment. Indeed, Bax was recruited to mitochondria and activated in 38 ± 6% of Q97-expressing neurons (mean ± range, two independent experiments, duplicate independent infections) (Bax activation shown in supplemental Fig. S4, available at www.jneurosci.org as supplemental material), and yet Q97-expressing neurons failed to die by apoptosis, the nuclei remaining intact. These data, together with the data shown in Figure 2, demonstrate that there is a strong correlation between the presence of Q97 IB and suppression of apoptosis, which may be mediated through at least two mechanisms: (1) by suppression of transcriptional upregulation of Puma, thereby preventing Bax activation, and (2) by suppressing caspase activation downstream of Bax activation.
Hsp70 induction is responsible for the resistance of Q97-expessing cells to apoptotic treatments
Hsp70 is known to inhibit apoptotic death by interfering with signals that lie both upstream and downstream of mitochondrial outer membrane permeabilization during induction of apoptosis (Beere and Green, 2001). To test whether Q97-mediated upregulation of Hsp70 is responsible for evoking antiapoptotic effects against A/-N treatment, we transfected the neurons with plasmids encoding Hsp70 shRNA constructs (#1 or #2) or a nonspecific shRNA as control and then infected neurons with Ad-Q97 the following day. We cotransfected H2B–GFP or H2B–monomeric red fluorescent protein (mRFP) together with the shRNAs to assess nuclear morphology of transfected cells (Goemans et al., 2008). Figure 6A shows that shRNA against Hsp70 reduced the amount of Hsp70 expressed in Q97+ neurons to below detectable levels compared with the strong induction of Hsp70 in Ad-Q97 infected cells transfected with the nonspecific shRNA construct. Some Q97+ neurons in which Hsp70 was downregulated contained apoptotic nuclei (Fig. 6A). Concomitant with the suppression of Hsp70 upregulation, treatment with Hsp70–shRNA resensitized neurons containing Q97 IB to undergo apoptosis when treated with A/-N for 24 h between days 4 and 5 (Fig. 6B), indicated by the significant increase of fragmented mRFP–H2B-labeled nuclei (to 32% for #1, 29% for #2) compared with neurons expressing the nonspecific shRNA (12%) (Fig. 6C). Hsp70 shRNA also induced apoptosis of 30–40% neurons when lactacystin, which elevated Hsp70 (Fig. 7C), was added 12 h before addition of A/-N (Fig. 6D,E). To demonstrate the efficacy of the shRNA, we transfected PC12 cells with the three constructs and probed for Hsp70 by immunoblotting (Fig. 6F,G). Approximately 50% of the cells were H2B–GFP positive, and, accordingly, there was ∼50% decrease in the intensity of Hsp70 in PC12 cells induced to express Hsp70 by treatment with β-lactone. Basal expression of Hsp70 was very low, but it was still possible to demonstrate a reduction in expression in cells expressing the specific shRNA constructs. Thus, elevation of Hsp70 in response to expression of Q97 is a major barrier to execution of apoptosis in response to A/-N.
Figure 7.

Q97 inhibits proteasomal activity and function. A, Measurement of protein degradation. SCG cultures were infected with Q25 or Q97 for 2 d, whereas uninfected cohort cultures were either left untreated or treated with 10 μm lactacystin for 12 h before the four sets of cultures were washed free of methionine and labeled with [35S]methionine and described in Materials and Methods. After 1 h, cells were washed, medium was replenished with unlabeled methionine, and the amount of TCA-precipitable 35S-labeled protein remaining in the neurons was determined after 1 h or 11 h of incubation. Results show the proportion of label retained in the neurons 11 h after onset of chase normalized to values obtained after 1 h [mean ± SD; n = 4, Q25, Q97; n = 3 uninfected (Uninf) or lactacystin (Lact) treated; p < 0.012 by ANOVA; pairwise comparisons by t test]. B, Proteasomal activity assay. Neurons expressing Q25 or Q97 for 2 d were extracted in proteasome assay buffer, normalized for the amount of protein, and incubated for 30 h with z-LLL-AMC. Control neurons were treated with 20 μm epoxomycin (epox) over the same period to measure maximal activity. Activity was measured in a fluorescent plate reader (mean ± range; n = 2; p < 0.05, one-sample t test). C, Proteasome inhibition causes accumulation of Q25 and Hsp70. Neurons were infected with Ad-Q25 or Ad-Q97 and after 90 min were treated with 4 μm β-lactone when indicated for 20 h. Soluble fractions were analyzed by immunoblotting for expression of Q25/Q97 with anti-GFP or Hsp70 expression. Loading was controlled with anti-tERK1/2. Note accumulation of Q25 and Hsp70 in the soluble fraction in β-lactone-treated neurons but no parallel increase in the amount of Q97. The insoluble fraction was probed with anti-GFP and anti-ubiquitin. Note no change in the amount of Q97 or ubiquitylated Q97 in β-lactone-treated neurons. D, Q97 causes accumulation of Q25 or GFP. SCG neurons were infected with Ad-Q25 or Ad-GFP alone or coinfected with Ad-Q97 and cultured without additional treatment for 24 h. Soluble fractions were analyzed by immunoblotting with anti-GFP antibodies. Note increased amounts of Q25 or EGFP only under conditions in which Q97 was coexpressed. Bottom panels show actin and Ponceau S staining as loading controls.
Hsp70 accumulation is attributable to inhibition of proteasome function by Q97
We next investigated why Hsp70 accumulated in Q97-expressing neurons. We suspected that Hsp70 accumulates as a result of proteasome inhibition because Kim et al. (2004) reported that inhibition of proteasomal activity is sufficient to increase Hsp70 expression in SCG neurons. Moreover, we found a coincident increase in Hsp70 accumulation, recruitment of proteasomes to Q97 IBs, and an increase in the proteasomal substrates P-ser63–c-Jun and BimEL under conditions when they are normally suppressed, i.e., in the presence of NGF. Furthermore, consistent with induction of Hsp70, Bim, and c-Jun mRNA by proteasome inhibition (Butts et al., 2005; Yew et al., 2005; Concannon et al., 2007), we found an induction of their mRNAs (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Notably, the relative elevation in P-ser63–c-Jun protein induced by Q97 was much higher than that of c-Jun mRNA, indicating that at least part of the protein accumulation was attributable to inhibition of proteasomal degradation. We tested whether proteasomal activity was inhibited by Q97 using two different assays (Fig. 7). We first investigated whether Q97 expression impairs general protein degradation after methionine starvation, a condition in which amino acids are generated during the first few hours via proteasome activity rather than autophagy (Vabulas and Hartl, 2005). Neurons expressing Q25 or Q97 for 2 d (when Hsp70 is first increased) were labeled with [35S]methionine in methionine-free medium for 1 h and chased in unlabeled medium containing methionine for 1 h (to provide a reference point) or 11 h (to observe the bulk fraction) (Franklin and Johnson, 1998). Uninfected and Q25-expressing neurons showed a similar loss of TCA-precipitable 35S-labeled proteins during the 11 h chase period of 48 or 40%, respectively (Fig. 7A). Q97 expression, however, clearly reduced loss of 35S-labeled proteins by approximately fourfold, to ∼10% (Fig. 7A), similar to the reduction caused by the proteasomal inhibitor lactacystin. We also assayed proteasomal activity using the proteasomal substrate z-LLL-AMC. Neurons expressing Q97 showed a significant 30% reduction in the rate of hydrolysis of z-LLL-AMC compared with Q25-expressing cells (Fig. 7B). Consistent with the idea that proteasome inhibition was mediating the increase in Hsp70 (Kim et al., 2004), the amount of soluble Hsp70 was increased by the inhibitor β-lactone in Q25- and Q97-expressing neurons within 1 d after infection (Fig. 7C), before the time (day 2) when Q97 causes Hsp70 induction on its own (Fig. 2C). It is interesting to note that the Hsp70 that accumulated as a result of treatment with β-lactone at day 1 was entirely soluble. Thus, the presence of Hsp70 in the insoluble fraction at days 2–4 (Fig. 2C) is likely attributable to the accumulation of insoluble Q97.
Given that Q97 inhibited proteasomal activity and Hsp70 has been shown to inhibit Q127–HA accumulation (Kim et al., 2004), we investigated whether deliberate inhibition of proteasomal activity would alter the accumulation of Q97 or Q25. Figure 7C shows that the amount of Q25 was increased approximately fourfold in response to β-lactone, indicating its turnover via the constitutive proteasome system. However, there was no increase in the amount of insoluble Q97. Rather, there was a small reduction in soluble Q97, possibly attributable to Hsp70 accumulation (Kim et al., 2004). Hence, degradation of insoluble Q97 is impervious to proteasome activity. To further demonstrate proteasome inhibition by Q97, we also coexpressed Q97 with Q25, or with Ad-Rous sarcoma virus promoter-driven GFP, reasoning that Q97 expression will prevent the degradation of the latter proteins via the proteasome. Figure 7D shows that the amount of Q25 or EGFP increased dramatically during coexpression with Q97, consistent with inhibition of proteasome by Q97. Thus, we propose that proteasomal inhibition by Q97 IBs is the likely cause of Hsp70 accumulation, which, in turn, subverts the death mechanism away from apoptosis, despite the release of cyt c from the mitochondria by as yet undefined mechanisms. Other apoptotic mechanisms upstream of cyt c release are also subverted by Q97, demonstrated by the lack of Bax recruitment to mitochondria.
Lysosomal activity can modify the amounts of Q25 and Q47 but not that of Q97
Because Q97-expressing neurons die despite the presence of Hsp70 and the proteasomal system was already inhibited, we wondered whether we could reduce the amount of insoluble Q97 accumulation by manipulating the lysosomal system (Williams et al., 2006). We first examined whether lysosomal activity regulates polyQ protein turnover. The H+ ATPase inhibitor bafilomycin A1 (BA1) disables lysosomal degradation by inhibiting lumen acidification (Dröse and Altendorf, 1997). Treatment with BA1 increased the amount of soluble Q25 by fivefold within 24 h after onset of expression, indicating that constitutive lysosomal activity contributes to Q25 degradation (Fig. 8A). Efficient lysosomal inhibition by BA1 was confirmed by the accumulation of LC3-II, a lysosomal-targeted autophagosome-associated protein whose degradation by lysosomes is blocked by BA1 (Tanida et al., 2005) (Fig. 8A). However, BA1 caused no marked change in the amount of soluble Q97 during the first 22 h after infection, nor did it induce a significant change in the percentage of neurons with Q97 IBs, although there was a notable trend toward more insoluble Q97 (Fig. 8B) and more cells with Q97-positive IBs (data not shown; 0.05 < p < 0.1). It may be that the rate at which Q97 IBs form exceeds the rate at which aggregates can be turned over by lysosomes during this brief period. In keeping with this idea, BA1 caused a large, more than fourfold increase in the amount of insoluble Q47 from near undetectable levels within 24 h (Fig. 8A). Likewise, whereas no Q47-positive IBs were detectable in the absence of BA1 after 24 h, ∼11% of neurons contained Q47-positive inclusions after treatment with BA1 (Fig. 8B). We next asked whether we could increase the degradation of Q97 by treatment with rapamycin, which activates the lysosomal degradation pathway (Williams et al., 2006). Figure 8 shows that rapamycin decreased the amount of Q25 within 3 d by 70%. However, there was no reduction in the amount of soluble or insoluble Q97. Evidence that rapamycin was active is shown by the lack of S6 phosphorylation (Fig. 8C) in both Q25- and Q97-expressing cells. Note also an increase in LC3-II in the rapamycin-treated samples, indicating the activation of autophagy. Hence, other mechanisms and/or longer periods of treatment may be required to clear Q97 and reduce its input and toxicity.
Figure 8.

Lysosomes participate in clearance of soluble polyQ proteins and contribute to prevention of IB formation. A, SCG neurons were infected with Ad-Q25, Ad-Q47, or Ad-Q97 and treated with 50 nm BA1 for 22 h immediately after removal of the virus. Soluble and insoluble (insol) fractions were probed with anti-GFP to indicate polyQ expression. Soluble fraction was also probed for LC3, in which the accumulation of LC3-II in the presence of BA1 demonstrates inhibition of lysosomal proteolytic activity by the drug. tERK indicates loading control. Note accumulation of soluble Q25 and insoluble Q47 and Q97 by treatment with BA1. The increase in insoluble Q97 in three experiments did not reach statistical significance. B, Fluorescent images of Q47-expressing control and BA1-treated cells. Arrows indicate cells with inclusions (images taken at 32× magnification; scale bar, 100 μm). Numbers show the percentage of GFP-positive neurons containing IBs (mean ± range; n = 2). Bottom panels indicate nuclei stained with Hoechst 33342. C, Ad-Q25- and Ad-Q97-infected neurons were treated with 200 nm rapamycin (Rap) for 3 d immediately after removal of the virus. Blot shows soluble Q25 and Q97, tERK, P-ser235/236–S6 (P-S6) (the inhibition of which demonstrates the efficacy of treatment with rapamycin), and insoluble Q97. D, Percentage change in the relative amount of soluble Q25, soluble Q97, and insoluble Q97 (mean ± SD; n = 3). Q25, p < 0.05, one-sample t test. Q97, No significant difference.
Discussion
This study sought to model mutant polyQ toxicity in a well defined neuronal system to delineate pathways involved in the pathogenesis and execution of neuronal death. Our results provide a novel molecular mechanism that explains how mutant polyQ forces neurons into necrotic-like death.
Previous studies using sympathetic neurons to study polyQ accumulation and toxicity used polyQ constructs that yielded nuclear inclusions (Kim et al., 2004; Suzuki and Koike, 2005). In our model, Q97 formed exclusively cytosolic aggregates, thereby providing a novel background on which to study polyQ toxicity. The difference in subcellular localization of inclusions between this and previous studies is likely attributable to the increased size of our protein and the rapidity of its aggregation into insoluble IB (Jana et al., 2001). Analysis of Q97 IB formation suggests that a small number of nucleation events per cell attract the remainder of newly synthesized Q97 into IBs (Colby et al., 2006). Consistent with features of cytosolic aggresomes, cytoplasmic Q97 IBs redistributed the neurofilament network, proteasomes, and lysosomes (Johnston et al., 1998; Taylor et al., 2003; Iwata et al., 2005a,b).
Mutant Q97, but not Q25, robustly induced Hsp70 from 2 d onward, which accumulated in the insoluble fraction together with massively ubiquitylated Q97. We propose that Hsp70 accumulated as a result of Q97-mediated proteasomal inhibition based on the following criteria. (1) Protein degradation was similarly inhibited by lactacystin and by Q97 after 2 d. (2) Proteasomal activity was inhibited in lysates from Q97-expressing cells by 30% compared with lysates from Q25-expressing neurons. (3) Some Q97-expressing cells formed ubiquitin-positive bodies that were dissociated from sites of polyQ aggregation, a feature noted after proteasome inhibition in other neuronal systems (Rideout et al., 2001). (4) Insoluble Hsp70 accumulated only after Q97 IBs became ubiquitylated. (5) The proteasomal substrates P-ser63–c-Jun and BimEL (Jariel-Encontre et al., 1995; Fuchs et al., 1996; Biswas and Greene, 2002; Ley et al., 2005) accumulated with the same kinetics as Hsp70, although Akt, ERK, and JNK phosphorylation were unchanged in Q25- and Q97-expressing cells (although increased mRNA may explain part of the protein accumulation). (6) Q97 prevented degradation of Q25 and EGFP, similarly to β-lactone. Q97 may cause proteasome inhibition by direct sequestration and inhibition of 26S proteasomes and/or indirectly by sequestering and preventing the recycling of ubiquitin. The finding that mutant polyQ in either the nuclear or cytoplasmic compartment reduces proteasome activity globally (Bennett et al., 2005) supports an indirect mode of inhibition.
Proteasome inhibition has diverse effects on different neuronal populations. Cultured mouse SCG neurons die apoptotically during proteasome inhibition, showing Bax activation, cyt c release, and caspase activation (Lang-Rollin et al., 2004). However, under similar conditions, rat SCG neurons may survive NGF deprivation during acute withdrawal (Sadoul et al., 1996) but ultimately die nonapoptotically (with some apoptotic features) (Lang-Rollin et al., 2008). In our hands, Q97 expression did not reproduce all the effects of proteasome inhibition because Lang-Rollin et al. (2008) found that CsA had no protective effect on proteasome-inhibition-mediated death, whereas we found that Q97-mediated cyt c release was delayed by treatment with cyclosporine A. Hence, Q97 causes additional signals in SCG neurons.
Q97-expressing neurons showed an enhanced rate of cell death compared with Q25-expressing cells from day 3 to day 6. This death was nonapoptotic, because death kinetics were unchanged in rat neurons treated with the pan-caspase inhibitor BAF or in Bax-null mouse neurons. Despite retaining robust NGF-mediated survival signaling up to day 4 after infection, Q97-expressing neurons accumulated the proapoptotic proteins P-ser63–c-Jun and BimEL (Ham et al., 2000; Putcha and Johnson, 2004; Ham et al., 2005), which have also been linked to the vulnerability of striatal neurons to mutant huntingtin (Garcia et al., 2004; García-Martínez et al., 2007). Normally, NGF signaling via Akt and ERK inhibits c-Jun and BimEL expression (Virdee et al., 1997; Whitfield et al., 2001; Besirli et al., 2005), but, by inhibiting proteasomal activity, these proteins can accumulate until they overpower the suppressive effects of NGF, just as p53 can overcome NGF/ERK-mediated neuroprotection (Anderson and Tolkovsky, 1999).
Despite c-Jun and Bim elevation, neither mitochondrial Bax translocation nor caspase activation was observed. Rather, uncoupling of the apoptotic pathway was demonstrated by the absence of apoptosis after cotreatment with NGF withdrawal and AraC (A/-N). This occurred despite induction of the expected death-signaling network (intense P-ser15–p53 and additional BimEL upregulation). Because Puma expression is a critical factor for AraC- and NGF-withdrawal-induced apoptosis (Wyttenbach and Tolkovsky, 2006) and can also mediate death during proteasome inhibition (Concannon et al., 2007), we investigated whether its expression was altered by Q97. We found that Q97-expressing neurons were specifically defective for the induction of Puma mRNA. A deficit in global transcription was excluded because RNA synthesis, measured by [3H]uridine incorporation, was not affected (King, 2007). Interestingly, Suhr et al. (2001) showed that p53 and SP1, a cofactor required for Puma induction, may be inhibited by sequestration into cytosolic or nuclear polyQ aggregates, perhaps reflecting a more widespread depression of specific transcriptional circuits. Indeed, a delayed, atypical form of nonapoptotic cell death attributable to transcriptional repression (TRIAD) was described in cortical, cerebellar, and striatal neurons and HD mice (Hoshino et al., 2006). The localization of p53-related factors remains to be studied in Q97-expressing neurons. However, even when overexpressed Puma overcame the block in Bax activation, Q97-positive neurons remained refractive to apoptosis, indicating the presence of an overwhelming survival factor in this system.
We propose that Hsp70 is this antiapoptotic factor, because shRNA-mediated Hsp70 knockdown facilitated the induction of apoptosis by A/-N in Q97-expressing neurons. Apoptosis by Hsp70 shRNA was also induced in the absence of A/-N (King, 2007). Hsp70 is pluripotent, protecting cells against apoptosis at multiple levels, including JNK activity suppression (Salehi et al., 2006; Bienemann et al., 2008), Bax translocation (Stankiewicz et al., 2005), cyt c release (Creagh et al., 2000), apoptosome assembly (Beere et al., 2000; Saleh et al., 2000), and caspase activation (Mosser et al., 1997). JNK activation remained intact in Q97-expressing neurons, eliminating its suppression by Hsp70 as the reason for apoptosis inhibition. However, the pluripotency of Hsp70 may explain why apoptosis remained inhibited even after enforced expression of Puma, Bax activation, and cytochrome c release. Previously, Kim et al. (2004) showed that overexpression of active Hsp70 or treatment of SCG neurons with lactacystin reduced the percentage of neurons with nuclear polyQ127–HA IBs, in keeping with the role of Hsp70 as a molecular chaperone. In our system, however, Q97 accumulation was likely exacerbated because of inhibition of proteasomal activity despite expression of Hsp70. Perhaps the chaperone capacity of Hsp70 is overwhelmed in Q97-expressing neurons, thereby leaving polyQ accumulation derepressed (Sakahira et al., 2002).
Given that Hsp70 accumulation failed to disaggregate Q97 IBs, we wondered whether lysosomal clearance could be stimulated with rapamycin, because inhibition of pathological polyQ accumulation, or enhanced clearance of mutant protein, may be paramount for long-term protection of CNS neurons in HD (Williams et al., 2006). Evidence that constitutive lysosomal activity controls the steady-state expression of these proteins was deduced from the finding that bafilomycin A1, a lysosomal inhibitor, increased soluble Q25 expression, and accelerated Q47 (and to a lesser extent Q97) IB formation. However, under conditions of continued polyQ synthesis, rapamycin decreased the amount of Q25 but not that of Q97, which may be attributable to the rapid insolubility of Q97.
The actual mechanism of Q97-induced death remains to be elucidated. Suzuki and Koike (2005) described a necrotic-like death after expression of penetratin–polyQ22–GFP and found mitochondrial dysfunction (also noted in the Q97-expressing neurons) and perturbed calcium homeostasis. Recently, a Drosophila model of HD with exclusively cytoplasmic inclusions showed calcium perturbations before degeneration (Romero et al., 2008). Whether proteasomal activities are inhibited and/or Hsp70 accumulates in these neuronal models remains to be investigated. However, in keeping with the importance of Hsp70 in defining neuronal susceptibility to HD, Tagawa et al. (2007) reported an inverse correlation between the relative amount of Hsp70 in neuronal subtypes and susceptibility to mutant huntingtin-induced cell death in a mouse HD model. In support, our preliminary experiments did not find a comparable rise in Hsp70 in Q97-expressing striatal neuron cultures versus SCG neurons (A. M. Tolkovsky, unpublished data). Apoptotic neuronal death in HD may therefore be restricted to those areas of the brain that fail to sufficiently upregulate Hsp70, underlining the characteristic neuronal vulnerability of the striatum, perhaps aided by calcium perturbations induced by excitotoxicity (Zeron et al., 2004; Tang et al., 2005). Because Q97-expressing SCG neurons released cytochrome c before death, studies reporting the protective effect of caspase inhibition or suppression of cytochrome c release in HD transgenic mice may reflect the benefit of preserving vulnerable neurons before the onset of death that proceeds by nonapoptotic mechanisms (Ona et al., 1999; Chen et al., 2000; Wang et al., 2008). Recently it was shown that Hsp70 overexpression and reduction of soluble input operate via separate mechanisms in an HD model and both are crucial for neuronal rescue (McLear et al., 2008). However, Hsp70 affords a window of opportunity to treat neurons with factors that reduce polyQ expression and toxicity.
Footnotes
C. G. Goeman's present address: Molecular Neurobiochemistry, Ruhr-University Bochum, D-44780 Bochum, Germany.
This work was supported by Wellcome Trust Prize Studentship 070430 (M.A.K.) and Programme Grant 064232 (A.M.T., C.G.G.). F.H. was supported by the Hereditary Disease Foundation, A.W. by a Career Development Award from the Medical Research Council, and M.A.K. and J.H.M.P. by Science Foundation Ireland Grant 03/RP/B344. We thank Helen Bye for looking after our mice and rats, Drs. J. P. Luzio and Y. Uchiyama for antibodies, and Andreas Strasser (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia) for providing the Puma mouse line. Stanley Korsmeyer, whose generosity will always be remembered, provided the original Bax mouse line.
References
- AdamsJM, Cory S (2007) Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 19:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AndersonCN, Tolkovsky AM (1999) A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J Neurosci 19:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BaeBI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A (2005) p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron 47:29–41. [DOI] [PubMed] [Google Scholar]
- BeereHM, Green DR (2001) Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol 11:6–10. [DOI] [PubMed] [Google Scholar]
- BeereHM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–475. [DOI] [PubMed] [Google Scholar]
- BennettEJ, Bence NF, Jayakumar R, Kopito RR (2005) Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell 17:351–365. [DOI] [PubMed] [Google Scholar]
- BesirliCG, Wagner EF, Johnson EM Jr (2005) The limited role of NH2-terminal c-Jun phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a potential target of the JNK pathway. J Cell Biol 170:401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BienemannAS, Lee YB, Howarth J, Uney JB (2008) Hsp70 suppresses apoptosis in sympathetic neurones by preventing the activation of c-Jun. J Neurochem 104:271–278. [DOI] [PubMed] [Google Scholar]
- BiswasSC, Greene LA (2002) Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J Biol Chem 277:49511–49516. [DOI] [PubMed] [Google Scholar]
- BuckmasterA, Nobes CD, Edwards SN, Tolkovsky AM (1991) Nerve growth factor is required for induction of c-Fos immunoreactivity by serum, depolarization, cyclic AMP or trauma in cultured rat sympathetic neurons. Eur J Neurosci 3:698–707. [DOI] [PubMed] [Google Scholar]
- BuddSL, Castilho RF, Nicholls DG (1997) Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett 415:21–24. [DOI] [PubMed] [Google Scholar]
- ButtsBD, Hudson HR, Linseman DA, Le SS, Ryan KR, Bouchard RJ, Heidenreich KA (2005) Proteasome inhibition elicits a biphasic effect on neuronal apoptosis via differential regulation of pro-survival and pro-apoptotic transcription factors. Mol Cell Neurosci 30:279–289. [DOI] [PubMed] [Google Scholar]
- ChangLK, Johnson EM Jr (2002) Cyclosporin A inhibits caspase-independent death of NGF-deprived sympathetic neurons: a potential role for mitochondrial permeability transition. J Cell Biol 157:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ChenM, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797–801. [DOI] [PubMed] [Google Scholar]
- ColbyDW, Cassady JP, Lin GC, Ingram VM, Wittrup KD (2006) Stochastic kinetics of intracellular huntingtin aggregate formation. Nat Chem Biol 2:319–323. [DOI] [PubMed] [Google Scholar]
- ConcannonCG, Koehler BF, Reimertz C, Murphy BM, Bonner C, Thurow N, Ward MW, Villunger A, Strasser A, Kögel D, Prehn JH (2007) Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene 26:1681–1692. [DOI] [PubMed] [Google Scholar]
- CreaghEM, Carmody RJ, Cotter TG (2000) Heat shock protein 70 inhibits caspase-dependent and -independent apoptosis in Jurkat T cells. Exp Cell Res 257:58–66. [DOI] [PubMed] [Google Scholar]
- DeckwerthTL, Elliott JL, Knudson CM, Johnson EM Jr, Snider WD, Korsmeyer SJ (1996) BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17:401–411. [DOI] [PubMed] [Google Scholar]
- DröseS, Altendorf K (1997) Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol 200:1–8. [DOI] [PubMed] [Google Scholar]
- FranklinJL, Johnson EM (1998) Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol 142:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FuchsSY, Dolan L, Davis RJ, Ronai Z (1996) Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene 13:1531–1535. [PubMed] [Google Scholar]
- GarciaM, Charvin D, Caboche J (2004) Expanded huntingtin activates the c-Jun terminal kinase/c-Jun pathway prior to aggregate formation in striatal neurons in culture. Neuroscience 127:859–870. [DOI] [PubMed] [Google Scholar]
- García-MartínezJM, Pérez-Navarro E, Xifró X, Canals JM, Díaz-Hernández M, Trioulier Y, Brouillet E, Lucas JJ, Alberch J (2007) BH3-only proteins Bid and Bim(EL) are differentially involved in neuronal dysfunction in mouse models of Huntington's disease. J Neurosci Res 85:2756–2769. [DOI] [PubMed] [Google Scholar]
- GillardonF, Zimmermann M, Uhlmann E, Krajewski S, Reed JC, Klimaschewski L (1996) Antisense oligodeoxynucleotides to bax mRNA promote survival of rat sympathetic neurons in culture. J Neurosci Res 43:726–734. [DOI] [PubMed] [Google Scholar]
- GoemansCG, Boya P, Skirrow CJ, Tolkovsky AM (2008) Intra-mitochondrial degradation of Tim23 curtails the survival of cells rescued from apoptosis by caspase inhibitors. Cell Death Differ 15:545–554. [DOI] [PubMed] [Google Scholar]
- GreeneLA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73:2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HackamAS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR (1998) The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol 141:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HamJ, Eilers A, Whitfield J, Neame SJ, Shah B (2000) c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol 60:1015–1021. [DOI] [PubMed] [Google Scholar]
- HamJ, Towers E, Gilley J, Terzano S, Randall R (2005) BH3-only proteins: key regulators of neuronal apoptosis. Cell Death Differ 12:1015–1020. [DOI] [PubMed] [Google Scholar]
- HawrotE, Patterson PH (1979) Long-term culture of dissociated sympathetic neurons. Methods Enzymol 58:574–584. [DOI] [PubMed] [Google Scholar]
- HazekiN, Tukamoto T, Goto J, Kanazawa I (2000) Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: applying to quantification of protein components of the aggregates. Biochem Biophys Res Commun 277:386–393. [DOI] [PubMed] [Google Scholar]
- HeTC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HoshinoM, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, Enokido Y, Marubuchi S, Harjes P, Arai N, Oyanagi K, Blandino G, Sudol M, Rich T, Kanazawa I, Wanker EE, Saitoe M, Okazawa H (2006) Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol 172:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IwataA, Riley BE, Johnston JA, Kopito RR (2005a) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280:40282–40292. [DOI] [PubMed] [Google Scholar]
- IwataA, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR (2005b) Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A 102:13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JacksonGR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL (1998) Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21:633–642. [DOI] [PubMed] [Google Scholar]
- JanaNR, Zemskov EA, Wang Gh, Nukina N (2001) Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 10:1049–1059. [DOI] [PubMed] [Google Scholar]
- Jariel-EncontreI, Pariat M, Martin F, Carillo S, Salvat C, Piechaczyk M (1995) Ubiquitinylation is not an absolute requirement for degradation of c-Jun protein by the 26 S proteasome. J Biol Chem 270:11623–11627. [DOI] [PubMed] [Google Scholar]
- JohnstonJA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KazantsevA, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A 96:11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KimWY, Horbinski C, Sigurdson W, Higgins D (2004) Proteasome inhibitors suppress formation of polyglutamine-induced nuclear inclusions in cultured postmitotic neurons. J Neurochem 91:1044–1056. [DOI] [PubMed] [Google Scholar]
- KingMA (2007) Neurotoxicity and autophagic turnover of huntingtin exon-1 containing expanded polyglutamine. PhD thesis, Cambridge University.
- KingMA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A (2008) Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol 73:1052–1063. [DOI] [PubMed] [Google Scholar]
- KirklandRA, Franklin JL (2003) Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid Redox Signal 5:589–596. [DOI] [PubMed] [Google Scholar]
- KisselevAF, Goldberg AL (2005) Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol 398:364–378. [DOI] [PubMed] [Google Scholar]
- Lang-RollinI, Vekrellis K, Wang Q, Rideout HJ, Stefanis L (2004) Application of proteasomal inhibitors to mouse sympathetic neurons activates the intrinsic apoptotic pathway. J Neurochem 90:1511–1520. [DOI] [PubMed] [Google Scholar]
- Lang-RollinI, Dermentzaki G, Vekrellis K, Xilouri M, Rideout HJ, Stefanis L (2008) A novel cell death pathway that is partially caspase dependent, but morphologically non-apoptotic, elicited by proteasomal inhibition of rat sympathetic neurons. J Neurochem 105:653–665. [DOI] [PubMed] [Google Scholar]
- LeyR, Ewings KE, Hadfield K, Cook SJ (2005) Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ 12:1008–1014. [DOI] [PubMed] [Google Scholar]
- MagranéJ, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed β-amyloid in neurons. J Neurosci 24:1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MartindaleD, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR (1998) Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet 18:150–154. [DOI] [PubMed] [Google Scholar]
- McLearJA, Lebrecht D, Messer A, Wolfgang WJ (2008) Combinational approach of intrabody with enhanced Hsp70 expression addresses multiple pathologies in a fly model of Huntington's disease. FASEB J 22:2003–2011. [DOI] [PubMed] [Google Scholar]
- MosserDD, Caron AW, Bourget L, Denis-Larose C, Massie B (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 17:5317–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MoulderKL, Onodera O, Burke JR, Strittmatter WJ, Johnson EM Jr (1999) Generation of neuronal intranuclear inclusions by polyglutamine-GFP: analysis of inclusion clearance and toxicity as a function of polyglutamine length. J Neurosci 19:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NobesCD, Tolkovsky AM (1995) Neutralizing anti-p21ras Fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cAMP. Eur J Neurosci 7:344–350. [DOI] [PubMed] [Google Scholar]
- OnaVO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399:263–267. [DOI] [PubMed] [Google Scholar]
- OrrHT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. [DOI] [PubMed] [Google Scholar]
- PierchalaBA, Ahrens RC, Paden AJ, Johnson EM Jr (2004) Nerve growth factor promotes the survival of sympathetic neurons through the cooperative function of the protein kinase C and phosphatidylinositol 3-kinase pathways. J Biol Chem 279:27986–27993. [DOI] [PubMed] [Google Scholar]
- PierchalaBA, Tsui CC, Milbrandt J, Johnson EM (2007) NGF augments the autophosphorylation of Ret via inhibition of ubiquitin-dependent degradation. J Neurochem 100:1169–1176. [DOI] [PubMed] [Google Scholar]
- PutchaGV, Johnson EM Jr (2004) Men are but worms: neuronal cell death in C. elegans and vertebrates. Cell Death Differ 11:38–48. [DOI] [PubMed] [Google Scholar]
- PutchaGV, Deshmukh M, Johnson EM Jr (1999) BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci 19:7476–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RideoutHJ, Larsen KE, Sulzer D, Stefanis L (2001) Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem 78:899–908. [DOI] [PubMed] [Google Scholar]
- RomeroE, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J (2008) Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SadoulR, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schröter M, Becherer JD, Irmler M, Tschopp J, Martinou JC (1996) Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J 15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- SakahiraH, Breuer P, Hayer-Hartl MK, Hartl FU (2002) Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci U S A 99 [Suppl 4]:16412–16418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SalehA, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2:476–483. [DOI] [PubMed] [Google Scholar]
- SalehiAH, Morris SJ, Ho WC, Dickson KM, Doucet G, Milutinovic S, Durkin J, Gillard JW, Barker PA (2006) AEG3482 is an antiapoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem Biol 13:213–223. [DOI] [PubMed] [Google Scholar]
- SánchezI, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J (1999) Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 22:623–633. [DOI] [PubMed] [Google Scholar]
- SathasivamK, Hobbs C, Mangiarini L, Mahal A, Turmaine M, Doherty P, Davies SW, Bates GP (1999) Transgenic models of Huntington's disease. Philos Trans R Soc Lond B Biol Sci 354:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SaudouF, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66. [DOI] [PubMed] [Google Scholar]
- SmithMI, Deshmukh M (2007) Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ 14:1011–1019. [DOI] [PubMed] [Google Scholar]
- SongC, Perides G, Liu YF (2002) Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem 277:6703–6707. [DOI] [PubMed] [Google Scholar]
- StankiewiczAR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD (2005) Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 280:38729–38739. [DOI] [PubMed] [Google Scholar]
- SuhrST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH (2001) Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 153:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SuzukiM, Koike T (2005) Early apoptotic and late necrotic components associated with altered Ca2+ homeostasis in a peptide-delivery model of polyglutamine-induced neuronal death. J Neurosci Res 80:549–561. [DOI] [PubMed] [Google Scholar]
- TagawaK, Marubuchi S, Qi ML, Enokido Y, Tamura T, Inagaki R, Murata M, Kanazawa I, Wanker EE, Okazawa H (2007) The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci 27:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TangTS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR, Bezprozvanny I (2005) Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A 102:2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TanidaI, Minematsu-Ikeguchi N, Ueno T, Kominami E (2005) Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1:84–91. [DOI] [PubMed] [Google Scholar]
- TaylorJP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH (2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 12:749–757. [DOI] [PubMed] [Google Scholar]
- Tsui-PierchalaBA, Putcha GV, Johnson EM Jr (2000) Phosphatidylinositol 3-kinase is required for the trophic, but not the survival-promoting, actions of NGF on sympathetic neurons. J Neurosci 20:7228–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TurmaineM, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW (2000) Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A 97:8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VabulasRM, Hartl FU (2005) Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310:1960–1963. [DOI] [PubMed] [Google Scholar]
- VirdeeK, Bannister AJ, Hunt SP, Tolkovsky AM (1997) Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J Neurochem 69:550–561. [DOI] [PubMed] [Google Scholar]
- VirdeeK, Xue L, Hemmings BA, Goemans C, Heumann R, Tolkovsky AM (1999) Nerve growth factor-induced PKB/Akt activity is sustained by phosphoinositide 3-kinase dependent and independent signals in sympathetic neurons. Brain Res 837:127–142. [DOI] [PubMed] [Google Scholar]
- VonsattelJP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384. [DOI] [PubMed] [Google Scholar]
- WangX, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, Kristal BS, Friedlander RM (2008) Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease. J Neurosci 28:9473–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WhitfieldJ, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29:629–643. [DOI] [PubMed] [Google Scholar]
- WilliamsA, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76:89–101. [DOI] [PubMed] [Google Scholar]
- WyttenbachA, Tolkovsky AM (2006) The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem 96:1213–1226. [DOI] [PubMed] [Google Scholar]
- WyttenbachA, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC (2000) Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc Natl Acad Sci U S A 97:2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XueL, Fletcher GC, Tolkovsky AM (2001) Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol 11:361–365. [DOI] [PubMed] [Google Scholar]
- YewEH, Cheung NS, Choy MS, Qi RZ, Lee AY, Peng ZF, Melendez AJ, Manikandan J, Koay ES, Chiu LL, Ng WL, Whiteman M, Kandiah J, Halliwell B (2005) Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J Neurochem 94:943–956. [DOI] [PubMed] [Google Scholar]
- ZeronMM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA (2004) Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci 25:469–479. [DOI] [PubMed] [Google Scholar]