

Abstract
Recently, dopamine (DA) neurons of the substantia nigra pars compacta (SNc) were found to exhibit sustained responses related to reward uncertainty, in addition to the phasic responses related to reward-prediction errors (RPEs). Thus, cue-dependent anticipations of the timing, magnitude, and uncertainty of rewards are learned and reflected in components of DA signals. Here we simulate a local circuit model to show how learned uncertainty responses are generated, along with phasic RPE responses, on single trials. Both types of simulated DA responses exhibit the empirically observed dependencies on conditional probability, expected value of reward, and time since onset of the reward-predicting cue. The model's three major pathways compute expected values of cues, timed predictions of reward magnitudes, and uncertainties associated with these predictions. The first two pathways' computations refine those modeled by Brown et al. (1999). The third, newly modeled, pathway involves medium spiny projection neurons (MSPNs) of the striatal matrix, whose axons corelease GABA and substance P, both at synapses with GABAergic neurons in the substantia nigra pars reticulata (SNr) and with distal dendrites (in SNr) of DA neurons whose somas are located in ventral SNc. Corelease enables efficient computation of uncertainty responses that are a nonmonotonic function of the conditional probability of reward, and variability in striatal cholinergic transmission can explain observed individual differences in the amplitudes of uncertainty responses. The involvement of matricial MSPNs and cholinergic transmission within the striatum implies a relation between uncertainty in cue–reward contingencies and action-selection functions of the basal ganglia.
Keywords: dopamine, substance P, neuropeptide, reinforcement learning, substantia nigra, computational model
Introduction
Firing patterns observed in the dopamine cells of the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA) are related to the occurrence and nonoccurrence of rewards and cues that predict reward (Schultz et al., 1997; Schultz, 1998). Dopamine (DA) cells, which fire tonically at moderate levels, respond immediately to unexpected rewards with a phasic burst. When a reward consistently follows a preceding conditioned stimulus (CS), the phasic burst “transfers” from the time of expected reward delivery to the time of CS onset. The amount of the “transfer” depends on the expectation of reward, R̂ = |R*| × p(R*|CS), that is, the conditional probability, p(R*|CS), that a reward of magnitude |R*| follows the CS (Schultz et al., 1997; Fiorillo et al., 2003; Tobler et al., 2005; Schultz, 1998, 2004). After learning, the omission of an expected reward induces a depression in firing rate to a below-baseline level at the time of expected reward delivery. Thus, dopamine cells are part of an adaptive system that uses learned expectations to filter reward-related signals. This filtering creates dopamine bursts and pauses that respectively signal positive and negative violations of reward-related predictions. Therefore, SNc/VTA dopamine signals, which are broadcast to the dorsal and ventral striatum as well as other brain regions, such as the amygdala and frontal cortex, can be conceptualized as internal teaching signals that foster rapid acquisition of goal-directed behavior (Schultz et al., 1997; Schultz, 1998; Doya, 2002).
Several proposals have been made to explain the adaptive computations that give rise to the “reward-prediction error” (RPE) responses of dopamine neurons (for review, see Wörgötter and Porr, 2005). Some have been implementations of the temporal difference (TD) model (Nakahara et al., 2004; Pan et al., 2005), whereas others have been based on local circuit anatomy and physiology (Houk et al., 1995; Brown et al., 1999). Brown et al. (1999) introduced and simulated a local circuit model that can explain most of the key results without predicting effects that are known not to occur. In particular, as recently shown by Pan et al. (2005), most parameterizations of the TD model incorrectly predict that during training the DA burst will gradually “slide” from the time of reward delivery to the time of CS onset, such that as learning progresses, the burst appears at a full range of successively earlier intermediate times within a trial, but never appears at both the time of the CS and the time of primary reward on a single trial. In contrast, the Brown et al. (1999) model correctly predicts that the burst never occurs at intermediate times between the CS onset and the reward delivery, and that instead there will be DA bursts at just two times on each trial. In particular, across early learning trials, a burst at the time of CS onset will wax (become gradually larger across trials) as the burst at the time of reward delivery wanes (becomes gradually smaller).
Recently, however, Fiorillo et al. (2003) discovered a new reward-related component of DA cell discharges in SNc, which they called an “uncertainty response.” This sustained component builds up during the interval from CS onset to the expected time of reward if the reward schedule is probabilistic, and contrary to published TD model predictions (Niv et al., 2005), uncertainty responses in SNc are robust in single-trial data (Fiorillo et al., 2005). The size of the buildup is a nonmonotonic, “inverted-U” function of p(R*|CS), and also depends on the amount of reward, |R*|, that is at risk. Neither the “uncertainty response” nor its functional dependencies were predicted by any basal ganglia (BG) learning model of the time. However, the Brown et al. (1999) model did imply a separate new observation of Fiorillo et al. (2003) and Tobler et al. (2005), namely that after asymptotic learning, both the degree of waxing of the DA burst to the CS onset and the degree of waning of the burst at the time of reward delivery are monotonic functions of |R*| × p(R*|CS).
The Brown et al. (1999) model, like others, omits many known features of the BG microcircuit that can be expected to play important roles in shaping DA responses, including uncertainty responses. The model SN had only one class of DA cells and lacked corelease of GABA and neuropeptides. The model striatum lacked cholinergic tonically active neurons as well as GABAergic interneurons. In this paper, we propose an extended model based on such features and corresponding computational hypotheses. The new model retains the explanatory successes of the prior model, but also incorporates an efficient basis for the uncertainty responses discovered by Fiorillo et al. (2003).
Materials and Methods
The proposed model is schematized in Figure 1, which also labels sites in the circuit with the names of corresponding variables in the formal model. The model for the genesis of phasic DA responses follows the proposal by Brown et al. (1999), with modest modifications (explained below). The model uses ordinary differential equations in a Hodgkin–Huxley type formulation, modified to emphasize key dynamical properties of cell types. The model is qualitative. Model neuron firing rates range from zero to one, and parameters were constrained to reflect empirically reported responses of neurons. No attempt was made to optimize curve fits. Parameter ranges across which the qualitative behavior of the model DA neuron's phasic responses are preserved and consistent with experimental reports were discussed by Brown et al. (1999). Therefore, we report and discuss only the sensitivity analyses for the key parameters governing the genesis of sustained DA responses. These analyses yield new predictions that are experimentally testable. Mathematical description of the model is specified in the Appendix (Eqs. 1–21). The model was implemented in Matlab. Numerical integration was performed with an adaptive step size, fourth-order, Runge-Kutta method. The model was simulated with three different reward magnitudes (|R*|; 0.05, 0.15, and 0.50; arbitrary units) intended to span a full range of magnitudes representable by a single neuron, and five different probabilistic schedules [p(R*|CS); 0.00, 0.25, 0.50, 0.75, and 1.00]. Parameter values used for simulations are given in Table 1. What follows below describes the assumptions and hypotheses about the biological substrates of the model. The next section (Results) reports real-time behavior of the model and describes how its operation leads to model DA neuron responses.
Figure 1.

Summary diagram of the interactions represented in the model. Arrowheads denote excitatory pathways, circles denote inhibitory pathways, and hemidisks denote synapses at which learning occurs. Thin lines at bottom left show the anatomical connections proposed by Brown et al. (1999). These control the phasic dopaminergic responses via one fixed and two adaptive pathways: an excitatory pathway via hypothalamus and the PPTN that relays primary reward information to dopaminergic cells; a pathway from CS representations via ventral striatum, ventral pallidum, and PPTN that can learn to excite DA cells at the onset of a reward-predicting CS; and an adaptive striosomal MSPN pathway by which a CS can inhibit dopaminergic neurons after a learned time delay. Thick lines show the pathways proposed in the current model. Striatal GABAergic interneurons [both nitric-oxide synthesizing interneuron (NOS-INs) and FS-INs], TANs, and matricial SP-D1-MSPNs (M) receive inputs from the CS representation, the latter via synaptic weights that adapt in the same way as at synapses onto ventral striatal cells (WiS). Both TANs and GABAergic NOS-INs are recipients of thalamic centromedian–parafascicular nuclei (CM–Pf) projections. NOS-INs inhibit TANs, whereas ACh released by TANs excites matricial SP-D1-MSPNs and GABAergic interneurons (both NOS-INs and FS-INs). The latter effect is mediated by nicotinic ACh receptors (nAChRs). ACh also inhibits GABAergic transmission between FS-INs and matricial SP-D1-MSPNs presynaptically, via muscarinic ACh receptors (mAChRs). Matricial SP-D1-MSPNs, in turn, send projections to both SNr cells (SR) and SNc dopaminergic cells, whose dendrites reach into SNr. SP-D1-MSPN terminals in the SN corelease GABA and SP, which act simultaneously on distal dendrites of dopaminergic neurons. GABA also acts presynaptically, via GABAB receptors, to oppose SP and GABA release. Dashed arrows indicate sites where axon terminals of DA cells release DA, which acts as a reinforcement signal at corticostriatal synapses onto ventral striatal, matricial, and striosome MSPNs. DA also modulates ACh release from TANs.
Table 1.
Parameter values used to simulate the model
| Parameter | Value | Parameter | Value | Parameter | Value |
|---|---|---|---|---|---|
| τP | 200 | ΓS | 0.20 | γg | 3.00 |
| τUP | 4.00 | Γ− | 0.50 | AS | 0.70 |
| τS | 30.0 | Γ+ | 0.50 | AZ | 5.00 |
| τWS | 20.0 | ΓK | 0.41 | AM | 0.70 |
| τD | 15.0 | ΓIN | 0.73 | AT | 1.00 |
| τM | 600 | ΓM | 0.035 | BM | 1.00 |
| τD̄ | 1.00 | αWS | 0.20 | BK | 0.35 |
| τg− | 5.00 | αr | 50.0 | BT | 0.00 |
| τg+ | 5.00 | αG | 5.00 | CWSmax | 3.50 |
| τK | 30.0 | αY | 1.00 | CT | 1.50 |
| τT | 50.0 | αZ | 100 | DT | 0.10 |
| τFS | 100 | αiS | 0.46 | hD | 0.50 |
| τS̄ | 2.00 | αR | 5.8 | ID | 0.15 |
| ΓG | 0.37 | αTh | 2.00 | WUP | 130 |
| ΓY | 0.18 | αSD | 0.40 | WSP | 2.20 |
| ΓP | 0.095 | βWS | 1.00 | WRP | 0.80 |
| ΓN | 0.05 | βr | 2.00 | WRS | 12.0 |
| ΓD2-dir | 0.10 | βG | 20.0 | WE | 0.20 |
| ΓD1 | 0.10 | βY | 80.0 | WC | 1.67 |
| ΓD2-mod | 0.09 | β | 15.0 | ||
| ΓDFS | 0.15 | βM | 0.29 |
Parameter values for the equations given in the supplementary material (available at www.jneurosci.org as supplemental material) (striatal circuit) are also provided in the table for completeness. τ: Time constants, Γ: thresholds; α, β, and γ: gains; A–D: firing rate/activation (upper or lower) bounds; W: synaptic weights.
Circuit for learned phasic dopamine responses.
A circuit basis for genesis of learned phasic dopamine responses was proposed and discussed in detail by Brown et al. (1999). We provide here a summary for completeness, and describe modifications to the original model. Formal specification is given in the Appendix. A model pedunculopontine tegmental nucleus (PPTN) stage relays signals generated by conditioned and unconditioned rewarding stimuli to DA cells in the SNc (Nakamura and Ono, 1986; Semba and Fibiger, 1992; Brown et al., 1999; Pan and Hyland, 2005). Accordingly, the major excitatory input to phasic DA cells is a signal from the PPTN. The CS is assumed to be relayed to the striatum via corticostriatal afferents (although the relay to ventral striatum may be via amygdala). Within the BG, CS-related inputs excite both ventral striatum, via a set of adaptive synaptic weights, and medium spiny projection neurons (MSPNs) in the striosome compartment of the striatum, via another set of adaptive synaptic weights. After learning, the former pathway is responsible for eliciting the phasic DA response that immediately follows CS onset. This adaptive excitatory pathway is complemented in the model by an adaptive inhibitory pathway from CSs to SNc DA cells via striosomes. An inhibitory projection from striosomes to DA cells is well established (Gerfen, 1992). To explain the data on timing of dopamine dips noted above, the inhibitory signals carried by this projection must be adaptively timed to arrive at DA cells at the expected time of primary reward. Therefore, model striosomal MSPN dendritic spines exhibit a spectrum of delayed calcium spikes mediated by second messengers (Fiala et al., 1996; Brown et al., 1999). When a delayed spike coincides with DA burst release engendered by primary reward (via lateral hypothalamus and PPTN, as shown in Fig. 1), learning specific to the corticostriatal synapse on that spine can occur. Such learning potentiates inhibitory outputs at the relevant delay, and such learning is self-terminating, because once the inhibition is strong enough, it precisely cancels the excitatory effect of PPTN inputs to DA cells.
The ensemble of mechanisms captured in Equations 1–10 (given in the Appendix) enables gradual learned transfer of scaled DA responses from the time of rewarding stimulus onset to the (earlier) time of CS onset (Schultz et al., 1997; Schultz, 1998, 2004). The original formulation of this model was shown to successfully explain the reward- and timing-related responses of phasic dopamine bursts (Brown et al., 1999), and as shown below, all those properties are preserved in the current formulation. However, the learning laws used in our model deviate from those in the report by Brown et al. (1999) in three respects. First, because of a misprint in the report by Brown et al. (1999), the published equation governing CS-striosomal synaptic weight, Zij, converged to zero when negative and positive reinforcement signals (N+ and N−) were both equal to zero. Our equation for the CS-striosomal weight (Eq. 9) corrects this problem while also setting an intrinsic upper bound on weight growth. Second, because DA-dependent learning in the striatum is mediated by second messenger-mediated calcium release (Kötter, 1994), which is slightly delayed relative to CS onset, we modified the learning rule (Eq. 4) to reflect calcium-gated adaptation of corticoventral striatal synapses. This more realistic learning rule helps explain classic observations that a CS must slightly precede a reward for behavioral learning to occur. At the same time, the improved rule eliminates the “self-learning” (i.e., reinforcement of a CS by the very DA burst that it induces) that O'Reilly et al. (2007) noted as a problematic aspect of the Brown et al. (1999) model. Third, the increment and decrement scalars, αWS and βWS, for the nonstriosomal striatal weights (Eq. 4), were adjusted to partially compensate for the very small range of RPEs represented by DA dips relative to the large range of RPE represented by DA bursts (Fiorillo et al., 2003; Nakahara et al., 2004). This adjustment improved the model's weights' sensitivities to conditional probability (treated in more detail in Results).
Circuit for genesis of learned sustained dopamine responses.
It is currently not known whether DA cells in VTA exhibit similar uncertainty responses. Therefore, we limit our discussion to SNc DA cells. Both GABAergic and dopaminergic cells in the SN exhibit (apparently) unlearned baseline activity levels. However, GABAergic MSPNs of the striatal matrix and striosome compartments can inhibit the firing of the GABAergic pars reticulata (SNr) and dopaminergic SNc cells, respectively (Ragsdale and Graybiel, 1990; Joel and Weiner, 2000). Moreover, the dendrites of GABAergic SNr cells are intermingled with distal dendrites from the dopaminergic cells of the SNc (Condé, 1992; Gerfen and Wilson, 1996). Thus, we hypothesize that matricial MSPNs of the direct pathway, whose terminals contact SNr dendrites, also contact invading dendrites of DA cells whose somata are in SNc (Fig. 1). In particular, the model embodies the hypothesis that such matricial MSPNs, which also release the neuropeptide substance P (SP) (Jessell, 1978; Iversen et al., 1980; Parent, 1986; Condé, 1992; Otsuka and Yoshioka, 1993), exert a net excitatory effect on dopaminergic SNc cells, and that this excitatory effect is responsible for the sustained uncertainty responses of such cells. This complements the treatment above, in which model MSPNs attributed to striosomes learned to regulate the phasic DA responses that are widely regarded as RPE signals (Schultz et al., 1997; Schultz, 1998; Brown et al., 1999).
The primary inputs to the matricial and striosomal MSPNs in striatum are relayed through glutamatergic cortical afferents via adaptive corticostriatal connections (Joel and Weiner, 2000). The model also captures the influence of cholinergic transmission on MSPNs both directly (Di Chiara et al., 1994) and indirectly through GABAergic interneurons (Koós and Tepper, 1999, 2002; de Rover et al., 2002; Ravel et al., 2003), via Equation 11. This treatment is consistent with reports showing that tonically active neurons (TANs; giant cholinergic interneurons) are preferentially located in, or near the border of, the matrix compartment of the striatum (Kubota and Kawaguchi, 1993; Prensa et al., 1999). Although both learned phasic and learned sustained responses of model DA neurons show qualitatively correct functional dependencies on conditional probability of reward in the absence of fast-spiking interneurons (FS-INs) and TANs in the striatal microcircuit, inclusion of the latter markedly enhances the sensitivity of the modeled sustained responses to probability (see Results).
As noted above, in addition to releasing GABA, the MSPNs of the direct pathway release the neuropeptide SP. Such SP release from the axon terminals of MSPNs projecting to SNr can excite the comingled dendrites of DA cells (Condé, 1992; Otsuka and Yoshioka, 1993; Hanson et al., 2002; Martorana et al., 2003; Betarbet and Greenamyre, 2004) via neurokinin receptors (Whitty et al., 1997; Lévesque et al., 2007). Beaujouan et al. (2004) reported activity-dependent release of SP from striatonigral terminals, and SP released intranigrally increases striatal dopamine release (Reid et al., 1990; Khan et al., 1996). The exact mechanisms and net effects of interactions between the coreleased peptide (SP) and nonpeptides (GABA) are currently unknown. However, “practically all combinations of peptide and non-peptide transmitter have been found. … The exact significance of cohabitation remains to be determined. It may be that the peptide and the non-peptide act synergistically or, in contrast, it could be that the peptide or non-peptide act presynaptically to inhibit the release of their companion” (Smith, 1996). Several studies point toward a presynaptic interaction between a peptide and nonpeptide transmitter, such that the nonpeptide transmitter inhibits both its own and its companion's release (Malcangio and Bowery, 1999; Salio et al., 2005, 2006). Notably, several studies clearly show a GABAB-autoreceptor mediated inhibition of corelease of peptides in the cerebral cortex (Bonanno et al., 1996), spinal cord (Malcangio and Bowery, 1999; Riley et al., 2001), and further regions of the nervous system (Bowery, 1993). Available data suggest a similar mechanism in the substantia nigra. Indeed, GABA acts via GABAB autoreceptors on presynaptic terminals in SN to progressively inhibit release from those terminals [for a detailed description of the data, see Paladini and Tepper (1999) and Tepper and Lee (2007)], and Humpel and Saria (1989) suggested that GABA receptors are involved in the presynaptic regulation of tachykinin release from striatonigral terminals. Direct evidence for presynaptic inhibition of SP by GABA was provided in seminal reports of Jessell (1978) and Iversen et al. (1980). They showed in vitro that higher GABA concentrations produced progressively greater inhibition of potassium-evoked SP release from striatonigral terminals, without affecting its spontaneous release. This regulation was mediated by GABA receptors. Thus, the weight of available evidence makes it likely that any accurate computational model must assume that GABA, coreleased with SP from striatonigral terminals, inhibits SP release from those terminals via presynaptic GABAB autoreceptors.
To represent these interactions in the model, the net SP release level in SN initially grows as matricial MSPN activation exceeds the firing threshold, but as MSPN activation increases further, the growth in SP release is eventually terminated, and then reversed, by GABA-dependent presynaptic inhibition. For this reason, the net SP release in the model is a nonmonotonic function of matricial MSPN firing rate, which because of learning is a monotonic function of conditional reward probability. Mathematical formulations of this case, as well as an alternative case in which GABA also acts postsynaptically, are provided in the Appendix (Eqs. 12–18). Whereas available data apparently require some presynaptic inhibition of SP release by GABA in models of substantia nigra, our model further assumes that such inhibition of SP release is stronger than presynaptic inhibition of GABA release itself. No empirical assessment of the relative sizes of these effects is known to us. Therefore, the Discussion clarifies how model predictions could be tested with new, in vivo, experiments. A statement of the hypothesis that a GABA–SP interaction of an appropriate mathematical form could enable “uncertainty responses,” together with results of one preliminary simulation (without any model equations), appeared earlier, in a brief letter (Tan and Bullock, 2008a). Since then, the model has been modified in several key respects, and this is the first report detailing model equations, parameter ranges, and alternative interactions that simulate learning and signal processing sufficient to yield both sustained and phasic DA responses as seen in vivo.
Some SNc neurons appear to respond (at statistically significant levels) only with phasic or sustained components to a predictive stimulus, whereas other SNc cells show both components in their responses (C. Fiorillo, personal communication) [see also Fiorillo et al., 2003 (Fig. 3D)]. Therefore, to simulate coexisting responses in a single neuron, the model includes a third set of DA neurons affected by all three pathways (PPTN-mediated excitatory projections and projections from both matricial and striosomal MSPNs).
Figure 3.

Bidirectional responses of model phasic dopamine neurons reflect deviations from the outcome expected after training with reward magnitudes |R*| = 0.05 (triangles) and |R*| = 0.15 (circles) with a deterministic schedule p(R*|CS) = 1.0 (black lines), and after training with probabilistic schedules p(R*|CS) = 0.25 (squares), p(R*|CS) = 0.50 (diamonds), and p(R*|CS) = 0.75 (crosses) with a reward magnitude |R*| = 0.15 (gray lines). Responses are normalized to the response after the unpredicted delivery of a reward of magnitude 0.15. Inset, The neurophysiological data [from Tobler et al. (2005), their Fig. 3B, reprinted with permission from AAAS] after training the monkey with a 0.15 ml reward with p(R*|CS) = 1.0, equivalent to the model response shown with black circles.
Dopamine level in the striatum.
In the model, the striatal DA level, D̄, tracks, and is a running average of, momentary DA cell firing rate. This corresponds well with observations (Floresco et al., 2003) that dopamine uptake is quite rapid in striatum, and so DA level will not be elevated above baseline levels for very long after termination of a DA neuron's burst. The model's positive reinforcement signal (which gates learning) reflects transient positive deviations from the running average, whereas the complementary negative reinforcement signal reflects transient negative deviations. Because slowly ramping changes in the DA cell firing rate, e.g., as seen in the model's sustained “uncertainty” responses, induce only negligible deviations from the running average in the normal striatum (with fast DA uptake), they do not induce reinforcement signals. However, such slow changes will be reflected in the striatal DA level (the running average).
The equations governing model striatal FS-INs and TANs, whose inclusion improves but was not critical for emergence of the basic DA responses (as shown below), are derived and discussed in detail by Tan and Bullock (2008b), and provided as supplemental Equations S1–S8 (available at www.jneurosci.org as supplemental material) for completeness. Operation of the model in real time is explained in more detail next.
Results
Scaled DA bursts induced by uncued unconditioned stimuli (primary rewards)
In the absence of any predictive stimulus, delivery of reward in the model induces a DA burst, at the onset of reward, that is a monotonically increasing function of the reward magnitude (Fig. 2), consistent with neurophysiological observations (Schultz et al., 1997; Schultz, 1998; Tobler et al., 2005). The primary reward input generates phasic firing in the lateral hypothalamus (LH) (Nakamura and Ono, 1986), which transiently excites the PPTN (Semba and Fibiger, 1992) and ventral striatum (Schultz et al., 1992). The PPTN signal, in turn, excites SNc dopaminergic cells (Scarnati et al., 1988; Condé, 1992; Futami et al., 1995) and leads to the phasic dopamine burst (Gerfen, 1992). The present model emphasizes LH cells that respond to a rewarding unconditioned stimulus (US). It does not treat LH cells that show learned responses to CS onsets. However, the network embedding and role of such cells were treated recently in a complementary model (Grossberg et al., 2008) of LH–amygdala–orbitofrontal interactions that mediate such processes as simultaneous visual discrimination, motivational/attentive enhancement of stimulus representations, and rapid selective devaluations of stimuli after satiety.
Figure 2.

Neural discrimination of reward magnitude in response to the delivery of an unpredicted reward by model dopaminergic neurons of the phasic type. Bursts in response to reward are a function of the magnitude of reward in the absence of any predictive stimulus. The responses are normalized to the response after delivery of an unpredicted reward of magnitude 0.5. Inset shows the corresponding neurophysiological data [from Tobler et al. (2005), their Fig. 2B, reprinted with permission from the American Association for the Advancement of Science (AAAS)].
Learned DA bursts and pauses that reflect reward-prediction errors
When an initially neutral conditioned stimulus arrives to striatum via cortical afferents, it excites both ventral striatum and striosomal MSPNs through trainable adaptive weights. Ventral striatum disinhibits PPTN through ventral pallidum (Yang and Mogenson, 1987). In the model, phasic DA signals induced by primary reward input act as a teaching signal on these two sets of adaptive weights (Wickens et al., 1996). As learning proceeds, the cortical CS representation learns to excite DA cells by itself through ventral striatum, while it also learns to inhibit DA cells at the expected time of arrival of the primary reward input. The timing of the latter inhibition depends on the internal calcium dynamics of model striosomal MSPNs (Gerfen, 1992; Fiala et al., 1996; Brown et al., 1999). The larger the magnitude of primary reward, the larger the weights (onto ventral striatal and striosomal MSPNs) become during learning. Hence, after learning, the DA burst induced by a predictive stimulus is a monotonically increasing function of the reward magnitude |R*|.
Because the strength of striatal inhibition of model DA cells by MSPNs is matched to the excitation that would be generated by the expected reward, after learning, a larger than expected reward elicits a DA burst, whereas a smaller than expected reward elicits a pause in DA cell firing. These bidirectional DA responses in the model reflect deviations from internal predictions (Fig. 3), i.e., are RPEs. This behavior was also achieved by the precursor dual-pathway model of Brown et al. (1999), and is consistent with many neurophysiological studies (Schultz et al., 1997; Schultz, 1998, 2004; Tobler et al., 2005).
Learning effects of probabilistic schedules of reward
During exposure to probabilistic cue–reward contingencies, adaptive weights are incremented by DA bursts induced by delivery of primary reward, but decremented by dips consequent to each omission of the expected reward. Therefore, the net potentiation of the model's striatal synaptic weights is a function not only of the reward magnitude, as noted above, but also of the conditional probability of reward given the CS, p(R*|CS). Given this dual dependence of the striatal synaptic strengths (Fig. 4A), model neuronal responses to a CS (Fig. 4B; see also Fig. 7) increase with that CS's expected reward value, R̂ = |R*| × p(R*|CS). This dual dependence of CS-induced striatal activations is consistent with experimental observations (Tobler et al., 2005).
Figure 4.
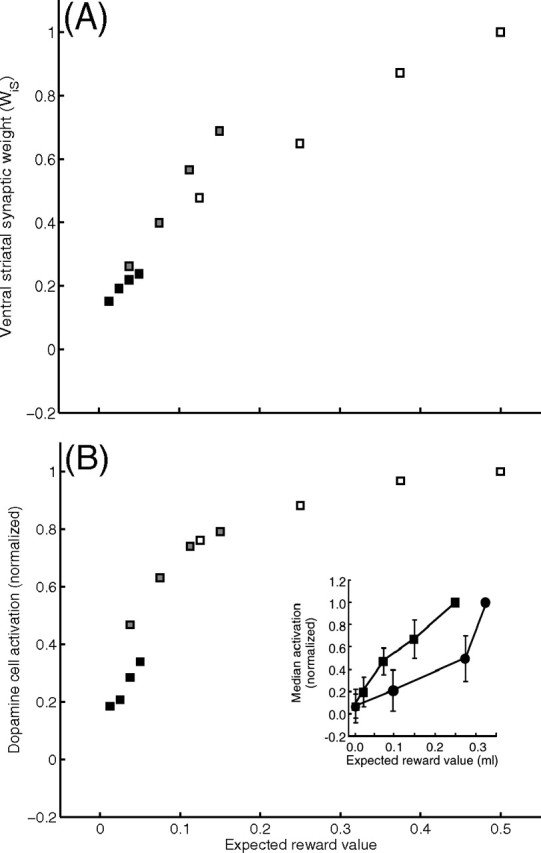
A, B, Model ventral striatal weights (WiS) (A) and phasic dopamine cell responses (above baseline) to conditioned stimuli (B) after learning. Activations are normalized by the activation elicited by the conditioned stimulus predicting the largest expected value. Responses to conditioned stimuli elicit cell activation as a function of expected reward value, p(R*|CS)|R*|. Model neurons' activations discriminate among expected reward values. Black, gray, and white squares show the outcomes of training with reward magnitudes, |R*|, of 0.05, 0.15, and 0.50, respectively. B, Inset, The corresponding neurophysiological data [from Tobler et al. (2005), their Fig. 1C, reprinted with permission from AAAS], where the two curves (squares and circles) represent data from two different monkeys.
Figure 7.

Dopaminergic cell responses under different probabilistic schedules (from top to bottom, p = 0, 0.25, 0.5, 0.75, 1), with a reward magnitude, if any, of 0.15 ml. A, Data are reprinted from the study by Fiorillo et al. (2003), their Fig. 3B, with permission from AAAS. B, C, Model phasic dopamine cell responses (B) and model sustained-type dopamine cell responses (C) after 26 trials. D, Model dopamine cells both with a burst and a sustained activation after 26 trials. Note that the DA burst occurs at both CS onset and reward onset, but at no other times, at this intermediate phase of learning. Learning in the model reaches its asymptote, and the residual phasic activation of DA cells in response to reward delivery disappears under a p = 1 schedule, after ∼65 classical conditioning trials. However, robust uncertainty responses appear after 18–24 conditioning trials (depending on the probabilistic schedule), and remain qualitatively the same during further trials. The figure shows model dopaminergic cell responses during intermediate stage of learning (26 trials), by which time robust uncertainty responses have emerged for each of the three probabilistic schedules.
Weights of corticostriatal synapses onto matricial MSPNs, striosomal MSPNs, and ventral striatal cells all express the same dual dependence. Thus, striatal inhibition of DA cells in the model is also an increasing function of the CS's expected reward value, R̂, rather than the absolute reward magnitude |R*|. This dependence implies that, for any probabilistic schedule, expected reward value associated with the CS after learning is smaller than the absolute reward magnitude (R̂ ≤ |R*|), because probability, by definition, must be between zero and one. Furthermore, the expected reward value of the CS will be equal to the absolute reward value only if the probability of reward given the CS is one [R̂ = |R*| only if p(R*|CS) = 1.00]. As a consequence, a primary reward elicits a residual DA burst in the model (see below, Fig. 7) even after asymptotic learning on a probabilistic schedule, consistent with the observations of Fiorillo et al. (2003) and Tobler et al. (2005). The latter study also observed that after a training schedule with a CS equiprobably followed by two potential outcomes (e.g., no reward or a cue-specific volume of liquid reward, |R*|), the sizes of residual phasic DA responses to the cue-specific reward volumes had become almost independent of the reward volume, in stark contrast to the continuing strong dependence of phasic DA responses on sizes of uncued rewards. A simulation of this paradigm (Fig. 5) showed that the model exhibits similar behavior. Furthermore, a short mathematical derivation by Tan et al. (2008) showed that such an outcome is a robust property of an entire class of RPE models defined by a simplified subset of the assumptions incorporated in the model developed here.
Figure 5.

Data and simulation indicating adaptation of the gain of phasic responses of dopamine neurons to rewards of two sizes after a CS that had been equiprobably followed by either the larger or smaller (possibly zero) reward. Insets, Data [from Tobler et al. (2005), their Fig. 4C, reprinted with permission from AAAS] for monkeys A and B. Monkey A had received many conditioning trials, on each of which it saw one of three CSs, each of which was followed by one of two outcomes with equal probability: a juice reward or a nonreward. The size of the reward, if given, was 0.05, 0.15, or 0.5 ml, and this size depended on the identity of the CS. All data points for monkey A reflect the median activation level changes (above or below baseline) of dopamine neurons in response to reward or nonreward. The activation levels were normalized to the activation in response to uncued delivery of a 0.15 ml reward. Each line connects a point, corresponding to a below-baseline activation (dip) observed in response to nonreward, to a second point corresponding to observed activation in response to whichever of the three reward sizes (0.05, 0.15, or 0.5 ml) could be expected given the CS presented just before. That all activation levels are <1.0 means that learning was sufficient for CS-dependent “canceling” of much of the activation that would otherwise be induced by the primary rewards. This postlearning plot also reveals that the “gain” of the response to reward size (indicated by the slope of the line) after a trained CS is a decreasing function of expected reward size, and this is reflected in the greatly compressed range of activations, relative to what would be observed in response to uncued rewards spanning the same range of sizes. The inset for monkey B, which was trained similarly (but with nonreward trials replaced by smaller-reward trials), shows a similar pattern, but with even less variation across responses to cued rewards of varying sizes. The main plot in the figure shows that a simulation of the model, for the monkey A protocol, produced results in accord with the data.
The adaptively weighted CS inputs to the model matricial MSPNs, and thus these neurons' CS-induced activities, become increasing functions of the expected reward during learning. Therefore, in the model as in the data, striatal MSPN activity is modulated by the expected amount of reward (Kawagoe et al., 1998; Watanabe et al., 2003; Samejima et al., 2005). Model simulations (see below) showed that the depth of this modulation is enhanced because matricial MSPNs also receive inputs related to the reward contingency indirectly, through the pathway from DA neurons to striatal TANs to FS-INs to MSPNs (Fig. 1), in accord with neurophysiological studies (Samejima et al., 2005).
Emergence of uncertainty responses on probabilistic schedules of reward
The distal dendrites of SNc dopaminergic cells are densely intermingled with SNr GABAergic cells (Condé, 1992; Gerfen and Wilson, 1996). Thus, we hypothesized that although matricial and striosomal MSPNs project to SNr and SNc, respectively (Flaherty and Graybiel, 1994), matricial MSPNs also affect SNc dopaminergic cells. During the time between CS onset and expected time of reward, the matricial MSPN projection of GABA and SP to the SNc is active, and model transmitter release depends on MSPN activation, in accord with Khan et al. (1996) and Beaujouan et al. (2004). In particular, the model's net SP release, and consequent excitation of SNc, is progressively gated off by presynaptic action of the coreleased GABA. The net effect of the proposed multiplicative gating interaction is an inverted-U-shaped excitation that is lowest for high, p(R*|CS) = 1.00, and low, p(R*|CS) = 0.00, conditional probabilities, and maximum at p(R*|CS) = 0.5. Thus, as shown in Figures 6 and 7, the model is able to explain the genesis of DA uncertainty responses and their nonmonotonic dependence on p(R*|CS), as reported by Fiorillo et al. (2003).
Figure 6.

Activity (percentage above baseline) of model sustained-response dopamine neurons as a function of reward probability, after training with reward magnitude |R*| = 0.15. Inset, The corresponding neurophysiological data [from Fiorillo et al. (2003), their Fig. 3C, reprinted with permission from AAAS].
Coexistence of sustained and phasic responses in single dopamine neurons
In the model, DA neurons responding with sustained or phasic activations constitute two of three subpopulations (Eqs. 10, 16). In a third subpopulation, as shown in Figure 7D, both sustained and phasic responses occur in single cells, and these components exhibit the same functional dependencies noted above.
Parameter ranges across which the qualitative behavior of the model DA neurons' phasic responses are preserved were discussed by Brown et al. (1999). Similarly, the choice of parameters for, and associated sensitivity analyses of, striatal TANs was delineated by Tan and Bullock (2008b). An additional key component in the current model is the overall effect of the striatal microcircuit. As the expected reward value [|R*| × p(R*|CS)] increases, so does the DA burst to CS onset, as does the depth of the TAN pause response. Thus, post-CS acetylcholine (ACh) release in the striatum is inversely related to the expected value signaled by the CS. As ACh release decreases, so does the feedforward inhibition exerted on the matricial MSPNs by FS-INs. Therefore, a CS-representing cortical input of a particular weighted value will induce higher activations of MSPNs with than without the DA-induced pause response by the TANs. Thus, although weights of corticostriatal synapses onto MSPN themselves reflect expected values, cholinergic modulation of MSPNs amplifies the contrast between matricial MSPN activations in response to different reward contingencies (Fig. 8).
Figure 8.

Effect of striatal circuit on sustained responses of model dopamine neurons. The black line shows the percentage sustained change in the dopamine activity as percentage above baseline as simulated by the complete model (same as Fig. 6), and gray lines indicate the percentage sustained change in the activity as simulated with selected elements of the striatal circuit (TANs and FS interneurons) excluded. Both simulations are performed with the same reward magnitude (|R*| = 0.15) and with the same parameter set. Although the qualitative behavior of the model is similar in both cases (inverted-U-shaped elevations in sustained responses as a function of conditional probability of reward), inclusion of the interneurons of the striatal circuit enhances the contrast between the responses to different probabilistic schedules.
Although Fiorillo et al. (2003) reported an increase in the sustained dopamine response with greater reward magnitude, they did not perform a parametric study to characterize the nature of the dependency. In the model, presynaptic inhibitory gating by GABA (G) of the excitatory substance-P effect, MRS [1 − G]+, on substantia nigra dopaminergic cell dendrites translates the monotonic relation between probability and matricial MSPN activation at the striatal level into a nonmonotonic relation between probability and sustained dopamine activity. The maximum of the resulting nonmonotonic function is determined by the activation threshold of matricial MSPNs for GABA and substance-P release in substantia nigra (Eq. 12). Sensitivity analysis reveals an inverted-U-shaped response to reward probabilities regardless of the reward magnitude if the threshold for GABA and substance-P release is proportional to the magnitude of DA burst in response to CS onset. To assess this, we assumed that after learning, a signal, proportional to the magnitude of DA burst, arrives near terminals of matricial MSPNs at the onset of the predictive stimulus, to scale the release threshold. If such a signal exists, Figure 9 (top) shows that the sustained response of dopamine cells would be approximately similar across a range of absolute reward magnitudes. If such a signal does not exist, and the threshold is constant regardless of the DAergic signal, then the model predicts that the sustained response of dopamine neurons would no longer be a function of reward probability alone (Fig. 9, bottom left). Rather, the sustained response will be a function of expected reward value, R̂ = |R*| × p(R*|CS). As shown in Figure 9 (bottom right), the response is maximum for moderate values of expected reward, and declines toward smaller and larger values. Thus, while also serving as a sensitivity analysis for key parameters regulating GABA and substance-P corelease, these results show that a DA-dependent threshold for GABA and substance-P release from the striatonigral terminals is required if the sustained response of dopaminergic cells in response to the reward probability is to be similar across a range of absolute reward magnitudes.
Figure 9.
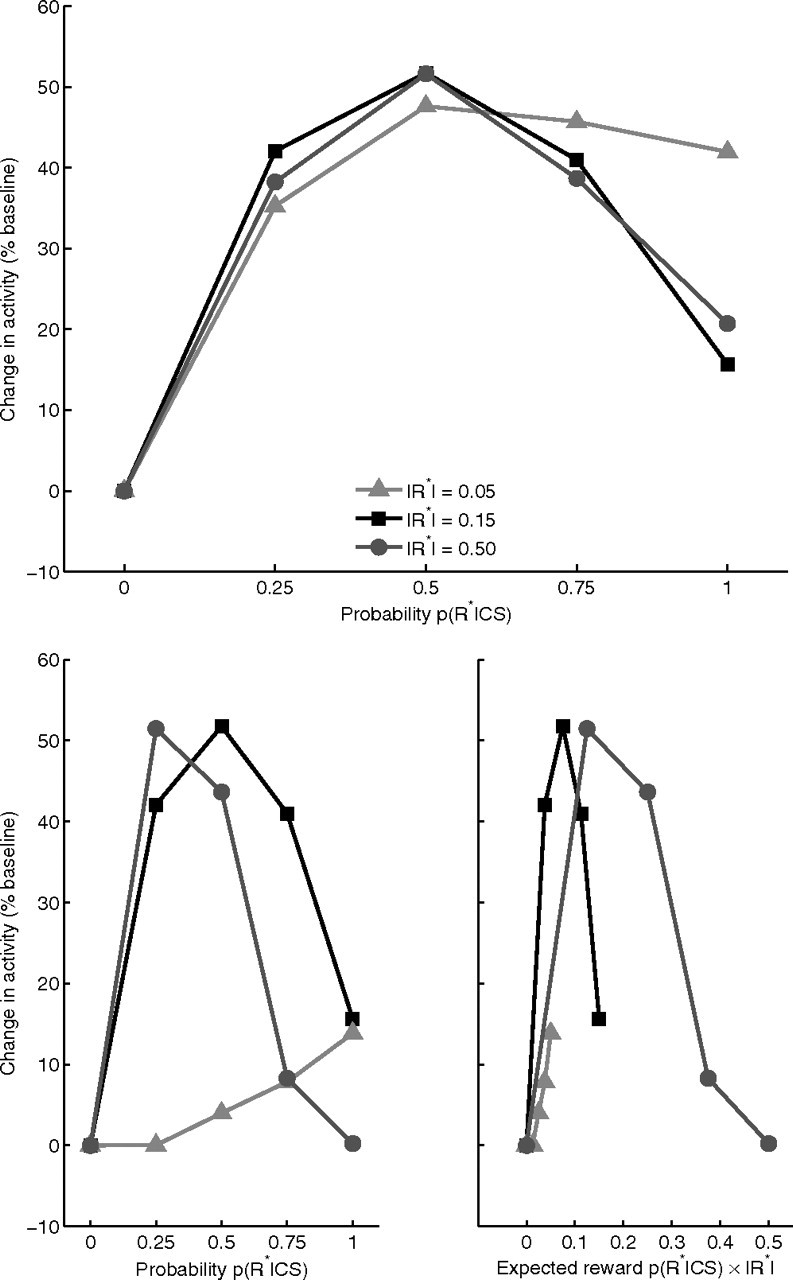
Top, Sustained activation of dopamine cells (percentage above baseline) is an inverted-U-shaped function of reward probability, qualitatively similar under moderate to large reward magnitudes, if and only if MSPN activation threshold for GABA and substance-P release from striatonigral terminals is a function of absolute reward magnitude. Plots are generated by assuming an arbitrary threshold at the time of predictive stimulus onset, proportional to the CS-induced DA signal. Bottom, Sustained activation of dopamine cells is an inverted-U-shaped function of expected reward value R̂ = p(R*|CS)|R*| (right) but not of conditional probability of reward p(R*|CS) (left), in contrast to Fiorillo et al. (2003), if MSPN activation threshold for GABA and substance-P release from striatonigral terminals is constant, ΓM = c in Equation 12.
Discussion
Many reports demonstrate the sensitivity of striatal and DA neurons to properties of cue–reward contingencies that are important for learning to predict outcomes and make decisions (Kawagoe et al., 1998; Watanabe et al., 2003; Samejima et al., 2005). This research helps unify these observations by showing how the sustained uncertainty responses (Fiorillo et al., 2003) that emerge in DA cells when animals experience probabilistic cue–reward contingencies can be explained by correcting and extending a circuit model that Brown et al. (1999) developed to explain phasic DA responses. The new model incorporates several conspicuous, interwoven features of the striatonigral architecture that were previously omitted. The simulations show that uncertainty responses can be computed efficiently via the corelease of GABA and SP by MSPN fibers that synapse not just on SNr cells but also on the comingled dendrites of DA cells whose somata are in SNc. This mechanism works even if the adaptive synapses on cue-excited inputs to the matricial MSPNs giving rise to the GABA/SP fibers obey the same learning law that enables the model to learn to generate phasic DA signals that represent RPEs (Schultz et al., 1997; Schultz, 1998). The model's uncertainty responses are robust on single trials. Because they do not arise by averaging across trials, the uncertainty computation can influence single-trial decision making. This distinguishes the current model from TD models, proponents of which (Niv et al., 2005) showed how TD models could generate uncertainty responses, but only as averaging artifacts. The present model's single-trial RPEs and single-trial uncertainty responses accord with findings (Fiorillo et al., 2005) that uncertainty responses appear in DA neurons on single trials.
Another model proposed as an alternative to TD models was the “primary value, learned value” (PVLV) model (O'Reilly et al., 2007). It incorporated core assumptions from Brown et al. (1999), but omitted others judged problematic, and added postulates that diverge from both the current model and more abstract RPE models. Although the PVLV model cannot explain sustained uncertainty responses, it can avoid a self-learning problem in the Brown et al. (1999) model. Whereas our model avoids that problem via a known biophysical constraint (namely, delayed, calcium-dependent gating of learning, which also explains why a CS must precede a US for robust conditioning), the PVLV model avoids self-learning via formal postulate, and this and further postulates produce a model that deviates in several consequential ways from most RPE-based learning theories. A common feature of the PVLV model and the new model presented here is that both exhibit the phenomenon known as blocking (Kamin, 1968). If a CS, CSA, predicts a reward, with p(US|CSA) = 1, and if CSA learning proceeds until the delayed inhibition of DA neurons (induced by CSA) cancels the US-induced excitation of DA neurons, then a newly introduced second CS, CSB, whose onset is simultaneous with CSA, will not “condition.” The predictively redundant CSB is blocked from becoming able to generate DA bursts, in accord with neural data (Waelti et al., 2001). Finally, O'Reilly et al. (2007) reported a TD model simulation in which, contrary to published observations, a CS failed to become able to induce a model DA burst when the reward followed the CS after unpredictable intervals. Although O'Reilly et al. (2007) mistakenly supposed otherwise, models such as Brown et al. (1999) and the one presented here readily explain CS conditioning with unpredictable CS–US intervals.
Perhaps the most novel feature of the new model is the role for GABA/SP corelease in genesis of sustained DA responses. If correct, this links two conspicuous BG features that are pivotal in neurodegenerative disease. SP-MSPN loss underlies Huntington's disease, whereas nigral DA cell loss underlies Parkinson's disease. The SP in fibers sent from “direct BG pathway” MSPNs (Kaneko et al., 2000; Beaujouan et al., 2004) to ventral SN make it the most SP-rich part of the brain (Otsuka and Yoshioka, 1993), with dramatic SP depletion in Huntington's disease (Buck et al., 1981; Cicchetti et al., 2000). As noted above, direct pathway activation causes MSPN corelease of SP and GABA at nigral terminals that are positioned to affect DA neurons, which SP excites. The model predicts that SP-mediated excitation is presynaptically gated by GABAergic inhibition of release, mediated by GABAB receptors. The simulated inverted-U interaction accords with observations (Fiorillo et al., 2003) that cue-induced sustained DA responses scale nonmonotonically with the conditional probability of reward, given the cue. Because in vivo data are scarce, validation of the main hypothesis awaits further experiments. One testable prediction is that local blockade of presynaptic GABAB receptors, in animals trained on a simple pavlovian task with a probabilistic schedule [similar to Fiorillo et al. (2003)], should eliminate the nonmonotonic relationship between probability and sustained responses, and leave sustained responses that are a monotonic function of expected value.
Reid et al. (1990) showed that SP delivery alone may have a nonmonotonic effect on DA cell activation. Part of this nonmonotonic effect may have resulted from SP activation of nigral GABAergic neurons with local collaterals. Whitty et al. (1997) reported that NKR1 mRNAs were found in some nonpigmented (i.e., non-DA) SN cells, and Mendez et al. (1993) reported that GABAergic cells in the SNr that receive SP terminals do synapse onto dendrites that DA cells of SNc extend ventrally into SNr. Thus, SP-excited GABAergic neurons of SNr may shunt the excitatory effect of SP onto SNc DAergic cells' dendrites. The Appendix shows that the model is robust in the presence of such a shunt.
Tobler et al. (2005) reported that “the gain of neural activity with respect to liquid volume appeared to adapt in proportion to the range … of the predicted reward outcomes” (Fig. 5), and noted that adaptation might “be achieved by subtracting the expected value from the absolute reward value and then dividing by the variance.” They also acknowledged that the apparent adaptation might not arise in DA neurons, but might instead arise “upstream.” The choice between these two attributions is theoretically important. If DA neurons divided the difference between expected and actual inputs by the variance, such neurons could not compute RPEs that could guide learning of synaptic efficacies that reflect absolute reward magnitude. Published observations suggest otherwise. Apparent adaptation arises in the current model from “upstream” striatal weight learning, and requires no normalization of the DA neurons' sensitivities to differences. Tan et al. (2008) proved this for a class of dual-path learning models that assume that DA neurons compute the (unnormalized) difference between an unlearned excitation scaled by primary-reward magnitude and a cue-conditioned, time-delayed inhibition, whose size is adjusted by the history of RPEs. One other mathematical constraint was needed: that negative RPEs (DA dips) have smaller dynamic ranges than positive RPEs (DA bursts). This model feature accords with data (Fiorillo et al., 2003; Nakahara et al., 2004).
Fiorillo et al. (2003) did not fully assess how the sustained component depends on reward magnitude: probability varied over its full range, but reward magnitude (|R*|) did not. Simulations of the current model provide novel predictions regarding this dependency. Although the observed function emerged for a full range of conditional probabilities when cues predicted moderate-sized rewards, the model predicts that the peak of the inverted U could shift away from p = 0.5 with very large or very small rewards, unless the threshold for activation of corelease is a function of expected reward magnitude (Eq. 12; ΓM). With fixed threshold, simulated sustained activation can be a nonmonotonic function of expected reward, R̂ = |R*| × p(R*|CS), rather than of probability alone. It is maximum for moderate values of R̂, and declines for smaller and larger values. If the relationship between p(R*|CS) and sustained DA response is to be qualitatively similar across at least moderate-to-large reward magnitudes, then the GABA and substance-P release threshold should depend on expected reward magnitude. In principle (although not modeled), CS-induced phasic release of dopamine from SNc dendrites in SNr might help create such a functional dependence.
Although both sustained and phasic responses of model DA neurons survive without the TANs and FS-INs in the model striatum, the amplitude of the inverted U was enhanced by these striatal interneurons. ACh release by model striatal TANs modulates the matricial MSPNs via a dual control of FS-INs by ACh (de Rover et al., 2002; Koós and Tepper, 2002), and via a direct inhibitory influence of ACh on SP-MSPNs (Bernard et al., 1993; Di Chiara et al., 1994). Interestingly, simulations (Tan and Bullock, 2008b) show that many data accord with the hypothesis that TANs' conditioned pause responses largely reflect learned phasic DA inputs. Cholinergic modulation of SP-MSPNs may shed light on the individual differences in uncertainty responses observed (Fig. 6, inset). For example, the model's uncertainty response to p = 1 is zero in the absence of interneurons, whereas the model responds with an ∼10% increase in baseline activity when the striatal circuit is intact (although the qualitative profiles of the uncertainty responses are similar) (Fig. 8). These model variations are consistent with the variations observed in vivo (Fig. 6, inset).
In SNc, it appears that ∼50% of dopaminergic cells respond to a CS with a phasic activation, 9% with a sustained activation, and 18% with both (C. Fiorillo, personal communication). However, neurons with no significant activation might show such activation, if a larger reward were tested. It remains to be discovered whether, in vivo, there are three DA subpopulations that correspond to the model's three distinct DA neurons (phasic only, sustained only, and mixed). Whereas learning is not affected by relative proportions of these cells, the size of the sustained component felt by target structures could be.
Uncertainty responses may be important components of DA signals to the forebrain. Its local effects may differ dramatically across signal recipients because of differing DA receptors, local circuits, DA uptake rates, etc. Predictions vary considerably across conceptualizations of the role of DA in forebrain function (Seamans and Yang, 2004). One hypothesis is that sustained DA release during an interval between CS onset and possible reward delivery might act in frontal cortex to facilitate vigilant attention and working memory storage during the delay, while acting in striatum to facilitate switches among alternative plans. A stimulus that is an uncertain predictor is often the leading edge of an information stream, later components of which resolve the uncertainty. Uncertainty-reducing information will be better detected and learned by a vigilant perceiver who remembers recent events, and better used by an actor ready to switch as further information favors one plan among several suggested by the initial information. Uncertainty responses may also facilitate behavioral switches during reversal learning (Pasupathy and Miller, 2005) (but see Tan and Bullock, 2008a).
Finally, many brain areas not modeled here are implicated in value-related computations. For example, orbitofrontal cortex mediates explicit cognitive “reframing” of costs and benefits (De Martino et al., 2006), operations beyond this model's scope. Model development was driven by data from experiments using pavlovian protocols, in which there is no required action. Therefore, there are no action costs to factor into value computations. Thus, the model does not address many issues in motivated behavior and foraging.
Appendix
Except where otherwise noted, all simulations reported in the main text used Equations 1–16 and 18–21, as described below.
Changes in the level of P are described by the following differential equation, which reflects two afferents to PPTN: a primary reward-induced input, IR, from lateral hypothalamus and a conditioned stimulus-induced input, S, from ventral striatum (via the interposed ventral pallidum):
 |
where WRP and WSP are synaptic weights that multiply IR and S, respectively, and UP is an afterhyperpolarization governed by the following equation:
 |
The learned ventral striatal activation level, S, is governed by
 |
where IR again is the primary reward signal, multiplied by fixed weight WRS, Ii is a signal coming from the ith CS representation to the ventral striatal cells, and WiS is the adaptive synaptic weight that multiplies this signal. Weight adaptation is governed by
 |
where CWSmax is the upper bound on each weight. Provided that the CS-induced signal Ii is positive, then synaptic weight potentiation and depression are induced, respectively, by phasic dopamine burst or dip signals, N+ and N− (defined below in Eqs. 20, 21). These signals are never on simultaneously. Learning at the model ventral striatal adaptive weights is gated by delayed release of a second messenger, and calcium signal GWS is governed by Equations 5 and 7 (below) at a rate r = 12.5.
The formal model approximates the spectrum of delayed calcium spikes (adaptive timing spectrum) as follows: a spectrum of striosomal MSPN second messenger activities xij respond to the ith input at rates rj:
 |
where the second messenger buildup rates are given by
 |
The activities xij induce intracellular calcium dynamics within a given spine (j) at delays determined by rj. The intracellular calcium spike is represented by the quantity [GijYij]+ (Brown et al., 1999), where
 |
and
 |
where fG(x) is a step function: 0 for x ≤ 0 and 1 for x > 0. Parameters ΓG and ΓY define signal thresholds for calcium accumulation (Eq. 7) and calcium decay (Eq. 8), respectively. In the brief interval when the calcium concentration at a particular spine exceeds a threshold activity ΓS, CS-striosomal weight Zij at that particular spine becomes eligible for adaptation that may be induced by dopaminergic bursts (N+) or dips (N−):
 |
Thus, phasic DA bursts and dips during learning trials respectively potentiate and depress two sets of adaptive weights, the Zij and the WiS (treated above). With the parameters chosen in Equations 4 and 9, the latter weights change somewhat faster during extinction than the former. Indeed, primate data reported by Ljungberg et al. (1992), and new rodent data reported by Pan et al. (2005), led the latter authors to conclude that “there are different time courses for acquisition of new conditioned responses to cues and the loss of responses to the rewards predicted by those cues”; notably, the data indicate that the learning responsible for controlling the cue-dependent inhibition of DA responses to primary rewards is slower than that controlling predictive excitatory responses to cues.
As learning progresses, the CS comes to immediately excite the DA neurons through the ventral striatal pathway and to inhibit the same DA cells after a learned delay. The equation governing activity of phasic DA cells is
 |
Here the thresholded PPTN signal [P − ΓP]+ excites DA cells, but the summed spectrum of striosomal MSPN signals Σi,j [GijYij − ΓS]+ Zij inhibits DA cells. In Equation 10, ID represents endogenous factors that control the low baseline activation (and associated firing rate) of phasic DA cells.
Matricial MSPNs are modeled by
 |
where WiSIi is adaptively weighted cortical input, and the inhibition, F (supplemental Eq. S8, available at www.jneurosci.org as supplemental material), from FS-INs, is gated by factor (1 − T), which reflects activation of presynaptic receptors on FS-IN axon terminals (Koós and Tepper, 2002) by ACh released from TANs (whence “T”). The last term, T, reflects the direct inhibitory action of ACh on MSPNs (Di Chiara et al., 1994). The model used to generate the ACh signal T is treated by Tan and Bullock (2008b) and summarized in Striatal Microcircuit in the supplemental material (Eq. S7, available at www.jneurosci.org as supplemental material). The synaptic strengths of cortical inputs to the matricial MSPNs are assumed to be governed by the same mechanism as ventral striatal cells (WiS in Eq. 4).
To model how axon terminals from such matricial MSPNs affect SNc cell firing, suppose that, in the absence of any synaptic feedbacks, the GABA release level, MRG, and the substance-P release level, MRS, in SN would be equal and proportional to the thresholded matricial MSPN activation:
If the release of both GABA and SP from the MSPN terminals is inhibited by released GABA acting via presynaptic GABAB receptors, net GABA (G) and SP (SP) release can be written as
where 0 < βM < 1. The latter term introduces an asymmetry to GABAB receptor-mediated inhibition of SP release relative to GABA release. Stronger inhibition of SP release than GABA release is necessary for the model's successful genesis of nonmonotonic uncertainty responses. Although data are apparently lacking to prove this assumed asymmetry, several studies in the cortex and spinal cord indicate that modulation of SP and GABA release via GABA receptors can be heterogeneous, and may even involve different receptor subtypes (Bonanno et al., 1996; Teoh et al., 1996). Therefore, the model's prediction of asymmetric effects is not implausible. Equations 13 and 14 imply that the potential levels MRS and MRG are gated by factors that reflect feedback inhibition of release. Because the multiplicative gating factor declines as corelease of GABA increases, the net SP release given by the product MRS[1 − G]+ is a nonmonotonic, inverted-U, function of matricial MSPN activation level M (see also Tan and Bullock, 2008a). In accord with data already noted, released SP excites the “sustained” type of model DA cell. The net SP level, S, that acts on postsynaptic neurokinin receptors can be approximated by
 |
The activity, (Dsust), of the subset of SNc DA cells that receive this net SP signal is modeled by
 |
where ID is the same endogenous excitation defined for Equation 10, and αSD is the gain of the SP excitation effect. Equation 16, which was used for all the simulations reported in the main text, does not include a postsynaptic effect of GABA release. This omission seems reasonable for present purposes, because DA neurons are relatively insensitive to GABA (Tepper and Lee, 2007), and because any postsynaptic effect of increased GABA release from matricial MSPN inputs would likely be fully offset by decreased GABA release from nearby terminals of tonically active SNr cells that would be inhibited by the same matricial MSPNs. This “offset hypothesis” is supported by data indicating that DA cells do not show responses that are time locked to the motor initiations gated by matricial MSPN outputs (DeLong et al., 1983; Romo and Schultz, 1990; Schultz, 1998). Nevertheless, it is important to show that the conclusions reached on the basis of simulations using Equation 16 are robust even if the GABA released by GABA/SP terminals has a postsynaptic effect on DA neurons that is not fully offset by reduced GABA release by fibers from SNr. To treat that case, we also studied the DA cell equation:
 |
where ID is a baseline excitation, E is the net excitatory input attributable to released SP, and I is the net inhibitory input attributable to GABA released by GABA/SP terminals. In particular, let I = kX, where X = αR [M − ΓM]+. The coefficient k (set to 0.5) scales the postsynaptic effect of GABA, as needed to reflect the relatively low sensitivity of DA cells to GABA (cf. Tepper and Lee, 2007). One more parameter is needed to allow a thorough study of the robustness of the system's ability to exhibit DA uncertainty responses that are nonmonotonic and approximately proportional to p × (1 − p). That parameter, c, scales the strength of the presynaptic GABAergic reduction of SP release, and 0 ≪ c < 1 allows for significant, but less than total, blockade. (For simplicity, we ignore, without loss of generality, the feedback effect of GABA on itself.) Thus, let E = X × (1 − cX). For three parameterizations, Figure 10 plots a full range of solutions of Equation 17 at equilibrium, for values of X ranging from 0 to 1. As can be seen, the qualitative shape of the response is nonmonotonic, with only small differences between the three. The black trace shows the result if the postsynaptic effect of GABA is negligible, i.e., if k = 0 and c = 1. Both the other curves reflect a postsynaptic (ultimately divisive) effect of GABA, and indeed both curves have a smaller amplitude than the black curve. If k = 0.5 and c = 1 (red curve), there is a slight leftward “peak shift.” Although the resultant nonmonotonic function peaks below p = 0.5, it is still compatible with the rank ordering probed in extant experiments: the response at p = 0.5 is greater that the responses at p = 0.25 or p = 0.75, and there is essentially no response at p = 0 or p = 1.
Figure 10.

Full range of solutions of Equation 17 at equilibrium for three parameterizations. Black curve, k = 0 and c = 1 (equivalent to the equilibrium solutions of Eq. 16). Red curve, k = 0.5 and c = 1. Blue curve, k = 0.5 and c = 0.9.
Furthermore, much of this peak shift disappears (blue curve) if k = 0.5 and c = 0.9, i.e., if the presynaptic gating of SP release by GABA remains proportionate, but with a slope less than one. This analysis indicates that the key property, a nonmonotonic function whose shape approximates the function p × (1 − p), is a very robust property of the system, even in the presence of a significant postsynaptic effect of GABA.
To simulate coexisting responses in a single neuron, the model includes a third set of DA neurons affected by all three pathways (PPTN-mediated excitatory projections and projections from both matricial and striosomal MSPNs):
 |
Striatal DA release is modeled by the following equation:
 |
where D = Dphasic + Dsust is the overall firing rate of DA cells as a population. Positive (N+) and complementary negative (N−) reinforcement signals are respectively derived from above- and below-baseline fluctuations from the overall DA level, D̄:
Footnotes
This work was supported by National Science Foundation Grant SBE-354378. C.O.T. was partly supported by the Higher Education Council of Turkey and Canakkale Onsekiz Mart University of Turkey. We thank the anonymous reviewers for careful readings and comments that inspired numerous improvements to this article.
References
- Beaujouan JC, Torrens Y, Saffroy M, Kemel M-L, Glowinski J. A 25 year adventure in the field of tachykinins. Peptides. 2004;25:339–357. doi: 10.1016/j.peptides.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Bernard V, Dumartin B, Lamy E, Bloch B. Fos immunoreactivity after stimulation or inhibition of muscarinic receptors indicates anatomical specificity for cholinergic control of striatal efferent neurons and cortical neurons in the rat. Eur J Neurosci. 1993;5:1218–1225. doi: 10.1111/j.1460-9568.1993.tb00976.x. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Greenamyre JT. Regulation of dopamine receptor and neuropeptide expression in the basal ganglia of monkeys treated with MPTP. Exp Neurol. 2004;189:393–403. doi: 10.1016/j.expneurol.2004.05.041. [DOI] [PubMed] [Google Scholar]
- Bonanno G, Gemignani A, Schmid G, Severi P, Cavazzani P, Raiteri M. Human brain somatostatin release from isolated cortical nerve endings and its modulation through GABAB receptors. Br J Pharmacol. 1996;118:1441–1446. doi: 10.1111/j.1476-5381.1996.tb15558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG. GABAB receptor pharmacology. Annu Rev Pharmacol Toxicol. 1993;33:109–147. doi: 10.1146/annurev.pa.33.040193.000545. [DOI] [PubMed] [Google Scholar]
- Brown J, Bullock D, Grossberg S. How the basal ganglia use parallel excitatory and inhibitory learning pathways to selectively respond to unexpected rewarding cues. J Neurosci. 1999;19:10502–10511. doi: 10.1523/JNEUROSCI.19-23-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck SH, Burks TF, Brown MR, Yamamura HI. Reduction in basal ganglia and substantia nigra substance P levels in Huntington's disease. Brain Res. 1981;209:464–469. doi: 10.1016/0006-8993(81)90171-2. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Rev. 2000;34:80–101. doi: 10.1016/s0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- Condé H. Organization and physiology of substantia nigra. Exp Brain Res. 1992;88:233–248. doi: 10.1007/BF02259099. [DOI] [PubMed] [Google Scholar]
- DeLong MR, Crutcher MD, Georgopoulos AP. Relations between movement and single cell discharge in the substantia nigra of the behaving monkey. J Neurosci. 1983;3:1599–1606. doi: 10.1523/JNEUROSCI.03-08-01599.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martino B, Kumaran D, Seymour B, Dolan RJ. Frames, biases, and rational decision-making in the human brain. Science. 2006;313:684–687. doi: 10.1126/science.1128356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rover M, Lodder JC, Kits KS, Schoffelmeer ANM, Brussaard AB. Cholinergic modulation of nucleus accumbens medium spiny neurons. Eur J Neurosci. 2002;16:2279–2290. doi: 10.1046/j.1460-9568.2002.02289.x. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Morelli M, Consolo S. Modulatory functions of neurotransmitters in the striatum—ACh/dopamine/NMDA interactions. Trends Neurosci. 1994;17:228–233. doi: 10.1016/0166-2236(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Doya K. Metalearning and neuromodulation. Neural Netw. 2002;15:495–506. doi: 10.1016/s0893-6080(02)00044-8. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Grossberg S, Bullock D. Metabotropic glutamate receptor activation in cerebellar Purkinje cells as substrate for adaptive timing of the classically conditioned eye-blink response. J Neurosci. 1996;16:3760–3774. doi: 10.1523/JNEUROSCI.16-11-03760.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W. Discrete coding of reward probability and uncertainty by dopamine neurons. Science. 2003;299:1898–1902. doi: 10.1126/science.1077349. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W. Evidence that the delay-period activity of dopamine neurons corresponds to reward uncertainty rather than backpropagating TD errors. Behav Brain Funct. 2005;1:7. doi: 10.1186/1744-9081-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty AW, Graybiel AM. Input-output organization of the sensorimotor striatum in the squirrel monkey. J Neurosci. 1994;14:599–610. doi: 10.1523/JNEUROSCI.14-02-00599.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Futami T, Takakusaki K, Kitai ST. Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci Res. 1995;21:331–342. doi: 10.1016/0168-0102(94)00869-h. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Gerfen C, Wilson CJ. The basal ganglia. In: Swanson LW, Bjorklund A, Hokfelt T, editors. Handbook of chemical neuroanatomy, Vol 12, Integrated systems of the CNS, Pt III. Amsterdam: Elsevier; 1996. pp. 371–468. [Google Scholar]
- Grossberg S, Bullock D, Dranias M. Neural dynamics underlying impaired autonomic and conditioned responses following amygdala and orbitofrontal lesions. Behav Neurosci. 2008 doi: 10.1037/a0012808. in press. [DOI] [PubMed] [Google Scholar]
- Hanson GR, Bush L, Keefe KA, Alburges ME. Distinct responses of basal ganglia substance P systems to low and high doses of methamphetamine. J Neurochem. 2002;82:1171–1178. doi: 10.1046/j.1471-4159.2002.01053.x. [DOI] [PubMed] [Google Scholar]
- Houk J, Adams J, Barto A. A model of how basal ganglia generate and use neural signals that predict reinforcement. In: Houk J, Davis J, Beiser D, editors. Models of information processing in the basal ganglia. Cambridge, MA: MIT; 1995. pp. 249–270. [Google Scholar]
- Humpel C, Saria A. Effects of GABA and L-glutamic acid on the potassium-evoked in vitro release of substance P- and neurokinin A-like immunoreactivities are different in the rat striatum and substantia nigra. Neurosci Lett. 1989;105:159–163. doi: 10.1016/0304-3940(89)90029-3. [DOI] [PubMed] [Google Scholar]
- Iversen LL, Lee CM, Gilbert RF, Hunt S, Emson PC. Regulation of neuropeptide release. Proc R Soc Lond B Biol Sci. 1980;210:91–111. doi: 10.1098/rspb.1980.0121. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Substance P release from the rat substantia nigra. Brain Res. 1978;151:469–478. doi: 10.1016/0006-8993(78)91080-6. [DOI] [PubMed] [Google Scholar]
- Joel D, Weiner I. The connections of the dopaminergic system with the striatum in rats and primates: an analysis with respect to the functional and compartmental organization of the striatum. Neuroscience. 2000;96:451–474. doi: 10.1016/s0306-4522(99)00575-8. [DOI] [PubMed] [Google Scholar]
- Kamin L. “Attention-like” processes in classical conditioning. In: Jones MR, editor. Miami symposium on the prediction of behavior: aversive simulation; Miami: University of Miami; 1968. pp. 9–31. [Google Scholar]
- Kaneko S, Hikida T, Watanabe D, Ichinose H, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Synaptic integration mediated by striatal cholinergic interneurons in basal ganglia function. Science. 2000;289:633–637. doi: 10.1126/science.289.5479.633. [DOI] [PubMed] [Google Scholar]
- Kawagoe R, Takikawa Y, Hikosaka O. Expectation of reward modulates cognitive signals in the basal ganglia. Nat Neurosci. 1998;1:411–416. doi: 10.1038/1625. [DOI] [PubMed] [Google Scholar]
- Khan S, Whelpton R, Michael-Titus AT. Evidence for modulatory effects of substance P fragments (1–4) and (8–11) on endogenous outflow in rat striatal slices. Neurosci Lett. 1996;205:33–36. doi: 10.1016/0304-3940(96)12363-6. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–535. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kötter R. Postsynaptic integration of glutamatergic and dopaminergic signals in the striatum. Prog Neurobiol. 1994;44:163–196. doi: 10.1016/0301-0082(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Kawaguchi Y. Spatial distributions of chemically identified intrinsic neurons in relation to patch and matrix compartments of rat neostriatum. J Comp Neurol. 1993;332:499–513. doi: 10.1002/cne.903320409. [DOI] [PubMed] [Google Scholar]
- Lévesque M, Wallman MJ, Parent R, Sík A, Parent A. Neurokinin-1 and neurokinin-3 receptors in primate substantia nigra. Neurosci Res. 2007;57:362–371. doi: 10.1016/j.neures.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Ljungberg T, Apicella P, Schultz W. Responses of monkey dopamine neurons during learning of behavioral reactions. J Neurophysiol. 1992;67:145–163. doi: 10.1152/jn.1992.67.1.145. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. Peptide autoreceptors: does an autoreceptor for substance P exist? Trends Pharmacol Sci. 1999;20:405–407. doi: 10.1016/s0165-6147(99)01388-7. [DOI] [PubMed] [Google Scholar]
- Martorana A, Fusco FR, D'Angelo V, Sancesario G, Bernardi G. Enkephalin, neurotensin and substance P immunoreactivity neurones of the rat GP following 6-hydroxydopamine lesion of the substantia nigra. Exp Neurol. 2003;183:311–319. doi: 10.1016/s0014-4886(03)00050-5. [DOI] [PubMed] [Google Scholar]
- Mendez I, Elisevich K, Flumerfelt BA. GABAergic synaptic interactions in the substantia nigra. Brain Res. 1993;617:274–284. doi: 10.1016/0006-8993(93)91095-a. [DOI] [PubMed] [Google Scholar]
- Nakahara H, Itoh H, Kawagoe R, Takikawa Y, Hikosaka O. Dopamine neurons can represent context-dependent prediction error. Neuron. 2004;41:269–280. doi: 10.1016/s0896-6273(03)00869-9. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Ono T. Lateral hypothalamus neuron involvement in integration of natural and artificial rewards and cue signals. J Neurophysiol. 1986;55:163–181. doi: 10.1152/jn.1986.55.1.163. [DOI] [PubMed] [Google Scholar]
- Niv Y, Duff MO, Dayan P. Dopamine, uncertainty and TD learning. Behav Brain Funct. 2005;1:6. doi: 10.1186/1744-9081-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly RC, Frank MJ, Hazy TE, Watz B. PVLV: the primary value and learned value pavlovian learning algorithm. Behav Neurosci. 2007;121:31–49. doi: 10.1037/0735-7044.121.1.31. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Yoshioka K. Neurotransmitter functions of mammalian tachykinins. Physiol Rev. 1993;73:229–308. doi: 10.1152/physrev.1993.73.2.229. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Tepper JM. GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse. 1999;32:165–176. doi: 10.1002/(SICI)1098-2396(19990601)32:3<165::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Pan W-X, Hyland BI. Pedunculopontine tegmental nucleus controls conditioned responses of midbrain dopamine neurons in behaving rats. J Neurosci. 2005;25:4725–4732. doi: 10.1523/JNEUROSCI.0277-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WX, Schmidt R, Wickens JR, Hyland BI. Dopamine cells respond to predicted events during classical conditioning: evidence for eligibility traces in the reward-learning network. J Neurosci. 2005;25:6235–6242. doi: 10.1523/JNEUROSCI.1478-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A. Wiley series in neurobiology. New York: Wiley; 1986. Comparative neurobiology of the basal ganglia. [Google Scholar]
- Pasupathy A, Miller EK. Different time courses of learning-related activity in the prefrontal cortex and striatum. Nature. 2005;433:873–876. doi: 10.1038/nature03287. [DOI] [PubMed] [Google Scholar]
- Prensa L, Giménez-Amaya JM, Parent A. Chemical heterogeneity of the striosomal compartment in the human striatum. J Comp Neurol. 1999;413:603–618. [PubMed] [Google Scholar]
- Ragsdale CW, Jr, Graybiel AM. A simple ordering of neocortical area established by the compartmental organization of their striatal projections. Proc Natl Acad Sci U S A. 1990;87:6196–6199. doi: 10.1073/pnas.87.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel S, Legallet E, Apicella P. Responses of tonically active neurons in the monkey striatum discriminate between motivationally opposing stimuli. J Neurosci. 2003;23:8489–8497. doi: 10.1523/JNEUROSCI.23-24-08489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Herrera-Marschitz M, Terenius L, Ungerstedt U. Intranigral substance P modulation of striatal dopamine: interaction with N-terminal and C-terminal substance P fragments. Brain Res. 1990;526:228–234. doi: 10.1016/0006-8993(90)91226-7. [DOI] [PubMed] [Google Scholar]
- Riley RC, Trafton JA, Chi SI, Basbaum AI. Presynaptic regulation of spinal cord tachykinin signaling via GABA(B) but not GABA(A) receptor activation. Neuroscience. 2001;103:725–737. doi: 10.1016/s0306-4522(00)00571-6. [DOI] [PubMed] [Google Scholar]
- Romo R, Schultz W. Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements. J Neurophysiol. 1990;63:592–606. doi: 10.1152/jn.1990.63.3.592. [DOI] [PubMed] [Google Scholar]
- Salio C, Lossi L, Ferrini F, Merighi A. Ultrastructural evidence for a pre- and postsynaptic localization of full-length trkB receptors in substantia gelatinosa (lamina II) of rat and mouse spinal cord. Eur J Neurosci. 2005;22:1951–1966. doi: 10.1111/j.1460-9568.2005.04392.x. [DOI] [PubMed] [Google Scholar]
- Salio C, Lossi L, Ferrini F, Merighi A. Neuropeptides as synaptic transmitters. Cell Tissue Res. 2006;326:583–598. doi: 10.1007/s00441-006-0268-3. [DOI] [PubMed] [Google Scholar]
- Samejima K, Ueda Y, Doya K, Kimura M. Representation of action-specific reward values in the striatum. Science. 2005;310:1337–1340. doi: 10.1126/science.1115270. [DOI] [PubMed] [Google Scholar]
- Scarnati E, Hajdu F, Pacitti C, Tömböl T. An EM and Golgi study on the connection between the nucleus tegmenti pedunculopontinus and the pars compacta of the substantia nigra in the rat. J Hirnforsch. 1988;29:95–105. [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz W. Neural coding of basic reward terms of animal learning theory, game theory, microeconomics and behavioral ecology. Curr Opin Neurobiol. 2004;14:139–147. doi: 10.1016/j.conb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Schultz W, Apicella P, Scarnati E, Ljungberg T. Neuronal activity in monkey ventral striatum related to the expectation of reward. J Neurosci. 1992;12:4595–4610. doi: 10.1523/JNEUROSCI.12-12-04595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Semba K, Fibiger HC. Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and anterograde transport and immunohistological study. J Comp Neurol. 1992;323:387–410. doi: 10.1002/cne.903230307. [DOI] [PubMed] [Google Scholar]
- Smith CUM. Elements of molecular neurobiology. Ed 2. New York: Wiley; 1996. [Google Scholar]
- Tan CO, Anderson E, Dranias M, Bullock D. Can the apparent adaptation of dopamine neurons' mismatch sensitivities be reconciled with their computation of reward prediction errors? Neurosci Lett. 2008;438:14–16. doi: 10.1016/j.neulet.2008.04.059. [DOI] [PubMed] [Google Scholar]
- Tan CO, Bullock D. Neuropeptide co-release with GABA may explain functional non-monotonic uncertainty responses in dopamine neurons. Neurosci Lett. 2008a;430:218–223. doi: 10.1016/j.neulet.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Tan CO, Bullock D. A dopamine-acetylcholine cascade: simulating learned and lesion-induced behavior of striatal cholinergic interneurons. J Neurophysiol. 2008b doi: 10.1152/jn.90486.2008. in press. [DOI] [PubMed] [Google Scholar]
- Teoh H, Malcangio M, Bowery NG. GABA, glutamate and substance P-like immunoreactivity release: effects of novel GABAB antagonists. Br J Pharmacol. 1996;118:1153–1160. doi: 10.1111/j.1476-5381.1996.tb15518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Lee CR. GABAergic control of substantia nigra dopaminergic neurons. Prog Brain Res. 2007;160:189–208. doi: 10.1016/S0079-6123(06)60011-3. [DOI] [PubMed] [Google Scholar]
- Tobler PN, Fiorillo CD, Schultz W. Adaptive coding of reward value by dopamine neurons. Science. 2005;307:1642–1645. doi: 10.1126/science.1105370. [DOI] [PubMed] [Google Scholar]
- Waelti P, Dickinson A, Schultz W. Dopamine responses comply with basic assumptions of formal learning theory. Nature. 2001;412:43–48. doi: 10.1038/35083500. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Lauwereyns J, Hikosaka O. Neural correlates of rewarded and unrewarded eye movements in the primate caudate nucleus. J Neurosci. 2003;23:10052–10057. doi: 10.1523/JNEUROSCI.23-31-10052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitty CJ, Paul MA, Bannon MJ. Neurokinin receptor mRNA localization in human midbrain dopamine neurons. J Comp Neurol. 1997;382:394–400. [PubMed] [Google Scholar]
- Wickens JR, Begg AJ, Arbuthnott GW. Dopamine reverses the depression of rat corticostriatal synapses which normally follows high-frequency stimulation of cortex in vitro. Neuroscience. 1996;70:1–5. doi: 10.1016/0306-4522(95)00436-m. [DOI] [PubMed] [Google Scholar]
- Wörgötter F, Porr B. Temporal sequence learning, prediction, and control: a review of different models and their relation to biological mechanisms. Neural Comput. 2005;17:245–319. doi: 10.1162/0899766053011555. [DOI] [PubMed] [Google Scholar]
- Yang C, Mogenson G. Hippocampal signal transmission to the pedunculopontine nucleus and its regulation by dopamine D2 receptors in the nucleus accumbens: an electrophysiological and behavioural study. Neuroscience. 1987;23:1041–1055. doi: 10.1016/0306-4522(87)90179-5. [DOI] [PubMed] [Google Scholar]