

Abstract
Altered glutamatergic and dopaminergic signaling has been proposed as contributing to the specific striatal cell death observed in Huntington's disease (HD). However, the precise mechanisms by which mutant huntingtin sensitize striatal cells to dopamine and glutamate inputs remain unclear. Here, we demonstrate in knock-in HD striatal cells that mutant huntingtin enhances dopamine-mediated striatal cell death via dopamine D1 receptors. Moreover, we show that NMDA receptors specifically potentiate the vulnerability of mutant huntingtin striatal cells to dopamine toxicity as pretreatment with NMDA increased D1R-induced cell death in mutant but not wild-type cells. As potential underlying mechanism of increased striatal vulnerability, we identified aberrant cyclin-dependent kinase 5 (Cdk5) activation. We demonstrate that enhanced Cdk5 phosphorylation and increased calpain-mediated conversion of the Cdk5 activator p35 into p25 may account for the deregulation of Cdk5 associated to dopamine and glutamate receptor activation in knock-in HD striatal cells. Moreover, supporting a detrimental role of Cdk5 in striatal cell death, neuronal loss can be widely prevented by roscovitine, a potent Cdk5 inhibitor. Significantly, reduced Cdk5 expression together with enhanced Cdk5 phosphorylation and p25 accumulation also occurs in the striatum of mutant HdhQ111 mice and HD human brain suggesting the relevance of deregulated Cdk5 pathway in HD pathology. These findings provide new insights into the molecular mechanisms underlying the selective vulnerability of striatal cells in HD and identify p25/Cdk5 as an important mediator of dopamine and glutamate neurotoxicity associated to HD.
Keywords: striatum, neurotoxicity, huntingtin, dopamine D1 receptors, glutamate, p25
Introduction
Huntington's disease (HD) is a progressive neurological disorder characterized by chorea, cognitive decline, and psychiatric disturbances (MacDonald et al., 2003; Pérez-Navarro et al., 2006). Dysfunction and death of striatal medium-spiny neurons is the major feature of neuropathological changes in HD (Martin and Gusella, 1986; de la Monte et al., 1988). Although it is not known why spiny neurons are preferentially targeted for degeneration, there is evidence to support the idea that mutant huntingtin toxicity may involve excitotoxicity caused by aberrant NMDA receptor activation (Pérez-Navarro et al., 2006; Fan and Raymond, 2007). In addition to glutamatergic afferents, the striatum also receives the densest dopaminergic innervation in the brain from the ventral midbrain neurons. Despite the high concentration of dopamine (DA) in the striatum, a number of studies have revealed a potential toxic role of DA in the nervous system (Jakel and Maragos, 2000). Thus, exposure of primary striatal neurons to DA causes significant neurotoxicity with increased free radical production and accelerated neuronal death (Wersinger et al., 2004). Moreover, there is increasing evidence that the dopaminergic system may contribute to HD neuropathology. Data from striatal primary neurons that express exon-1 mutant huntingtin indicates that DA exacerbates mutant huntingtin toxicity via reactive oxygen species production and D2 receptor activation (Charvin et al., 2005; Benchoua et al., 2008). Furthermore, in double mutant mice expressing endogenous levels of exon-1 mutant huntingtin and knock-out for the dopamine transporter, DA accelerates protein aggregation and motor dysfunction (Cyr et al., 2006). These data suggest that disturbed DA and NMDA signaling may participate in the enhanced sensitivity of striatal neurons to huntingtin toxicity. In this line, Tang et al. (2007) have reported that dopamine potentiates glutamate-induced apoptosis in cultured striatal neurons from YAC128 HD mice via activation of D1 receptors. However, little is known about the molecular mechanisms that underlie the increased vulnerability of mutant huntingtin striatal cells to dopamine and glutamate inputs.
Cyclin-dependent kinase 5 (Cdk5) is a serine/threonine kinase whose activity is primarily restricted to the nervous system where its main activator p35 is expressed. Although Cdk5 is essential for brain development, many neurodegenerative diseases involve sustained activation of Cdk5 in neurons (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Several studies indicate that Cdk5 becomes a cell death inductor when it binds to p25, the calpain-mediated cleaved product of p35 (Lee et al., 2000). Thus, accumulation of p25 has been observed in neurons treated with glutamate or oxidative stress as well as in animal models of several neurodegenerative diseases (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Notably, treatment with 3-nitropropionic, the systemic administration of which produces selective striatal degeneration with symptoms resembling those of HD (Beal et al., 1993), induces striatal but not cortical Cdk5 activation (Crespo-Biel et al., 2007; Akashiba et al., 2008). More recently, it has been postulated for Cdk5 a role in dopaminergic neurodegeneration. Thus, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic cell loss involves enhanced Cdk5 activity and increased conversion of p35 to the pathogenic p25 form (Smith et al., 2003, 2006).
The above observations prompted us to explore the involvement of aberrant glutamatergic and dopaminergic signaling in striatal neurodegeneration in HD to determine whether Cdk5 might account for this neurotoxic effect. Our data suggest that mutant huntingtin alters NMDAR and D1R signaling via Cdk5 to increase the vulnerability of striatal cells to neurotoxicity.
Materials and Methods
Reagents and antibodies.
NMDA, (±)-SKF-38393 hydrochloride, (±)-Quinpirole dihydrochloride, R(+)-SCH-23390 hydrochloride, (-)-MK-801 hydrogen maleate, EGTA, roscovitine, and SP600125 were obtained from Sigma-Aldrich. Hoechst 33258 pentahydrate was from Invitrogen. Anti-p35 (C-19), anti-phospho-Cdk5 (Tyr15), and anti-Cdk5 (J-3) antibodies were purchased from Santa Cruz Biotechnology. Anti-phospho-stress-activated protein kinase (SAPK)/c-Jun N-terminal protein kinase (JNK) (Thr183/Tyr185) and anti-SAPK/JNK were from Cell Signaling Technologies. Paired helical filament (PHF)-1 and TG5 anti-tau antibodies were a generous gift from Peter Davis (Albert Einstein College of Medicine, Bronx, NY). Anti-green fluorescent protein (GFP) was obtained from Clontech. Anti-α-spectrin was purchased from Millipore. Anti-α-tubulin was from Sigma-Aldrich.
Cell cultures.
Conditionally immortalized wild-type STHdhQ7 and mutant STHdhQ111 striatal neuronal progenitor cell lines expressing endogenous levels of normal and mutant huntingtin with 7 and 111 glutamines, respectively, have been described previously (Trettel et al., 2000). Striatal cells were grown at 33°C in DMEM (Sigma-Aldrich), supplemented with 10% fetal bovine serum (FBS), 1% streptomycin-penicillin, 2 mm l-glutamine, 1 mm sodium pyruvate, and 400 μg/ml G418 (Geneticin; Invitrogen).
Genetic Huntington's disease mouse model and postmortem brain tissues.
HdhQ111 knock-in mice expressing mutant huntingtin with 111 glutamine residues have been described previously (Wheeler et al., 1999). Animals were deeply anesthetized with CO2 and decapitated. Striatum was dissected from 9-month-old homozygous mutant HdhQ111/Q111 and wild-type HdhQ7/Q7 littermate offspring, obtained from the mating of male and female HdhQ111/Q7 heterozygotes. The animals were housed under a 12 h light/dark cycle with food and water ad libitum. All procedures were performed in accordance with the National Institutes of Health and were approved by the local animal care committee of the Universitat de Barcelona (99/01) and the Generalitat de Catalunya (00/1094).
Samples of the caudate and putamen nucleus from three patients with HD (with death at end-stage disease at 71, 68, and 65 years, postmortem intervals of 4–15 h) and three control cases (42, 77, and 74 years, postmortem intervals of 4–23 h) were supplied by the Banc de Teixits Neurològics (Servei Científico-Tècnic, Universitat de Barcelona, Barcelona, Spain). All of the ethical guidelines contained within the latest Declaration of Helsinki were taken into consideration, and informed consent was obtained from all subjects under study.
Drug treatments of striatal cell lines.
To induce NMDA excitotoxicity, wild-type STHdhQ7 and mutant knock-in STHdhQ111 striatal cells were exposed to 500 or 50 μm NMDA in Locke's solution (154 mm NaCl, 5.6 mm KCl, 2.3 mm CaCl2, 3.6 mm NaHCO3, 5 mm HEPES, 5.6 mm glucose, and 10 μm glycine) for 30 min. After treatment, the medium was replaced by fresh DMEM medium with 2.5% FBS.
To assess the effect of dopamine receptor activation, wild-type STHdhQ7 and mutant STHdhQ111 striatal cells were treated for 24 h in DMEM medium (2.5% FBS) with different concentrations of the D1 receptor agonist SKF38393 (30, 60, 80, or 100 μm) or the D2 receptor agonist quinpirole (30, 60, or 150 μm).
In some experiments, cultures were treated with the D1 receptor antagonist (SCH-23390, 10 μm), the JNK inhibitor (SP600125, 10 μm), or the Cdk5 inhibitor (roscovitine, 20 μm) 1 h before NMDA or SKF38393 treatments.
To study Cdk5 and p35/p25 protein levels, wild-type STHdhQ7 and mutant knock-in STHdhQ111 striatal cells were first placed in DMEM serum-free medium for 3 h. For the NMDA treatment, cells were exposed to 500 μm NMDA in Locke's solution for 30 min. After treatment, the medium was replaced by fresh DMEM serum-free medium, and total cell extracts were obtained at different time periods (5, 15, 30, and 60 min). For the SKF38393 treatment, cells were exposed to Locke's solution for 30 min before the addition of SKF38393 (100 μm) in fresh DMEM serum-free medium. Total extracts were obtained at different time periods (5, 15, 30, and 60 min) after SKF38393 treatment. Finally, to assess the combined treatment, cells were pre-exposed to 500 μm NMDA in Locke's solution for 30 min. After treatment, the medium was replaced by fresh DMEM serum-free medium containing 100 μm SKF38393 and total cell extracts were obtained at different time periods (5, 15, 30, and 60 min) after SKF38393 treatment.
To test the role of calcium influx in the p35/p25 protein levels, mutant STHdhQ111 striatal cells were first placed in DMEM serum-free medium for 3 h and then exposed to 25 μm MK-801 or 400 μm EGTA in DMEM serum-free medium for 6 h. Total cell extracts were obtained after treatment.
Cell survival.
Cell survival was assessed by nuclear DNA staining with Hoechst 33258. Cells were washed twice with PBS, fixed with 4% paraformaldehyde in PBS for 10 min, washed twice in PBS, and stained with Hoechst 33258 (1 μg/ml) for 5 min. Stained cells were then washed twice with PBS and mounted under glass coverslips with Mowiol. Cell survival is represented as the proportion of Hoechst-stained nuclei counted in treated cells compared with the number of control (vehicle-treated) cells (100%). Forty fields were counted per condition and experiment, comprising at least 30–40 cells. Data are given as mean ± SD of values obtained in three independent experiments performed in triplicate.
Cell transfection.
For the study of polyQ length-dependent Cdk5 and p-Cdk5 expression, wild-type STHdhQ7 striatal cells were transfected using Lipofectamine 2000 as described by the manufacturer (Invitrogen). Wild-type STHdhQ7 at 50% confluence were transfected with three different constructs containing the mutant exon 1 of the huntingtin gene with different CAG/CAA repeats and with enhanced GFP: 23, Q23htt; 72, Q72htt and 103, Q103htt (Kazantsev et al., 1999) (generously provided by Dr. George M. Lawless, Cure HD Initiative, Reagent Resource Bank of the Hereditary Disease Foundation, New York, NY). The protein extracts were prepared 24 h after transfection.
Western blot analysis.
Total cellular extracts were collected in lysis buffer containing 50 mm Tris base, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.1 mm phenylmethylsulphonyl fluoride, 1% NP-40, and supplemented with 1 mm sodium orthovanadate and protease inhibitor mixture (Sigma-Aldrich). Samples were centrifuged at 10,000 × g for 10 min and the protein contents determined by (detergent-compatible) protein assay (Bio-Rad). Protein extracts (20–40 μg) were mixed with 5× SDS sample buffer, boiled for 5 min, resolved by 8–10% SDS-PAGE, and transferred to nitrocellulose membranes (Whatman Schleicher and Schuell). Blots were blocked in 10% nonfat powdered milk in TBS-T (50 mm Tris-HCl, 150 mm NaCl, pH 7.4, 0.05% Tween 20) for 30 min at room temperature. The membranes were then incubated overnight at 4°C with primary antibodies [phospho-JNK (Thr183/Tyr185) (1:1000), total JNK (1:1000), p35 (1:1000), phospho-Cdk5 (Tyr15) (1:5000), total Cdk5 (1:1000), GFP (1:500), α-spectrin (1:1000), TauG5 (1:500), PHF-1 (1:500), or α-tubulin (1:50,000)]. The membranes were then rinsed three times with Tris-buffered saline–Tween 20 (TBS-T) and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. After washing for 30 min with TBS-T, the membranes were developed using the enhanced chemiluminescence substrate kit (Santa Cruz Biotechnology). The Gel-Pro densitometry program (Gel-Pro Analyzer for Windows, version 4.0.00.001) was used to quantify the different immunoreactive bands relative to the intensity of the α-tubulin band in the same membranes. We ensured signals were within a linear range of detection for the ECL reagent on Hyperfilm ECL (GE Healthcare) by probing increasing amounts of lysate with the relevant antibody. Data are expressed as the mean ± SD of band density obtained in three independent experiments.
Cdk5 kinase activity.
Cdk5 was immunoprecipitated with 1 μg of anti-Cdk5 agarose conjugated antibody (C-8; Santa Cruz Biotechnology) from 500 μg of total cell lysates obtained from wild-type STHdhQ7 and mutant STHdhQ111 striatal cells treated with 100 μm SKF38393 alone or pre-exposed (30 min) to NMDA before SKF38393 treatment. The immunoprecipitates were washed three times with lysis buffer and twice with kinase buffer (50 mm Tris, pH 7.5, 0.1% β-mercaptoethanol, 0.1 mm EGTA, 0.1 mm EDTA). The standard assay contained the following (50 μl total volume): washed agarose immunoprecipitate, 50 mm Trs-HCl, pH 7.5, 0.1 mm EGTA, 0.1% β-mercaptoethanol, 10 mm magnesium acetate, 0.1 mm [γ-32P]-ATP (5 μCi), and 200 μm of substrate (peptide histone 1; Jena Bioscience). The assays were performed for 60 min at 30°C, and the assay tubes were agitated continuously to keep the immunoprecipitate in suspension. Incorporation of [γ-32P]-phosphate into peptide substrate was determined using p81 phosphocellulose paper. Papers were washed in 0.5% ortophosphoric acid, dried, and Cherenkov radiation was counted. Control experiments were performed in parallel without the peptide substrate.
Statistical analysis.
All of the data were analyzed with the program Graph Pad Prism version 4.0 (GraphPad Software). Results are expressed as mean ± SD. Experimental data were analyzed either by a one- or two-way ANOVA followed by the post hoc Bonferroni's multiple comparison test or by Student's t test. A value of p < 0.05 was accepted as denoting statistical significance.
Results
D1 receptor activation increases neuronal death in STHdhQ111 striatal cells
To determine whether mutant huntingtin enhances the vulnerability of striatal cells to dopamine, immortalized striatal cells expressing endogenous levels of full-length wild-type STHdhQ7 or mutant STHdhQ111 huntingtin were treated with increasing concentrations of the D1 receptor agonist SKF38393 (0–100 μm) or the D2 receptor agonist quinpirole (0–150 μm) followed by the determination of cell survival. SKF38393 treatment caused a dose-dependent increase in neuronal cell death in both wild-type and mutant striatal cells (Fig. 1A). Two-way ANOVA analysis for drug treatment and genotype showed that the effect was statistically significant (F(4,15) = 444.30, p < 0.001; F(1,15) = 192.04, p ≤ 0.001, respectively). Importantly, the interaction between genotype and treatment was also statistically significant (F(4,15) = 31.69; p < 0.001), indicating that the two cell genotypes responded differently to the SKF38393 treatment. Thus, at all SKF38393 concentrations tested, the reduction in cell survival was significantly higher in mutant STHdhQ111 than it was in wild-type STHdhQ7 cells, with a maximal difference detected at 100 μm SKF38393 (wild type, 64 ± 11% vs mutant, 39 ± 6%; p ≤ 0.001). It should be noted that (1) at the lower tested concentration (SKF38393 30 μm) only mutant huntingtin STHdhQ111 cells exhibited a reduction in cell survival (∼20%; p ≤ 0.001), and (2) the decrease in cell survival in STHdhQ7 wild-type cells reached a maximum at 80 μm SKF38393 (64 ± 11%; p ≤ 0.001), whereas STHdhQ111 mutant cells still exhibited higher cell death at 100 μm SKF38393 (∼8% more compared with 80 μm SKF38393; p ≤ 0.05). Importantly, coincubation with the D1-selective antagonist SCH23390 significantly reduced SKF38393-induced cell death near to that of control levels in both wild-type and mutant huntingtin cells (∼85 ± 10% and 79 ± 11%; p ≤ 0.001, respectively) (Fig. 1B). No effect on neuronal survival was observed after stimulation of D2 receptors with quinpirole (Fig. 1A). Thus, our results imply that D1 receptor activation mediates the dopaminergic toxic effect observed in wild-type and mutant knock-in striatal cells and that full-length mutant huntingtin expression potentiates dopamine-cell death through D1 but not D2 receptors.
Figure 1.

Increased dopamine D1-receptor-mediated cell death in STHdhQ111 striatal cells. A, Wild-type (ST7/7Q) and mutant (ST1111/111Q) striatal cells were treated with different concentrations of the D1 receptor agonist SKF38393 (30, 60, 80, or 100 μm) or the D2 receptor agonist quinpirole (30, 60, or 150 μm) during 24 h. After treatment, cell survival was measured by scoring the percentage of Hoechst-stained nuclei. Forty fields were counted per condition per experiment, comprising at least 30–40 cells. Results are expressed as percentage of control (vehicle-treated) cells, and data are the mean ± SD of three independent experiments performed in triplicate. A full statistical analysis by the two-way ANOVA is described in the text. ***p < 0.001 treated versus vehicle-treated wild-type cells; +++p < 0.001 treated versus vehicle-treated mutant cells; ##p < 0.01, ###p < 0.001 treated mutant cells versus treated wild-type cells (Bonferroni's multiple comparison test). CNT, Control. B, Wild-type and mutant striatal cells were treated (1 h) with the D1 receptor antagonist SCH23390 (10 μm) before SKF38393 treatment (100 μm). Cell survival was assessed 24 h later by scoring the percentage of Hoechst-stained nuclei. Forty fields were counted per condition per experiment, comprising at least 30–40 cells. Results are expressed as percentage of control (vehicle-treated) cells, and data are the mean ± SD of three independent experiments performed in triplicate. ***p < 0.001 treated versus vehicle-treated wild-type cells; +++p < 0.001 treated versus vehicle-treated mutant cells; $$$p < 0.001 SCH23390 plus SKF38393-treated wild-type cells versus SKF38393-treated wild-type cells; &&&p < 0.001 SCH23390 plus SKF38393-treated mutant cells versus SKF38393-treated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test.
NMDA enhances dopamine D1 receptor-mediated cell death selectively in STHdhQ111 cells
Previous results from our laboratory have demonstrated that mutant STHdhQ111 striatal cells display enhanced sensitivity to NMDA receptor activation (Xifró et al., 2008). Therefore, we next analyzed whether NMDA treatment potentiates the neurotoxic effect of dopamine. We first analyzed the role of a toxic concentration of NMDA (500 μm) on cell death induced by D1R activation. Wild-type STHdhQ7 and mutant STHdhQ111 striatal cells were exposed for 30 min to NMDA before SKF38393 treatment, and cell survival was analyzed 24 h later. Pretreatment with NMDA failed to increase SKF38393-induced cell death in wild-type STHdhQ7 cells (Fig. 2A). Two-way ANOVA for SKF38393 treatment and wild-type group (control and pretreated with NMDA) showed that the effect was statistically significant for drug treatment (F(3,12) = 285.6; p ≤ 0.001) but not for wild-type group (F(1,12) = 0.97; p = 0.33). Two-way ANOVA analysis also revealed lack of interaction between factors (F(3,12) = 0.38; p = 0.77). In contrast, mutant STHdhQ111 striatal cells exhibited a significant decrease in cell survival rates when pre-exposed to NMDA compared with SKF38393 treatment alone (Fig. 2A). Statistical analysis revealed a main effect of NMDA pretreatment in SKF38393-induced cell death in mutant huntingtin striatal cells [F(3,12) = 740.23, p < 0.001 for SKF38393 treatment; F(1,12) = 97.07, p ≤ 0.001 for mutant group (control and pretreated with NMDA)]. Notably, two-way ANOVA analysis also revealed a statistically significant mutant group × SKF38393 treatment interaction (F(3,12) = 13.4; p ≤ 0.001), indicating that mutant huntingtin cells responded differently to SKF38393 treatment when pre-exposed to NMDA toxic concentrations (Fig. 2A). It is important to remark that this effect was observed at concentrations of both SKF38393 and NMDA that induced moderate cell death.
Figure 2.

NMDA potentiates the toxic effect of D1-receptor activation in STHdhQ111 striatal cells. A, Wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells were treated with a toxic concentration of NMDA (500 μm) for 30 min before treatment with different concentrations of SKF38393 (60, 80, or 100 μm). Cell survival was assessed 24 h later by scoring the percentage of Hoechst-stained nuclei. B, Wild-type and mutant striatal cells were exposed to a subtoxic concentration of NMDA (50 μm) for 30 min before treatment with different concentrations of SKF38393 (30, 60, or 100 μm). Cell survival was measured 24 h later by scoring the percentage of Hoechst-stained nuclei. Forty fields were counted per condition per experiment, comprising at least 30–40 cells. Results are expressed as percentage of control (vehicle-treated) cells, and data are the mean ± SD of three independent experiments performed in triplicate. A full statistical analysis by the two-way ANOVA is described in the text. **p < 0.01, ***p < 0.001 treated versus vehicle-treated wild-type cells; ++p < 0.01, +++p < 0.001 treated versus vehicle-treated mutant cells; #p < 0.05, ##p < 0.01, ###p < 0.001 NMDA plus SKF38393-treated mutant cells versus SKF38393-treated mutant cells (Bonferroni's multiple comparison test).
To analyze further the effect of NMDA and SKF38393 on striatal cell death, we next evaluated whether a subtoxic concentration of NMDA (50 μm) modulates D1R-mediated cell death. Consistent with the lack of an additive effect at toxic NMDA concentrations, wild-type STHdhQ7 striatal cells pretreated with 50 μm NMDA did not show increased cell death to that reported for SKF38393 treatment alone (Fig. 2B). Two-way ANOVA for SKF38393 treatment and wild-type group (control and pretreated with NMDA) showed that the effect was statistically significant for drug treatment (F(3,12) = 814.7; p ≤ 0.001) but not for wild-type group (F(1,12) = 0.92; p = 0.35). No interaction between factors was demonstrated by two-way ANOVA analysis (F(3,12) = 0.30; p = 0.82). Interestingly, NMDA pre-exposure was still found to potentiate the cell death induced by SKF38393 treatment in mutant STHdhQ111 striatal cells [F(1,12) = 481.68, p < 0.001 for SKF38393 treatment; F(1,12) = 49.6, p ≤ 0.001 for mutant group (control and pretreated with NMDA)]. Moreover, two-way ANOVA analysis showed that the interaction between factors was statistically significant (F(3,12) = 9.07; p ≤ 0.001), which indicates that the curve of dose-dependent cell death induced by SKF38393 was different in mutant cells when pre-exposed to subtoxic NMDA concentrations. Together, these data indicate that NMDA enhances the sensibility of mutant but not wild-type striatal cells to D1 receptor activation.
JNK activation is not associated with NMDA and SKF38393-mediated neurotoxicity in STHdhQ111 cells
Activation of the JNK may contribute to neuronal cell death after excitotoxicity (Brecht et al., 2005; Centeno et al., 2007). To investigate the potential role of the JNK pathway in the neurotoxic effect of NMDA and SKF38393, wild-type STHdhQ7 and mutant STHdhQ111 striatal cells were treated with 500 μm NMDA or 100 μm SKF38393, and the levels of total and phospho-JNK were determined by Western blot analysis. The immunoblot results revealed that neither NMDA nor SKF38393 exposure induced phosphoactivation of JNK (Fig. 3A). Moreover, analysis of wild-type and mutant cells exposed to 500 μm NMDA for 30 min before SKF38393 treatment revealed that a combination of both drugs did not increase JNK phosphorylation either (Fig. 3A). In line with the lack of phospho-JNK activation, pre-exposure of wild-type STHdhQ7 and mutant STHdhQ111 huntingtin cells to 10 μm SP600125, a JNK inhibitor, did not rescue wild-type or mutant cells from neurotoxic cell death mediated by NMDA or SKF38393 or both (Fig. 3B). These results indicate that JNK does not participate in the observed neurotoxicity induced by NMDA or D1R activation in STHdhQ7 and STHdhQ111 cells.
Figure 3.

JNK activation is not associated with NMDA and dopamine-mediated neurotoxicity in wild-type STHdhQ7 and mutant STHdhQ111 striatal cells. A, Representative Western blot showing levels of p-JNK, JNK, and α-tubulin as a loading control, from wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells treated with NMDA (500 μm) or SKF38393 (100 μm) alone, or preincubated with NMDA (30 min) before SKF38393 treatment. Total cell extracts were obtained 30 min after treatment. The histogram represents the relative p-JNK/JNK ratio expressed as fold increase versus control cells. Values are given as mean ± SD of three independent experiments. B, Wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells pretreated (1 h) with the JNK inhibitor SP600125 (10 μm) were exposed to NMDA (500 μm) or SKF38393 (100 μm) alone, or preincubated with NMDA before SKF38393 exposure. Cell survival was evaluated 24 h later by scoring the percentage of Hoechst-stained nuclei. Forty fields were counted per condition per experiment, comprising at least 30–40 cells. Results are expressed as percentage of control (vehicle-treated) cells, and data are the mean ± SD of three independent experiments performed in triplicate. ***p < 0.001 treated versus vehicle-treated wild-type cells; +++p < 0.001 treated versus vehicle-treated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test.
NMDA potentiates D1R-induced phosphorylation of Cdk5 in mutant STHdhQ111 cells
Deregulation of Cdk5 activity has been associated to neuronal cell death in several neurodegenerative disorders (Cruz and Tsai, 2004). To determine whether Cdk5 activation was involved in the increased vulnerability of mutant STHdhQ111 cells to NMDAR and D1R activation, we first analyzed the levels of total and phosphorylated Cdk5 (Tyr 15) in extracts obtained from wild-type STHdhQ7 and mutant STHdhQ111 striatal cells (Fig. 4A). Immunoblot analysis revealed that total Cdk5 levels were lower in mutant cells than they were in wild-type cells (∼2-fold; p ≤ 0.001), whereas the levels of p-Tyr15 Cdk5 were slightly increased (∼1.2-fold; p ≤ 0.05). Therefore, a significant ∼3-fold increase in the p-Tyr15 Cdk5/Cdk5 ratio (p ≤ 0.001) was apparent in mutant cells compared with wild-type cells.
Figure 4.
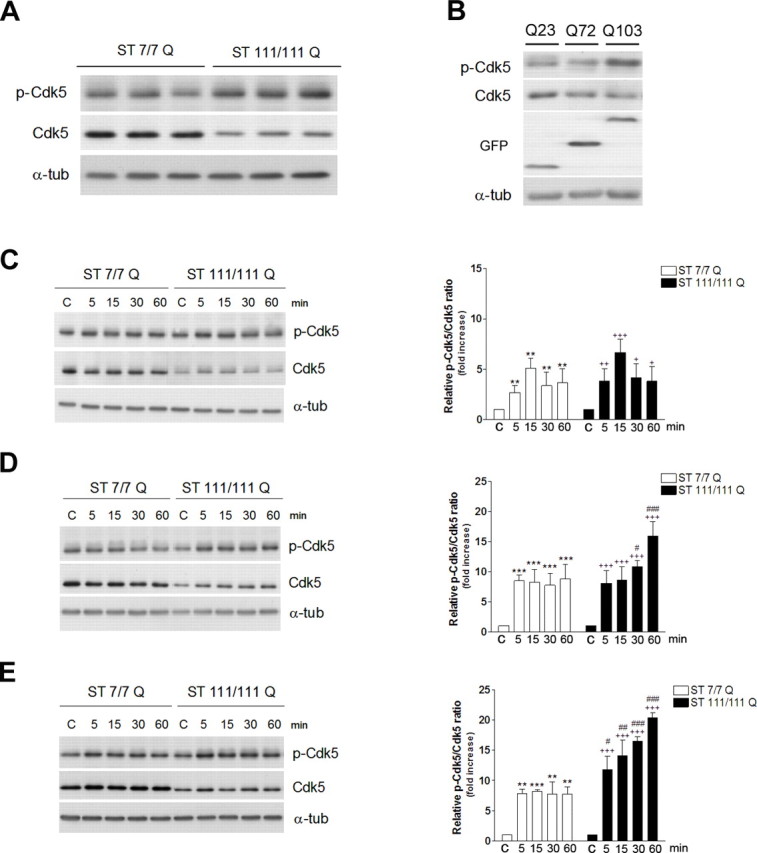
NMDA receptor activation potentiates D1R-induced Cdk5 phosphorylation in mutant STHdhQ111 cells. A, Representative Western blot showing levels of p-Cdk5, Cdk5, and α-tubulin as a loading control from wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells. B, Representative Western blot showing levels of p-Cdk5, Cdk5, and GFP and α-tubulin as a loading control from wild-type striatal cells transfected with different constructs encoding for enhanced fluorescent protein-tagged exon-1 mutant huntingtin protein with 23 (Q23), 72 (Q72), or 103 (Q103) CAG repeats. C–E, Representative Western blot showing levels of p-Cdk5, Cdk5, and α-tubulin as a loading control from wild-type (ST7/7Q) and mutant (ST111/111Q) cell extracts obtained after treatment with NMDA 500 μm (C), SKF38393 100 μm (D), or a pretreatment with NMDA before SKF38393 exposure (E). Total cell extracts were obtained at different time periods (5, 15, 30, and 60 min) after treatment. The histograms represent the relative p-Cdk5/Cdk5 ratio expressed as fold increase versus control (vehicle-treated) cells. Values are given as mean ± SD of five independent experiments. A full statistical analysis by the two-way ANOVA is described in the text. **p < 0.01, ***p < 0.001 treated versus vehicle-treated wild-type cells; +p < 0.05, ++p < 0.01, +++p < 0.001 treated versus vehicle-treated mutant cells; #p < 0.05, ##p < 0.01, ###p < 0.001 treated mutant cells versus treated wild-type cells (Bonferroni's multiple comparison test).
To determine whether deregulation of Cdk5 pathway was dependent on mutant huntingtin expression, wild-type striatal cells were transfected with different constructs encoding for green fluorescent protein-tagged exon-1 mutant huntingtin protein with 23, 72, or 103 CAG repeats (Q23, Q72, and Q103), and total extracts were analyzed by Western blot. The results, shown in Figure 4B, revealed an inverse relationship between changes in Cdk5 and the length of the CAG repeat. Thus, reduced Cdk5 expression along with higher Tyr15-Cdk5 phosphorylation was found in wild-type striatal cells transfected with 72 or 103 poly-Q containing exon-1 mutant huntingtin compared with those transfected with the normal poly-Q length (Q23). These data suggest that changes on Cdk5 pathway are directly dependent on mutant huntingtin expression.
We next analyzed whether activation of NMDA and D1 receptors was associated with changes in Cdk5 expression and phosphorylation. Exposure of wild-type STHdhQ7 and mutant STHdhQ111 striatal cells to NMDA resulted in a similar time-dependent increase in Cdk5 phosphorylation. Statistical analysis using two-way ANOVA revealed a significant time interval effect (F(4,25) = 16.03; p < 0.001) without differences between genotypes (F(1,25) = 3.57; p = 0.08) (Fig. 4C). The analysis also demonstrated no interaction between factors (F(4,25) = 0.57; p = 0.7). Thus, the increase in Cdk5 phosphorylation was already evident at 5 min, maximal at 15 min, and remained high even 60 min after treatment. In contrast, in the presence of 100 μm SKF38393, the kinetic of Cdk5 phosphorylation differed in mutant STHdhQ111and wild-type STHdhQ7 cells (Fig. 4D). Statistical analysis using two-way ANOVA revealed a significant effect of genotype (F(1,25) = 39.36; p < 0.001) and time (F(4,25) = 11.85; p < 0.001) and a significant genotype × treatment interaction (F(4,25) = 20.16; p < 0.001), which demonstrates that the time-dependent phosphorylation of Cdk5 induced by SKF38393 differs between wild-type and mutant huntingtin striatal cells. Thus, wild-type STHdhQ7 cells and mutant STHdhQ111 cells showed a significant increase in Cdk5 phosphorylation induced by SKF38393 at 5 min (∼8-fold; p ≤ 0.001). However, at longer periods, no higher increase in p-Cdk5 was observed in wild-type STHdhQ7 cells, whereas mutant STHdhQ111 cells exhibited a sustained increase in Cdk5 phosphorylation that was maximal at 60 min after treatment (∼17-fold; p ≤ 0.001) (Fig. 4D). These data suggest a dual role of mutant huntingtin on the Cdk5 pathway by reducing the total expression of Cdk5 and enhancing D1R-mediated Cdk5 phosphorylation.
Finally, we investigated whether pretreatment with NMDA modified the D1R-mediated phosphorylation of Cdk5 in wild-type STHdhQ7 and mutant STHdhQ111 striatal cells. Exposure of wild-type STHdhQ7 cells to NMDA before SKF38393 treatment did not change the pattern of Cdk5 phosphorylation compared with SKF38393 alone (F(1,25) = 0.89; p = 0.35) (Fig. 4E). However, mutant STHdhQ111 cells pre-exposed to NMDA exhibited higher Cdk5 phosphorylation compared with those treated with SKF38393 alone (F(1,25) = 147.07; p < 0.001) without significant interaction between treatment and time (F(4,25) = 2.21; p = 0.092). These results demonstrate that in the presence of mutant huntingtin, activation of Cdk5 by D1R is potentiated by NMDAR activation.
STHdhQ111 mutant cells exhibited higher p25 levels
Conversion of p35 into p25 by Ca2+-dependent calpain activation has been suggested to deregulate Cdk5 activity in several neurodegenerative disorders (Dhariwala and Rajadhyaksha, 2008). Indeed, calpain cleavage turns p25 into a more stable protein, which results in prolonged Cdk5 activity (Patrick et al., 1999). To examine whether mutant huntingtin altered the levels of p35 or its cleaved product p25, we performed immunoblot analysis using an antibody that recognizes either p35 or p25 forms (Fig. 5A). Our results showed a significant decrease in p35 levels in mutant STHdhQ111 cells compared with those observed in wild-type STHdhQ7 cells (∼2-fold; p ≤ 0.001). In contrast, the levels of p25, which were almost undetectable in wild-type STHdhQ7 striatal cells, were significantly higher in mutant STHdhQ111 cells (∼2.5-fold; p ≤ 0.001). Because p25 results from calpain activity, using Western blot we next measured the breakdown products of spectrin (SBDP), a well known calpain substrate that yields specific products at 150/160 kDa after calpain cleavage. Consistent with higher p25 accumulation, mutant STHdhQ111 cells exhibited increased levels of SBDP at 150/160 kDa compared with wild-type cells (∼2-fold; p ≤ 0.05) (Fig. 5B). Given that calpain activity is calcium dependent, and because of the fact that mutant STHdhQ111 cells exhibit at basal levels enhanced NMDAR activation (Gines et al., 2003; Seong et al., 2005), we next evaluated whether p25 accumulation might be a consequence of increased NMDAR-mediated calcium influx. To test this hypothesis, mutant huntingtin cells were treated with MK-801, a specific NMDAR antagonist that blocks calcium influx associated to NMDAR activation or with the calcium chelator EGTA. Immunoblot analysis revealed that p25 levels in mutant cells treated with MK-801 or EGTA were lower than those observed in untreated mutant cells (∼1.2-fold decrease; p ≤ 0.01), although still significantly higher compared with wild-type cells (∼2.4-fold increase; p ≤ 0.001) (Fig. 5C). Importantly, this reduction in p25 levels parallels the increase of p35 levels (∼1.1-fold increase; p ≤ 0.01) (Fig. 5C). These data suggest that endogenous activation of NMDAR in mutant striatal cells is not the major contributor to p25 accumulation.
Figure 5.

Increased p25/p35 levels in mutant STHdhQ111 striatal cells. A, Representative Western blot showing levels of p35, p25, and α-tubulin as a loading control from wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells. B, Representative Western blot showing α-spectrin and SDBP (150–160 kDa) and α-tubulin as a loading control from wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells. C, Representative Western blot showing levels of p35, p25, and α-tubulin as a loading control from wild-type and mutant striatal cells treated with MK-801 25 μm or EGTA 400 μm for 6 h. The histograms represent the relative p35 and p25 levels expressed as fold increase versus wild-type cells. Values are given as mean ± SD of three independent experiments. ***p < 0.001 mutant cells versus wild-type cells; ##p < 0.01, ###p < 0.001 treated versus vehicle-treated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test. D–F, Representative Western blot showing p35 and p25 levels and α-tubulin as a loading control from wild-type and mutant striatal cells treated with NMDA 500 μm alone (D), SKF38393 100 μm alone (E), or pretreated with NMDA before SKF38393 exposure (F). Total extracts were obtained at different time periods (5, 15, 30, and 60 min) after treatment. (n = 3 independent experiments).
We next evaluated whether either costimulation of NMDAR and D1R or single activation induced higher accumulation of p25. Surprisingly, compared with untreated cells, all treatments failed to increase the accumulation of p25 any further in both wild-type and mutant striatal cells (Fig. 5D–F). These findings suggest that mutant huntingtin expression is sufficient to induce the conversion of p35 into p25 and support the idea that accumulation of p25 in parallel with higher Cdk5 activity may contribute to the increased vulnerability of mutant STHdhQ111 striatal cells to NMDA and D1R activation.
Increased Cdk5 activity and tau phosphorylation in STHdhQ111 mutant cells
Our previous results have demonstrated higher phosphorylation of Cdk5 induced by D1R activation alone or combined by pre-exposure to NMDA in mutant compared with wild-type striatal cells. To determine whether increased Cdk5 phosphorylation and accumulation of p25 was associated to higher Cdk5 activation, kinase activity was determined in vitro by using Cdk5 immunoprecipitated from untreated or treated (SKF38393 alone or combined with NMDA) wild-type STHdhQ7 and mutant STHdhQ111 striatal cells. Our results showed that at basal conditions mutant striatal cells exhibited a significant increase in Cdk5 activity compared with wild-type striatal cells (∼2.5-fold; p ≤ 0.01) (Fig. 6A). Interestingly, whereas NMDA exposure before SKF38393 treatment did not further increase Cdk5 activity in wild-type striatal cells compared with SKF38393 alone (∼1.2-fold and ∼1.3-fold, respectively; p = 0.53), in mutant striatal cells NMDA significantly potentiates D1R-induced Cdk5 activity (∼1.9-fold, p ≤ 0.001 and ∼2.8-fold, p ≤ 0.001, respectively) (Fig. 6A). In support of enhanced Cdk5 activity in mutant striatal cells, Western blot analysis using antibodies specific for phosphorylated tau (PHF-1, Ser396 and Ser404) showed increased tau phosphorylation in basal mutant striatal cells compared with wild-type cells without changes on total tau levels (∼1.5-fold increase; p ≤ 0.05). Consistent with our results on Cdk5 activity, the levels of tau phosphorylation in wild-type cells pretreated with NMDA were similar than those detected in wild-type cells treated with SKF38393 alone (∼1.2-fold increase and ∼1.3-fold increase, respectively; p = 0.97) (Fig. 6B). In contrast, in mutant cells, higher levels of tau phosphorylation were detected when cells were pre-exposed to NMDA (∼2.6-fold and ∼2.1-fold, respectively; p ≤ 0.05) (Fig. 6B). These data suggest that mutant huntingtin induces enhanced Cdk5 activity that results in increased phosphorylation of tau, an effect that is exacerbated by activation of D1R alone or in combination with NMDAR.
Figure 6.
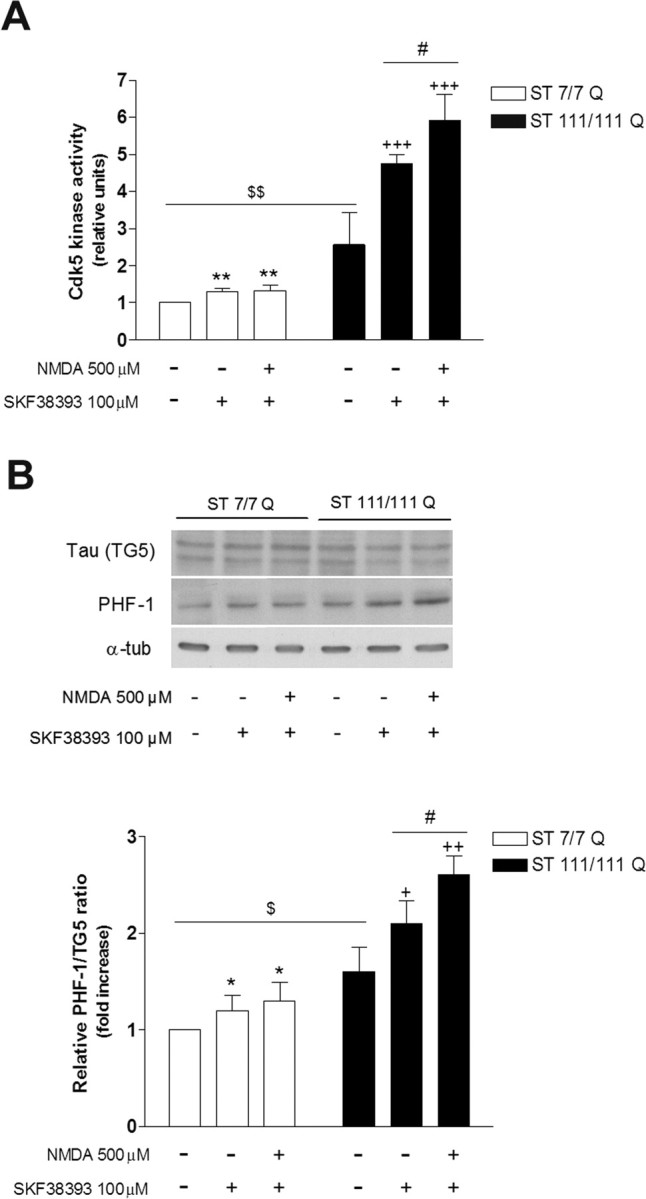
Mutant STHdhQ111 striatal cells exhibit increased Cdk5 activity associated with higher tau phosphorylation. Wild-type (ST7/7Q) and mutant (ST111/111Q) striatal cells were treated with SKF38393 (100 μm) or pretreated with NMDA (500 μm) before SKF38393 exposure, and total cell extracts were obtained 30 min after treatment. A, In vitro kinase assay of immunoprecipitated Cdk5 from 500 μg of whole-cell lysates was performed using H1 peptide as substrate. The histogram represents the Cdk5 kinase activity in relative units corrected for the protein levels of Cdk5 for each sample and refereed to control (vehicle-treated) wild-type cells. Values are given as mean ± SD of three independent experiments. **p < 0.01 treated versus vehicle-treated wild-type cells; +++p < 0.001 treated versus vehicle-treated mutant cells; $$p < 0.01 vehicle-treated mutant cells versus vehicle-treated wild-type cells; #p < 0.05 NMDA plus SKF38393-treated mutant cells versus SKF38393-treated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test. B, Representative Western blot showing total Tau (TG5) levels, phosphorylated Tau (PHF-1) levels, and α-tubulin as a loading control from wild-type and mutant striatal cells. The histogram represents the relative PHF-1/TG5 ratio expressed as fold increase versus wild-type cells. Values are given as mean ± SD of three independent experiments. *p < 0.05 treated versus vehicle-treated wild-type cells; +p < 0.05, ++p < 0.01 treated versus vehicle-treated mutant cells; $p < 0.05 vehicle-treated mutant cells versus vehicle-treated wild-type cells; #p < 0.05 NMDA plus SKF38393-treated mutant cells versus SKF38393-treated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test.
Roscovitine ameliorates cell death evoked by SKF38393 and NMDA treatment in STHdhQ111 cells
The previous data suggest that Cdk5 may act as a down-stream mediator of NMDA and SKF38393 neurotoxicity in striatal cells. To test this hypothesis, we next evaluated the effect of roscovitine, a Cdk5 inhibitor, on NMDA and SKF38393-induced cell death. We found that roscovitine was able to significantly reverse the cell death induced by NMDA, SKF38393, or both in either wild-type and mutant huntingtin cells (Fig. 7). These findings demonstrate that a blockade of Cdk5 by roscovitine correlates with a reduction of cell death induced by NMDAR and D1R activation in striatal cells and supports the involvement of aberrant Cdk5 activation in the increased vulnerability of mutant STHdhQ111 striatal cells to NMDA and SKF38393 treatment.
Figure 7.
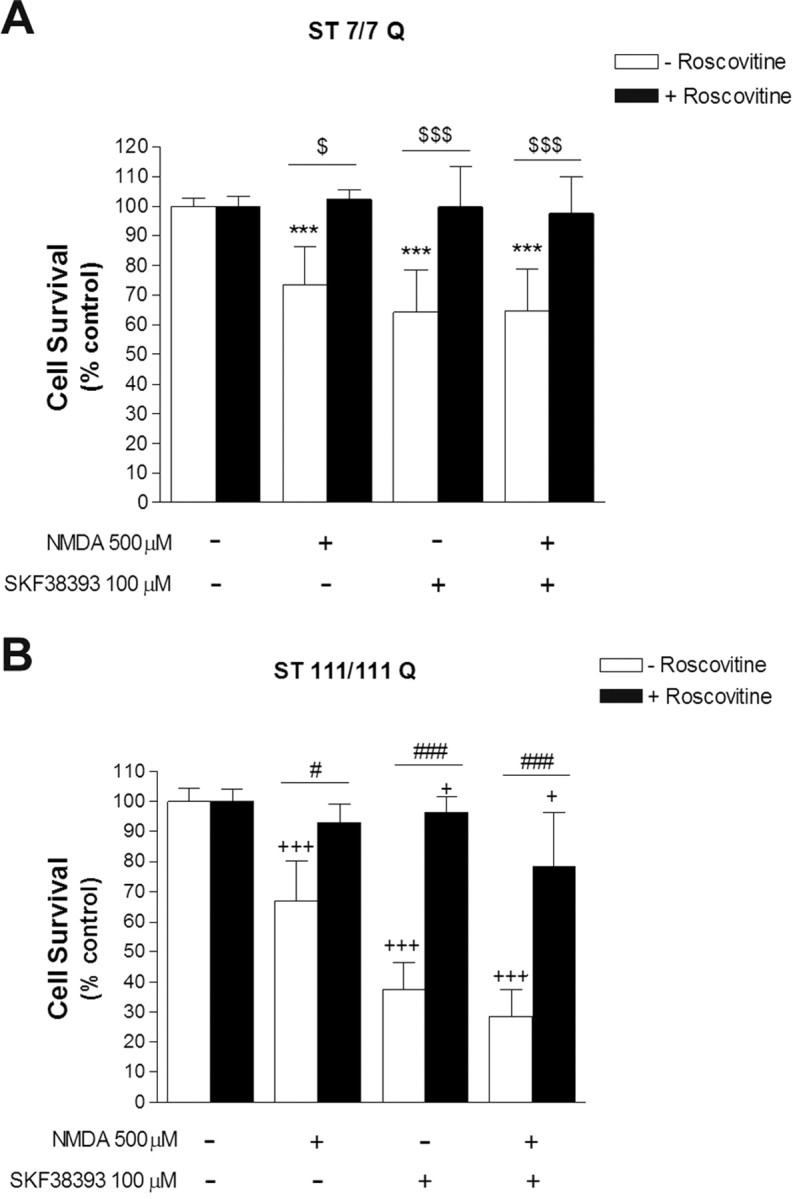
Inhibition of Cdk5 kinase activity by roscovitine ameliorates STHdhQ111 cells from cell death induced by SKF38393 and NMDA treatment. A, B, Wild-type (ST7/7Q; A) and mutant (ST111/111Q; B) striatal cells pretreated (1 h) with the Cdk5 inhibitor roscovitine (20 μm) were exposed to NMDA (500 μm) or SKF38393 (100 μm) alone, or preincubated with NMDA before SKF38393 exposure. Cell survival was evaluated 24 h later by scoring the percentage of Hoechst-stained nuclei. Forty fields were counted per condition per experiment, comprising at least 30–40 cells. Results are expressed as percentage of control (vehicle-treated) cells, and data are the mean ± SD of three independent experiments performed in triplicate. ***p < 0.001 treated versus vehicle-treated wild-type cells; $p < 0.05, $$$p < 0.001 roscovitine-treated versus roscovitine-untreated wild-type cells; +p < 0.05, +++p < 0.001 treated versus vehicle-treated mutant cells; #p < 0.05,###p < 0.001 roscovitine-treated versus roscovitine-untreated mutant cells as determined by one-way ANOVA followed by Bonferroni's multiple comparison test.
Cdk5 pathway is deregulated in HdhQ111 knock-in mice and in HD human brain
To determine the relevance of deregulation of Cdk5 pathway in an in vivo HD model, we next analyzed whether Cdk5 alteration was also detected in the striatum of mutant HdhQ111 mice. We first analyzed the levels of Cdk5 in striatal brain extracts obtained from mutant HdhQ111 mice and wild-type HdhQ7 littermate mice at 9 months of age. In agreement with our results on mutant striatal cells, immunoblot analysis revealed a significant increase in the ratio p-Tyr15 Cdk5/Cdk5 in the striatum of mutant HdhQ111 mice compared with wild-type HdhQ7 mice (∼2-fold; p ≤ 0.05) (Fig. 8A). Moreover, decreased p35 expression (∼2-fold; p ≤ 0.01) and higher accumulation of p25 forms (∼2-fold; p ≤ 0.01) were detected in mutant HdhQ111 striatal extracts compared with wild-type HdhQ7 striatal extracts (Fig. 8A). Finally, to determine whether increased Cdk5 phosphorylation and p25 expression involved higher Cdk5 activity, we next examined the levels of tau phosphorylation. Western blot analysis revealed a significant increase of tau phosphorylation (PHF-1) in mutant HdhQ111 striatal brain compared with wild-type HdhQ7 striatal brain (∼2-fold; p ≤ 0.01) (Fig. 8B). These results suggest that mutant huntingtin expression enhances Cdk5 activity in vivo by increasing Cdk5 phosphorylation and the conversion of p35 to p25, which results in aberrant phosphorylation of tau protein. Finally, we also investigated whether aberrant Cdk5 pathway was manifested in HD human brain. Western blot analysis confirmed a decrease of Cdk5 in human striatal brain samples from HD patients compared with control brains (Fig. 8C). Unfortunately, we failed to detect Cdk5 phosphorylation (Tyr15) in human brain extracts probably because of the sensitivity of this phosphorylation site to postmortem intervals (range from 4 to 15 h for HD samples and from 4 to 23 h for control samples). Importantly, an increase of p25/p35 levels were also detected in HD human striatal brains compared with control striatal brains. Thus, HD striatal brains exhibited a significant decrease in p35 expression (∼3-fold; p ≤ 0.05) and increased p25 levels (∼4-fold; p ≤ 0.001) (Fig. 8C) compared with control striatal brains. These data support the view that aberrant Cdk5 pathway is also manifested in HD patients and suggest a role of p25/Cdk5 in HD pathology.
Figure 8.

Cdk5 pathway is altered in HD knock-in mice and HD human brain. A, Representative Western blot showing levels of p-Cdk5, Cdk5, p35, p25, and α-tubulin as a loading control from striatal extracts of wild-type HdhQ7 and mutant HdhQ111 mice at 9 months of age. B, Representative Western blot showing levels of total Tau (TG5), phosphorylated Tau (PHF-1), and α-tubulin as a loading control from striatal extracts of wild-type HdhQ7 and mutant HdhQ111 mice at 9 months of age. Values are given as mean ± SD of three independent samples. **p < 0.01 as determined by Student's t test. C, Representative Western blot showing levels of total Cdk5, p35, p25, and α-tubulin as a loading control from human brain striatal samples of control (n = 3) and HD patients (n = 3). Scatter plots represent the relative levels of Cdk5, p35, and p25. The higher value obtained in control brain extracts was set as 1.
Discussion
Degeneration of medium spiny neurons in the striatum is the main hallmark of HD neuropathology. However, the molecular mechanism by which mutant huntingtin toxicity causes such specific cell death remains unknown. Our present work demonstrates that full-length mutant huntingtin enhances the sensitivity of striatal cells to dopamine leading to increased neuronal death. This effect on neurotoxicity is mediated by D1 but not D2 receptors as demonstrated by using specific dopamine-receptor agonists. Thus, the direct activation of D1R by SKF38393 revealed a higher dose–response reduction in cell survival in mutant STHdhQ111 than in wild-type STHdhQ7 cells, an effect that was mostly reverted by the selective D1 antagonist SCH23390. The lack of cell death in the presence of the D2 agonist quinpirole indicates that the neurotoxic effect of dopamine in knock-in mutant striatal cells is mainly associated with D1R activation. These results agree with previous reports indicating the role of D1R in striatal neuronal loss. It has been proposed that dopamine, by acting through activation of D1R and auto-oxidation, causes striatal neurotoxicity by inducing the neuronal expression and activity of inducible nitric oxide synthase (Wersinger et al., 2004). Moreover, dopamine through D1R potentiates glutamate-induced toxicity in cultured striatal cells from YAC128 HD mice (Tang et al., 2007). However, the involvement of D2R in HD pathology cannot be completely ruled out because dopamine via D2 receptors enhances mutant huntingtin aggregation and mitochondrial dysfunction in exon-1 HD mouse models (Charvin et al., 2005; Benchoua et al., 2008). Therefore, it is possible that at early disease stages that is in the absence of mutant huntingtin cleavage and aggregate formation, dopamine toxicity acts primarily via D1R whereas later in the disease progress altered D1 and D2 receptor signaling contribute to HD pathology.
It is well known that glutamate and dopamine receptors regulate striatal neuronal function by interacting and modulating each other. Specifically, an association between NMDAR and D1R has been reported, although the nature of this interaction remains unclear (Scott et al., 2002; Salter, 2003; Cepeda and Levine, 2006; Missale et al., 2006). Here, we have demonstrated that NMDA potentiates D1R-induced cell death in mutant STHdhQ111 cells but not in wild-type STHdhQ7 cells. We have tested toxic NMDA concentrations, which induce similar cell death in wild-type and mutant cells and low NMDA concentrations, which, per se, are not toxic (Xifró et al., 2008). Notably, either toxic or subtoxic NMDA concentrations enhanced the effect in cell death mediated by SKF38393 only in mutant STHdhQ111 cells. These results agree with previous studies showing that glutamate and dopamine act synergistically to induce degeneration of striatal cells in YAC128 HD mouse models (Tang et al., 2007). Altogether, our data suggest that mutant huntingtin alters the dopaminergic and glutamatergic signaling not only by affecting NMDAR and D1R but also by altering the cross-talk between both pathways. Consequently, to gain a better understanding of how mutant huntingtin increases the vulnerability of striatal cells to neurotoxicity, we focused on defining the molecular mechanisms that underlie the toxic effect of NMDAR and D1R activation on striatal cell survival.
Recent studies have identified Cdk5 as mediator of dopaminergic neuronal loss in mouse models of Parkinson's disease because inhibition of Cdk5 reduces neurodegeneration and behavior disturbances in these mouse models (Smith et al., 2003, 2006; Przedborski, 2007). Moreover, it has been demonstrated that Cdk5 can regulate NMDAR signaling by direct phosphorylation of NR2A and NR2B subunits or indirectly via phosphorylation of postsynaptic density-95 (Morabito et al., 2004; Zhang et al., 2008). Therefore, it is reasonable to hypothesize a role for Cdk5 in the increased vulnerability of mutant huntingtin striatal cells to NMDAR and D1R activation.
We have examined two important features of Cdk5 activation related to its pro-apoptotic function: the levels of Tyr15 phosphorylation (Lin et al., 2007) and the accumulation of p25 as a result of calpain-mediated p35 cleavage (Kusakawa et al., 2000; Lee et al., 2000). Our results in knock-in striatal cells define a new role of mutant huntingtin as modulator of the Cdk5 pathway by (1) decreasing Cdk5 expression and increasing the levels of phosphorylated Tyr15-Cdk5 and (2) by increasing the conversion of p35 into p25, which in turn results in enhanced Cdk5 activity. Moreover, we have demonstrated a polyQ-dependent reduction of Cdk5 expression together with an increase in Tyr15-Cdk5 phosphorylation in striatal cells. These data support the idea of a direct role of mutant huntingtin in Cdk5 changes rather than a secondary consequence of mutant huntingtin toxicity. Consistent with the results obtained in the knock-in striatal cell line, we also found deregulation of Cdk5 in HdhQ111 knock-in mice and HD human brain. Our studies demonstrate an increased of p-Tyr15-Cdk5/Cdk5 ratio and a significant accumulation of p25 together with reduced p35 expression in the striatum of HdhQ111 mutant mice and HD human brain. In agreement with these results, Anne et al. (2007) have reported reduced levels of Cdk5 and p35 in HD human brain, although no data on p25 accumulation was reported. These data are relevant because it has been demonstrated a protective role of p35/Cdk5 complexes in HD. Phosphorylation of mutant huntingtin by p35/Cdk5 complexes reduces huntingtin cleavage by caspases resulting in attenuated aggregate formation and toxicity (Luo et al., 2005). Moreover, phosphorylation of wild-type huntingtin by Cdk5 protects striatal cells against DNA damage (Anne et al., 2007). Both reports speculate that loss of p35/Cdk5 function may have detrimental consequences for HD pathology. Therefore, our novel results demonstrating an accumulation of p25 in HD mouse brain and striatal cell models and in HD human brain might be relevant to explain the apparent dual role of Cdk5 in HD pathology. Indeed, it is well established the role of p25 as a cell death inductor (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Thus, neurotoxicity induced by β-amyloid, ischemia, or MPTP causes aberrant Cdk5 activity through the generation and accumulation of p25 (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Furthermore, this truncated p25 form relocalizes Cdk5 activity with the consequent alteration of Cdk5 substrate specificity (Patrick et al., 1999; Ko et al., 2001; Bian et al., 2002). Actually, elevated Cdk5 activity accumulation of p25 and hyperphosphorylated tau have been described in brain of AD patients (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Notably, our studies have identified tau as a downstream target of aberrant Cdk5 activity in HD mouse and striatal cell models. The specific role of tau in HD pathology remains to be elucidated. However, given evidence of impaired axonal trafficking in HD (Truant et al., 2006), it seems reasonable to speculate that altered tau may be involved in the vesicular trafficking deficiencies associated to HD pathology.
Collectively, our results clearly demonstrate that mutant huntingtin deregulates Cdk5 pathway. However, one important question is to determine whether aberrant Cdk5 activity plays a role in the increased sensitivity of mutant huntingtin cells to NMDAR and D1R activation. Indeed, our data indicate that deregulated Cdk5 pathway might be instrumental in the HD neurodegenerative process by increasing the vulnerability of striatal cells to dopamine and glutamate inputs. This conclusion is supported by the following observations: first, SKF38393 induced higher Cdk5 phosphorylation and activity in mutant cells than in wild-type cells. Second, exposure of mutant cells to NMDA before SKF38393 induced higher levels of Cdk5 phosphorylation and activity than in wild-type cells or mutant cells treated with SKF38393 alone. Third, compared with wild-type cells, increased levels of phosphorylated tau were detected in mutant striatal cells treated with SKF38393 alone or pre-exposed to NMDA. Therefore, these data demonstrate that mutant huntingtin increases the sensitivity of striatal cells to dopamine and glutamate inputs by altering a common NMDAR and D1R downstream pathway such is Cdk5. Supporting this conclusion are the findings that the inhibition of Cdk5 activity by roscovitine mainly prevented striatal cell death induced by NMDA, SKF38393, or both. However, because neuronal loss induced by NMDA and SKF38393 cotreatment was not fully prevented by roscovitine in mutant cells, we can speculate that additional mechanism independent of Cdk5 might be involved in the toxicity associated to NMDAR and D1R activation.
Based on these findings, an attractive model of striatal cell death is one in which aberrant Cdk5 activity (Fig. 6) as a result of enhanced Cdk5 phosphorylation at Tyr15 (Fig. 4) and p25 accumulation (Fig. 5) might sensitize mutant huntingtin striatal cells to NMDAR and D1R activation. Therefore, once NMDAR and D1R were activated, both receptors might act coordinately to promote calcium homeostasis deregulation (Cepeda et al., 2001; Starling et al., 2005; Tang et al., 2007) with the consequent mitochondrial depolarization and caspase activation (Zeron et al., 2002, 2004). In addition, as our data demonstrate, the activation of NMDAR may further potentiate D1R-mediated phosphorylation of Cdk5, leading to the generation of p-Cdk5/p25 complexes and enhanced Cdk5 activity. Sustained Cdk5 activity may now target for phosphorylation different substrates such as tau or additional recognized Cdk5 effectors (Gong et al., 2003; Lee et al., 2007) and contribute to striatal cell death in HD.
In conclusion, our results provide evidence that aberrant Cdk5 activity is involved in the increased sensitivity of mutant huntingtin striatal cells to dopamine and glutamate inputs and reveal p25/Cdk5 complexes as potential candidates for future pharmacological intervention in HD (Fig. 9).
Figure 9.

Increased sensitivity of mutant huntingtin striatal cells to glutamate and dopamine receptor activation involves enhanced Cdk5 activity. Hypothetical model to depict the link between neurotoxicity induced by dopamine and glutamate receptor activation and the Cdk5 pathway in HD. Enhanced activation of NMDAR and D1R induced by mutant huntingtin leads to deregulation of calcium homeostasis (Cepeda et al., 2001; Seong et al., 2005; Starling et al., 2005; Tang et al., 2007). Increased intracellular calcium in turn leads to higher calpain activity that results in enhanced cleavage of p35 into p25 (Lee et al., 2000). In addition, as our data demonstrate, activation of NMDAR may potentiate D1R-mediated phosphorylation of Cdk5 at Tyr15, which has been shown to regulate Cdk5-induced neurotoxicity (Lin et al., 2007). The generation of p-Cdk5/p25 complexes implies Cdk5 hyperactivation that is responsible for the phosphorylation of new substrates such as tau protein. Now, hyperphosphorylation of tau in concert with unknown yet down-stream p25/Cdk5 effectors might contribute to striatal cell death and dysfunction and therefore to HD neuropathology. The model is supported by the capacity of roscovitine, a Cdk5 inhibitor, to prevent cellular death induced by NMDAR and D1R activation.
Footnotes
This work was supported by grants from Ministerio de Educación y Ciencia, Centro de Investigaciones Biomédicas en Red sobre Enfermedades Neurodegenerativas, Fundació La Marató de TV3, and HighQ Foundation. P.P. and M.R. are fellows of the HighQ Foundation. We thank Dr. M. Macdonald for the knock-in striatal cell lines and knock-in HD mice and Dr. P Davis for tau antibodies. We are very grateful to Cristina Herranz, Ana Lopez, and M. Teresa Muñoz for technical assistance. We thank Banc de Teixits Neurològics (University of Barcelona, Barcelona, Spain) for human tissue samples. We thank C. A. Saura and members of our laboratory for helpful discussion.
References
- Akashiba H, Ikegaya Y, Nishiyama N, Matsuki N. Differential involvement of cell cycle reactivation between striatal and cortical neurons in cell death induced by 3-nitropropionic acid. J Biol Chem. 2008;283:6594–6606. doi: 10.1074/jbc.M707730200. [DOI] [PubMed] [Google Scholar]
- Anne SL, Saudou F, Humbert S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J Neurosci. 2007;27:7318–7328. doi: 10.1523/JNEUROSCI.1831-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Diguet E, Malgorn C, Gaillard MC, Dufour N, Elalouf JM, Krajewski S, Hantraye P, Déglon N, Brouillet E. Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet. 2008;17:1446–1456. doi: 10.1093/hmg/ddn033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian F, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KK, Walker LC. Axonopathy, tau abnormalities and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol. 2002;446:257–266. doi: 10.1002/cne.10186. [DOI] [PubMed] [Google Scholar]
- Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, Pearse D, Kuan CY, Vaudano E, Behrens A, Wagner E, Flavell RA, Davis RJ, Herdegen T. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- Centeno C, Repici M, Chatton JY, Riederer BM, Bonny C, Nicod P, Price M, Clarke PG, Papa S, Franzoso G, Borsello T. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007;14:240–253. doi: 10.1038/sj.cdd.4401988. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;333:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernández J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Charvin D, Vanhoutte P, Pagès C, Borrelli E, Borelli E, Caboche J. Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci U S A. 2005;102:12218–12223. doi: 10.1073/pnas.0502698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Biel N, Camins A, Pelegrí C, Vilaplana J, Pallàs M, Canudas AM. 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci Lett. 2007;421:77–81. doi: 10.1016/j.neulet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol. 2004;14:390–394. doi: 10.1016/j.conb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Cyr M, Sotnikova TD, Gainetdinov RR, Caron MG. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 2006;20:2541–2543. doi: 10.1096/fj.06-6533fje. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Vonsattel JP, Richardson EP., Jr Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol. 1988;47:516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- Dhariwala FA, Rajadhyaksha MS. An unusual member of the cdk family: cdk5. Cell Mol Neurobiol. 2008;28:351–369. doi: 10.1007/s10571-007-9242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME. Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem. 2003;278:50514–50522. doi: 10.1074/jbc.M309348200. [DOI] [PubMed] [Google Scholar]
- Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003;38:33–46. doi: 10.1016/s0896-6273(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci. 2001;21:6758–6771. doi: 10.1523/JNEUROSCI.21-17-06758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolitic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim HS, Lee SJ, Kim KT. Stabilization and activation of p53 induced by Cdk5 contributes to neuronal cell death. J Cell Sci. 2007;120:2259–2271. doi: 10.1242/jcs.03468. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lin H, Lin TY, Juang JL. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death Differ. 2007;14:607–615. doi: 10.1038/sj.cdd.4402033. [DOI] [PubMed] [Google Scholar]
- Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Gines S, Gusella JF, Wheeler VC. Huntington's disease. Neuromolecular Med. 2003;4:7–20. doi: 10.1385/NMM:4:1-2:7. [DOI] [PubMed] [Google Scholar]
- Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- Missale C, Fiorentini C, Busi C, Collo G, Spano PF. The NMDA/D1 receptor complex as a new target in drug development. Curr Top Med Chem. 2006;6:801–808. doi: 10.2174/156802606777057562. [DOI] [PubMed] [Google Scholar]
- Morabito MA, Sheng M, Tsai LH. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pérez-Navarro E, Canals JM, Ginés S, Alberch J. Cellular and molecular mechanisms involved in the selective vulnerability of striatal projection neurons in Huntington's disease. Histol Histopathol. 2006;21:1217–1232. doi: 10.14670/HH-21.1217. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Peroxiredoxin-2 links Cdk5 to neurodegeneration. Nat Med. 2007;13:907–909. doi: 10.1038/nm0807-907. [DOI] [PubMed] [Google Scholar]
- Salter MW. D1 and NMDA receptor hook up: expanding on an emerging theme. Trends Neurosci. 2003;26:235–237. doi: 10.1016/S0166-2236(03)00081-X. [DOI] [PubMed] [Google Scholar]
- Scott L, Kruse MS, Forssberg H, Brismar H, Greengard P, Aperia A. Selective up-regulation of dopamine D1 receptors in dendritic spines by NMDA receptor activation. Proc Natl Acad Sci U S A. 2002;99:1661–1664. doi: 10.1073/pnas.032654599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RH, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Mount MP, Shree R, Callaghan S, Slack RS, Anisman H, Vincent I, Wang X, Mao Z, Park DS. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2006;26:440–447. doi: 10.1523/JNEUROSCI.2875-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling AJ, André VM, Cepeda C, de Lima M, Chandler SH, Levine MS. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington's disease. J Neurosci Res. 2005;82:377–386. doi: 10.1002/jnr.20651. [DOI] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Truant R, Atwal R, Burtnik A. Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem Cell Biol. 2006;84:912–917. doi: 10.1139/o06-181. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Chen J, Sidhu A. Bimodal induction of dopamine-mediated striatal neurotoxicity is mediated through both activation of D1 dopamine receptors and autoxidation. Mol Cell Neurosci. 2004;25:124–137. doi: 10.1016/j.mcn.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, Joyner AL, MacDonald ME. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- Xifró X, García-Martínez JM, Del Toro D, Alberch J, Pérez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J Neurochem. 2008;105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Zhang S, Edelmann L, Liu J, Crandall JE, Morabito MA. Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J Neurosci. 2008;28:415–424. doi: 10.1523/JNEUROSCI.1900-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]