

Abstract
Although the microglial activation is concomitant to the Alzheimer's disease, its precise role (neuroprotection vs neurodegeneration) has not yet been resolved. Here, we show the existence of an age-dependent phenotypic change of microglial activation in the hippocampus of PS1xAPP model, from an alternative activation state with Aβ phagocytic capabilities (at 6 months) to a classic cytotoxic phenotype (expressing TNF-α and related factors) at 18 months of age. This switch was coincident with high levels of soluble Aβ oligomers and a significant pyramidal neurodegeneration. In vitro assays, using astromicroglial cultures, demonstrated that oligomeric Aβ42 and soluble extracts from 18-month-old PS1xAPP hippocampus produced a potent TNF-α induction whereas monomeric Aβ42 and soluble extract from 6- or 18-month-old control and 6-month-old PS1xAPP hippocampi produced no stimulation. This stimulatory effect was avoided by immunodepletion using 6E10 or A11. In conclusion, our results show evidence of a switch in the activated microglia phenotype from alternative, at the beginning of Aβ pathology, to a classical at advanced stage of the disease in this model. This change was induced, at least in part, by the age-dependent accumulation of extracellular soluble Aβ oligomers. Finally, these cytotoxic activated microglial cells could participate in the neuronal lost observed in AD.
Keywords: Alzheimer, transgenic model, neuroinflammation, hippocampus Aβ plaques, oligomers, hippocampus
Introduction
As proposed by the inflammation hypothesis of Alzheimer's disease (AD), the neurodegenerative process could be exacerbated by a chronic inflammatory response to β-amyloid (Aβ) peptides (for review, see Griffin et al., 1998; Wyss-Coray, 2006; Heneka and O'Banion, 2007). Secondary to Aβ accumulation, there is an inflammatory response characterized by activated microglia and reactive astrocytes. Activated inflammatory cells could mediate neuronal damage by producing toxic products, such as inflammatory cytokines, excitatory amino acids, reactive oxygen intermediates and other factors (Mrak and Griffin, 2005; Craft et al., 2006; Ralay Ranaivo et al., 2006; Zipp and Aktas, 2006). This potential cytotoxic effect was further emphasized by clinical studies demonstrating that the symptoms of AD could be attenuated by nonsteroidal antiinflammatory drugs (Aisen, 2000; McGeer and McGeer, 2007). However, recent trials have not confirmed this positive effect (Aisen et al., 2003; Reines et al., 2004).
Although a deleterious inflammatory reaction could indeed mediate the neurodegeneration in AD, a completely different possibility is just beginning to be considered, supporting a trophic, proregenerative role of the inflammatory response. Activated glial cells are also capable to secrete anti-inflammatory cytokines (Butovsky et al., 2006), as well as neuroprotective factors that may protect against AD pathology (Streit, 2005). In this sense, vaccination against the Aβ peptides led to activation of microglia and successfully decreased amyloid load (Wilcock et al., 2003, 2004a,b). Similarly, stimulating the immune system with lypopolisaccharide (LPS) led to a reduction in Aβ plaques (DiCarlo et al., 2001; Herber et al., 2004). Nevertheless, at present little information is known about the balance between procytotoxic and anticytotoxic events occurring in AD or about the cellular and temporal induction of inflammatory cascade by Aβ.
Transgenic (tg) mice models are widely used to study AD pathology. We have previously characterized a double PS1xAPP tg mouse. These transgenic mice developed early (3–4 months) hippocampal Aβ plaques (Blanchard et al., 2003; Ramos et al., 2006; Caballero et al., 2007). In parallel with Aβ deposition, we also demonstrated the existence of degeneration of a particular subset of hippocampal GABAergic neurons (O-LM and HIPP cells; Ramos et al., 2006). However, despite the age-dependent accumulation of extracellular Aβ (Blanchard et al., 2003; Ramos et al., 2006; Caballero et al., 2007), no significant pyramidal degeneration was detected until 17–18 months of age (Schmitz et al., 2004; Ramos et al., 2006; unpublished results). Thus, it is possible that in the AD tg models the Aβ pathology could be attenuated until relatively old ages. In this work, we determined the in vivo inflammatory response in the hippocampus of PS1xAPP tg mice from a wide age range (from 2 to 18 months). At early ages (6 months), we have observed the activation of the microglial cells to an alternative phenotype, exclusively, surrounding the Aβ plaques. However, at 18 months of age, expanded microglial activation throughout all hippocampal layers displaying a classic cytotoxic phenotype was observed. Finally, we also investigated the reasons that could determine this age-dependent microglial phenotypic change.
Materials and Methods
Transgenic mice.
The generation and initial characterization of the PS1M146L (PS1) and PS1xAPP751sl (PS1xAPP) tg mice have been reported previously (Blanchard et al., 2003). PS1 tg mice (C57BL/6 background) overexpressed the mutated PS1M146L form under the control of the HMGCoA-reductase promoter. PS1xAPP double tg mice (C57BL/6 background) were generated by crossing homozygotic PS1 tg mice with heterozygotic Thy1-APP751SL mice (all tg mice were provided by Transgenic Alliance-IFFA-Credo). Mice represented filial generation 10–15 (F10–F15) offspring of heterozygous tg mice. Only male mice were used in this work. Age-matched non-transgenic male mice of the same genetic background (C57BL/6) were used as controls (WT).
Anesthetized mice were killed by decapitation, and both hippocampi were dissected, frozen in liquid N2, and stored at −80°C until use. All animal experiments were performed in accordance with the guidelines of the Committee of Animal Research of the University of Seville (Spain) and the European Union Regulations.
RNA and total protein extraction.
Total RNA was extracted using the Tripure Isolation Reagent (Roche) as described previously (Ramos et al., 2006; Caballero et al., 2007). The contaminating DNA in the RNA samples was removed by incubation with DNAase (Sigma-Aldrich) and confirmed by PCR analysis of total RNA samples prior reverse transcription (RT). After isolation, the integrity of the RNA samples was assessed by agarose gel electrophoresis. The yield of total RNA was determined by measuring the absorbance (260 of 280 nm) of ethanol-precipitated aliquots of the samples. The recovery of RNA was comparable in all groups (1.2–1.5 μg/mg tissue).
The protein pellets, obtained using the Tripure Isolation Reagent, were resuspended in 4% SDS and 8 m urea in 40 mm Tris-HCl, pH 7.4, and rotated overnight at room temperature (Ramos et al., 2006; Caballero et al., 2007).
Retrotranscription and real-time RT-PCR.
The retrotranscription was done using random hexamers, 3 μg of total RNA as template and High-Capacity cDNA Archive Kit (Applied Biosystems) following the manufacturer recommendations (Ramos et al., 2006; Caballero et al., 2007). For real time RT-PCR, each specific gene product was amplified using commercial Taqman probes, following the instruction of the manufacturer (Applied Biosystems), using an ABI Prism 7000 sequence detector (Applied Biosystems). For each assay, a standard curve was first constructed, using increasing amounts of cDNA. In all cases, the slope of the curves indicated optimal PCR conditions (slope 3.2–3.4). The cDNA levels of the different mice were determined using two different housekeepers [i.e., glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin]. The amplification of the housekeepers was done in parallel with the gene to be analyzed. Similar results were obtained using both housekeepers. Thus, the results were normalized using only the GAPDH expression.
Independently of the gene analyzed, the results were always expressed using the comparative Ct method, after the Bulleting number 2 from Applied Biosystems. As a control condition, we selected 6-month-old WT mice. In consequence, the expression of all tested genes, for all ages and mice types, was referenced to the expression levels observed in 6-month-old WT mice.
Peptide preparation.
To prepare the Aβ42 peptides, we allowed synthetic lyophilized Aβ1–42 peptide (human sequence; AnaSpec) to equilibrate, at 20−23°C, for 30 min before it was resuspended and diluted to 1 mm in 1,1′,1“,3,3′,3”-hexafluoro-2-propanol. After evaporation, peptide films were dried in a Speed Vacuum and stored at −40°C. Peptide films were resuspended to 5 mm in dimethyl sulfoxide (DMSO) for 10 min. To form the ADDLs (Lambert et al., 2001), we diluted the 5 mm DMSO solution to 100 μm in cold PBS, vortexed for 30 s, and incubated overnight at 4°C. Before use, the Aβ-PBS solution was further diluted in culture media. The presence of ADDLs was tested by Western blots using 6E10 (data not shown).
To form the monomers, immediately before use, we diluted the 5 mm DMSO solution in PBS (to a final concentration of 100 μm), followed by ultrafiltration through 5 kDa cutoff device (Vivaspin 2; Sartorius Biolab Products). The presence of the monomeric Aβ42 peptide (Mr 4.5 kDa) was verified by Western blots (data not shown).
Soluble protein extraction.
The soluble fractions (S1) were obtained by ultracentrifugation of the homogenates as described previously (Kayed et al., 2003). Briefly, tissue samples were homogenized (using a Teflon-glass homogenizer) in cold PBS [containing a mixture of protease inhibitors (Sigma-Aldrich)] and ultracentrifuged (Optima MAX Preparative Ultracentrifuge; Beckman Coulter) at 120,000 × g, 4°C, during 60 min. Immediately after centrifugation, the samples were aliquoted and stored at −81°C until use. The protein content in the soluble fractions was determined by Lowry.
Western blot and dot blot.
Western blots were performed as described previously (Araujo et al., 1996). Briefly, 20 μg of protein from the different samples were loaded on 16% SDS-Tris-Tricine-PAGE and transferred to nitrocellulose (Hybond-C Extra; Amersham). After blocking, the membranes were incubated overnight, at 4°C, with the appropriate antibody (monoclonal 6E10, Sigma-Aldrich; dilution 1:2000). The membranes were then incubated with anti-mouse horseradish-peroxidase-conjugated secondary antibody (Dako) at a dilution of 1:8000. The blots were developed using the ECL-plus detection method (Amersham).
Dot-blots were done as described previously (Araujo et al., 1996; Lambert et al., 2007). One microgram of protein from the different soluble fractions was directly applied to dry nitrocellulose in a final volume of 2 μl. Blots were air-dried, blocked for 1 h, and incubated overnight at 4°C, with either Nu-1 (courtesy of Dr. W. Klein, Northwestern University, Evanston, IL; 1 μg/ml) or A11 (1:5000 dilution; Biosource) antibodies. After the incubation, the blots were washed and visualized as described above. For quantification, the scanned (Epson 3200) images were analyzed using PCBAS program. In each experiment, the intensity of dots from WT mice were averaged and considered as background of the corresponding age group. Data were always normalized by the specific signal observed in 6-month-old PS1xAPP group.
Tissue preparation.
After deep anesthesia with sodium pentobarbital (60 mg/kg), 2-, 4-, 6-, 12-, and 18-month-old WT and PS1xAPP tg male mice were perfused transcardially with 0.1M PBS, pH 7.4 followed by 4% paraformaldehyde, 75 mm lysine, 10 mm sodium metaperiodate in 0.1 m phosphate buffer (PB), pH 7.4. Brains were then removed, postfixed overnight in the same fixative at 4°C, cryoprotected in 30% sucrose, sectioned at 40 μm thickness in the coronal plane on a freezing microtome and serially collected in wells containing cold PBS and 0.02% sodium azide. Each experiment was composed of 3–6 sets of animals (each one containing one WT and one PS1xAPP tg mice). All animal experiments were approved by the Committee of Animal Use for Research of the Malaga University (Spain) and the European Union Regulations.
Immunohistochemistry.
Coronal free-floating sections (40 μm thick) from WT and PS1xAPP hippocampus were first treated with 3% H2O2/3% methanol in PBS and with avidin-biotin Blocking Kit (Vector Labs). For single immunolabeling, sections were incubated overnight at room temperature with one of the following primary antibodies: mouse monoclonal anti-Aβ 6E10 (1:1500 dilution; Sigma), rat monoclonal anti-CD11b (1:150,000; Serotec), chicken polyclonal anti-GFAP (1:10,000; Dako), hamster monoclonal anti-CD3 (1:100; BD PharMingen), rat monoclonal anti-TNFalpha (1:100; Abcam), rabbit polyclonal anti-iNOS (1:1000; Transduction Laboratories), goat polyclonal anti-IL4 (1:250 dilution; SantaCruz), and goat polyclonal anti-AMCase (YM-1; 1:100 dilution; SantaCruz). The tissue-bound primary antibody was detected by incubating with the corresponding biotinylated secondary antibody (1:500 dilution; Vector Laboratories), and then followed by streptavidin-conjugated horseradish peroxidase (Sigma-Aldrich), diluted 1:2000. The peroxidase reaction was visualized with 0.05% 3-3′-diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich), 0.03% nickel ammonium sulfate, and 0.01% hydrogen peroxide in PBS. Some immunolabeled sections were then incubated for 3 min in a solution of 20% Congo red. Sections were then mounted on gelatin-coated slides, air dried, dehydrated in graded ethanols, cleared in xylene and coverslipped with DPX (BDH) mounting medium. Specificity of the immune reactions was controlled by omitting the primary antiserum.
For double immunofluorescence labeling (GFAP-6E10, GFAP-iNOS, GFAP-IL4, or CD3-IL4), sections were first sequentially incubated with the primaries antibodies (see antibodies above listed), followed by the corresponding Alexa488/Alexa568-conjugated secondary antibodies (1:1000 dilution, Invitrogen) or by biotinylated secondary antibody (1:500; Vector Laboratories) and streptavidin-conjugated Alexa488/568 (1:2000 dilution; Invitrogen). For double CD11b-Thioflavin-S, YM1-Tomato lectin, 6E10-Tomato lectin or IL4-Tomato lectin, fluorescence labeling, sections were first incubated with the primary antibody followed by Alexa568 or Alexa488 conjugated secondary antibody. Then, sections were washed and processed for thioflavin-S staining (see below) or incubated for 1 h with a solution of 5 μg/ml biotinylated Tomato lectin (Sigma) followed by streptavidin-conjugated Alexa 568 (1:1000; Invitrogen). Sections were mounted onto gelatin-coated slides, coverslipped with 0.01 m PBS containing 50% glycerin and 2.5% triethylenediamine and then examined under a confocal laser microscope (Leica TCS-NT).
Thioflavin S staining.
Free-floating sections were incubated for 5 min with 0.015% Thio-S (Sigma) in 50% ethanol, and then washed in 50% ethanol, mounted onto gelatin-coated slides and coverslipped with 0.01M PBS containing 50% glycerin and 2.5% triethylenediamine.
Plaque loading and plaque size distribution.
Hippocampal 6E10 immunostaining from 2-, 4-, 6-, 12-, and 18-month-old PS1xAPP mice was observed under a Nikon Eclipse 50i microscope using a 4× objective and images acquired with a Nikon DS-5M high-resolution digital camera. The camera settings were adjusted at the start of the experiment and maintained for uniformity. Digital images (seven sections per mouse and six mice per age group) were analyzed using Visilog 6.3 analysis program (Noesis). The plaque area within the hippocampus was identified by bright-level threshold, the level of which was maintained throughout the experiment for uniformity. The gray-scale image was converted to a binary image with plaque and hippocampal field areas identified. Plaque loading was defined as percentage of total hippocampal area stained for Aβ, excluding principal cell layers intracellular labeling that was removed by manual editing. The hippocampal area in each 4×image was manually outlined. The plaque loading (percentage) for each tg mouse was estimated and defined as (sum plaque area measured/sum hippocampal area analyzed) ×100. The sums were taken over all slides sampled and a single plaque burden was computed for each mouse. The mean and SD of the plaque loading were determined using all the available data. Quantitative comparisons were performed on sections processed at the same time.
For β-amyloid plaque morphometric analysis (surface area), three coronal sections immunostained with 6E10 from 6- (n = 3), 12 (n = 3) and 18-month-old (n = 3) PS1xAPP mice were analyzed using the nucleator method with isotropic probes by the NewCast software package from Olympus sterological system. Hippocampal CA1 subfield (9 sections per age) was analyzed using a counting frame of 7154.7 μm2 and step lengths of 299.55 × 225.54 μm. For individual plaque measurement we used the 40× objective. Number of plaques/mm2 falling into four surface categories (ranging from <200 μm2 to >2000 μm2) was calculated. Each analysis was done by a single examiner blinded to sample identities.
Stereology.
Cresyl-violet stained CA1 principal cell nuclei belonging to 6-, 12-, and 18-month-old WT and PS1xAPP mice (n = 6/group/age; 10–15 sections per animal) were quantified according to the optical fractionator method, using an Olympus BX51 microscope (Olympus), interfaced with a computer and a color JVC digital videocamera. The CAST-Grid software package (Olympus) generated sampling frames with a known area (aframe) and directed the motorized X-Y stage (Prior Proscan; Prior Scientific Instruments), and a microcator (MT12; Heidenheim), which monitored the movements in the z-axis with a resolution of 0.5 μm. The number of neurons was quantified in every seventh section (with a distance of 280 μm) through the entire anteroposterior extent of the hippocampus (between −0.94 mm anterior and 3.64 mm posterior to Bregman according to the atlas of Paxinos and Watson). This selection criteria prevented counting neurons from contiguous sections. CA1 subfield was defined using a 10× objective and the number of principal cells was counted using a 100×/1.35 objective. Each counting frame was 2342.8 μm2. We used the optical 3 μm from the upper surfaces as look-up, and those 3–13 μm from the surfaces as reference sections. The software calculated the estimated total number of cresyl-violet stained nuclei in the CA1 region using the optical fractionator formula (West et al., 1991; Schmitz and Hof, 2005), N = 1/bsf.1/ssf.1/asf.1/hsf.ΣQ −, where bsf is the block sampling fraction, ssf represents the section sampling fraction, asf is the area sampling fraction, which is calculated by dividing the area sampled with the total area of the layer, hsf stands for the height sampling fraction, which is calculated by dividing the height sampled (10 μm in this study) with the section thickness, and ΣQ − is the total count of nuclei sampled for the entire layer. The precision of the individual estimations is expressed by the coefficient of error (CE) (Gundersen et al., 1999) and here we have estimated the total CE (CE group value) that was calculated using the CEs in each individual animal. The CEs ranged between 0.03 and 0.07.
The numerical density (Nv) of activated microglial cells (number of cells per mm3) was determined in the CA1 subfield using the NewCast Grid Stereological System from Olympus. For each animal from 6- (n = 3) and 18- (n = 3) month-old PS1xAPP, activated microglial cells were quantified with the optical dissector method in two slices immunostained with anti-CD11b at −1.82 mm and 2.30 mm from the Bregman according to the atlas of Paxinos and Watson. Each section was analyzed using a systematically random manner, the counting frame was 29,031 μm2 and step lengths of 121.06 × 91.15 μm. CA1 subfield was defined using a 4× objective and the number of activated microglia cells was counted using a 100×/1.35 objective. We used the optical 3 μm from the upper surfaces as look-up, and that 3–13 μm from the surface as references sections and optical dissector height was 10 μm. The Nv of microglial cells per mm3 was calculated by using the following equation: Nv = ΣQ/Σa × h. Where Q is the number of activated microglial cells per counting box, a is the area of the counting frame, and h is the height of the optical dissector. The average number of cells per mm3 was derived for each section. Animal means were derived by averaging the Nv from two sections from each animal in the CA1 subfield.
Astro-microglial cultures.
Mixed astromicroglial primary cultures were prepared from newborn C57BL/6 mice (1–3 d). Briefly, dissected brains were treated, for 5 min, with trypsin-DMEM-EDTA medium (Biowhittaker, Cambrex). The treatment was stopped using complete DMEM-F12 plus 10% FBS and the cells were mechanically dissociated. After mechanical dissociation, the debris were eliminated by filtration (40 μm; BD Falcon) and the cells were seeded (at a density of 250.000 cells/ml) in DMEM-F12 plus 10% FBS medium (containing glutamine, nonessential amino acids, 1% penicillin-streptomycin and gentamycin) on poly-d-lysine (Sigma-Aldrich) -treated Nunc 12-well plates. The cells were cultured at 37°C, in humidified 5% CO2/95% atmosphere. Medium was replaced every 4 d. After 13–15 d in culture, the mixed glial cultures were treated with different concentrations (ranging from 1 to 50 μm) of ADDLs or monomeric Aβ42, prepared as described above. The cells were then incubated for 3 h. Control cultures (two wells per plate) were treated with equivalent volume of sterilized PBS (negative control) or 1 μg/ml LPS (E. coli O26:B6; Sigma-Aldrich) as positive control. The S1 from the different mice and ages were thawed immediately before use, diluted with DMEM-F12 (without FBS), sterilized by filtration (through 0.22 μm filters; Millipore), and added to the cultures (ranging from 5 to 100 μg of protein). For each experiment, duplicate wells were stimulated under the same experimental condition.
The immunodepletion experiments were done basically as described previously (Araujo et al., 1996). Briefly, 10 μg of protein from S1 fractions from 18-month-old PS1xAPP mice (n = 3) were subjected to three sequential incubations (8–12 h at 4°C) with either 6E10 (2 μg)-Protein G-Sepharose or A11(2 μg)-Protein-A-Sepahrose immunocomplexes. After immunodepletion, the S1 fractions were treated as above. As control, the different S1 fractions were sequentially incubated with either Protein G-Sepharose or Protein A-Sepharose and tested, in parallel experiments, with the immunodepleted samples.
After incubation, the cultures were treated with Tripure and RNA was isolated and retrotranscribed as described above.
Statistical analysis.
Data were expressed as mean ± SD. The comparison between two mice groups (WT and PS1xAPP tg mice) was done by two-tailed t test. For comparison between several age groups, we used one-way ANOVA followed by Tukey's post hoc multiple comparisons test (Statgraphics plus 3.1). The significance was set at 95% of confidence.
Results
Phenotypic characterization of microglial cells in 6- and 18-month-old PS1xAPP
Coincident with the apparition of the extracellular Aβ plaques (4 months of age; data not shown), we observed a remarkable microglial activation in the hippocampus of PS1xAPP mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Numerous CD11b-positive activated microglial cells, showing a marked cellular hypertrophy and thicker and shorter processes, were concentrated surrounding and infiltrating the Aβ plaques (Fig. 1 A; supplemental Figs. 1C, 2, available at www.jneurosci.org as supplemental material). However, at these early ages (4–6 months) most microglial cells no associated to Aβ plaques, which were also CD11b-positive, displayed a quiescent or resting morphology, with small compact somata bearing many long thin ramified processes (Fig. 1 A, a1, inset).
Figure 1.

Phenotypic characterization of the activated microglial cells in PS1xAPP mice hippocampus at 6 and 18 months of age. A, High magnification of CD11b positive microglial cell in 6 (a1) and 18 month (a2) PS1xAPP mice. At 6 months of age (a1), the activated microglial cells were mostly restricted to the Aβ plaques (asterisks) whereas the interplaque microglia displayed a resting morphology (a1, inset). At 18 months of age (a2), both plaque-associated and interplaque microglia displayed an activated morphology. The inset displays an interplaque activated microglial cell. B, C, The expression of classic (B) activation markers of microglial cells and the mRNA expression of genes considered markers of the alternative activation (C) were quantitatively determined (by real time RT-PCR) in 6- and 18-month-old WT and PS1xAPP mice (10 mice per mice group and age). The expression of the different genes was normalized by GAPDH or β-actin with identical results. Data are expressed in reference to 6-month-old WT mice. Significance was analyzed by one-way ANOVA followed by Tukey's test (*p < 0.05). D, The mRNA expression levels of TNF-α and iNOS were quantitatively determined in 6-, 12-, and 18-month-old WT and PS1xAPP hippocampi. For each age and mice group, 10 animals were used. Data (mean ± SD) between mice groups (WT and PS1xAPP) and ages were compared by one-way ANOVA (TNF-α, F (5,54) = 91.42; p < 0.0001; iNOS F (5,54) = 57.03, p < 0.0001) followed by Tukey's test. Significance (p < 0.05) was indicated in the figure. Scale bars: a1, a2, 100 μm; insets, 20 μm.
Microglia could adopt several different phenotypes. The microglial activation by Aβ peptides has been associated with the production of proinflammatory and potentially toxic cytokines (Heneka and O'Banion, 2007). Thus, we quantitatively determined the mRNA expression of proinflammatory factors, including Il-1β; TNF-α and TNF-α related factors (TRAIL and FASL); iNOS; Cox2 and Nox1. As shown (Fig. 1 B), in 6-month-old PS1xAPP mice, none of the classic proinflammatory and cytotoxic markers were significantly altered. Only the expression of Il-1β was moderately increased at this age. This absence of induction in the expression of cytotoxic factors occurred despite clear microglial activation (Fig. 1 A, a1; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, we next tested whether these early activated microglial cells could display a different phenotype. We determined the expression of YM-1 and Arg-1 genes, considered markers of the alternative differentiation in peripheral macrophages (Edwards et al., 2006). Results (Fig. 1 C) demonstrated the existence of a clear induction in the expression of YM-1 mRNA at 6 months of age. The YM-1 positive cells were exclusively located surrounding and infiltrating the Aβ plaques (supplemental Fig. 2A, a1, available at www.jneurosci.org as supplemental material) and were identified as microglial cells, demonstrated by its colocalization with Tomato Lectin (supplemental Fig. 2A, a3–5, available at www.jneurosci.org as supplemental material). However, the expression of Arg-1 was not altered at any age (Fig. 1 C).
However, at this early age, confocal laser microscopy demonstrated the existence of Aβ phagocytosis, as judged by the presence of intracellular 6E10 immunostaining in Tomato Lectin positive cells, surrounding the Aβ plaques (supplemental Fig. 2B, b1–3, available at www.jneurosci.org as supplemental material). Pseudo-3D reconstruction of 6E10-Tomato Lectin labeled confocal images confirmed this observation (supplemental Fig. 2B, b4, available at www.jneurosci.org as supplemental material).
A complete different scenario was observed in 18-month-old PS1xAPP hippocampus (Fig. 1 A). At this old age, we have observed a patent further increase in the expression and density of activated CD11b-positive cells (supplemental Fig. 1A,C, available at www.jneurosci.org as supplemental material). These CD11b positive cells showed a widespread distribution, around plaques and also in areas free of Aβ plaques (Fig. 1 A, a2; supplemental Fig. 1C, c3,c6, available at www.jneurosci.org as supplemental material). Importantly, these inter-Aβ plaques microglial cells also exhibited an activated morphology with marked cellular hypertrophy and thicker and shorter processes (Fig. 1 A, compare a2, inset, a1, inset). This widespread interplaque microglial activation was also verified by quantifying (using stereology) the activated CD11b-positive microglia (morphologically discriminated) in 6 and 18 month PS1xAPP CA1 region. As expected, few interplaque activated microglial cells were detected at 6 months of age whereas, at 18 months, a highly significant increase was observed (199.80 ± 54.60 cell/mm3 vs 1020.51 ± 143.22 cell/mm3, n = 3, for 6- and 18-month-old, respectively; p < 0.05). Furthermore, this widespread activation of microglial cells was coincident with a prominent increase in the expression of TNF-α and TNF-α related factors (TRAIL and FASL), compared with 6-month-old Ps1xAPP mice. Similarly, the expression of iNOS mRNA and, more attenuated Cox2 and Nox1 mRNAs, was also significantly increased at this advanced age (Fig. 1 B). However, the expression of the alternative marker YM-1 remained elevated in 18-month-old tg mice (Fig. 1 C) and YM-1 positive microglia could be observed surrounding the Aβ plaques at this old age (supplemental Fig. 2A, a2, available at www.jneurosci.org as supplemental material).
The expression of TNF-α and iNOS was also quantified in 6-, 12- and 18-month-old PS1xAPP, to more precisely determine the age of the microglial phenotypic switch. As shown (Fig. 1 D), the expression of TNF-α and iNOS was not significantly altered (compared with WT) in 6- and 12-month-old tg mice (although at 12 months a moderate increase in the TNF-α expression was observed). However, a remarkable induction in the expression of both TNF-α and iNOS was detected in 18-month-old PS1xAPP mice (Fig. 1 D). Thus, the classic microglial activation seemed to be predominantly restricted to old ages.
The cellular origin of TNF-α, in 18-month-old PS1xAPP, was then determined by immunocytochemistry (Fig. 2 A, a2; compare a4, a1,a3). TNF-α positive cells, displaying a clear microglial morphology (Fig. 2 A, a5), were located in all hippocampal layers except in the vicinity of Aβ plaques, that appeared clearly immunonegative (Fig. 2 A, a2). In fact, as shown in Figure 2 A–a6, Congo red positive plaques were surrounded by a TNF-α immunonegative perimeter. As described in Figure 1 and supplemental Fig. 1C–c6 (available at www.jneurosci.org as supplemental material), there was a cluster of activated microglial cells in close contact with Aβ plaques; however, TNF-α positive microglia was mainly nonassociated to the amyloid plaques. This suggests the existence of, at least, two different activated microglial populations in aged PS1xAPP mice. To test this proposition, we performed Tomato Lectin labeling on sections previously immunostained for TNF-α and Congo Red. In this particular case, we did not use confocal microscopy because of the high tissue autofluorescence in aged animals. In any case, as shown (Fig. 2 A, a7), the Congo Red stained plaques, as well as cells immediately in contact with the plaques, were TNF-α-negative. However, these TNF-α negative cells surrounding plaques were stained with Tomato Lectin, demonstrated their microglial origin (Fig. 2 A, a8). Furthermore, a close inspection of the Congo Red-TNF-α double labeled sections revealed the absence of TNF-α immunopositive processes infiltrating the Aβ plaques (Fig. 2 A, a7) whereas the same plaque, counterstained with Tomato Lectin displayed a clear infiltration by microglial prolongations (Fig. 2 A, a8). Similar results were obtained using YM-1 (data not shown)
Figure 2.
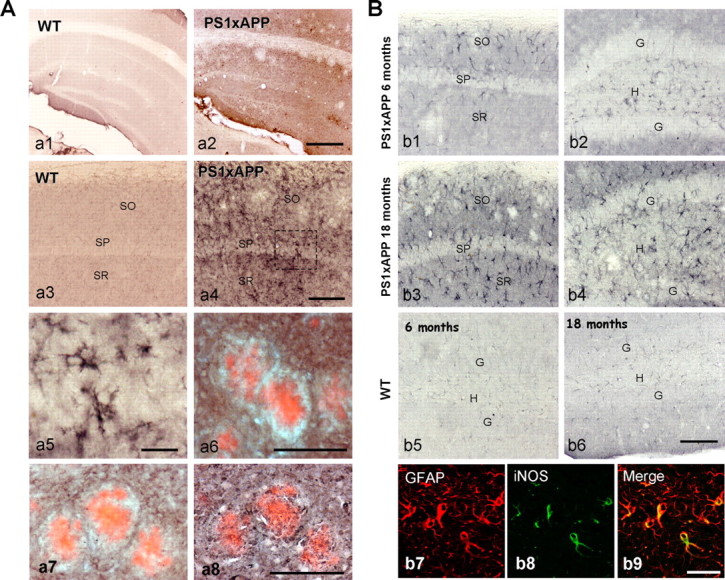
Expression of TNF-α and iNOS in 18-month-old PS1xAPP hippocampus. A, The expression of TNF-α, in 18-month-old WT and PS1xAPP mice, was assessed by immunohistochemistry. TNF-α immunoreactivity was low in WT mice (a1, a3), whereas a prominent immunoreactivity was observed in PS1xAPP (a2, a4). The TNF-α positive cells displayed a microglial phenotype (a5). Importantly, the TNF-α immunoreactivity was widespread found in all layers of the hippocampus; however, the Aβ plaques border zone appeared immunonegative for TNF-α (a6). Sections from 18-month-old PS1xAPP mice were first immunostained with anti-TNF-α antibody followed by Congo Red staining (a7). These double labeled sections were photographed and then counterstained with Tomato Lectin (a8). As shown, the Aβ plaques (stained by Congo-red) were surrounded and infiltrated by TNF-α immunonegative and Tomato-lectin immunopositive microglia cells (a8). Scale bars: a1, a2, 500 μm; a3, a4, 100 μm; a5, 20 μm; a6–a8, 100 μm. B, Immunostaining of iNOS expressing cells in 6- (b1, b2) and 18-month-old (b3, b4) PS1xAPP mice. At 6 months of age, a limited expression, restricted to stratus oriens and hilus, was observed. In 18-month-old PS1xAPP, the iNOS positive cells were observed in all hippocampal layers. A faint immunostaining was observed in WT mice of 6 months and, relatively more intense, 18 months of age (b5, b6). Double iNOS-GFAP immunofluorescence labeling and confocal laser microscopy (b7–b9) demonstrated the localization of iNOS in astroglial cells. Scale bars: b1–b6, 100 μm; b7–b9, 20 μm.
Concerning to iNOS expression, immunohistochemical experiments demonstrated the existence of few iNOS immunopositive cells in 6-month-old PS1xAPP hippocampus, restricted to certain areas, such as stratum oriens and hilus (Fig. 2 B, b1,b2). The number of iNOS immunopositive cells increased markedly at 18 months, compared with age-matched WT (Fig. 2 B, compare b3, b4, b6) or 6 month WT (Fig. 2, b5) or PS1xAPP mice (Fig. 2 B, b1,b2). As also shown in Figure 2 B, the iNOS-positive cells displayed an astroglial appearance. The astroglial origin of iNOS was confirmed by double GFAP-iNOS labeling and confocal microscopy (Fig. 2 B, b7–b9). Furthermore, at 18 months of age, iNOS containing astroglial cells were widely distributed in all layers of the hippocampus and not restricted to the Aβ plaques (Fig. 2 B, b3). The induction of iNOS expression by astrocytes, because of TNF-α and/or TRAIL, has been previously reported (Akama and Van Eldik, 2000; Cantarella et al., 2008).
Together, our data demonstrated that the apparition of the Aβ plaques (at early ages in this model) determined the microglial activation restricted to the Aβ plaques. These active microglial cells adopted a, perhaps incomplete, alternative phenotype (YM-1 positive) and were TNF-α negative. This alternative activated microglial cells displayed Aβ phagocytic capabilities. However, at 18 months of age, microglial activation was expanded into hippocampal areas free of plaques showing, in this case, a classic proinflammatory phenotype, with the expression of potential cytotoxic factors. This cytotoxic environment was also coincident with a significant loss of pyramidal cells in this model (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Nevertheless, the microglia surrounding plaques seemed to keep expressing the alternative phenotype. The possible Aβ phagocytosis, at old ages, was not assessed because of the high tissue autofluorescence in these animals. However, we also observed an age-dependent increase in both the number and size of Aβ plaques (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). This increase in the plaque size might reflect a diminution in the microgial phagocytic capability.
Infiltration of CD3 positive T-cells in aged PS1xAPP mice
We next investigated the possible cause(s) that determine the microglial activation at old ages. The generalized glial activation, observed at 18 months of age in this AD model, could be attributable to the infiltration of peripheral T-cells into the hippocampal parenchyma. The recruitment of CD4+ Th1-cells in experimental autoimmune encephalomyelitis (EAE; a disease similar to human multiple sclerosis) induced the production of, among other cytotoxic factors, TNF-α (Dhib-Jalbut et al., 2006; Weaver et al., 2007). In consequence, we first determined the possible infiltration of T cells in PS1xAPP mice by examining the presence CD3-positive cells. As shown (Fig. 3 A, a3) in 6-month-old PS1xAPP mice few CD3-positive cells were observed. On the contrary, in 18-month-old PS1xAPP, numerous CD3 immunopositive cells were clearly detected (Fig. 3 A, a2). The presence of CD3-positive cells was observed in all layers of the hippocampus, although they were more abundant around some plaques and close to the hippocampal fissure (Fig. 3 A, a2,a4). However, in WT mice very few CD3-positive cells were observed even at advanced ages (Fig. 3 A, a1).
Figure 3.

Infiltrated CD3 positive cells in 18-month-old PS1xAPP hippocampus predominantly developed a Th2 response. A, Double labeling for anti-CD3 and Congo Red immunohistochemistry demonstrated the infiltration of CD3-positive cells in hippocampus of 18 month PS1xAPP mice (a2, a4), whereas the presence of CD3 cells was scarce in 18 month WT (a1) or in 6 month PS1xAPP mice (a3). The infiltrated CD3 cells were preferential, but not exclusively located around some Aβ plaques (a4). B, The expression of different interleukin mediators, IFN-gamma and TGF-β1, considered representative of the different adaptive T-mediated immune responses, were determined in 6- and 18-month-old WT and PS1xAPP mice (n = 10 per group). Compared with WT (using one-way ANOVA), only the expression of IL-10, TGF-β and, more prominently, IL-4 were significantly induced in PS1xAPP mice. Interestingly, the expression of IL-4 also displayed a significant increased in 6-month-old PS1xAPP. C, At 6 months of age (c1), IL-4 punctate immunostaining was concentrated around Aβ plaques. Double GFAP-IL4 immunolabeling and confocal laser microscopy (c2–c4) revealed that activated astroglial cells expressed this interleukin. In 12-month-old PS1xAPP mice, all infiltrating CD3-positive cells were also immunoreactive for anti-IL4 (c5–c7). Scale bars: a1–a3, 200 μm; a4, 100 μm; c1, 25 μm; c2–c4, 5 μm; c5-c7, 5 μm.
Because the T-cells could modify the microglial phenotype, as it was observed in 18-month-old PS1xAPP mice, we also determined the mRNA expression of interleukins, or key factors, implicated in the different polarization linage of the T cells. In this sense, we have quantified the expression of IL-12p35 and IFN-gamma (proinflammatory Th1 cells); IL-23 and IL-17 (proinflammatory Th17 cells); IL-10 and TGF-β1 (Treg) and IL-4 (characteristic of anti-inflammatory Th2 response). Unexpectedly (Fig. 3 B), absolutely no differences (compared with WT mice) were observed in the mRNA expression of any of the Th1/Th17 cell response interleukins. However, the expression of IL-10, TGF-β-1 and, more patently, IL-4 was highly increased in PS1xAPP mice. This response, in conjunction with the moderate increase in the expression of IL-10 and TGF-β1 could represent a strong adaptive anti-inflammatory response (Th2 and Treg). Thus, it is unlikely that this response could mediate the induction in the expression of cytotoxic factors by the activated microglial cells. It is noteworthy that the expression of IL-4 mRNA was also upregulated in 6-month-old PS1xAPP mice.
The expression of IL4 was further studied, by immunohistochemistry, in 6- and 18-month-old PS1xAPP mice. At 6 months of age (Fig. 3 C, c1), IL-4-positive punctated structures were localized predominantly around amyloid deposits whereas, in 18-month-old tg mice (data not shown), IL-4 positive cells were located around plaques and as isolated small rounded cells. Double immunofluorescence and confocal laser microscopy, in 6-month-old PS1xAPP mice, determined that neither activated microglia surrounding plaques or APP positive principal cells expressed IL-4 (data not shown). Instead the IL-4 immunoreactivity colocalized with GFAP positive (Fig. 3 C, c2–c4) and reactive (as judged by the hypertrophic cell body and prolongations) astroglial cells. These reactive astrocytes were located in close association with Aβ deposits (data not shown).
Concerning to the CD3 infiltration observed at older ages (18 months), we cannot directly assess the coexpression with IL-4 by confocal microscopy. However, we have indeed identified CD3-IL-4 coexpressing cells in 12-month-old PS1xAPP mice (Fig. 3 C, c5–c7). In these middle age tg mice, the infiltration of CD3 positive cells was lower than that observed at 18 months (data not shown). However, and despite this limitation, all identified CD3-positive cells infiltrating the hippocampal parenchyma were also IL-4 positive.
Soluble oligomeric Aβ could be responsible for the generalized microglial activation at advanced ages
Finally, we investigated the presence of extracellular soluble Aβ species [monomers or amyloid–beta-derived diffusible ligands (ADDLs)/oligomers] as putative inductors of the age-dependent glial generalized activation. We have first determined, by sandwich ELISA, the Aβ42 content in the soluble fractions (S1 fractions, see Materials and Methods). The Aβ42 levels in 6-month-old PS1xAPP hippocampus was maintained at very low levels (6.7 ± 1.3 pmol/mg protein, n = 4), despite abundant Aβ deposits in this model (Fig. 1 A; supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). However, at 18 months we observed a dramatic increase in the Aβ42 content. In fact, this soluble Aβ42 increased 15-fold (86.9 ± 54.2 pmol/mg protein, n = 4; p < 0.05), in average, compared with 6-month-old PS1xAPP. This increase was notably higher than the twofold increase in the plaque loading, observed between 6- and 18-month-old PS1xAPP (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material).
We next tested the presence of Aβ oligomeric forms in the extracellular, soluble fractions. It is well known that diffusible aggregated Aβ forms (ADDLs/oligomers) are highly toxic (De Felice et al., 2007). Thus, we determined the presence of these Aβ forms using the specific monoclonal antibody Nu-1 in dot blots (Lambert et al., 2007). As shown in Figure 4 A, quantitatively in Figure 4 B, the presence of soluble ADDLs was barely detectable in 6- and 12-month-old PS1xAPP mice, as well as in WT mice. However, at 18 months, the soluble extracts displayed an intense immunoreactivity with Nu-1. Although it was difficult to quantify this age-dependent increase, because of the low specific signal observed at 6 and 12 months, our estimative quantification indicated the existence of 10-folds of increase at 18 months of age. Moreover, we also used A11 to corroborate the presence of oligomeric Aβ in these soluble extracts. As shown in Figure 4 A and quantitatively in Figure 4 B, the soluble extract from these 18 month PS1xAPP mice displayed a clear immunoreactivity with A11, compared with age-matched WT mice or 6 and 12 month PS1xAPP. It is also interesting that, using A11, we detected a small increase (although nonsignificant) in the oligomeric content in 12-month-old PS1xAPP. In conclusion, the amount of Nu-1 and A11 immunopositive oligomeric Aβ displayed a clear age-dependent increase in the soluble extract of PS1xAPP mice. At 18 months of age, the presence of these immunopositive Nu-1 and A11 oligomers was clearly patent.
Figure 4.

The increase in the soluble oligomeric Aβ, in 18 month PS1xAPP hippocampus. A, Representative dot blots demonstrating the presence of oligomeric Aβ in the soluble fractions. In these experiments we used the conformational-specific antibodies Nu-1 and A11. For each blot, the soluble extracts form the different age and group (WT and PS1xAPP) mice (n = 4 per age and group) were used in different combinations. These experiments were repeated twice. B, Quantitative analysis of PS1xAPP dot blots using Nu-1 and A11. The immunoreactivity of the different PS1xAPP mice was normalized by 6-month-old PS1xAPP mice and displayed individually or as mean ± SD. C, Representative Western blot, using 6E10, of the different soluble fractions from 6-, 12-, and 18-month-old PS1xAPP mice. These experiments were repeated three times with similar results.
Although SDS-PAGE is a reductive technique and as such cannot provide an accurate reflection of the noncovalently associated Aβ oligomers (Hepler et al., 2006), the presence of the soluble oligomeric Aβ forms was also assayed in Western blots using 6E10. As shown (Fig. 4 C), a prominent αAPPs band was clearly detected in 6-, 12-, and 18-month-old PS1xAPP mice. At 6 and 12 months, very small amount of low-n aggregated Aβ was detected, under the conditions used for these experiments. A faint band, corresponding to 5- or 6-mer Aβ could be barely observed in 6- and 12-month-old PS1xAPP mice. However, as expected from the dot blot analysis, in 18 month PS1xAPP mice multiple Aβ forms were clearly distinguished. The observed 6E10-positive bands corresponded with the reported Mr for the oligomeric Aβ forms, detected by Western blots (see Lambert et al., 2007), and also similar to soluble oligomers identified in other aged tg models (Lesné et al., 2006). In fact, according with the Mr, three predominant Aβ forms were identified in the soluble extracts; monomer, 6-mer and 12-mer. Although the 12-mer could also be present, in lower amount, in 6 and 12 month mice (Fig. 4 C), these low Mr oligomers were barely detectable in the soluble extract from 6- and 12-month-old PS1xAPP. We cannot discard that other high molecular weight forms were also increased in 18-month-old tg mice. Independently of the oligomeric state of Aβ, these data clearly demonstrated the existence of a marked increase in the soluble Aβ forms in our 18-month-old PS1xAPP mice population. Therefore, the presence of these ADDLs/oligomeric Aβ forms could be implicated in the classic microglial activation, observed in our 18-month-old PS1xAPP cohort.
To validate this hypothesis, we next tested, in vitro using astromicroglial cultures, the effect of monomeric and oligomeric Aβ42 forms in the expression of TNF-α. LPS was used as a positive control. The results (Fig. 5 A) demonstrated that LPS and, dose-dependently, the oligomeric Aβ42 strongly stimulated the expression of the proinflammatory TNF-α. Interestingly, equivalent concentrations of monomeric Aβ42 were totally ineffective stimulating the TNF-α expression in these cultures. Based on these data, we predicted that the soluble extracts from 18-month-old PS1xAPP mice should also stimulate the expression of TNF-α in the glial cultures. Thus, we tested the stimulatory effect of increasing amounts of soluble proteins (ranging from 5 to 100 μg of protein), obtained from 6- and 18-month-old WT and PS1xAPP mice. As shown, the soluble fractions from young WT and PS1xAPP mice produced no apparent effect on the expression of TNF-α (Fig. 5 B). Similarly, the soluble fractions from 18-month-old WT mice were also ineffective stimulating the TNF-α production. However, the soluble fractions derived from 18-month-old PS1xAPP mice produced a potent dose-dependent stimulation in the expression of TNF-α in these astromicroglial cultures (Fig. 5 B).
Figure 5.

The oligomeric Aβ induced the TNF-α expression in glial primary cultures. A, Dose–response stimulation of the TNF-α expression by oligomeric and monomeric Aβ42 assessed in vitro in astromicroglial cultures. PBS and LPS (1 μg/ml) were used as negative and positive controls, respectively. Data are mean ± SD from three independent cultures and Aβ42 preparations. B, Stimulation of the glial cultures using the S1 soluble fractions. Increasing protein amounts (from 5 to 100 μg) of the different S1 fractions (6 and 18 months of age; WT and PS1xAPP; n = 3 per age and group) was added to the cultures. In parallel, PBS, monomeric (20 μm), oligomeric Aβ42 (20 μm) and LPS (1 μg/ml) were included as negative and positive controls, respectively. For each experiment, duplicate culture wells were used. This experiment was repeated twice, using independent cultures. Only the soluble extract from 18-month-old PS1xAPP produced the stimulation of the TNF-α expression in these experiments (asterisk; p < 0.05, Tukey's test). C, Immunodepletion experiments. In these experiments, the Aβ content from the S1 fractions (10 μg of protein) of three different 18-month-old PS1xAPP mice was immunodepleted by three sequential immunoprecipitations using either 6E10-Protein G-Sepharose or A11-Protein A-Sepharose complexes. After immunodepletion, the S1 fractions were used in stimulation experiments. In parallel, the different S1 fractions were treated with Protein G-Sepharose or Protein A-Sepharose. We observed no differences in the TNF-α stimulation between these control S1 fractions and the results were pooled. The immunodepletion of Aβ, by any of the antibodies used, precluded the stimulatory effect of the S1 fractions. PBS, oligomeric Aβ42 (20 μm) and LPS (1 μg/ml) were included as negative and positive controls, respectively.
Finally, if the soluble oligomeric Aβ in the S1 fractions was indeed the causative agent of the TNF-α stimulation, this effect should be avoided by immunodepletion of the supernatant using specific antibodies. To test this point, the Aβ content of the S1 fractions, derived from 18-month-old PS1xAPP mice (n = 3), was immunodepleted by three sequential immunoprecipitations using either the mAb 6E10 (that should recognized the total soluble Aβ) or the conformation specific polyclonal A11 (specific of the oligomeric Aβ). As shown (Fig. 5 C), after immunodepletion using 6E10, the induction in the TNF-α expression by these immunodepleted S1 fractions was deeply reduced (from 94.60 ± 6.02–2.87 ± 0.90, n = 3, before and after immunodepletion, relative units normalized by PBS effect, 1.01 ± 0.24; p < 0.05, Tukey's test). Moreover, the immunodepletion of the oligomeric Aβ using A11 (Fig. 5 C), also produced an almost completed reduction in the TNF-α induction by the S1 fractions (2.33 ± 0.45, n = 3, after immunodepletion; p < 0.05, Tukey's test).
Discussion
Neuroinflammation is a key feature of AD pathology (Meda et al., 2001; Dudal et al., 2004; Craft et al., 2006). However, its role is still conflictive. Here, we have analyzed the microglial response associated to the age-dependent amyloid pathological progression in the hippocampus of PS1M146LxAPP751SL mice. The principal findings are as follows. (1) At early ages (4–6 months), activated microglia was restricted to Aβ deposits and characterized by the absence of induction of cytotoxic factors and by the expression of one alternative marker (YM-1). (2) In 18-month-old, microglial activation was expanded throughout the hippocampus, displaying a classical potentially cytotoxic phenotype (expressing TNF-α, FASL, TRAIL, Cox2 and Nox1). The expression of iNOS was restricted to astrocytes. (3) Although a clear hippocampal T-cell infiltration was detected at 18 months, these T-cells were polarized to Th2 phenotype. (4) There was an age-dependent increase in extracellular soluble Aβ oligomers, which were potent microglial stimulators as assessed by in vitro studies.
We observed a close spatial and temporal parallelism between Aβ deposits and microglial activation (Simard et al., 2006). At early ages, activated microglia was concentrated in clusters surrounding and infiltrating Aβ plaques. In fact, few extra-plaque microglial activation was observed. Thus, according with recent in vivo observations (Meyer-Luehmann et al., 2008), plaques attracted and stimulated microglial cells. However, activated microglia could adopt different phenotypes. Our data demonstrated that plaque-associated microglia displayed an alternative state. In peripheral macrophages, this phenotype was characterized by the absence of expression of cytotoxic factors and the expression of alternative markers (YM-1 and Arg-1) (Edwards et al., 2006). Although we have not observed the induction in Arg-1, the YM-1 expression was highly elevated in microglia surrounding plaques. These results, together with the absence of significant TNF-α, TRAIL, FASL or iNOS expression led us to conclude that the activated microglia could adopt an incomplete alternative phenotype. Furthermore, this alternative phenotype, associated to Aβ plaques, seemed to be maintained also at relative old ages. In fact, at 18-months, the microglia surrounding Aβ plaques was TNF-α negative and YM-1 positive. Therefore, activated microglia in direct apposition with Aβ plaques adopted an alternative phenotype, regardless of the age of the animal. This proposition was consistent with the increased expression of IL-4. Multiple in vitro reports have probed the influence of IL-4 in the development of a non-proinflammatory alternative phenotype (Iribarren et al., 2005; Butovsky et al., 2005; Ponomarev et al., 2007; Lyons et al., 2007a,b). Furthermore, at early ages, the expression of this interleukin was restricted to reactive astrocytes, closely associated to Aβ plaques. Similarly, at 18 months, plaques were also surrounded by IL-4 positive astrocytes and, probably, CD3 cells. In consequence, we proposed that the alternative phenotype, associated to Aβ plaques, could be because of the expression of IL-4 by activated astrocytes and, when present, CD3-cells. We cannot discard that Aβ plaques could directly produce this microglial differentiation. The factor(s) that determine the astroglial activation remains to be investigated.
Concerning to the physiological role, the alternative activated microglia could exert a neuroprotective function. In presence of IL-4, microglia produces growth factors (i.e., IGF-1) (Butovsky et al., 2006; Zhao et al., 2006). The IL-4 reduced Aβ toxicity, in vitro and in vivo (Butovsky et al., 2005, 2006; Iribarren et al., 2005; Lyons et al., 2007a,b) and enhanced Aβ phagocytosis (Koenigsknecht-Talboo and Landreth, 2005). In agreement, we also observed the expression of IGF-1, in the vicinity of Aβ deposits (our unpublished results) and Aβ phagocytosis by microglia surrounding plaques (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). In this sense, Bolmont et al. (2008) have recently demonstrated the internalization of Aβ peptides by plaque-associated microglia and proposed an active role in the maintenance of plaque size. Furthermore, the decrease in the early recruitment of the microglial cells, by genetic ablation of either TLR2 receptor, in a PS1/APP model (Richard et al., 2008) or CCR2 in Tg2576 (El Khoury et al., 2007), increased Aβ42 levels, accelerated memory impairments and increased vascular amyloid pathology. Thus, we propose that the plaque-associated alternative activated microglia could limit Aβ toxicity directly, by phagocytosis (see also Simard et al., 2006), maintaining low extracellular Aβ levels. In fact, our data demonstrated low levels of soluble Aβ42 and oligomeric forms until advanced ages.
Furthermore, this alternative activated microglia could also indirectly exert a neuroprotective role by releasing growth factors, such as IGF-1. This proposition is also consistent with the positive effects of vaccination with the Th2 adjuvant glatiramer acetate, the concomitant increase in IL-4 expressing cells and the reduction of Aβ plaques (Butovsky et al., 2006).
At 18 months of age, microglia located between, not in direct contact with Aβ plaques, was clearly activated. Furthermore, these interplaque microglial cells expressed TNF-α (and probably other TNF-α related factors), whereas the microglia closely associated to Aβ plaques remained TNF-α negative and YM-1 positive. Thus, at old age, two functionally different activated microglial populations should coexist, indicating the existence of different microglial activators. In this sense, even at this old age, there was a clear IL-4 expression surrounding plaques. Thus, and similar to the situation observed in 6-month-old PS1xAPP mice, plaques (directly and/or indirectly through IL-4) might stimulate microglia to a noncytotoxic phenotype. This observation let us to propose that the generalized microglial activation should be induced by a diffusible agent.
As mentioned, the general microglial activation could reflect a Th1/Th17 CD3-mediated response. According with previous reports in AD and AD models (Itagaki et al., 1988; Togo et al., 2002; Stalder et al., 2005) we observed CD3-cells in 18-month-old PS1xAPP hippocampus. However, the expression of IL-12, IL-17 and IL-23 was unchanged, compared with age-matched WT. Instead, we observed a considerable induction of IL-4 and IL-10 expression. Moreover, these CD3-cells also expressed IL-4. Together, data strongly suggest that the infiltrated T-cells were predominantly polarized to Th2 and/or Treg states. These anti-inflammatory Th2/Treg cells could explain the absence of encephalitis signs in AD models (Stalder et al., 2005). Thus, the Th2 polarization could limit the potential neurotoxic effect of the activated microglia and it is highly unlikely that CD3 infiltration mediated the expanded microglial activation, observed in this model.
Multiple evidences suggested that diffusible Aβ oligomers (ADDLs) were the toxic agents in AD. These ADDLs were exclusively present in AD patients (Gong et al., 2003; Lacor et al., 2004) and their content increased with the disease severity (Lambert et al., 2007). In fact, our results demonstrated the increase in soluble Aβ42 in 18-month-old PS1xAPP. Furthermore, our data also probed the presence of Nu-1 and A11 immunopostive oligomers in these soluble fractions (barely detectable at early ages). Thus, we hypothesized that the oligomeric Aβ was also the causative agent of the generalized microglial response at 18 months. Our in vitro experiments strongly supported this proposal. In fact, oligomeric Aβ42 highly stimulated the TNF-α expression in cultures, whereas monomeric Aβ42 was ineffective. More relevant, the soluble S1 fractions from 6-month-old WT and PS1xAPP mice and 18-month-old WT, that lack detectable levels of oligomers, were also ineffective stimulating TNF-α production in culture. However, S1 fractions from 18-month-old PS1xAPP produced a strong TNF-α induction. Moreover, the immunodepletion of Aβ, using 6E10 or A11, completely abolished the stimulatory effect of the S1 fractions. In consequence, we proposed that the generalized microglial activation with a classic and potentially cytotoxic phenotype, observed at 18 months, could be because of the accumulation of soluble oligomeric Aβ in the hippocampal parenchyma. Furthermore, it has been reported that aging and/or Aβ could decrease the neuronal expression of microglial inhibitory factors, such as CD200, fractalkine or neurotransmitters (Acetylcholine or noradrenaline) (Cardona et al., 2006; Chitnis et al., 2007; Heneka and O'Banion, 2007; Lyons et al., 2007a; Duan et al., 2008). Furthermore, damaged neurons could release microglial stimulants, such as ATP, UDP or CCL21 (de Jong et al., 2005; Inoue et al., 2007). Therefore, it is possible that the classic microglial activation in our model could reflect a synergic effect of Aβ oligomers, acting directly on microglia and, indirectly, by affecting neuronal-microglial interplay.
The role of the classical microglial activation was unknown. The neuronal toxicity to TNF-α, TNF-α related factors and iNOS has been extensively probed (Cantarella et al., 2003; Lee et al., 2004; Li et al., 2004; Medeiros et al., 2007; Uberti et al., 2007). Indeed, in our model, we (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and others (Schmitz et al., 2004) have observed a significant decrease in pyramidal cell number, coincident with the classical microglial activation (17–18 months). Although present results do not allow establishing a direct relationship between these events, the production of TNF-α, TRAIL, FASL and NO derivates, together with the presence of soluble Aβ oligomers, could directly contribute to the observed pyramidal degeneration.
In conclusion, at early ages in our AD model, the apparition of Aβ plaques determined the microglia activation to an alternative phenotype with, apparently, a neuroprotective role. At older ages, the accumulation of extracellular oligomeric Aβ produced marked widespread microglial activation toward a classic phenotype and the production of cytotoxic factors. The reasons that determined this age-dependent increase in the ADDL/oligomers content remained unknown.
Footnotes
This work was supported by Fondo de Investigación Sanitaria (FIS) from Instituto de Salud Carlos III of Spain through Grants PI060567 (J.V.), PI060556 (A.G.), and PI060781 (D.R.), and by Junta de Andalucia, Proyecto de Excelencia CVI-902. S.J. and I.M.-G. are the recipients of a contract from CIBERNED. D.B.-V. and M.T. were recipients of PhD fellowships from Junta de Andalucia and R.S.-V. and C.C. from Ministerio de Educación y Ciencia (MEC) of Spain. We thank Dr. I. Torres-Aleman for his valuable critical reading of this manuscript, Drs. W. Klein and M. Lambert for their generous gift of Nu-1 antibody, and Dr. D. Pozo for his help with tissue culture experiments.
References
- Aisen PS. Anti-inflammatory therapy for Alzheimer's disease. Neurobiol Aging. 2000;21:447–448. doi: 10.1016/s0197-4580(00)00140-8. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, Grudman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- Akama KT, Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappa B-inducing kinase-dependent signaling mechanism. J Biol Chem. 2000;275:7918–7924. doi: 10.1074/jbc.275.11.7918. [DOI] [PubMed] [Google Scholar]
- Araujo F, Tan S, Ruano D, Schoemaker H, Benavides J, Vitorica J. Molecular and pharmacological characterization of native cortical gamma-aminobutyric acid(A) receptors containing both alpha(1) and alpha(3) subunits. J Biol Chem. 1996;271:27902–27911. doi: 10.1074/jbc.271.44.27902. [DOI] [PubMed] [Google Scholar]
- Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol. 2003;184:247–263. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero C, Jimenez S, Moreno-Gonzalez I, Baglietto-Vargas D, Sanchez-Varo R, Gavilan MP, Ramos B, Del Rio JC, Vizuete M, Gutierrez A, Ruano D, Vitorica J. Inter-individual variability in the expression of the mutated form of hPS1M146L determined the production of Abeta peptides in the PS1xAPP transgenic mice. J Neurosci Res. 2007;85:787–797. doi: 10.1002/jnr.21172. [DOI] [PubMed] [Google Scholar]
- Cantarella G, Uberti D, Carsana T, Lombardo G, Bernardini R, Memo M. Neutralization of TRAIL death pathway protects human neuronal cell line from beta-amyloid toxicity. Cell Death Differ. 2003;10:134–141. doi: 10.1038/sj.cdd.4401143. [DOI] [PubMed] [Google Scholar]
- Cantarella G, Di Benedetto G, Pezzino S, Risuglia N, Bernardini R. TRAIL-related neurotoxicity implies interaction with the Wnt pathway in human neuronal cells in vitro. J Neurochem. 2008;105:1915–1923. doi: 10.1111/j.1471-4159.2008.05291.x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chitnis T, Imitola J, Wang Y, Elyaman W, Chawla P, Sharuk M, Raddassi K, Bronson RT, Khoury SJ. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am J Pathol. 2007;170:1695–1712. doi: 10.2353/ajpath.2007.060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Van Eldik LJ. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia. 2006;53:484–490. doi: 10.1002/glia.20306. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhib-Jalbut S, Arnold DL, Cleveland DW, Fisher M, Friedlander RM, Mouradian MM, Przedborski SA, Trapp BD, Wyss-Coray T, Yong VW. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. J Neuroimmunol. 2006;176:198–215. doi: 10.1016/j.jneuroim.2006.03.027. [DOI] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Duan RS, Yang X, Chen ZG, Lu MO, Morris C, Winbland B, Zhu J. Decreased fractalkine and increased IP-10 expression in aged brain of APP(swe) transgenic mice. Neurochem Res. 2008;33:1085–1089. doi: 10.1007/s11064-007-9554-z. [DOI] [PubMed] [Google Scholar]
- Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol Aging. 2004;25:861–871. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric Abeta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie J, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kiêu K, Nielsen J. The efficiency of systematic sampling in stereology–reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, Kinney G, Joyce JG. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN. Time-dependent reduction in A[beta] levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Inoue K, Koizumi S, Tsuda M. The role of nucleotides in the neuron-glia communication responsible for the brain functions. J Neurochem. 2007;102:1447–1458. doi: 10.1111/j.1471-4159.2007.04824.x. [DOI] [PubMed] [Google Scholar]
- Iribarren P, Chen K, Hu J, Zhang X, Gong W, Wang JM. IL-4 inhibits the expression of mouse formyl peptide receptor 2, a receptor for amyloid beta1–42, in TNF-alpha-activated microglia. J Immunol. 2005;175:6100–6106. doi: 10.4049/jimmunol.175.9.6100. [DOI] [PubMed] [Google Scholar]
- Itagaki S, McGreer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett. 1988;91:259–264. doi: 10.1016/0304-3940(88)90690-8. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem. 2004;91:568–578. doi: 10.1111/j.1471-4159.2004.02780.x. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 Ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007a;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem. 2007b;101:771–781. doi: 10.1111/j.1471-4159.2006.04370.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28:639–647. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Meda L, Baron P, Scarlato G. Glial activation in Alzheimer's disease: the role of Abeta and its associated proteins. Neurobiol Aging. 2001;22:885–893. doi: 10.1016/s0197-4580(01)00307-4. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Prediger RD, Passos GF, Pandolfo P, Duarte FS, Franco JL, Dafre AL, Di Giunta G, Figueiredo CP, Takahashi RN, Campos MM, Calixto JB. Connecting TNF-{alpha} Signaling Pathways to iNOS Expression in a Mouse Model of Alzheimer's Disease: Relevance for the Behavioral and Synaptic Deficits Induced by Amyloid- beta Protein. J Neurosci. 2007;27:5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-plaques in a mouse model of Alzheimer/'s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B, Baglietto-Vargas D, del Rio JC, Moreno-Gonzalez I, Santa-Maria C, Jimenez S, Caballero C, Lopez-Tellez JF, Khan ZU, Ruano D, Gutierrez A, Vitorica J. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1 x APP transgenic model of Alzheimer's disease. Neurobiol Aging. 2006;27:1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, Watterson DM. Glia as a therapeutic target: selective suppression of human amyloid-beta-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–670. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC. Rofecoxib: No effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–831. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schäfer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA. Hippocampal Neuron Loss Exceeds Amyloid Plaque Load in a Transgenic Mouse Model of Alzheimer's Disease. Am J Pathol. 2004;164:1495–1502. doi: 10.1016/S0002-9440(10)63235-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- Uberti D, Ferrari-Toninelli G, Bonini SA, Sarnico I, Benarese M, Pizzi M, Benussi L, Ghidoni R, Binetti G, Spano P, Facchetti F, Memo M. Blockade of the tumor necrosis factor-related apoptosis inducing ligand death receptor DR5 prevents beta-amyloid neurotoxicity. Neuropsychopharmacology. 2007;32:872–880. doi: 10.1038/sj.npp.1301185. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Abeta plaque removal following intracranial anti-A[beta] antibody administration. Neurobiol Disease. 2004a;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Zhao W, Xie W, Xiao Q, Beers DR, Appel SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem. 2006;99:1176–1187. doi: 10.1111/j.1471-4159.2006.04172.x. [DOI] [PubMed] [Google Scholar]
- Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends in Neurosciences. 2006;29:518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]