

Abstract
Although vascular endothelial growth factor-B (VEGF-B) is a homolog of the angiogenic factor VEGF, it has only minimal angiogenic activity, raising the question of whether this factor has other (more relevant) biological properties. Intrigued by the possibility that VEGF family members affect neuronal cells, we explored whether VEGF-B might have a role in the nervous system. Here, we document that the 60 kDa VEGF-B isoform, VEGF-B186, is a neuroprotective factor. VEGF-B186 protected cultured primary motor neurons against degeneration. Mice lacking VEGF-B also developed a more severe form of motor neuron degeneration when intercrossed with mutant SOD1 mice. The in vitro and in vivo effects of VEGF-B186 were dependent on the tyrosine kinase activities of its receptor, Flt1, in motor neurons. When delivered intracerebroventricularly, VEGF-B186 prolonged the survival of mutant SOD1 rats. Compared with a similar dose of VEGF, VEGF-B186 was safer and did not cause vessel growth or blood–brain barrier leakiness. The neuroprotective activity of VEGF-B, in combination with its negligible angiogenic/permeability activity, offers attractive opportunities for the treatment of neurodegenerative diseases.
Keywords: ALS, VEGF-B, Flt1, motor neuron degeneration, intracerebroventricular delivery, therapy
Introduction
Although originally discovered as an endothelial-specific angiogenic factor, vascular endothelial growth factor (VEGF) also has effects on neurons. Indeed, mice expressing reduced VEGF levels suffer motor neuron degeneration, reminiscent of amyotrophic lateral sclerosis (ALS) (Oosthuyse et al., 2001). Furthermore, transgenic overexpression of VEGF or its receptor Flk1 in neurons or intracerebroventricular delivery of VEGF prolongs the survival of ALS models (Azzouz et al., 2004; Storkebaum et al., 2005). VEGF induces many of its neurovascular activities through Flk1 (Sondell et al., 2000; Oosthuyse et al., 2001; Lambrechts and Carmeliet, 2006), but also binds VEGFR1 (Flt1); the latter is also a receptor for two other VEGF family members, i.e., placental growth factor (PlGF) and VEGF-B (Olofsson et al., 1998). In contrast to VEGF, little is known about the role of VEGF-B (Nash et al., 2006). It is expressed in the heart, skeletal muscle, nervous system, and other tissues as two alternatively spliced isoforms, i.e., VEGF-B167 and VEGF-B186 (Olofsson et al., 1996). Mice lacking VEGF-B are viable and fertile, and exhibit minor cardiac abnormalities (Bellomo et al., 2000; Aase et al., 2001). Apart from a selective angiogenic activity in ischemic myocardium (Tirziu et al., 2007; X. Li et al., 2008), the angiogenic role of VEGF-B in other tissues and conditions remains controversial and is, overall, weaker than that of VEGF or PlGF.
The functional relevance of Flt1 has also been subject of intense debate. Flt1 binds to VEGF, PlGF, and VEGF-B with at least 10-fold higher affinity than Flk1 binds to VEGF (Shibuya, 2001). However, Flt1 has a weaker tyrosine kinase activity (Park et al., 1994), and has been proposed to act as a decoy, regulating the availability of VEGF (Hiratsuka et al., 1998). Other studies indicated, however, that Flt1 interacts with various signal-transducing proteins and generates signals (Luttun et al., 2002; Autiero et al., 2003).
The precise role of VEGF-B and Flt1 in the nervous system is unclear. Flt1 is upregulated in astrocytes after administration of VEGF (Mani et al., 2005) or after injury (Choi et al., 2007), and stimulates migration of microglial cells in vitro (Forstreuter et al., 2002). Loss of VEGF-B enlarges stroke, whereas VEGF-B stimulates proliferation of neuronal cultures in vitro (Sun et al., 2004) and neurogenesis in vivo (Sun et al., 2006), and is protective in injury models, such as axotomy- and NMDA-induced cell death in the retina (Y. Li et al., 2008). VEGF-B has been presumed to exert direct neuroprotective effects, but this evidence was based on anti-apoptotic effects on smooth muscle cells and retinal pericyte cell lines (Y. Li et al., 2008). However, evidence that VEGF-B has direct neuroprotective effects on primary neurons has not been provided yet, nor has long-term delivery been shown to improve the disease course or outcome. We therefore investigated the role of VEGF-B and Flt1 in motor neuron degeneration in rodent models of ALS.
Materials and Methods
Transgenic mice.
Knock-in mice expressing a tyrosine kinase-dead Flt1 receptor (Flt1-TK−/− mice) were kindly provided by Dr. Shibuya and backcrossed to a FvB background. Mice lacking VEGF-B (VEGF-B−/− mice) were purchased from Jackson Laboratories and maintained onto a C57BL/6 background for eight generations.
Animal model for ALS.
Mice overexpressing human SOD1G93A backcrossed for more than 10 generations onto a FvB background were provided by Dr. C. Kunst (University of Colorado, Denver, CO). Rats expressing high levels of the human SOD1G93A transgene (line 26H; Sprague Dawley background) were kindly provided by Dr. Itoyama (Graduate School of Medicine, Tohoku University, Sendai, Japan) and backcrossed for five generations onto the HAN Wistar background (Charles River). The local animal ethical committee approved all animal experiments.
Surgical procedures.
Recombinant carrier-free mouse VEGF-B186 (767-VE-010/CF; R&D Systems) was infused into the left lateral ventricle of rats, using Alzet osmotic pumps, model 2004, connected with a catheter to a brain infusion cannula. The brain infusion assembly was filled with a solution containing 10 μg of recombinant carrier-free mouse VEGF-B186 dissolved in artificial CSF or with CSF alone, and primed during 48 h in saline. The composition of the CSF was 150 mm Na+, 3 mm K+, 1.4 mm Ca2+, 0.8 mm Mg2+, 1 mm PO4 3−, and 155 mm Cl−. After isoflurane anesthesia, the osmotic pump was subcutaneously inserted in the midscapular area on the back of the rat. After exposing the skull through a midline sagittal incision, a hole was drilled through the skull, and the cannula was placed using the following stereotaxic coordinates: 0.8 mm posterior and 1.6 mm lateral to bregma and 4.5 mm ventral from skull surface. The osmotic minipumps, delivering VEGF-B186, were replaced every 25 d.
Rotarod test.
Three times a week, an investigator who was blind to the treatment measured motor performance. Measurements were started in rats aged 60 d to obtain baseline values before disease onset at 85 d. A rat-specific rotarod (Ugo Basile), constantly rotating at a speed of 15 rotations per minute, was used to score rats for their ability to stay on the rotarod during a 3 min period. Rats were judged to fail when, on average in five separate trials, they fell off the rotarod before 1 min. Paralysis of the hindlimb was scored when the rat dragged one of its hindlimbs, and paralysis of a forelimb was scored when the rat failed to use its forelimbs for walking or righting. If rats displayed onset in forelimbs and hindlimbs at the same time, they were not categorized in one of these categories. Time of death was scored as the day when the rats had lost 40% of their presymptomatic body weight. Mice were analyzed using similar criteria, as described previously (Storkebaum et al., 2005).
Histology.
Animals were killed with Nembutal. Unfixed L3–L5 spinal cord were embedded in tissue freezing-medium (Tissue-Tek); 10 μm serial sections were stained using antibodies to VEGF-B (AF-590; 1/20; R&D Systems) or Flt1 (s.c.-316 and s.c.-9029; 1/50; Santa-Cruz, CA) and GFAP (G3893; 1/400; Sigma-Aldrich) or NeuN (AB 9337; 1/1000; Millipore Bioscience Research Reagents). Paraformaldehyde (4%)-fixed brain of rats, treated intracerebroventricularly with VEGF-B186 or artificial CSF, was embedded in paraffin; 7 μm serial brain sections were stained using antibodies to glucose transporter 1 (Glut1) (AB1341; 1/1000; Millipore Bioscience Research Reagents), endothelial barrier antigen (EBA) (SMI71; 1/2000; Sternberger Monoclonals) and GFAP (G3893; 1/400; Sigma-Aldrich). Glut1-, EBA-, and GFAP-positive areas were quantified around the site of injection and at the contralateral site at a magnification of 40× (AxioPlan Microscope; Zeiss). Sections were subsequently incubated with fluorescently conjugated secondary antibodies (Alexa 488 or 568; Invitrogen) or with biotin-labeled IgGs (Dako), followed by amplification with the signal amplification system (streptavidin-HRP-Cy3/fluorescein; PerkinElmer Life and Analytical Sciences) for VEGF-B, Flt1, and GFAP. Twenty-micrometers-thick fixed, paraffin-embedded mouse L3–L5 spinal cords and rat brainstem sections were stained with Nissl; the number of motor neurons in the entire spinal cord and brainstem was quantified in every fifth section by quantifying healthy α-motor neurons defined as intensely stained and having an area >200 μm2.
RNA extraction and cDNA preparation.
The RNeasy kit (Qiagen) was used for RNA extraction from animal cells. RNA from animal tissues was extracted using Trizol reagent (Invitrogen) and further purified using the RNeasy RNA cleanup protocol with on-column DNase digestion (Qiagen). One microgram of RNA was transcribed to cDNA using the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's instructions.
Quantitative real-time PCR.
Gene expression was analyzed using the 7500 Fast Real-Time PCR system (Applied Biosystems) and normalized to β-actin expression levels. The following forward (for) and reverse (rev) primers and probes (pro) labeled with a fluorescent dye (FAM) and a quencher (TAMRA) were used. Murine β-actin: for, 5′-AGA-GGG-AAA-TCG-TGC-GTG-AC-3′; rev, 5′-CAA-TAG-TGA-TGA-CCT-GGC-CGT-3′; pro, 5′-CAC-TGC-CGC-ATC-CTC-TTC-CTC-CC-3′. Murine VEGF-B total: for, 5′-TGC-CAT-GGA-TAG-ACG-TTT-ATG-C-3′; rev, 5′-TGC-TCA-GAG-GCA-CCA-CCA-C-3′; pro, 5′-CGT-GCC-ACA-TGC-CAG-CCC-A-3′. Murine VEGF-B167: for, 5′-GCC-AAT-GTG-AAT-GCA-GAC-CAA-3′; rev, 5′-TGC AAG GCG GGC AGA-3′; pro, 5′-TGT-GAA-GCC-AGA-CAG-CCC-CAG-GAT C-3′. Murine VEGF-B186: for, 5′-TCT-GTT-CCG-GGC-TGG-GA-3′; rev, 5′-AGT-GGG-ATG-GAT-GAT-GTC-AGC-T-3′; pro, 5′-TCT-ACC-CCG-GGA-GCA-TCC-TCC-C-3′. Murine Flt1: for, 5′-GGG-AAG-ACA-TCC-TTC-GGA-AGA-3′; rev, 5′-CCT-CGT-AGT-CAC-TGA-GGT-TTT-GAA-3′; pro, 5′-CGT-TAG-AGA-TTC-GGA-AGC-GCC-ACA-CC-3′. Murine GFAP: for, 5′-GGA-GGT-CCG-CTT-CCT-GGA-A-3′; rev, 5′-GGC-TCG-AAG-CTG-GTT-CAG-TT-3′; pro, 5′-AGC-TGC-CAG-CGC-CTT-GTT-TTG-CT-3′. Murine NF-L: Mm01315666_m1 (TaqMan gene expression assay; Applied Biosystems).
Protein extraction.
Protein extractions were obtained by placing freshly obtained neuronal cell pellets in a Lysing Matrix A tubes (MP Biomedicals) containing 200 μl of mammalian cell lysis extraction buffer containing detergents, NaCl, and protease inhibitor mixture solutions. (MCL1; Sigma-Aldrich). Samples were vortexed in a Hybaid RiboLyser for 40 s: the supernatant was collected for ELISA.
ELISA for Flt1.
ELISA for Flt1 was performed using the Quantikine ELISA kit for mouse sFlt1 (MVR100; R&D Systems), using the quantitative sandwich enzyme immunoassay technique.
Isolation of embryonic motor neurons and astrocytes.
Spinal motor neurons and astrocytes from embryonic day 14 (E14) Wistar rats, VEGF-B−/− mice, Flt1-TK−/− mice, and wild-type (WT) control mice were prepared by dissociating freshly dissected ventral spinal cords enzymatically and mechanically (Van Den Bosch et al., 2004). A motor neuron-enriched neuronal pellet was obtained by centrifugation on a 6.5% Optiprep cushion, which separates cells according to size. Motoneuron-enrichment was estimated by measuring ChAT activity: ChAT activity per 1000 cells was 110.2 ± 18.4 μU for the enriched pellet compared with 22.1 ± 4.8 (Vandenberghe et al., 1998). This motor neuron-enriched fraction was used for cDNA preparation and for the generation of E14 spinal motor neuron cultures. Motor neuron cultures were plated in culture dishes (3000 cells/cm2) or 96-well plates (10,000 cells per well) coated with poly-l-ornithine and laminin. The culture medium consisted of L15 supplemented with sodium bicarbonate (0.2%), glucose (3.6 mg/ml), progesterone (20 nm), insulin (5 μg/ml), putrescine (0.1 mm), conalbumin (0.1 mg/ml), sodium selenite (30 nm), penicillin (100 U/ml), streptomycin (100 μg/ml), and chick embryo extract. The survival of cultures of E14 spinal motor neurons was quantified by direct counting of unfixed cells under a phase-contrast microscope. Neurons in a marked area of 1 cm2 were counted before (on day 1 after seeding) application of recombinant protein or control. The relative survival was calculated as the number of cells on day 2 or 6 in culture, divided by the number of cells on day 1.
Alternatively, cultures of spinal motor neurons from E14 mice (CD1 strain) were prepared by a panning technique using a monoclonal rat anti-p75 antibody (Millipore Bioscience Research Reagents) (Bender et al., 2007). The ventrolateral parts of individual lumbar spinal cords were dissected and transferred to HBSS containing 10 mm 2-mercaptoethanol. After treatment with trypsin (0.05%, 10 min), single-cell suspensions were generated by titration. The cells were plated on a rat anti-p75 coated culture dish (24 well; Greiner) and left at room temperature for 30 min. The individual wells were subsequently washed with HBSS (three times), and the attaching cells were then isolated from the plate with depolarizing saline (0.8% NaCl, 35 mm KCl, and 1 mm 2-mercaptoethanol) and plated at a density of 1500 cells per well on laminin-coated coverslips in Greiner four-well culture dishes. Cells were grown in Neurobasal medium containing B27 supplement, 10% heat-inactivated horse serum, and 500 μm GlutaMax (Invitrogen). Medium was first replaced after 24 h and then every second day. Surviving cells were counted under a phase-contrast microscope (Leica). Initial counting of plated cells was done when all cells were attached to the culture dish at 4 h after plating.
Statistics.
Results are expressed as mean values ± SEM. We used SPSS version 13 for statistical calculations. Unless stated otherwise, standard t tests were used to assess statistical differences between groups. Cumulative analysis of rotarod and survival were calculated using Kaplan–Meier statistics. Log-rank p values were calculated to compare Kaplan–Meier curves. Significance was defined as two-tailed p < 0.05.
Results
Expression of VEGF-B in mouse spinal cord
For VEGF-B to affect motor neurons, it should be expressed in the ventral horn of the spinal cord. We therefore determined its expression in WT mice. Real-time (RT)-PCR analysis revealed that the VEGF-B167 and VEGF-B186 isoforms were detectable in a spinal motor neuron-enriched neuronal pellet isolated from E14 WT mouse embryos (Fig. 1 A) and from adult mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Double immunohistochemistry for VEGF-B and the neuronal marker NeuN revealed that VEGF-B was detectable in large neurons that, because of their large size and position in the ventral horn of the spinal cord, were considered to be motor neurons (Fig. 1 B–D); VEGF-B was also detectable in other neurons, e.g., DRG neurons (J. Dhondt, E. Peeraer, A. Verheyen, R. Nuydens, K. Poesen, I. Buysschaert, K. Van Geyte, M. Beerens, K. Raes, U. Eriksson, M. Shibuya, J. Haigh, T. Meert, P. Carmeliet, and D. Lambrechts, unpublished work) and blood vessels (data not shown). Specificity of the VEGF-B staining was confirmed in mice lacking VEGF-B (VEGF-B−/− mice).
Figure 1.
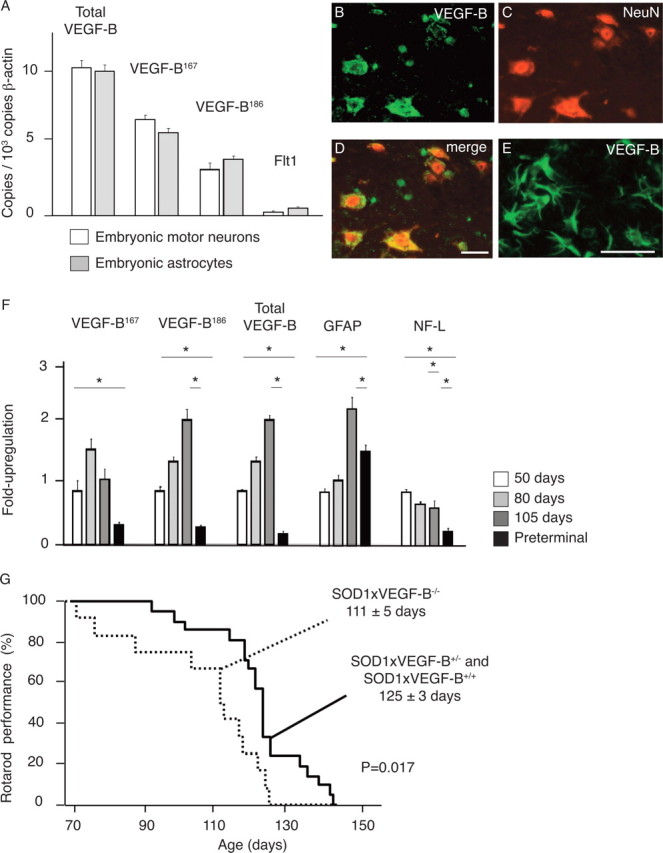
The role of VEGF-B in motor neuron degeneration. A, Real-time PCR measurements for total VEGF-B, the VEGF-B 167 (VEGF-B167) and 186 (VEGF-B186) isoforms, and the Flt1 receptor on cDNA preparations from primary embryonic motor neurons and primary embryonic astrocytes, showing that both VEGF-B isoforms are expressed in motor neurons and astrocytes. Primary embryonic motor neurons were derived from a spinal motor neuron-enriched neuronal pellet isolated from E14 WT mouse embryos. B–D, VEGF-B expression is detected on lumbar (L3–L5) spinal cord motor neurons as revealed by double immunostaining (D) on cryo-unfixed sections for endogenous VEGF-B (B) and the neuronal marker NeuN (C). E, In SOD1 mice aged 105 d and characterized by activated glial cells, VEGF-B expression is also clearly detected on activated astrocytes. F, Expression profile in cDNA preparations from whole lumbar spinal cords (L3–L5) for VEGF-B167, VEGF-B186, total VEGF-B, GFAP (as a marker for astrocyte activation), and NF-L (as a marker for the neurons) during SOD1-mediated motor neuron degeneration. G, Kaplan–Meier curve showing that VEGF-B deficiency accelerates the onset of motor impairment in SOD1 mice by 14 d (p = 0.017). Mice were considered to have a normal motor performance when they were capable of running >1 min on a rotarod. *p < 0.05. Scale bars: (in D) B–D, 50 μm; E, 20 μm.
We also considered the possibility that VEGF-B might be important in disease, and therefore analyzed its expression in mice expressing a SOD1G93A transgene (hereafter referred to as SOD1 mice), a standard mouse model of ALS (Lambrechts et al., 2007). Double immunohistochemistry for VEGF-B and NeuN confirmed that VEGF-B was detectable in motor neurons in this disease model as well (data not shown). VEGF-B was not detectable in quiescent glial cells in presymptomatic SOD1 mice (data not shown), but its expression became prominent and progressively upregulated in activated glial cells in symptomatic SOD1 mice (Fig. 1 E). RT-PCR analysis confirmed that both VEGF-B isoforms were also measurable in primary astrocyte cultures isolated from E14 mouse embryos (Fig. 1 A). Quantification of VEGF-B expression by RT-PCR in lumbar spinal cords of SOD1 mice at different stages of the disease revealed that, compared with preonset (day 50), VEGF-B transcript levels were increased twofold to threefold during the initial stages of the disease at day 80 and 105 (Fig. 1 F); this increase in VEGF-B expression is likely derived from the activated glial cells. At a preterminal disease stage (i.e., when SOD1 mice were completely paralyzed), expression of VEGF-B mRNA in lumbar spinal cords became reduced, presumably because motor neurons, that otherwise produce VEGF-B in healthy conditions (see above), had started to disappear (Fig. 1 F). Loss of motor neurons was indeed revealed by the disappearance of neurofilament-light (NF-L) expression at this stage of the disease (Fig. 1 F).
We also studied whether expression of VEGF-B is regulated by hypoxia or by the presence of mutant SOD1. Because of technical reasons, neuronal N2A cells stably transfected with WT or mutant SOD1 expression plasmids were used, as described previously (Dewil et al., 2007). The expression of VEGF-B in these cells was, however, not regulated by hypoxia [consistent with previous reports in other cell types (Gunningham et al., 2001)], and was also not affected by the expression of WT or mutant SOD1 protein (supplemental Note S1, available at www.jneurosci.org as supplemental material).
VEGF-B is dispensable for motor neuron survival in healthy mice
We then analyzed whether VEGF-B regulates motor neuron development. Despite its expression by embryonic motor neurons, survival and neurite outgrowth of primary cultured motor neurons from E14 VEGF-B−/− embryos were normal, indicating that endogenous production of VEGF-B by motor neurons was not critical (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). VEGF-B was also dispensable for motor neuron development in vivo. Indeed, when determining the number of adult motor neurons with a perikaryal area >200 μm2 in the lumbar spinal cord, we counted 2990 ± 46 neurons in VEGF-B−/− mice compared with 3075 ± 47 neurons in VEGF-B+/+ mice (n = 3 and 4, respectively; p = NS). Consistent with previous reports, we confirmed that VEGF-B−/− mice were healthy and fertile and had a normal longevity (Bellomo et al., 2000; Aase et al., 2001). Additional inspection further revealed that they also did not exhibit any spontaneous signs of abnormal motor performance, muscle weakness, or paralysis (data not shown). When testing open-field explorative behavior, adult VEGF-B−/− mice also exhibited a normal motor performance (supplemental Note S2, available at www.jneurosci.org as supplemental material). Overall, VEGF-B is dispensable for motor neuron development in the embryo, and for motor neuron survival in the adult mouse, at least in healthy conditions (see also below).
Deficiency of VEGF-B aggravates motor neuron degeneration
To investigate whether VEGF-B might regulate motor neuron survival in disease conditions, we intercrossed VEGF-B−/− mice with SOD1 mice. The resulting SOD1 × VEGF-B+/− mice were mated to obtain SOD1 × VEGF-B−/−, SOD1 × VEGF-B+/−, and SOD1 × VEGF-B+/+ littermates with matched genetic backgrounds. Because SOD1 × VEGF-B+/− and SOD1 × VEGF-B+/+ genotypes exhibited indistinguishable phenotypes, they were pooled. SOD1 × VEGF-B+/− and SOD1 × VEGF-B+/+ mice were able to stay on the rotarod until 125 ± 3 d of age (126 ± 3 d for SOD1 × VEGF-B+/− mice and 125 ± 6 d for SOD1 × VEGF-B+/+ mice; p = NS; n = 21) and died after 150 ± 2 d of age. In contrast, SOD1 × VEGF-B−/− mice performed significantly worse on the rotarod: they failed to stay on the rotarod beyond 111 ± 5 d (p = 0.017; n = 12) (Fig. 1 G), and died after 141 ± 2 d (p = 0.05). Nissl staining also revealed an increased loss of motor neurons in preterminal SOD1 × VEGF-B−/− mice at 115 d (α-motor neurons with a perikaryal area >200 μm2 in the lumbar spinal cord: 1593 ± 180 in SOD1 × VEGF-B−/− mice vs 2563 ± 70 in SOD1 × VEGF-B+/+ mice; n = 3 and 3, respectively; p = 0.026). Loss of VEGF-B did, however, not cause any vascular defects or amplify gliosis, as revealed by immunohistochemistry and RT-PCR analysis for endothelial cell and glial cell activation markers (supplemental Note S3, available at www.jneurosci.org as supplemental material). Thus, loss of VEGF-B itself did not cause muscle paralysis, but aggravated motor neuron degeneration in a transgenic mouse model of ALS.
VEGF-B is a neuroprotective factor
Compared with its homolog VEGF, VEGF-B has only minimal angiogenic activity (Y. Li et al., 2008); this prompted us to analyze whether the survival activity of VEGF-B for motor neurons might be attributable to a neuroprotective activity of VEGF-B. We therefore isolated primary motor neurons from E14 rat embryos and investigated whether addition of mouse recombinant VEGF-B could protect primary motor neuron cultures from cell death, which normally occurs when these neurons are deprived of growth factor supplements. The more diffusible isoform VEGF-B186 was used for these experiments, because the shorter VEGF-B167 isoform might get trapped in the extracellular matrix via its heparin-binding domain. When determining the percentage of surviving motor neurons relative to the initial number of motor neurons, VEGF-B186 dose-dependently increased survival (r 2 = 0.97 after a sigmoidal fit in 2 d cultures; EC50 = 73.6 ng/ml) (Fig. 2 A) at concentrations similar to those reported on other cell cultures (Sun et al., 2006; Y. Li et al., 2008). For instance, at day 2, only 55.3 ± 8.0% of motor neurons were still viable in the control-treated cultures, whereas 78.3 ± 2.3% survived after 100 ng/ml VEGF-B186 (p = 0.033 for VEGF-B186) (Fig. 2 A).
Figure 2.
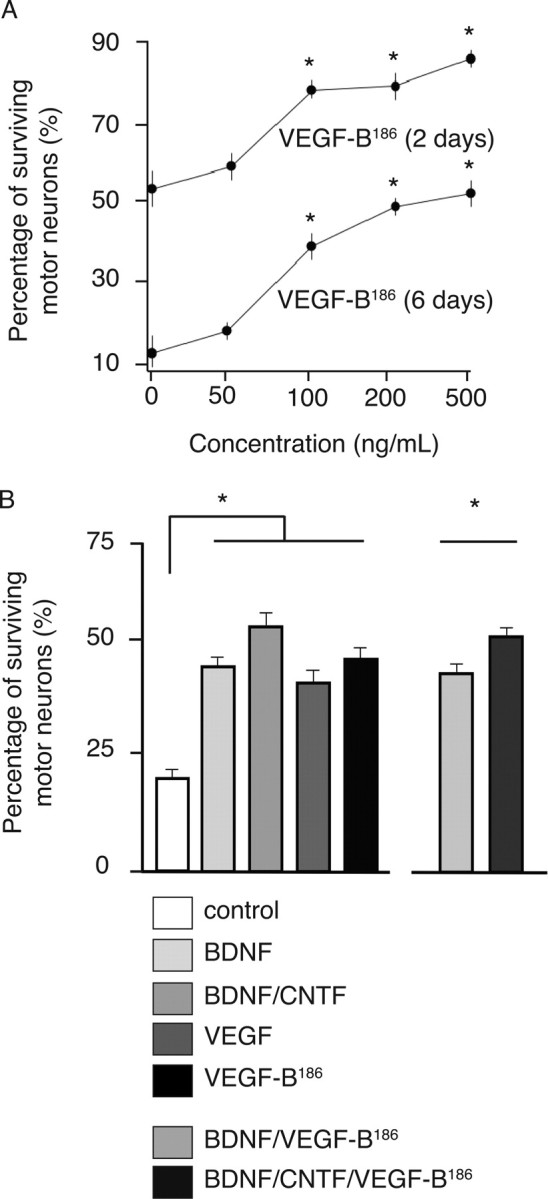
VEGF-B is a neuroprotective factor. A, Dose–response survival effects elicited by recombinant VEGF-B186 proteins on primary motor neuron cultures from E14 rat embryos. A typical sigmoidal dose–response relationship was observed at 2 and 6 d after culture: r 2 = 0.97 and 0.997 after a sigmoidal fit in 2 and 6 d cultures (n = 4 for every concentration tested). B, When applied to primary motor neuron cultures from E14 mouse embryos, VEGF-B186 (100 ng/ml) exerts survival effects similar to VEGF (100 ng/ml) or BDNF (1 ng/ml). p values versus untreated cultures are 5.7 × 10−7, 2.7 × 10−6, and 4.6 × 10−8 for VEGF-B186, VEGF, and BDNF, respectively (n = 8 per condition). CNTF exerted some additional survival effects: a combination of BDNF/CNTF was more effective than VEGF-B186 alone (p = 0.030). Likewise, BDNF/CNTF/VEGF-B186 was slightly more effective than BDNF/VEGF-B186 (p = 0.0022). Motor neuron counts relative to the initial values after plating are shown. *p < 0.05.
In a second series of experiments, we compared the survival effect of VEGF-B186 to that of archetypical growth factors, i.e., brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), or VEGF. To compare their effects, the concentration of each of these proteins was well above those of previously reported EC50 values (Arakawa et al., 1990; Metzger et al., 1998; Oosthuyse et al., 2001; Van Den Bosch et al., 2004). To further confirm our above findings in rat motor neuron cultures, we used an independent method of isolating motor neurons, via which mouse embryonic cultures were enriched to pure motor neuron cultures by panning with an antibody against the p75NTR receptor (Wiese et al., 1999; Bender et al., 2007). At 6 d after isolation, up to 44.4 ± 2.4% of the motor neurons treated with 100 ng/ml recombinant mouse VEGF-B186 had survived compared with only 19.4 ± 1.6% in the untreated cultures (n = 8 and 8, respectively; p = 5.7 × 10−7) (Fig. 2 B). Notably, the magnitude of the trophic effect of VEGF-B186 was comparable to that of other neurotrophic factors, such as VEGF or BDNF: the percentage of surviving motor neurons was 39.5 ± 2.1% after VEGF and 42.9 ± 1.5% after BDNF administration (p = 0.15 and p = 0.61, respectively, vs VEGF-B186; n = 8 and 8, respectively). CNTF exerted, however, a small additional survival effect as 49.3 ± 1.5% of the motor neurons survived with a combination of VEGF-B186/BDNF/CNTF (p = 0.0022 vs VEGF-B186/BDNF). The neuroprotective effect of VEGF-B186 was specific, because VEGF-B186 failed to stimulate neurite outgrowth (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). VEGF-B186 thus differs from BDNF and CNTF, which stimulate neurite outgrowth of primary motor neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), consistent with previous reports (Wiese et al., 1999).
Expression of Flt1 in mouse spinal cord
Because Flt1 is the receptor for VEGF-B, we also examined its expression in the spinal cord of WT and SOD1 mice. Analysis of Flt1 transcript levels by RT-PCR revealed that this receptor was expressed in a spinal motor neuron-enriched neuronal pellet isolated from WT mouse embryos (Fig. 1 A) or from adult mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). ELISA on the motor neuron-enriched pellet isolated from adult motor neurons confirmed that Flt1 was expressed in embryonic and adult motor neurons (respectively, 260 ± 85 and 305 ± 137 pg of Flt1 per mg of protein; n = 3 and 5 per condition). Double immunohistochemistry for NeuN and Flt1, using two different antibodies directed against the C- or N-terminal extracellular domain of Flt1, also revealed that Flt1 was detectable in spinal motor neurons of WT and presymptomatic SOD1 mice (Fig. 3 A–C). In symptomatic SOD1 mice (105 d of age), Flt1 was also expressed by activated GFAP+ glial cells (Fig. 3 D–F). As expected, immunoreactive Flt1 was also detected on blood vessels in healthy and diseased mice (data not shown).
Figure 3.
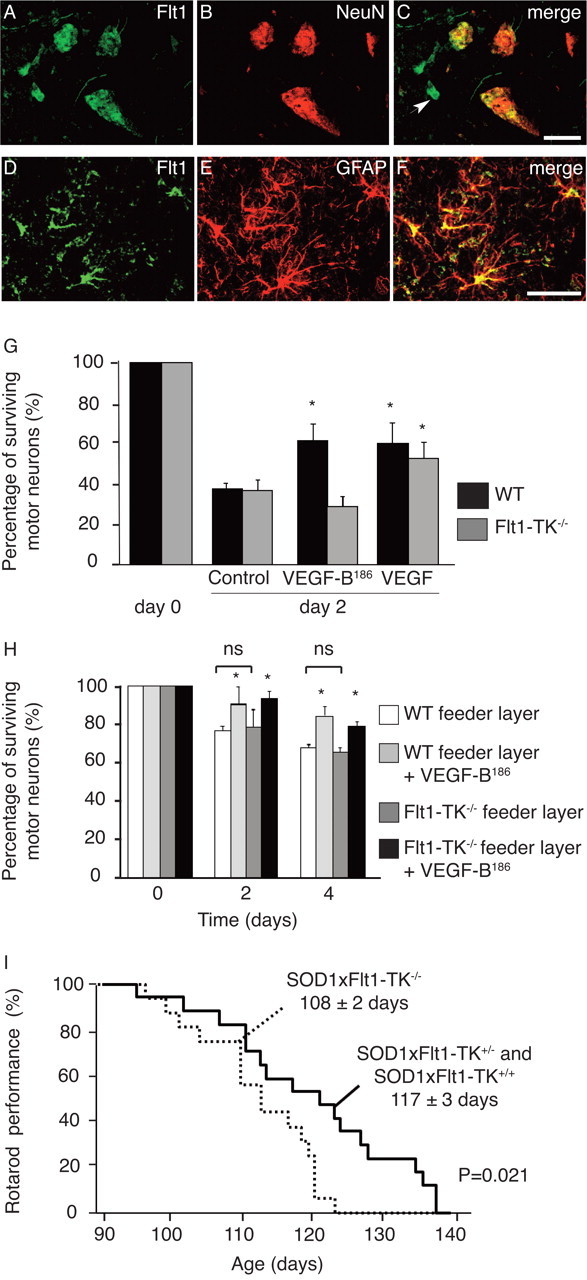
Flt1 is a neuroprotective receptor. A–C, Flt1 expression is detected on lumbar (L3–L5) spinal cord WT motor neurons as revealed by double immunostaining (C) on cryo-unfixed sections for the Flt1 receptor (as shown here using the s.c.-316 antibody) (A) and the neuronal marker NeuN (B). In addition, Flt1 is also expressed on blood vessels (as indicated by the arrow). D–F, Flt1 expression is detected on activated spinal cord astrocytes from symptomatic SOD1 mice as revealed by double immunostaining (F) for the Flt1 receptor (D) and the astrocyte marker GFAP (E). G, After 2 d, the survival of WT and Flt-TK−/− primary embryonic motor neurons was not different. VEGF-B and VEGF recombinant proteins also exert neuroprotective effects on primary motor neurons of WT mouse embryos, compared with untreated cultures (n = 6 for each condition; p = 0.012 and p = 0.034 for VEGF-B and VEGF, respectively). Primary motor neurons isolated from Flt-TK−/− mouse embryos only respond to stimulation by VEGF (p = 0.019) and not to VEGF-B (p = 0.65; n = 6 for each condition). H, Cultures from primary motor neurons of WT mouse embryos were prepared on a feeder layer of astrocytes from WT or Flt-TK−/− mice (n = 4 per condition). The survival of WT motor neurons was indistinguishable, regardless of the genotype of the cocultured astrocytes (p = NS). Supplementation of exogenous VEGF-B to these cocultures improved the survival of WT motor neurons, regardless of the genotype of the astrocytes used (p < 0.05). I, Kaplan–Meier curve showing that SOD1 mice lacking Flt1 tyrosine kinase activity exhibit more severe motor neuron degeneration, because they fall from the rotarod 9 d earlier than their WT littermates (p = 0.021). *p < 0.05. Scale bars: (in C) A–C, (in F) D–F, 50 μm.
Flt1 mediates the neuroprotective effect of VEGF-B
To investigate whether the neuroprotective effect of VEGF-B186 was mediated through Flt1 signaling, we used mice expressing a tyrosine kinase-dead Flt1 (Flt1-TK−/− mice), which cannot transmit a biological effect of VEGF-B (Hiratsuka et al., 1998). We used a maximal dose of 500 ng/ml VEGF-B186 (and, for comparison, VEGF) to avoid the possibility that a lack of effect might be attributable to underdosing. As expected, survival of primary motor neurons of WT mouse embryos was stimulated by VEGF-B186 or VEGF. The percentage of surviving motor neurons after 2 d was 39.0 ± 3.7% in untreated cultures versus 63.8 ± 8.7% and 62.7 ± 11.1% after VEGF-B186 or VEGF, respectively (p = 0.012 and p = 0.034, respectively; n = 6 per condition) (Fig. 3 G). In contrast, VEGF-B186 failed to protect embryonic Flt1-TK−/− motor neurons, whereas VEGF was still effective (percentage of surviving motor neurons: 38.6 ± 5.5% in controls vs 30.0 ± 5.6% after VEGF-B186 and 55.1 ± 8.5% after VEGF; p = 0.65 and p = 0.019; n = 6 per condition) (Fig. 3 G). Thus, VEGF-B186 exerts its survival signals through Flt1, whereas VEGF is still capable of exerting neuroprotective activity through Flk1.
To further explore whether VEGF-B186 stimulated motor neuron survival directly or, indirectly, through effects on Flt1+ astrocytes, we cultured primary motor neurons of WT mouse embryos on a feeder layer of astrocytes, harvested from WT or Flt-TK−/− mice. Survival of WT motor neurons was improved by exogenous VEGF-B, regardless of the genotype of the astrocytes used (Fig. 3 H), indicating that expression of Flt1 on motor neurons, but not on astrocytes, determined the survival of WT motor neurons in these cocultures. The absolute effect of VEGF-B186 in cocultures was somewhat smaller, which is likely because of the fact that astrocytes also release a number of other neuroprotective signals, which already provide substantial trophic support and therefore reduce the relative neuroprotective effect of exogenous neuroprotective factors.
Deficiency of Flt1 signaling aggravates motor neuron degeneration
The in vivo relevance of the Flt1 tyrosine kinase activity for motor neuron degeneration was further studied by intercrossing Flt1-TK−/− mice with SOD1 mice. SOD1 × Flt1-TK+/− and SOD1 × Flt1-TK+/+ genotypes developed muscle weakness at the same age (they were able to stay on a rotarod until 117 ± 3 and 118 ± 3 d of age, respectively; n = 6 and 10, respectively; p = NS). When analyzed as a single group, SOD1 × Flt1-TK+/− and SOD1 × Flt1-TK+/+ mice stayed on the rotarod until 117 ± 3 d and died after 143 ± 2 d of age. In contrast, SOD1 × Flt1-TK−/− mice performed significantly worse: they were unable to stay on the rotarod beyond 108 ± 2 d (n = 17 and 16, respectively; p = 0.021) (Fig. 3 I). However, SOD1 × Flt1-TK−/− mice did not survive for shorter periods as they died at 137 ± 2 d (n = 17 and 16, respectively; p = 0.071). Consistent with the analysis in SOD1 × VEGF-B−/− mice, loss of Flt1 signaling had a greater effect on accelerating disease onset (rotarod performance) than on decreasing overall survival (see Discussion).
Intracerebroventricular VEGF-B delivery prolongs the survival of ALS rats
To explore whether the neuroprotective effects of VEGF-B could be exploited therapeutically, we delivered recombinant VEGF-B protein intracerebroventricularly to SOD1 rats, using similar techniques that we previously used to continuously deliver recombinant VEGF (Storkebaum et al., 2005). This delivery route was chosen because previous studies demonstrated that intracerebroventricularly delivered VEGF diffuses from the CSF into the neural parenchyma, where it reaches its target motor neurons (Storkebaum et al., 2005). Because the VEGF-B186 isoform is more diffusible than VEGF-B167, we used VEGF-B186 for these experiments. Briefly, osmotic minipumps, preloaded with protein, were implanted subcutaneously, and a connected catheter was inserted in the left lateral ventricle and securely fixed to avoid movements. Because these pumps deliver protein continuously during only 4 weeks, we replaced them every 25 d, paying great care that the catheter remained patent until the end of the 100 d treatment period. For these experiments, SOD1 rats crossed on a HAN Wistar genetic background were used. These rats exhibit a considerable interlitter variability of the age at disease onset ranging from 103 to 151 d (as determined by their capacity to perform on a rotarod), and an aggressive disease course: they died on average within 6 ± 1 d of disease onset. SOD1 rats also displayed two types of disease onset, with onset of paralysis in either the hindlimbs or forelimbs. In general, disease symptoms occurred earlier and progressed more rapidly in SOD1 rats with forelimb disease onset (Storkebaum et al., 2005).
We first confirmed that the VEGF-B186 protein remained active in osmotic pumps for up to 4 weeks (supplemental Note S4, available at www.jneurosci.org as supplemental material) and then filled these pumps with 10 μg of recombinant mouse VEGF-B186, a dose equivalent to 0.2 μg/kg/d, which has previously been shown to stimulate neurogenesis in adult rats when delivered during 3 d (Sun et al., 2006). At the age of 60 d (i.e., ∼30 d before disease onset), catheters were implanted intracerebroventricularly into SOD1 rats, delivering a continuous influx of VEGF-B186 at a rate of 0.2 μg/kg/d. This dose was well tolerated without inducing any obvious side effects, such as weight loss or reduced spontaneous cage activity (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Compared with SOD1 rats receiving control artificial CSF, SOD1 rats treated with VEGF-B186 did not perform better on the rotarod than the CSF-treated rats (131 ± 4 d for CSF-treated rats vs 142 ± 4 d for VEGF-B186-treated rats; n = 11–13; p = 0.073) (Fig. 4 A). In contrast, VEGF-B186-treated rats survived significantly longer (137 ± 5 d for CSF-treated rats vs 152 ± 3 d for VEGF-B186-treated rats; p = 0.016) (Fig. 4 B). When counting the number of motor neurons in the facial nucleus on Nissl-stained brainstem sections of preterminal SOD1 rats, VEGF-B186-treated rats also contained a larger number of motor neurons than CSF-treated control rats: 3352 ± 73 motor neurons per facial nucleus after VEGF-B186 versus 2915 ± 74 after CSF; n = 8 and 6, respectively; p = 0.029 (Fig. 4 C,D). In addition, VEGF-B186 treatment changed the disease subtype from a severe to a milder form, as was observed for VEGF (Storkebaum et al., 2005). Indeed, after VEGF-B186 delivery, significantly fewer SOD1 rats suffered forelimb disease than after control CSF (3 of 11 VEGF-B186-treated rats vs 7 of 10 CSF-treated rats developed forelimb onset; p = 0.047 by likelihood ratio analysis). This suggests that especially motor neurons in the brainstem and cervical spinal cord were protected, presumably because intracerebroventricular delivery of VEGF-B186 results in a spatial gradient, with higher levels of VEGF-B186 in the rostral than caudal spinal cord, similar to what has been observed after intracerebroventricular delivery of VEGF (Storkebaum et al., 2005).
Figure 4.
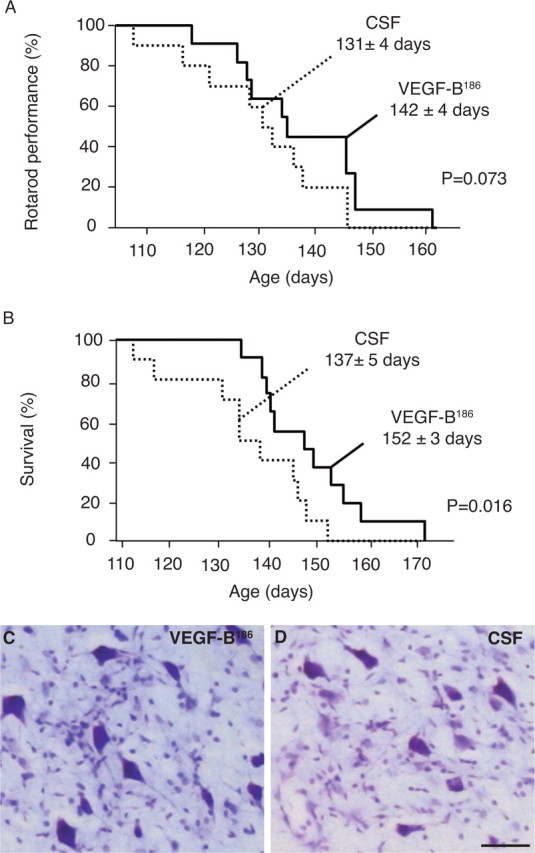
Therapeutic effect of VEGF-B in motor neuron degeneration. A, B, Intracerebroventricular delivery of VEGF-B to SOD1 rats delays motor neuron degeneration only slightly (11 d; p = 0.073) (A), but prolongs survival significantly in SOD1 rats by 15 d (p = 0.016) (B). C, D, Nissl staining in the facial nucleus showing that preterminal CSF-treated SOD1 rats (D) have more severe motor neuron loss than preterminal SOD1 rats treated with recombinant VEGF-B186 (C). Scale bar, 50 μm.
Safety profile of VEGF-B186 after intracerebroventricular delivery
When administered at low quantities (0.02 μg/kg/d, i.c.v.), VEGF was neuroprotective without causing adverse vascular effects (Storkebaum et al., 2005). In contrast, intracerebroventricular delivery of higher amounts (0.2 or 2 μg/kg/d) of VEGF induced angiogenesis, edema, and ventriculomegaly (Harrigan et al., 2003). Because VEGF-B is a much weaker angiogenic and permeability factor (Y. Li et al., 2008), and in fact may only have some cardioselective angiogenic activity in the ischemic myocardium (Tirziu et al., 2007), it might have a greater safety profile than VEGF. We therefore compared the effects of a similar dose of VEGF and VEGF-B186 (0.2 μg/kg/d VEGF-B186 was therapeutic) on astrogliosis and angiogenesis at 1 week after delivery; this analysis was performed in the brain parenchyma around the left lateral ventricle, close to where the needle was positioned (ipsilateral site), and at a site more distant from the needle implantation (i.e., around the right lateral ventricle; contralateral site). Angiogenesis and astrogliosis were scored by immunostaining for blood vessel markers (Glut1) and glial markers (GFAP), respectively.
Delivery of 0.2 μg/kg/d VEGF induced angiogenesis, both at the ipsilateral and contralateral site, with some effects on astrogliosis (Table 1). Remarkably, VEGF induced a substantial decrease in immunoreactivity for EBA, a marker, which only stains vessels with an intact blood–brain barrier (BBB), indicating that this dose of VEGF also increased vessel permeability (p < 0.05 vs CSF and VEGF-B186; Table 1). In contrast, delivery of 0.2 μg/kg/d VEGF-B186 (which was therapeutic; see above) did not induce any of such angiogenic or astrogliosis side effects (Table 1).
Table 1.
In contrast to VEGF, intracerebroventricular delivery of a similar VEGF-B186 dose does not induce angiogenesis or BBB permeability
| Site of injection |
Contralateral site |
|||||
|---|---|---|---|---|---|---|
| CSF (n = 7) | VEGF (n = 7) | VEGF-B (n = 7) | CSF (n = 7) | VEGF (n = 7) | VEGF-B (n = 7) | |
| Glut1 | 1.68 ± 0.08 | 2.28 ± 0.23* | 1.89 ± 0.21 | 1.56 ± 0.19 | 2.02 ± 0.29 | 1.43 ± 0.16 |
| EBA | 1.28 ± 0.13 | 0.84 ± 0.12*,# | 1.10 ± 0.11 | 1.35 ± 0.13 | 1.41 ± 0.21 | 1.52 ± 0.11 |
| GFAP | 3.15 ± 0.50 | 4.00 ± 0.73 | 3.75 ± 0.7 | 2.02 ± 0.25 | 2.72 ± 0.36 | 2.15 ± 0.26 |
Percentages of Glut1, EBA, and GFAP immunoreactivity relative to a representative area around the injection site are shown. *p < 0.05 versus CSF; # p < 0.05 versus VEGF-B analysis.
Overall, these findings implicate VEGF-B186 as a potent and safe therapeutic candidate for the treatment of motor neuron degeneration.
Discussion
The realization that some angiogenic factors, such as VEGF, might have evolved from the nervous system, prompted us to explore whether other factors with presumed angiogenic activity might have a role in the nervous system as well (Zacchigna et al., 2008). In this study, we analyzed the role of VEGF-B, a structural homolog of VEGF, which has been traditionally classified as an angiogenic factor. However, VEGF-B has only weak angiogenic activity, and only in a restricted set of conditions. A cardinal finding of our study is therefore that VEGF-B, rather than acting as an angiogenic factor in the nervous system, exerts neuroprotective effects.
Although VEGF-B was discovered as a putative angiogenic factor, it might have arisen in evolution as a neuronal factor. Indeed, a genomic survey of Caenorhabditis elegans, which is devoid of a vascular system, predicts four genes resembling the VEGF receptors, named ver-1 to ver-4, and a single ancestral VEGF/PDGF ligand (Popovici et al., 2002). Intriguingly, these receptors are expressed in specialized cells of neural origin, such as support glial cells of amphid and phasmid neurons, as well as chemosensorial neurons. Vessels, which developed later in evolution than nerves, thus seem to have coopted the ancestral VEGF/PDGF family as a critical regulator of their formation (Zacchigna et al., 2008). Because VEGF family members, during evolution, originated from a common VEGF ancestor, it is possible, if not likely, that VEGF-B has kept the original function of the ancestral VEGF/PDGF ligand in the nervous system. Interestingly, the neuroprotective effects of VEGF-B were restricted to pathological conditions. Indeed, mice lacking VEGF-B displayed a normal motor behavior, indicating that VEGF-B is not required for the survival of (motor) neurons in healthy conditions. However, when challenged in a model of neurodegenerative disease, VEGF-B-deficient mice displayed a more severe form of motor neuron degeneration, thus illustrating that VEGF-B plays a role in diseases of the nervous system.
How does VEGF-B protect against neurodegeneration? Although it is possible that VEGF-B might exert some of its neuroprotective effects indirectly via effects on the vasculature, several findings indicate that VEGF-B affects neurons directly. First, loss or delivery of VEGF-B did not cause any vascular defects or abnormalities. Second, adult motor neurons express Flt1, indicating that they can respond to VEGF-B. Third, VEGF-B186 stimulated the survival of primary motor neurons in vitro. Remarkably, the magnitude of the observed neuroprotective effects was comparable to that of VEGF or other classical neurotrophic factors for motor neurons (BDNF and CNTF). Fourth, Flt1 is expressed on motor neurons, and its signaling through the tyrosine kinase domain is essential to protect these neurons against degeneration. The finding that loss of VEGF-B or of the TK activity of Flt1 had a greater impact on disease onset than on overall survival may suggest that endogenous VEGF-B is important for protection of motor neurons early in the disease, but that its survival signaling becomes insufficient or redundant when motor neurons are severely crippled in a more preterminal stage of the disease.
Another intriguing observation is that VEGF, in contrast to VEGF-B186, is still capable of exerting its neuroprotective effects on motor neurons isolated from Flt1-TK−/− mice. This indicates that VEGF exerts its neuroprotection primarily through Flk1, whereas VEGF-B186, as expected, acts via Flt1. In contrast to VEGF-B186, VEGF also has effects on vascular permeability and endothelial cell proliferation and migration through Flk1 (Shibuya, 2001). Indeed, when delivering comparable quantities of VEGF and VEGF-B186 intracerebroventricularly, VEGF induced angiogenesis and BBB leakiness, as indicated by the reduced expression of EBA (a marker of vessels with an intact BBB). We previously demonstrated that VEGF can be delivered intracerebroventricularly at a therapeutic dose that prolongs the survival of SOD1 rats, without causing major vascular side effects (Storkebaum et al., 2005). However, the angiogenic and permeability-promoting activity of VEGF precludes unrestricted delivery of high doses, which might, otherwise, perhaps have increased the therapeutic benefit even more. In contrast, VEGF-B186 did not induce any such vascular side effects, but protected SOD1 rats from motor neuron degeneration; the greater safety of VEGF-B may, therefore, provide an attractive alternative to VEGF. Although delivery of VEGF-B (at the used dose of 0.2 μg/kg/d) already significantly prolonged overall survival, its beneficial effects on delaying the onset of paralysis would very likely have been more significant, if we could have used a higher dose of VEGF-B. However, financial restraints prevented us from testing this appealing strategy.
Because Flt1 expression is also detectable on glial cells, VEGF-B186 might also promote motor neuron survival through indirect effects on glia (e.g., by mediating glial proliferation and/or the secretion of neuroprotective factors by glia). However, when added to cocultures of motor neurons and astrocytes, VEGF-B186 exerted its neuroprotective effects on motor neurons independently of the Flt1 tyrosine kinase activity in astrocytes, suggesting that VEGF-B primarily affected neurons, rather than astrocytes. The role of Flt1 in the nervous system therefore still remains unknown: Flt1 expression on astrocytes is prominently induced after nervous system injury, but the biological significance of this upregulation, and whether it might contribute to neuroprotection, remains enigmatic so far. Similar to Flt1, VEGF-B is expressed in normal motor neurons and is upregulated in activated astrocytes from mutant SOD1 mice. It is therefore possible that under healthy conditions, motor neurons express VEGF-B to maintain neuroprotection in an autocrine manner, whereas astrocytes may express VEGF-B after injury to maintain survival in a paracrine manner. Another possibility, however, is that VEGF-B also signals intracellularly, as recently reported for VEGF (Lee et al., 2007).
Our findings reveal that the VEGF-B186 isoform plays an important role in the nervous system. Previous studies addressing the functional relevance of VEGF-B have mainly focused on the biological effects of the VEGF-B isoform 167 (VEGF-B167). The finding that VEGF-B186 exerts neuroprotective effects on cultured neurons in vitro and in a murine model of neurodegeneration is therefore novel. In addition, our findings may also be relevant from a therapeutic perspective. Indeed, delivery of recombinant neuroprotective factors for the treatment of neurodegenerative disease has been hampered by the limited capacity of these factors to cross the BBB, or to diffuse from the CSF into the neural parenchyma. We therefore anticipate that VEGF-B167, which binds to heparin sulfate proteoglycans and is partly sequestered in the extracellular matrix (Olofsson et al., 1996), would diffuse less after intracerebroventricular delivery and might have smaller therapeutic effects, compared with the more soluble and diffusible VEGF-B186. Overall, delivery of VEGF-B may offer therapeutic opportunities for neurodegenerative diseases that develop spontaneously.
Footnotes
D.L., P.V.D., E.B., and M.T. were supported by the Research Foundation Flanders (FWO), Belgium. K.P., J.D., B.C., and A.V. were supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). D.L. was supported by the “Schenking Rimaux-Bartier.” P.C. was supported by grants from the Geneeskundige stichting Koningin Elisabeth, the Geconcerteerde Onderzoeksacties–Katholieke Universiteit Leuven 2006/11, the FWO G 0210.07, Methusalem, and the Muscular Dystrophy Association. W.R. was supported by the ALS Association, by the Interuniversity Attraction Poles program P5/19 and P6/43 of the Belgian Federal Science Policy Office. We acknowledge Ann Bouché, Annemie Van Den Broeck, Martine Nijs, Joke Ver Elst, Ann Manderveld, An Carton, Maria De Mol, Sandra Jansen, Leen Notebaert, Sabine Wyns, and Susanne Knorr for technical assistance.
P.C. is named as an inventor on a patent application regarding the use of VEGF to treat amyotrophic lateral sclerosis. The Flanders Institute for Biotechnology (VIB) is one of the joint owners of this patent application, and the said patent application has been licensed to an outside company. Neither VIB nor P.C. has equity stakes in the company. However, VIB and P.C. stand to eventually receive royalties.
References
- Aase K, von Euler G, Li X, Pontén A, Thorén P, Cao R, Cao Y, Olofsson B, Gebre-Medhin S, Pekny M, Alitalo K, Betsholtz C, Eriksson U. Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation. 2001;104:358–364. doi: 10.1161/01.cir.104.3.358. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Sendtner M, Thoenen H. Survival effect of ciliary neurotrophic factor (CNTF) on chick embryonic motoneurons in culture: comparison with other neurotrophic factors and cytokines. J Neurosci. 1990;10:3507–3515. doi: 10.1523/JNEUROSCI.10-11-03507.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9:936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Bellomo D, Headrick JP, Silins GU, Paterson CA, Thomas PS, Gartside M, Mould A, Cahill MM, Tonks ID, Grimmond SM, Townson S, Wells C, Little M, Cummings MC, Hayward NK, Kay GF. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res. 2000;86:E29–E35. doi: 10.1161/01.res.86.2.e29. [DOI] [PubMed] [Google Scholar]
- Bender FL, Fischer M, Funk N, Orel N, Rethwilm A, Sendtner M. High-efficiency gene transfer into cultured embryonic motoneurons using recombinant lentiviruses. Histochem Cell Biol. 2007;127:439–448. doi: 10.1007/s00418-006-0247-5. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim HY, Cha JH, Choi JY, Park SI, Jeong CH, Jeun SS, Lee MY. Upregulation of vascular endothelial growth factor receptors Flt-1 and Flk-1 following acute spinal cord contusion in rats. J Histochem Cytochem. 2007;55:821–830. doi: 10.1369/jhc.6A7139.2007. [DOI] [PubMed] [Google Scholar]
- Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W, Van den Bosch L. Vascular endothelial growth factor counteracts the loss of phospho-Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2007;33:499–509. doi: 10.1111/j.1365-2990.2007.00850.x. [DOI] [PubMed] [Google Scholar]
- Forstreuter F, Lucius R, Mentlein R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J Neuroimmunol. 2002;132:93–98. doi: 10.1016/s0165-5728(02)00315-6. [DOI] [PubMed] [Google Scholar]
- Gunningham SP, Currie MJ, Han C, Turner K, Scott PA, Robinson BA, Harris AL, Fox SB. Vascular endothelial growth factor-B and vascular endothelial growth factor-C expression in renal cell carcinomas: regulation by the von Hippel-Lindau gene and hypoxia. Cancer Res. 2001;61:3206–3211. [PubMed] [Google Scholar]
- Harrigan MR, Ennis SR, Sullivan SE, Keep RF. Effects of intraventricular infusion of vascular endothelial growth factor on cerebral blood flow, edema, and infarct volume. Acta Neurochir (Wien) 2003;145:49–53. doi: 10.1007/s00701-002-1035-1. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: therapeutic implications for motor neuron disease. Biochim Biophys Acta. 2006;1762:1109–1121. doi: 10.1016/j.bbadis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Robberecht W, Carmeliet P. Heterogeneity in motoneuron disease. Trends Neurosci. 2007;30:536–544. doi: 10.1016/j.tins.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tjwa M, Van Hove I, Enholm B, Neven E, Paavonen K, Jeltsch M, Diez Juan T, Sievers RE, Chorianopoulos E, Wada H, Vanwildemeersch M, Noel A, Foidart JM, Springer ML, von Degenfeld G, Dewerchin M, Blau HM, Alitalo K, Eriksson U, et al. Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler Thromb Vasc Biol. 2008;28:1614–1620. doi: 10.1161/ATVBAHA.107.158725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, Hua J, Matsuo O, Fong GH, Ding H, Cao Y, Becker KG, Nash A, Heldin CH, Li X. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. 2008;118:913–923. doi: 10.1172/JCI33673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- Mani N, Khaibullina A, Krum JM, Rosenstein JM. Astrocyte growth effects of vascular endothelial growth factor (VEGF) application to perinatal neocortical explants: receptor mediation and signal transduction pathways. Exp Neurol. 2005;192:394–406. doi: 10.1016/j.expneurol.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Metzger F, Wiese S, Sendtner M. Effect of glutamate on dendritic growth in embryonic rat motoneurons. J Neurosci. 1998;18:1735–1742. doi: 10.1523/JNEUROSCI.18-05-01735.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash AD, Baca M, Wright C, Scotney PD. The biology of vascular endothelial growth factor-B (VEGF-B) Pulm Pharmacol Ther. 2006;19:61–69. doi: 10.1016/j.pupt.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Olofsson B, Pajusola K, von Euler G, Chilov D, Alitalo K, Eriksson U. Genomic organization of the mouse and human genes for vascular endothelial growth factor B (VEGF-B) and characterization of a second splice isoform. J Biol Chem. 1996;271:19310–19317. doi: 10.1074/jbc.271.32.19310. [DOI] [PubMed] [Google Scholar]
- Olofsson B, Korpelainen E, Pepper MS, Mandriota SJ, Aase K, Kumar V, Gunji Y, Jeltsch MM, Shibuya M, Alitalo K, Eriksson U. Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad Sci U S A. 1998;95:11709–11714. doi: 10.1073/pnas.95.20.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646–25654. [PubMed] [Google Scholar]
- Popovici C, Isnardon D, Birnbaum D, Roubin R. Caenorhabditis elegans receptors related to mammalian vascular endothelial growth factor receptors are expressed in neural cells. Neurosci Lett. 2002;329:116–120. doi: 10.1016/s0304-3940(02)00595-5. [DOI] [PubMed] [Google Scholar]
- Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct. 2001;26:25–35. doi: 10.1247/csf.26.25. [DOI] [PubMed] [Google Scholar]
- Sondell M, Sundler F, Kanje M. Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci. 2000;12:4243–4254. doi: 10.1046/j.0953-816x.2000.01326.x. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA. Increased severity of cerebral ischemic injury in vascular endothelial growth factor-B-deficient mice. J Cereb Blood Flow Metab. 2004;24:1146–1152. doi: 10.1097/01.WCB.0000134477.38980.38. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA. Vascular endothelial growth factor-B (VEGFB) stimulates neurogenesis: evidence from knockout mice and growth factor administration. Dev Biol. 2006;289:329–335. doi: 10.1016/j.ydbio.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, Roncal C, Eriksson U, Fu Q, Elfenbein A, Hall AE, Carmeliet P, Moons L, Simons M. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest. 2007;117:3188–3197. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe W, Van Den Bosch L, Robberecht W. Glial cells potentiate kainate-induced neuronal death in a motoneuron-enriched spinal coculture system. Brain Res. 1998;807:1–10. doi: 10.1016/s0006-8993(98)00569-1. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Storkebaum E, Vleminckx V, Moons L, Vanopdenbosch L, Scheveneels W, Carmeliet P, Robberecht W. Effects of vascular endothelial growth factor (VEGF) on motor neuron degeneration. Neurobiol Dis. 2004;17:21–28. doi: 10.1016/j.nbd.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Wiese S, Metzger F, Holtmann B, Sendtner M. The role of p75NTR in modulating neurotrophin survival effects in developing motoneurons. Eur J Neurosci. 1999;11:1668–1676. doi: 10.1046/j.1460-9568.1999.00585.x. [DOI] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]