

Abstract
Ischemic preconditioning (PC) of the brain is a phenomenon by which mild ischemic insults render neurons resistant to subsequent strong insults. Key steps in ischemic PC of the brain include caspase-3 activation and poly(ADP-ribose) polymerase-1 (PARP-1) cleavage, but upstream events have not been clearly elucidated. We have tested whether endogenous zinc is required for ischemic PC of the brain in rats. Mild, transient zinc accumulation was observed in certain neurons after ischemic PC. Moreover, intraventricular administration of CaEDTA during ischemic PC abrogated both zinc accumulation and the protective effect against subsequent full ischemia. To elucidate the mechanism of the zinc-triggered PC (Zn PC) effect, cortical cultures were exposed to sublethal levels of zinc, and 18 h later to lethal levels of zinc or NMDA. Zn PC exhibited the characteristic features of ischemic PC, including caspase-3 activation, PARP-1 cleavage, and HSP70 induction, all of which are crucial for subsequent neuroprotection against NMDA or zinc toxicity. HSP70 induction was necessary for protection, as it halted caspase-3 activation before apoptosis. Interestingly, in both Zn PC in vitro and ischemic PC in vivo, p75NTR was necessary for neuroprotection. These results suggest that caspase-3 activation during ischemic PC, a necessary event for subsequent neuroprotection, may result from mild zinc accumulation and the consequent p75NTR activation in neurons.
Keywords: poly(ADP-ribose) polymerase, caspase-3, heat shock protein 70, ischemia, zinc toxicity, excitotoxicity
Introduction
Whereas mechanisms such as calcium-overload excitotoxicity and oxidative injury may concertedly contribute to ischemic neuronal death, a growing body of evidence suggests that neurotoxicity triggered by zinc dyshomeostasis is a key contributing factor under certain conditions (Choi and Koh, 1998; Weiss et al., 2000; Koh, 2001; Capasso et al., 2005; Galasso and Dyck, 2007). For example, labile zinc has been shown to accumulate in degenerating neuronal cell bodies and nuclei after ischemia, and inhibition of zinc accumulation with chelators has been found to protect against ischemic neuronal death (Koh et al., 1996; Lee et al., 2002).
Ischemic preconditioning (PC) of the brain is an interesting phenomenon, in which mild ischemic insults render neurons more resistant to subsequent strong insults. Although ischemic PC results in the reduction of neuronal cell death during subsequent insults, qualitatively the same injury mechanisms as in ischemic neuronal death are likely to underlie this phenomenon of endogenous neuroprotection (Andoh et al., 2002; Dirnagl et al., 2003; Gidday, 2006).
The cascade of events leading to ischemic PC of the brain is not completely understood, although significant progress has been made in understanding its mechanism. Among the steps shown to be involved in the mechanism of ischemic PC are poly(ADP-ribose) polymerase (PARP) inhibition (Liaudet et al., 2001), caspase-3 activation (McLaughlin et al., 2003), and PARP-1 cleavage by caspase-3 (Garnier et al., 2003). In addition, heat shock protein 70 (HSP70), which regulates apoptotic cell death (Beere et al., 2000; Saleh et al., 2000; Steel et al., 2004; Stankiewicz et al., 2005), may be involved in ischemic PC (McLaughlin et al., 2003; Oksala et al., 2004).
A key question in ischemic PC is the mechanism by which sublethal caspase-3 activation and PARP-1 cleavage occur, but not severe, full-blown caspase-3 activation and apoptosis. Zinc has been found to activate caspase-3-dependent apoptosis through the p75NTR pathway (Park et al., 2000), or to induce PARP-dependent oxidative necrosis (Kim and Koh, 2002), depending on the degree of fulminance. Because ischemic PC may share the same mechanisms as ischemic injury (Andoh et al., 2002; Dirnagl et al., 2003; Gidday, 2006), we hypothesized that endogenous zinc plays a key role, not only in ischemic neuronal death (Koh et al., 1996) but also in ischemic PC. Hence, a mild degree of ischemia may induce sublethal zinc toxicity in neurons, triggering caspase-3 activation and PARP-1 cleavage. Because fulminant zinc toxicity requires PARP activation, the absence of PARP may protect against zinc toxicity.
Materials and Methods
Ischemic preconditioning and transient middle cerebral artery.
Male Sprague Dawley rats (300 ± 20 g, 8–9 weeks) were obtained from Charles River Laboratories. All animal experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (University of Ulsan, Seoul, South Korea). Each rat was anesthetized with an intramuscular injection of 50 mg/kg Zoletil, a 1:1 (weight:weight) mixture of tiletamine and zolazepam (Virbac). The left femoral artery was cannulated for monitoring arterial blood pressure (Stoelting) and sampling blood. An Opti critical care analyzer (Roche Diagnostics) was used to analyze hemoglobin, hematocrit, pH, arterial oxygen pressure (pO2) and partial pressure of carbon dioxide (pCO2) in arterial blood samples. Rectal and temporalis muscle temperatures (Physitemp) were maintained at 36.0 ± 0.5°C with a temperature control unit (Harvard). Physiological parameters before and after middle cerebral artery occlusion (MCAO) did not differ significantly. Transient ischemia in the MCA territory was induced as described previously (Longa et al., 1989). Under a surgical microscope, the right common (CCA), external (ECA), and internal carotid (ICA) arteries were identified and separated from the vagus nerve. The ECA and CCA were ligated with 4-0 silk sutures, and the ICA was temporarily clipped. A small incision in the CCA was made 1 mm proximal to the bifurcation, and a 4-0 suture (Ethylon surgical monofilament polyamide; Ethicon), the tip of which was fire polished to a diameter of 0.38–0.40 mm, was inserted into the ICA. After releasing the clip, the suture was advanced into the proximal portion of the MCA to ∼20 mm from the carotid bifurcation. Each surgical operation was completed within 15 min.
Ischemic preconditioning (IPC) was induced by 10 min of transient MCAO (tMCAO), followed 72 h later by 1 h tMCAO. Physiological parameters immediately before and after IPC and before and after 1 h MCA occlusion did not differ significantly between groups (Table 1). The experimental protocol is summarized in Figure 1A.
Table 1.
Physiological parameters (mean ± SEM, n = 4) immediately before and after 10 min (PC) or 1 h MCA occlusion
| Temporalis temperature (°C) | MABP (mmHg) | pH | pCO2 (mmHg) | pO2 (mmHg) | Hematocrit (%) | ||
|---|---|---|---|---|---|---|---|
| Ischemic preconditioning | |||||||
| Saline | Pre | 34.8 ± 0.3 | 138.3 ± 4.7 | 7.43 ± 0.0 | 46.8 ± 1.8 | 101.5 ± 1.3 | 37.3 ± 1.0 |
| Post | 35.0 ± 0.3 | 135.5 ± 4.1 | 7.38 ± 0.0 | 46.5 ± 0.5 | 88.0 ± 4.7 | 39.3 ± 1.2 | |
| CaEDTA | Pre | 33.9 ± 0.4 | 146.0 ± 5.5 | 7.42 ± 0.0 | 44.0 ± 2.6 | 105.7 ± 3.4 | 34.7 ± 2.3 |
| Post | 34.0 ± 0.4 | 146.0 ± 8.0 | 7.42 ± 0.0 | 40.7 ± 3.9 | 98.0 ± 1.5 | 35.3 ± 1.2 | |
| ZnEDTA | Pre | 33.4 ± 0.6 | 148.5 ± 15.5 | 7.32 ± 0.0 | 43.0 ± 2.0 | 117.5 ± 0.4 | 42.5 ± 0.6 |
| Post | 33.8 ± 0.6 | 152.5 ± 16.5 | 7.28 ± 0.0 | 50.5 ± 3.5 | 86.5 ± 8.5 | 44.0 ± 2.0 | |
| ap75NTR | Pre | 36.1 ± 0.1 | 145.0 ± 15.0 | 7.35 ± 0.0 | 48.0 ± 1.0 | 85.5 ± 8.5 | 40.0 ± 4.0 |
| Post | 35.9 ± 0.0 | 140.0 ± 14.0 | 7.34 ± 0.1 | 52.5 ± 5.5 | 75.0 ± 10.0 | 42.0 ± 1.0 | |
| PC plus 1 tMCAO | |||||||
| Saline | Pre | 35.8 ± 0.3 | 153.3 ± 6.9 | 7.44 ± 0.0 | 47.0 ± 2.0 | 96.0 ± 3.6 | 39.7 ± 0.3 |
| Post | 35.7 ± 0.3 | 148.5 ± 9.0 | 7.45 ± 0.0 | 44.7 ± 1.5 | 95.7 ± 5.5 | 42.0 ± 4.5 | |
| CaEDTA | Pre | 35.9 ± 0.1 | 147.3 ± 7.0 | 7.40 ± 0.0 | 47.7 ± 1.8 | 88.7 ± 2.9 | 45.0 ± 0.6 |
| Post | 36.2 ± 0.2 | 136.0 ± 8.5 | 7.54 ± 0.1 | 38.3 ± 4.5 | 89.3 ± 3.9 | 44.0 ± 0.6 | |
| ZnEDTA | Pre | 36.0 ± 0.5 | 135.0 ± 6.0 | 7.40 ± 0.0 | 46.0 ± 1.0 | 82.0 ± 22.0 | 35.5 ± 0.5 |
| Post | 35.5 ± 0.5 | 139.0 ± 0.0 | 7.39 ± 0.0 | 44.5 ± 0.5 | 91.5 ± 2.5 | 42.0 ± 0.0 | |
| ap75NTR | Pre | 36.1 ± 0.8 | 145.7 ± 6.4 | 7.43 ± 0.0 | 46.3 ± 1.8 | 95.3 ± 3.8 | 40.0 ± 1.0 |
| Post | 35.8 ± 0.2 | 141.0 ± 9.5 | 7.46 ± 0.0 | 39.5 ± 3.5 | 85.5 ± 8.5 | 39.0 ± 1.0 | |
| Sham plus 1 tMCAO | Pre | 36.7 ± 0.5 | 140.3 ± 10.8 | 7.44 ± 0.0 | 47.3 ± 0.0 | 103.0 ± 4.6 | 40.7 ± 0.9 |
| Post | 35.9 ± 0.3 | 141.7 ± 8.4 | 7.46 ± 0.0 | 42.0 ± 2.1 | 87.3 ± 3.8 | 39.7 ± 1.7 | |
Rats were treated with intraventricular injections of saline, CaEDTA, ZnEDTA, or anti-p75NTR function-blocking antibody (ap75NTR).
Figure 1.

Diagram for in vivo ischemic PC and in vitro Zn PC. A, The protocol for in vivo IPC experiments. IPC was induced by a period of 10 min MCAO. Drugs were injected into the lateral ventricle 30 min before IPC. One hour tMCAO to induce ischemic injury was performed 72 h after IPC. Infarct volume was assessed with TTC staining 24 h later. B, The protocol for Zn PC. Cultures were exposed for 12–24 h to a sublethal concentration of zinc (30 μm). In some cultures, Western blots were performed after Zn PC. In others, after washout, cultures were exposed for 24 h to lethal concentrations of toxins (40 μm Zn, 30 μm NMDA, 50 μm glutamate, or 30 μm Fe2+). Neuronal cell death was assessed by LDH release 18 h later.
Laser-Doppler flowmetry.
Laser-Doppler flowmetry (LDF) (BLF21D; Transonic Instrument) was performed as described (Dirnagl et al., 1989). For placement of the LDF probe, a burr hole (2 mm diameter) was drilled 2 mm posterior and 6 mm lateral to the bregma, with care taken not to injure the underlying dura mater. Animals were placed in the supine position, and the skull was firmly immobilized in a stereotaxic frame. Cerebral blood flow (CBF) was recorded 10 min before MCAO or PC, 5 min after PC, 20 min after MCAO and 30 min after reperfusion or PC. The CBF values were expressed as a percentage of the baseline value. LDF differed little between groups, before and after MCA occlusion (Table 2).
Table 2.
Laser-Doppler flowmetry data (mean ± SEM, n = 8) immediately before and after 10 min (PC) or 1 h MCA occlusion
| Preconditioning (% of baseline) |
tMCAO (% of baseline) |
|||
|---|---|---|---|---|
| Occlusion | Reperfusion | Occlusion | Reperfusion | |
| Sham | 22.0 ± 6.7 | 95.8 ± 19.8 | ||
| PC plus saline | 25.4 ± 5.5 | 99.5 ± 2.7 | 22.3 ± 1.5 | 114.2 ± 13.9 |
| PC plus CaEDTA | 25.6 ± 3.5 | 104.9 ± 13.1 | 24.4 ± 4.4 | 103.1 ± 16.0 |
| PC plus ZnEDTA | 22.7 ± 3.4 | 101.7 ± 10.8 | 21.8 ± 3.7 | 96.5 ± 4.4 |
| PC plus ap75NTR | 25.4 ± 4.0 | 91.6 ± 12.0 | 25.5 ± 4.0 | 108.5 ± 2.9 |
Rats were treated with intraventricular injections of saline, CaEDTA, ZnEDTA, or anti-p75NTR function-blocking antibody (ap75NTR). For comparison, data before and after MCA occlusion in sham operated controls (with no intraventricular injection) are shown.
Measurement of cerebral infarction.
At 24 h after ischemia induction, the rats were killed and their brains were collected for analysis. Two mm-thick brain slices (RBM-4000C, ASI, Warren, MI) were incubated in 2% 2,3,5-triphenyl tetrazolium chloride (TTC; Sigma) in normal saline at 37°C for 30 min, and stored in 4% paraformaldehyde (Longa et al., 1989). Digital images of brain slices were obtained with a flatbed digital scanner. Infarct volumes were quantified using image analysis software (ImagePro; Media Cybernetics), and total infarct volume was estimated by adding the infarct volume of each coronal slice along the AP axis. To correct for possible contribution by edema, infarct volumes were normalized to the volume of the intact contralateral hemisphere.
Intracerebroventricular injection.
A 10 μl microsyringe (Hamilton) was stereotactically introduced through a 1 mm burr hole into the right lateral ventricle, 0.8 mm posterior and 1.2 mm lateral from the bregma and 3.8 mm deep. CaEDTA, ZnEDTA (Sigma; 300 mm, 4 μl), or normal saline was injected through this microsyringe 15 min before IPC, and the microsyringe was removed 5 min later. To neutralize p75NTR, three aliquots of a p75NTR function-blocking antibody (Millipore Bioscience Research Reagents, 10 μg in 15 μl) were injected into the lateral ventricle, 30 min before, and 120 min and 270 min after, IPC.
TFL-Zn staining.
Brains were harvested 3 and 24 h after IPC, immediately frozen in isopentane and stored at −70°C. Coronal brain sections (10 μm thick) were prepared using a cryostat and stained for 90 s with a zinc specific fluorescent dye, N-(6-methoxy-8-quinolyl)-p-carboxybenzoylsulphonamide (TFL-Zn; Calbiochem), in Tris buffer (0.1 mm, pH 8.0). After washing with saline, the sections were examined under a fluorescence microscope with an ultraviolet filter (excitation, 355–375 nm; dichroic, 380 nm; barrier, 420 nm).
Mouse cortical cultures.
Cortical cultures were isolated from embryonic day 14–15 mice as described (Kim and Koh, 2002). Briefly, dissociated cortical cells were plated onto poly-l-lysine/laminin-coated plates (Nunc) at 10 hemispheres per 24-well or 6-well plate (450,000 cells/cm2) in DMEM (Invitrogen), supplemented with 20 mm glucose, 38 mm sodium bicarbonate, 2 mm glutamine, 5% fetal bovine serum (FBS), and 5% horse serum (HS). Cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. After 7 d in vitro (DIV 7), cultures were shifted to growth medium, which was identical to plating medium but contained 2.5% FBS and 2.5% HS. Cytosine arabinoside (10 μm) was also added at DIV 7 to halt the growth of non-neuronal cells, including oligodendrocytes and microglia. Mixed cultures of cortical neurons and astrocytes at DIV 10–14 were used in all experiments.
In vitro preconditioning and exposure to toxins.
Cortical cultures were washed-out with Eagle's MEM (supplied glutamine-free; Invitrogen) and PC was induced by exposure to 30 μm zinc (ZnCl2), 20 μm glutamate, 50 μm FeCl3, 5 μm CuCl2, or 50 nm staurosporine (STSP). Inhibitors of caspases or HSP induction, N-formyl-3,4-methylenedioxy-benzylidene-γ-butyrolactam (KNK437; Kaneka), or p75NTR function-blocking antibody (Millipore Bioscience Research Reagents) were added during PC treatment.
After PC, neuronal death was induced by continuous exposure of cultures to lethal concentrations of zinc, NMDA, glutamate, or Fe2+ in MEM, with PARP inhibitors added during toxic zinc or NMDA exposure. The experimental protocol is summarized in Figure 1B.
Knockdown of HSP70 or p75NTR with siRNA.
Three different HSP70 (GeneBank accession number EF100780) or p75NTR (GeneBank accession number NM_033217) siRNA sequences were designed and synthesized (Invitrogen): HSP70; stealth_358 siRNA (5′-UUAUCUUCGUCAGCACCAUGGACGA-3′), stealth_362 siRNA (5′-UCCUUUAUCUUCGUCAGCACCAUGG-3′), and stealth_1055 (5′-UUCAGGUCGCGCCCGUUAAAGAAGU-3′): p75NTR; stealth_566 siRNA (5′-UUCACACACUGUGUUCUGUUUGUCC-3′), stealth_935 siRNA (5′-AAUAUAGGCCACAAGGCCCACAACC-3′), and stealth_1245 siRNA (5′-UAAAGGAGUCUAUAUGCUCCG GCUG-3′). For delivery to cortical cultures, 128 pmol of each siRNA duplex was mixed with 1.5 μl of Lipofectamine RNAiMAX (Invitrogen) and incubated for 48 h with 2 × 104 cortical cells. Although each siRNA alone caused low-degree toxicity (<20%), it did not affect Zn toxicity or neuroprotection by Zn PC.
Estimation of cell death.
After morphological assessment, cell death was quantified by measuring lactate dehydrogenase (LDH) released from irreversibly damaged cells into the medium 18 h after the onset of lethal toxin exposure (Koh and Choi, 1987), unless specified otherwise. Each LDH value was normalized relative to the maximal LDH release (= 100) from cells exposed for 18 h to 100 μm NMDA or 40 μm zinc, both of which induced near-complete neuronal damage. All experiments were repeated at least three times using cultures from different platings.
In addition, propidium iodide (PI) staining was used to assess cell death in mixed cortical cultures. Five μg/ml PI was added to the medium and cell membrane disruption was assessed by fluorescence microscopy (IX70; Exciter filter BP510–550/Barrier filter B590; Olympus).
Western blots.
For poly(ADP-ribose) polymer (PAR) immunoblots, cell lysates were prepared by solubilizing cells in sample buffer (200 μl per well/6-well plate), consisting of 62.5 mm Tris, pH 6.8, 6 m urea, 10% (v/v) glycerol, 2% SDS, 5% β-mercaptoethanol, 20 mm dithiothreitol, 0.005% bromophenol blue, and freshly prepared protease and phosphatase inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm phenylmethylsulfonyl fluoride, 5 mm NaF, and 2 mm Na3VO4). After sonication for 30 s and incubation at 95°C for 10 min, total proteins (70 μl) were separated by SDS-PAGE (7%) under reducing conditions, transferred to nitrocellulose filters, and incubated with anti-PAR (Cell Signaling Technology), anti-actin or anti-ERK (Sigma) antibody.
For other immunoblots, cells lysates were prepared by solubilizing cells in lysis buffer [(in mm) 50 HEPES, 150 NaCl, 1.5 MgCl2, 5 EDTA, 2 EGTA, 1% Triton X-100, 0.5% SDS, pH 7.4]. Thirty microgram aliquots of total proteins were separated by SDS-PAGE, transferred to nitrocellulose filters, and immunoblotted with antibody to inducible HSP70, Mn SOD, Cu/Zn SOD (Stressgene), p75NTR (Santa Cruz Biotechnology), procaspase-3, cleaved caspase-3, PARP (Cell Signaling Technology), ERK or actin. Enhanced chemiluminescence (Intron) was used to visualize the immunoreactive bands.
Immunocytochemistry.
Tissues were fixed in 4% paraformaldehyde and blocked with 0.1% Triton X-100. After overnight incubation with primary antibody at 4°C, the samples were incubated with Alexa Fluor 488-conjugated secondary antibody (1:500; Alexa), and the stained tissues were examined and photographed under a fluorescence microscope.
Statistics.
All statistical comparisons were made using two-tailed t tests with Bonferroni's correction for multiple comparisons. A p value <0.05 was considered statistically significant.
Results
The role of zinc in ischemic preconditioning in rats
To assess whether endogenous zinc plays a role in ischemic PC, adult rats were subjected to 10 min of MCA occlusion. Three hours later, zinc accumulation was observed in neurons in the striatum and neocortex (Fig. 2A). In contrast to zinc accumulation in dying or dead neurons, zinc accumulation after ischemic PC was modest in degree and transient, largely disappearing 24 h later. Moreover, these neurons were not stained with TUNEL (data not shown). To determine whether endogenous zinc plays a causal role in ischemic PC, we used CaEDTA and ZnEDTA to reduce zinc-mediated effects and as a negative control, respectively (Koh et al., 1996; Lee et al., 2002). Whereas ischemic PC reduced subsequent infarct formation, this effect was completely reversed by intraventricular injections of CaEDTA, but not ZnEDTA, during PC (Fig. 2B). In addition, CaEDTA, but not Zn EDTA, during PC reversed the protective effect of ischemic PC on weight reduction after MCA occlusion (Fig. 2C).
Figure 2.
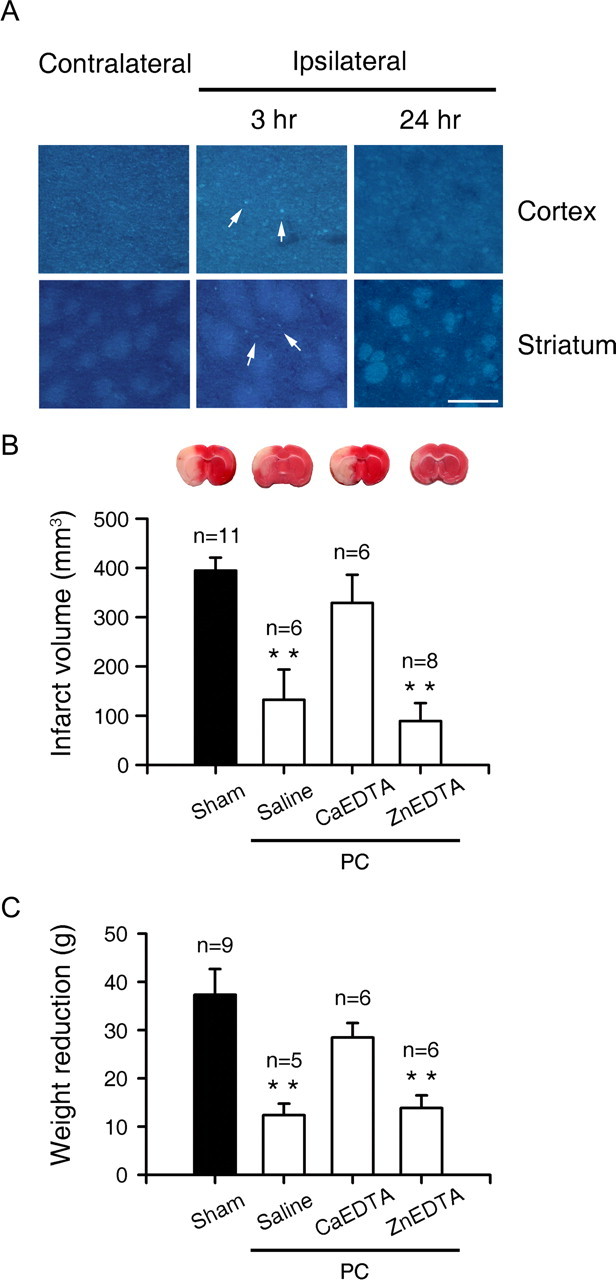
The crucial role of zinc in ischemic PC of the brain. A, TFL-Zn fluorescence of the contralateral (left) and ipsilateral (middle, right) cortices and striata 3 and 24 h after 10 min MCA occlusion for ischemic PC. Neurons in the cortex and striatum exhibited TFL-Zn fluorescence (arrows) 3 h after occlusion, but zinc fluorescence largely disappeared 24 h later. Scale bar, 100 μm. B, TTC-stained brain sections 24 h after unilateral MCA occlusion. Rats were subjected to 1 h MCA occlusion 72 h after sham operation (first) or IPC with the intracerebroventricular injection of saline (second), CaEDTA (third), or ZnEDTA (last). Bars denote infarct volume (cubic millimeters). C, Weight reduction (grams) 24 h after 1 h MCA occlusion in rats that underwent sham operation or 10 min MCA ischemia 72 h before full MCA ischemia. Rats that underwent 10 min IPC were intraventricularly injected with saline, CaEDTA, or ZnEDTA 15 min before ischemic PC.
Protective effects of zinc preconditioning in cortical cell culture
Consistent with the possible role of zinc in ischemic PC, treatment of cortical cultures with 20 or 30 μm zinc for 24 h (zinc preconditioning or Zn PC), which alone did not cause cell death in cultures (Fig. 3C), markedly attenuated neuronal death induced by subsequent exposure to 40 or 50 μm zinc (Fig. 3A–C). Furthermore, Zn PC reduced neuronal death induced by excitotoxins (NMDA and glutamate) or the metal oxidant Fe2+ (Fig. 3D). In contrast, 24 h exposure to 20 μm glutamate, at just below the lowest lethal concentrations, failed to protect against subsequent lethal exposure to zinc, NMDA or Fe2+ (Fig. 3E). In addition, other metal ions, Fe and Cu, did not show a PC protective effect (Fig. 3F).
Figure 3.
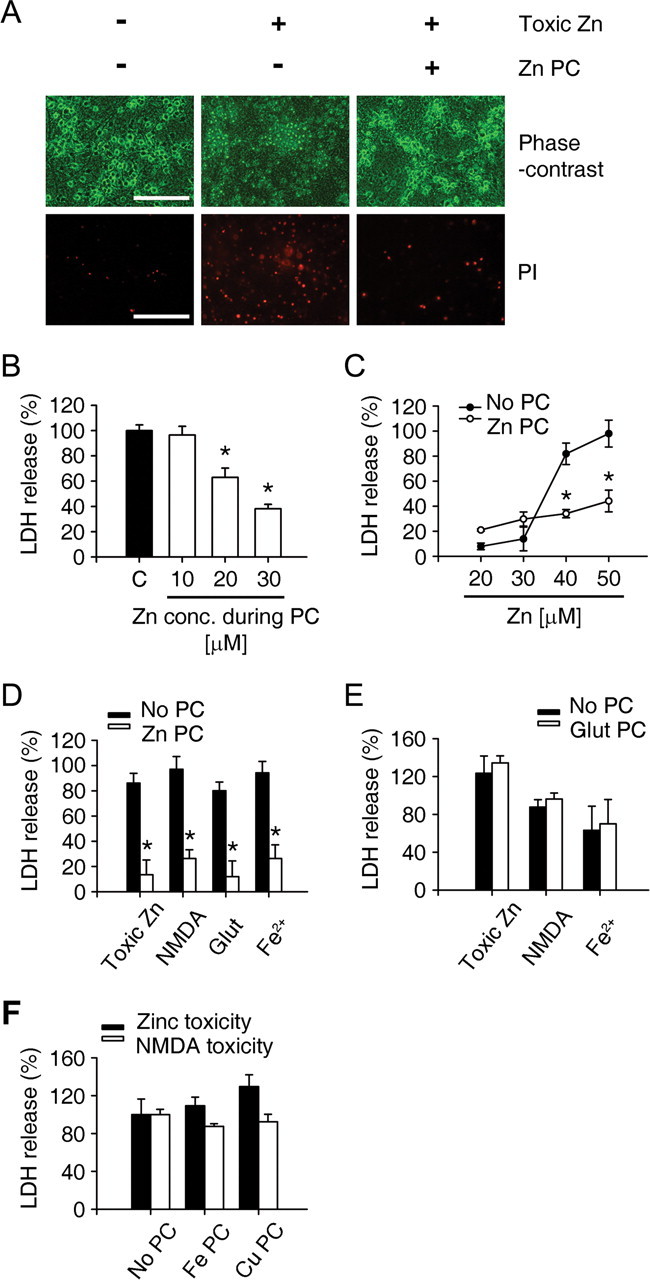
In vitro PC with sublethal zinc exposure. A, Phase-contrast and PI-stained cortical cultures. PC with 30 μm zinc for 24 h markedly reduced the number of PI-stained dead neurons after subsequent exposure to 40 μm zinc for 18 h. Scale bar, 100 μm. B, Concentration dependence of Zn PC, as measured by LDH release (mean ± SEM, n = 4) from cortical cultures. Mouse mixed cortical cultures were pretreated with the indicated concentrations of zinc (10 ∼ 30 μm) for 24 h and then substituted by toxic zinc (40 μm). *p < 0.05 compared with sham controls (2-tailed t test with Bonferroni correction for 3 comparisons). PC with 10 μm zinc did not show the protection against zinc toxicity. C, Zinc concentration-toxicity relationships in control cultures (no PC) and cultures incubated for 24 h with 30 μm zinc (Zn PC), as measured by LDH release (mean ± SEM, n = 4). Zn PC markedly reduced subsequent neuronal death induced by 18 h exposure to 40 or 50 μm zinc. D, LDH release after 18 h exposure to indicated toxins, without (no PC) or with 24 h pretreatment with 30 μm zinc (Zn PC). Zn PC significantly attenuated not only zinc toxicity, but also glutamate-, NMDA-, or Fe2+-induced cell death. *p < 0.05 compared with respective controls (2-tailed t test). E, LDH release from cortical cultures that underwent sham wash (No PC) or glutamate PC (20 μm, 24 h) and then treated with toxic levels of Zn, NMDA, or Fe2+. Glutamate PC did not protect against the toxicity of any of these agents. F, LDH release 18 h after exposure to 40 μm zinc or 30 μm NMDA from cortical cultures that previously underwent sham wash (No PC) or PC with 50 μm FeCl3 or 5 μm CuCl2. Neither Fe nor Cu showed PC protection against Zn or NMDA toxicity.
The role of PARP-1 cleavage in Zn PC
PARP-1 overactivation has been implicated in zinc and NMDA toxicity as a key step in neuronal cell death (Zhang et al., 1994; Eliasson et al., 1997; Lo et al., 1998; Kim and Koh, 2002). Consistently, we found that chemical inhibitors of PARP markedly attenuated zinc- or NMDA-induced neuronal death (Fig. 4A). However, PARP-1 overactivation is also regarded as a key step in ischemic PC (Liaudet et al., 2001; Garnier et al., 2003). Therefore, we assessed whether the neuroprotective effects of Zn PC resulted from the reduced activation of PARP-1 during subsequent toxic exposure. Robust PARP-1 activation was detected 4 h after the onset of zinc exposure and 30–60 min after the onset of NMDA exposure (Fig. 4B). Interestingly, Zn PC attenuated PARP-1 activation caused by subsequent exposure to zinc or NMDA, to a similar degree as that caused by the addition of PARP inhibitors during exposure to toxins (Fig. 4C). PARP inhibitors significantly reduced Zn- or NMDA-induced neuronal death (Fig. 4A), suggesting that the attenuation of PARP-1 overactivation may be the major protective mechanism of Zn PC. To further examine this possibility, we tested Zn PC in cortical cultures obtained from PARP-1 knock-out (KO) mouse embryos. We found that PARP-1 deficient neurons exhibited lessened vulnerability than did wild-type (WT) neurons (Fig. 4D), suggesting the importance of PARP-1 in zinc and NMDA toxicities. However, Zn PC did not have additional protective effects on zinc- or NMDA-induced death in PARP-1 deficient neuronal cultures (Fig. 4D), further suggesting that PARP-1 is a necessary element for Zn PC.
Figure 4.
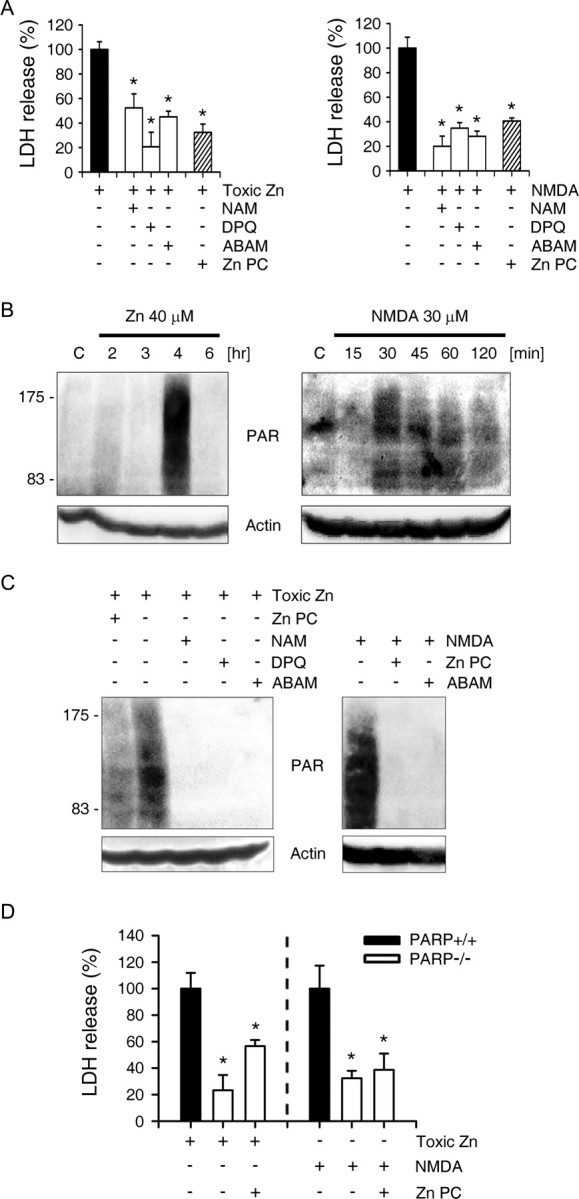
Reduced PARP-1 activation by Zn PC. A, LDH release after 18 h exposure to zinc (40 μm) or NMDA (30 μm) and the PARP inhibitors 20 mm nicotinamide (NAM), 30 μm 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone) (DPQ), or 20 mm 3-aminbenzamide (ABAM) or Zn PC. PARP inhibitors and Zn PC markedly attenuated neuronal death after exposure to Zn or NMDA. *p < 0.05 compared with respective controls (2-tailed t tests). B, Western blots with anti-PAR antibody of cells exposed to toxic concentrations of zinc (40 μm) or NMDA (30 μm) for the indicated time points. Actin was used as a loading control in this and the following Western blots. C, Western blots with anti-PAR antibody of cells exposed for 4 h to 40 μm zinc or 30 min to 30 μm NMDA, in the presence or absence of PARP inhibitors (20 mm NAM, 30 μm DPQ, or 20 mm ABAM) or Zn PC. D, LDH release from PARP-1 WT and KO cortical neuronal cultures after 18 h exposure to toxic concentrations of zinc (40 μm) or NMDA (30 μm) without or with Zn PC.
Mediation of PARP-1 cleavage by caspase-3 in Zn PC
The cleavage of PARP-1 by caspase-3 has been implicated in ischemic PC (Garnier et al., 2003). Western blot analysis showed that caspase-3 activation began 6 h after the onset of Zn PC, followed 3 h later by PARP-1 cleavage (Fig. 5A). This time course of PARP-1 cleavage correlated well with our finding that Zn PC for <9 h did not show any protective effect (Fig. 5B). The caspase inhibitors zVAD (nonselective) and zDEVD (selective for caspase-3) almost completely blocked caspase-3 activation and PARP-1 cleavage (Fig. 5C), suggesting that PARP-1 is cleaved predominantly by caspase-3. Caspase-3 activation was observed in PARP-1 KO cells (Fig. 5D), further indicating that caspase-3 activation is upstream of PARP-1 cleavage. As expected, caspase inhibitors added during Zn PC largely restored PARP-1 activation induced by subsequent toxic zinc exposure (Fig. 5E), and reversed cytoprotection against subsequent toxic zinc or NMDA exposure (Fig. 5F).
Figure 5.
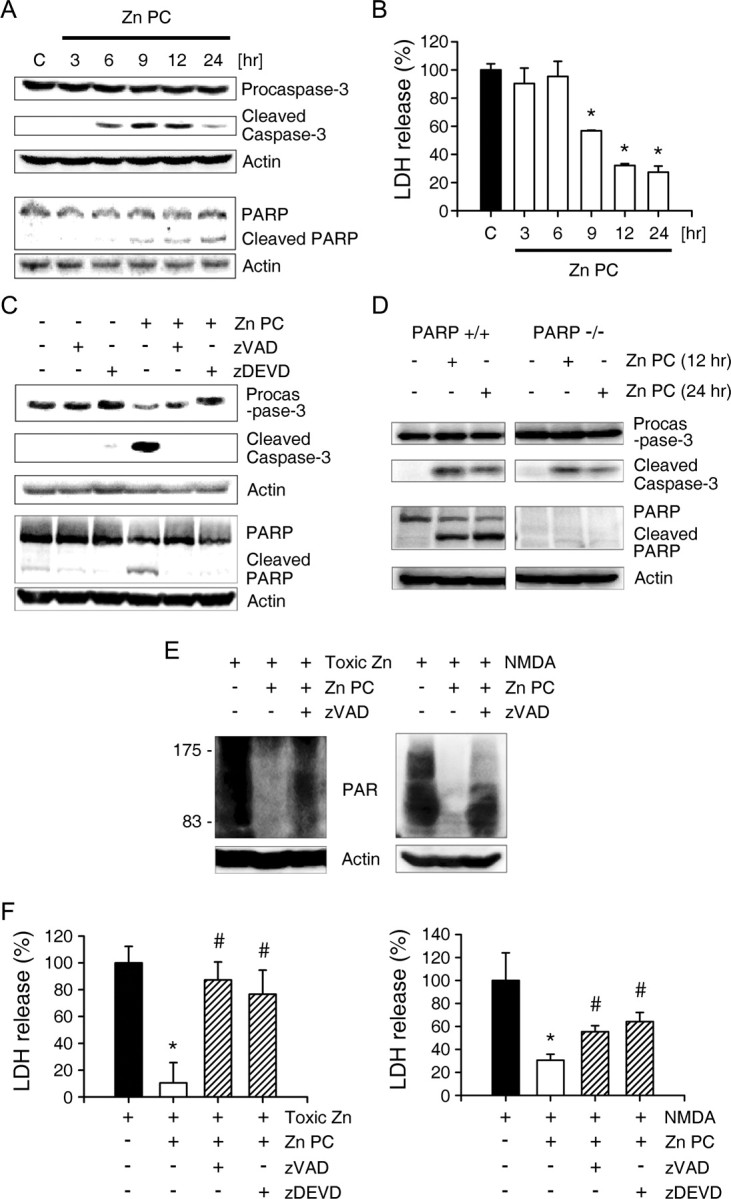
Caspase-3 mediates PARP-1 cleavage in Zn PC. A, Western blots using antibodies specific for procaspase-3, cleaved caspase-3 and PARP-1, of cells at the indicated times after the onset of Zn PC. B, Time dependence of the Zn PC effect. Bars represent LDH release (mean ± SEM, n = 4) from cortical cultures exposed to toxic (40 μm) zinc for 18 h after Zn PC for the indicated times. *p < 0.05 compared with zinc controls (2-tailed t test with Bonferroni correction for multiple comparisons). At least 9 h was required for Zn PC to show protection against zinc toxicity. C, Western blots using antibodies specific for procaspase-3, cleaved caspase-3 and PARP-1, of cells 12 h after the onset of Zn PC, without or with zVAD or zDEVD. D, Western blots using antibodies specific for procaspase-3, cleaved caspase-3 or PARP-1 of cells from PARP-1 WT and KO cortical cultures after exposure to sublethal zinc (30 μm) for 12 or 24 h. E, Western blots with anti-PAR antibody of cells exposed to toxic zinc (40 μm) for 4 h or to NMDA (30 μm) for 30 min, without or with Zn PC alone, or Zn PC plus zVAD. A pan-caspase inhibitor, zVAD, reversed the Zn PC attenuation of zinc- or NMDA-induced PARP-1 overactivation. F, LDH release (mean ± SEM, n = 4) from cortical cultures after 18 h exposure to 40 μm zinc (left) or 30 μm NMDA (right) without or with Zn PC alone or Zn PC plus caspase inhibitors. *p < 0.05 compared with toxin controls; #p < 0.05 compared with Zn PC controls (2-tailed t tests). The protective effects of Zn PC were reversed by zVAD or zDEVD.
A sublethal degree of caspase-3 activation may be sufficient to induce PARP-1 cleavage and PC protection. Exposure of cortical cultures for 12 h to 50 nm staurosporine, which induced caspase-3 activation and PARP-1 cleavage without apoptotic neuronal death (Fig. 6A,C), reduced both PARP-1 overactivation and the neuronal vulnerability to subsequent zinc, NMDA, or glutamate toxicity (Fig. 6B,C).
Figure 6.
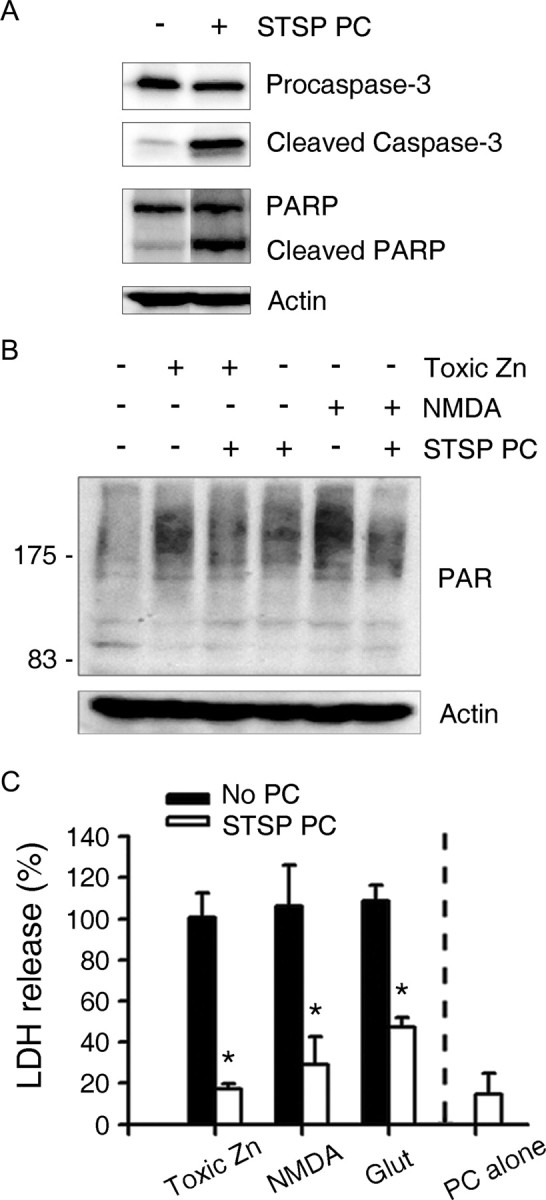
Sublethal caspase-3 activation with staurosporine mimics Zn PC. A, Western blots using antibodies to procaspase-3, active caspase-3, and PARP-1, of cells 12 h after the onset of STSP PC. STSP induced caspase-3 activation and PARP-1 cleavage. B, Western blots with anti-PAR antibody of cells exposed to 40 μm zinc for 4 h or to 30 μm NMDA for 30 min, without or with STSP PC. STSP PC reduced anti-PAR immunoreactivity caused by exposure to neurotoxic zinc or NMDA. C, LDH release by cortical cultures that underwent sham wash (No PC) or STSP PC, followed by treatment with toxic concentrations of Zn, NMDA, or glutamate. STSP PC reduced zinc, NMDA and glutamate neurotoxicity. STSP PC alone (12 h exposure to 50 nm STSP) did not show any toxicity in mouse cortical cultures.
The role of HSP70 in Zn PC
Under most conditions, caspase-3 activation usually signifies an irreversible event leading to apoptosis. However, caspase-3 activation during Zn PC seemed just adequate to induce PARP-1 cleavage for neuroprotection, but not potent enough to induce significant neuronal apoptosis. Hence, an endogenous break mechanism may be activated concurrently. We found that addition of cycloheximide (CHX) or actinomycin-D (ActD) abrogated the protective effect of Zn PC (Fig. 7A), indicating the requirement for new protein synthesis. Although various potential protective proteins were induced by Zn PC, including HSP70, Mn SOD, and Cu/Zn SOD (Fig. 7B), we focused on HSP70 because it has been found to inhibit caspase-9 and -3 through inhibition of apoptosome formation or cytochrome C release (Beere et al., 2000; Saleh et al., 2000; Steel et al., 2004; Stankiewicz et al., 2005). Addition of KNK437, a chemical inhibitor of HSP70 induction, during Zn PC markedly reduced HSP70 induction, while causing more robust and prolonged caspase-3 activation than in control PC cultures (Fig. 7C) (Yokota et al., 2000; Koike et al., 2006). Consistently, KNK437 treatment largely reversed the protective effect of Zn PC (Fig. 7D). This reversal may be brought about by unhindered caspase activation and apoptosis, because KNK437 added during zinc PC itself greatly potentiated neuronal death induced by otherwise minimally lethal zinc PC (Fig. 7D). Next, we confirmed the protective role of HSP70 in Zn PC using siRNAs to HSP70. Consistent with the effects of chemical inhibitor, knock-down of HSP70 by 48 h pretreatment with three different siRNAs significantly increased the neurotoxicity of Zn PC, although the degree of knock-down by siRNAs were variable (Fig. 7E,F). From these results, we suggest that HSP70 play a key role in Zn PC by inhibiting the sustained caspase-3 activation, resulting in apoptotic neuronal death.
Figure 7.

HSP70 induction halts caspase-3 activation in Zn PC. A, LDH release after 18 h exposure to 40 μm zinc without or with Zn PC alone, or Zn PC plus CHX (1 μg/ml) or ActD (300 nm). CHX or ActD largely reversed the zinc PC attenuation of neurotoxicity. B, Western blots using antibodies to HSP70, Mn SOD, and Cu/Zn SOD of cells at the indicated times after the onset of Zn PC. C, Top, Western blots using anti-HSP70 antibody of cells 12 h after the onset of Zn PC, showing that 100 μm KNK437 markedly attenuated HSP70 induction. Bottom, Western blots using antibody to caspase-3 of cells 12 or 24 h after the onset of Zn PC, showing that KNK437 inhibition of HSP70 induction during Zn PC resulted in greater and more prolonged activation of caspase-3. D, LDH release after 18 h exposure to 40 μm zinc or sham wash, without or with Zn PC alone or Zn PC plus KNK437. *p < 0.05 compared with toxin controls, #p < 0.05 compared with Zn PC controls (2-tailed t tests). Inhibition of HSP70 induction with KNK437 abrogated the protective effect of Zn PC and potentiated neurotoxicity by Zn PC. E, LDH release after 18 h exposure to sham wash, without or with Zn PC from cortical cultures pretreated with Stealth RNAi negative control (NC) or three different specific siRNAs to HSP70 (stealth_358, 362, or 1055) for 48 h. F, Western blots using anti-HSP70 antibody of cell lysates obtained 12 h after Zn PC, without or with 48 h exposure to siRNAs of nonspecific NC or three different specific sequences for HSP70 knock-down (358, 362, or 1055).
The necessary role of p75NTR in Zn PC in vitro and ischemic PC in vivo
Although excitotoxic exposure of our cortical cultures did not induce caspase-3 activation (Gottron et al., 1997) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), exposure to zinc, especially at lower toxic concentrations, induced expression of p75NTR, which then caused caspase-3 activation and neuronal apoptosis (Park et al., 2000). Induction of p75NTR expression began 3–6 h after the onset of Zn PC (Fig. 8A), somewhat earlier than caspase-3 activation. An antibody that blocked p75NTR function (Bamji et al., 1998; Park et al., 2000) almost completely blocked caspase-3 activation and PARP-1 cleavage during Zn PC (Fig. 8B), indicating that p75NTR plays a key role in caspase-3 activation during Zn PC. Moreover, this anti-p75NTR antibody completely reversed the protective effect of Zn PC on both zinc and NMDA neurotoxicity (Fig. 8C). In addition, p75NTR siRNAs that significantly downregulated p75NTR (Fig. 8E) almost completely reversed the protective effect of Zn PC (Fig. 8D), providing further evidence for the importance of the p75NTR pathway in caspase-3 activation and neuroprotection in Zn PC.
Figure 8.
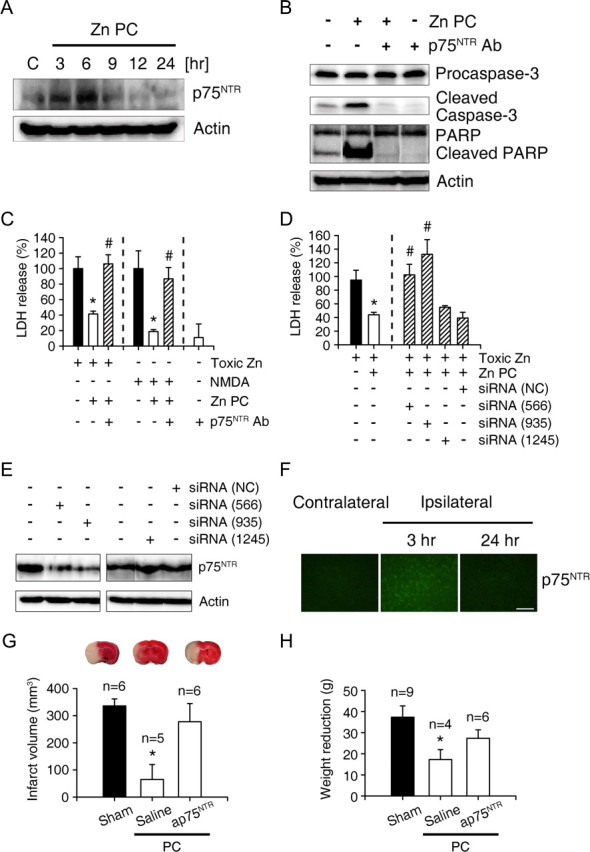
Induction of p75NTR and its role in Zn PC in vitro and ischemic PC in vivo. A, Western blots for using antibody to p75NTR of cells at the indicated times after onset of Zn PC. Levels of p75NTR increased 3–6 h after the onset of Zn PC. B, Western blots using antibodies to pro-caspase-3, caspase-3, and PARP-1 of cells 12 h after the onset of sham wash or Zn PC. Addition of an antibody that blocked the function of p75NTR (AB1554, 1:100 dilution) completely blocked cleavage of both caspase-3 and PARP-1. C, LDH release after 18 h exposure to 40 μm zinc or 30 μm NMDA, without or with Zn PC alone or Zn PC plus p75NTR function-blocking antibody. The protective effects of Zn PC were completely reversed by addition of antibody to p75NTR. D, LDH release after 18 h exposure to 40 μm zinc without or with Zn PC from cortical cultures pretreated with vehicle alone, Stealth RNAi negative control (NC) or respective 3 different specific siRNAs to p75NTR (566, 935, or 1245) for 48 h. Both stealth_566 and stealth_935 but not steath_1245 and nonspecific Steath RNAi negative control almost completely reversed the protective effects of Zn PC on zinc toxicity. E, Western blots using anti-p75NTR antibody of cell lysates obtained 6 h after Zn PC, without or with pretreatment of Stealth RNAi NC or specific stealth siRNAs to p75NTR for 48 h. Note that only stealth_566 and stealth_935, which blocked the protection by Zn PC, showed the knock-down effects ofp75NTR. F, Immunoreactivity to p75NTR in the cortex. Shown are control cortices and cortices 3 and 24 h after ischemic PC. Scale bar, 100 μm. G, TTC-stained brains 24 h after MCA occlusion. Ischemic PC greatly reduced infarcts, whereas administration of AB1554 during ischemic PC completely blocked the protective effect of PC. Bars denote the infarct volume in the above conditions. H, Weight reduction (grams) 24 h after 1 h MCA occlusion in rats that underwent sham operation or 10 min MCA ischemia 72 h before full MCA ischemia. Rats that underwent 10 min ischemic PC were intraventricularly injected with saline or anti-p75NTR antibody 30 min before ischemic PC.
Because endogenous zinc is a key mediator of ischemic PC in the rat brain (Fig. 2), we examined the role of p75NTR in vivo. We found that p75NTR immunoreactivity substantially increased in neurons 3 h after ischemic PC, but returned to baseline 24 h after PC (Fig. 8F). Furthermore, injection of the anti-p75NTR antibody into the lateral ventricle during and after ischemic PC almost completely abrogated the protective effects of ischemic PC (Fig. 8G,H).
Discussion
Our in vivo results have demonstrated the importance of endogenous zinc in ischemic PC as well as in ischemic neuronal death (Koh et al., 1996). Whereas dense zinc accumulation in neurons is highly correlated with ischemic neuronal death, more transient low-level zinc accumulation may provide protection against subsequent neuronal death. Mild zinc accumulation has been found to correlate with HSP70 induction in surviving hippocampal neurons after seizures (Lee et al., 2000). Among endogenous metals, only zinc induced PC protection in cortical cultures, providing further evidence for the importance of zinc in ischemic PC. Furthermore, CaEDTA, but not ZnEDTA, abrogated the protective effect of ischemic PC, strongly reinforcing the role of zinc in ischemic PC. Although CaEDTA effects are likely mediated by removal of free zinc, we cannot completely rule out the possibility that calcium release from CaEDTA after zinc binding additionally contributes to the reversal of ischemic PC. However, this change may not have significant pathophysiological effects because the upper limit of extracellular calcium concentration change may be <30 μm (1.5% of 2 mm normal calcium), based on the estimation that the peak zinc concentration after release is at most 30 μm, probably much less (Ueno et al., 2002; Frederickson et al., 2006).
Although calcium overload excitotoxicity and zinc toxicity share several common features, such as PARP-1 activation, we found that zinc was much more effective than excitotoxins in inducing PC in cortical cultures. This may be largely attributable to the ability of zinc to induce both caspase-dependent apoptosis and caspase-independent necrosis, whereas calcium overload, at least in cortical cultures, induces only caspase-independent necrosis (Gottron et al., 1997). In addition, in cultured cortical neurons, zinc is more effective than excitotoxins in inducing HSP70 (Lee et al., 2000). Whereas present results suggest that HSP70 induction by zinc is involved in ischemic or Zn PC mainly by limiting caspase-3 activation, other effects such as stabilization of hypoxia-inducible factor and activation of downstream neuroprotective genes may also contribute to PC protection (Siddiq et al., 2008).
Whereas caspase-3 activation has been demonstrated to be a critical event in ischemic PC (Garnier et al., 2003; McLaughlin et al., 2003), specific pathways leading to caspase-3 activation in PC have not been elucidated. Our results indicate that p75NTR is the key missing piece. Although some reports have shown that extracellular zinc inhibit p75NTR-mediated apoptosis via the modulation of structural changes of neurotrophins such as NGF (Ross et al., 1997; Allington et al., 2001), we previously reported that low level zinc toxicity that involves intracellular zinc action, as well as transient ischemia in vivo, but not calcium overload excitotoxicity, induced p75NTR and NADE, which in turn induced neuronal apoptosis via caspase-3 activation. Hence, our finding, that blockade of p75NTR with a function-blocking antibody or siRNA completely reversed ischemic PC in an MCA occlusion model, strongly supports the role of zinc rather than calcium excitotoxicity in ischemic PC.
Ischemic PC is an intriguing phenomenon that may shed light on endogenous neuroprotective mechanisms. Although various hypotheses have been raised (Andoh et al., 2002; Dirnagl et al., 2003; Gidday, 2006), recent studies have demonstrated the critical roles of caspases and PARP-1 in ischemic PC. The results presented here extend our understanding of this mechanism, providing evidence that sublethal zinc dyshomeostasis and p75NTR activation are the upstream events leading to caspase-3 activation, PARP-1 cleavage, and neuroprotection (Fig. 9). Although our present results suggest that these mechanisms are likely crucial for zinc PC and ischemic PC, it is possible that zinc may have other neuroprotecive mechanisms such as the induction of prosurvival genes (Dirnagl et al., 2003; Gidday, 2006). In addition, sublethal zinc may reduce calcium overload excitotoxicity by limiting glutamate release (Kitamura et al., 2006). Further studies may be warranted to fully investigate mechanisms of zinc-mediated neuroprotection and neuronal injury.
Figure 9.

Diagram for mechanism of ischemic PC. Ischemic PC appears to involve sublethal zinc accumulation, which induces p75NTR expression in neurons. P75NTR activates caspase-3, which mediates PARP-1 cleavage. The action of caspase-3 is terminated by the induction of HSP70 before neuronal apoptosis develops. The resultant reduction in the level of PARP-1 provides broad-spectrum protection against insults that require PARP-1 for the induction of neuronal death.
Finally, the mechanism underlying the increase in labile zinc in degenerating neurons is still unexplained. It may involve entry of zinc from the extracellular space, or more likely in situ zinc release from zinc-binding proteins, such as metallothioneins, or from zinc-containing organelles. Further investigations are needed to address this issue.
Footnotes
This work was supported by Korea Science and Engineering Foundation Grants M10412000012-07N1200-01210 and R01-2006-000-10771-0(2007) (Y.-H.K.), and M10600000181-06J0000-18110 (J.-Y.K.), and by the Star Faculty program of the Korean Ministry of Education and Human Resources Development (KRF-2005-084-C00026 to J.-Y.K.).
References
- Allington C, Shamovsky IL, Ross GM, Riopelle RJ. Zinc inhibits p75NTR-mediated apoptosis in chick neural retina. Cell Death Differ. 2001;8:451–456. doi: 10.1038/sj.cdd.4400831. [DOI] [PubMed] [Google Scholar]
- Andoh T, Chock PB, Chiueh CC. Preconditioning-mediated neuroprotection: role of nitric oxide, cGMP, and new protein expression. Ann N Y Acad Sci. 2002;962:1–7. doi: 10.1111/j.1749-6632.2002.tb04051.x. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Capasso M, Jeng JM, Malavolta M, Mocchegiani E, Sensi SL. Zinc dyshomeostasis: a key modulator of neuronal injury. J Alzheimers Dis. 2005;8:93–108. doi: 10.3233/jad-2005-8202. discussion 209–215. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Kaplan B, Jacewicz M, Pulsinelli W. Continuous measurement of cerebral cortical blood flow by laser-Doppler flowmetry in a rat stroke model. J Cereb Blood Flow Metab. 1989;9:589–596. doi: 10.1038/jcbfm.1989.84. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, Krezel A, McAdoo DJ, Mueller RN, Zeng Y, Balaji RV, Masalha R, Thompson RB, Fierke CA, Sarvey JM, de Valdenebro M, Prough DS, Zornow MH. Concentrations of extracellular zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol. 2006;198:285–293. doi: 10.1016/j.expneurol.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Galasso SL, Dyck RH. The role of zinc in cerebral ischemia. Mol Med. 2007;13:380–387. doi: 10.2119/2007-00044.Galasso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier P, Ying W, Swanson RA. Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J Neurosci. 2003;23:7967–7973. doi: 10.1523/JNEUROSCI.23-22-07967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci. 1997;9:159–169. doi: 10.1006/mcne.1997.0618. [DOI] [PubMed] [Google Scholar]
- Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol. 2002;177:407–418. doi: 10.1006/exnr.2002.7990. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Iida Y, Abe J, Ueda M, Mifune M, Kasuya F, Ohta M, Igarashi K, Saito Y, Saji H. Protective effect of zinc against ischemic neuronal injury in a middle cerebral artery occlusion model. J Pharmacol Sci. 2006;100:142–148. doi: 10.1254/jphs.fp0050805. [DOI] [PubMed] [Google Scholar]
- Koh JY. Zinc and disease of the brain. Mol Neurobiol. 2001;24:99–106. doi: 10.1385/MN:24:1-3:099. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Koike T, Uno S, Ishizawa M, Takahashi H, Ikeda K, Yokota S, Makishima M. The heat shock protein inhibitor KNK437 induces neurite outgrowth in PC12 cells. Neurosci Lett. 2006;410:212–217. doi: 10.1016/j.neulet.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Park KH, He YY, Hsu CY, Choi DW. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience. 2002;115:871–878. doi: 10.1016/s0306-4522(02)00513-4. [DOI] [PubMed] [Google Scholar]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, Koh JY. Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol. 2000;161:433–441. doi: 10.1006/exnr.1999.7297. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Yang Z, Al-Affar EB, Szabó C. Myocardial ischemic preconditioning in rodents is dependent on poly (ADP-ribose) synthetase. Mol Med. 2001;7:406–417. [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Bosque-Hamilton P, Meng W. Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke. 1998;29:830–836. doi: 10.1161/01.str.29.4.830. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci U S A. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksala N, Alhava E, Paimela H. Heat shock preconditioning and eicosanoid pathways modulate caspase 3-like activity in superficially injured isolated guinea pig gastric mucosa. Eur Surg Res. 2004;36:67–73. doi: 10.1159/000076645. [DOI] [PubMed] [Google Scholar]
- Park JA, Lee JY, Sato TA, Koh JY. Co-induction of p75NTR and p75NTR-associated death executor in neurons after zinc exposure in cortical culture or transient ischemia in the rat. J Neurosci. 2000;20:9096–9103. doi: 10.1523/JNEUROSCI.20-24-09096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GM, Shamovsky IL, Lawrance G, Solc M, Dostaler SM, Jimmo SL, Weaver DF, Riopelle RJ. Zinc alters conformation and inhibits biological activities of nerve growth factor and related neurotrophins. Nat Med. 1997;3:872–878. doi: 10.1038/nm0897-872. [DOI] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Ratan RR. Prolyl 4-hydroxylase activity-responsive transcription factors: from hydroxylation to gene expression and neuroprotection. Front Biosci. 2008;13:2875–2887. doi: 10.2741/2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–38739. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- Steel R, Doherty JP, Buzzard K, Clemons N, Hawkins CJ, Anderson RL. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J Biol Chem. 2004;279:51490–51499. doi: 10.1074/jbc.M401314200. [DOI] [PubMed] [Google Scholar]
- Ueno S, Tsukamoto M, Hirano T, Kikuchi K, Yamada MK, Nishiyama N, Nagano T, Matsuki N, Ikegaya Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J Cell Biol. 2002;158:215–220. doi: 10.1083/jcb.200204066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Yokota S, Kitahara M, Nagata K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000;60:2942–2948. [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]