

Abstract
Emotionally arousing events are particularly well remembered. This effect is known to result from the release of stress hormones and activation of β adrenoceptors in the amygdala. However, the underlying cellular mechanisms are not understood. Small conductance calcium-activated potassium (SK) channels are present at glutamatergic synapses where they limit synaptic transmission and plasticity. Here, we show that β adrenoceptor activation regulates synaptic SK channels in lateral amygdala pyramidal neurons, through activation of protein kinase A. We show that SK channels are constitutively recycled from the postsynaptic membrane and that activation of β adrenoceptors removes SK channels from excitatory synapses. This results in enhanced synaptic transmission and plasticity. Our findings demonstrate a novel mechanism by which β adrenoceptors control synaptic transmission and plasticity, through regulation of SK channel trafficking, and suggest that modulation of synaptic SK channels may contribute to β adrenoceptor-mediated potentiation of emotional memories.
Keywords: fear, fluorescence microscopy, noradrenergic, norepinephrine, learning and memory, potassium channels
Introduction
Our own experiences, as well as a wealth of experimental studies (McGaugh, 2004; Anderson et al., 2006), show that emotionally salient experiences are much better remembered than those that are emotionally neutral. Both animal and human studies have shown that this enhanced memory formation results from the release of stress hormones, adrenaline and corticosterone, and activation of β adrenoceptors within the basolateral amygdala (McGaugh, 2004). Thus, for example, augmenting the release of noradrenaline immediately after learning improves memory formation, whereas inhibition of β adrenoceptors within the basolateral amygdala blocks the memory enhancing effects of emotive experiences. However, although activation of β adrenoceptors in neurons is known to have a variety of physiological actions (Nicoll et al., 1990), the cellular mechanisms that underlie the memory enhancing actions of β adrenoceptor activation are not known.
The amygdaloid complex is a temporal lobe structure that plays a central role in the processing of emotional information. In particular, the amygdala plays a key role in the analysis of fear and fear related-learning (Davis and Whalen, 2001; LeDoux, 2003). Sensory information arising from both cortical and subcortical locations enters the amygdaloid complex at the level of the basolateral nucleus (Sah et al., 2003), and after local processing, projections from the central nucleus initiate the physiological responses seen during fear. A large body of evidence has shown that long-term synaptic plasticity at inputs to the basolateral amygdala contributes to fear learning (Sah et al., 2003; Paré et al., 2004). These sensory afferents are glutamatergic and make typical dual component synapses on principal neurons that that express both AMPA and NMDA receptors (Mahanty and Sah, 1999; Weisskopf and LeDoux, 1999). Electrophysiological studies in vitro have shown that repetitive stimulation of cortical or thalamic afferents to principal neurons in the lateral amygdala leads to long-term potentiation (LTP) at these synapses (Huang and Kandel, 1998; Bauer et al., 2002; Humeau et al., 2003). Although there is still debate regarding the site of plasticity that underlies this learning, the requirement for calcium influx via NMDA receptors in the induction of LTP is widely accepted.
Calcium-activated potassium channels are present in most neurons where they mediate the post-action potential afterhyperpolarization, thereby modulating the frequency of action potential discharge (Sah and Faber, 2002). Early in vivo experiments suggested that these channels contribute to synaptic transmission in the lateral amygdala (Lang and Paré, 1997). Recent experiments in vitro have confirmed that small conductance calcium-activated potassium (SK) channels, which mediate the medium afterhyperpolarization, are present at excitatory synapses on principal neurons in the lateral amygdala (Faber et al., 2005) and hippocampus (Ngo-Anh et al., 2005), where they are activated by calcium influx through NMDA receptors (Faber et al., 2005; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007). Activation of these synaptic SK channels acts as a postsynaptic shunt and effectively reduces that amplitude of EPSPs (Faber et al., 2005). Recent studies in hippocampal pyramidal neurons have suggested that postsynaptic SK channels are modulated during the induction of long-term synaptic potentiation at Schaffer collateral synapses (Lin et al., 2008). In this study, we have examined the effects of β adrenoceptor receptor stimulation on synaptic activity and plasticity in lateral amygdala principal neurons. Our results show that activation of β adrenoceptors enhances excitatory synaptic transmission in the lateral amygdala by modulating synaptic SK channels. We show that SK channels present at excitatory synapses undergo constitutive trafficking and activation of β adrenoceptors removes synaptic SK channels by a postsynaptic mechanism mediated by protein kinase A (PKA). Functionally, β adrenoceptor modulation of SK channels enhances LTP. Our findings demonstrate a novel mechanism by which β adrenoceptors control synaptic transmission and plasticity, through regulation of SK channel trafficking, and suggest that modulation of synaptic SK channels may be a mechanism that underlies β adrenoceptor-mediated potentiation of emotional memories.
Materials and Methods
Electrophysiological recordings.
Experiments were performed on rat brain slices maintained in vitro. Wistar rats (of either sex, 21–28 d old) were anesthetized with halothane and were killed by decapitation. These procedures were conducted in accordance with the guidelines of the University of Queensland Animal Ethics Committee. Coronal brain slices (400 μm thick) containing the amygdala were prepared using standard techniques (Faber et al., 2005) and maintained in artificial CSF (aCSF) containing (in mm) 118 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 1.2 NaH2PO4, 2.5 CaCl2, and 1.3 MgCl2. For LTP experiments, extracellular CaCl2 and MgCl2 were increased to 4 mm. Within the recording chamber, slices were continuously perfused with oxygenated aCSF maintained at 30–32°C.
Whole-cell recordings were made from neurons in the lateral amygdala using infrared differential interference contrast techniques. Electrodes (3–5 MΩ) were filled with pipette solution containing (in mm) 135 KMeSO4, 8 NaCl, 10 HEPES, 2 Mg2ATP, 0.3 Na3GTP, and 0.3 ethylene glycol bis(β-aminoethyl)ether-N,N,N′,N′-tetra-acetic acid, pH 7.3 with KOH, osmolarity 280 mOsm/kg. On some occasions, drugs were diluted to the required concentration in this solution for intracellular dialysis or a caesium-based solution was used containing (in mm) 135 CsMeSO4, 8 NaCl, 10 HEPES, 2 Mg2ATP, 0.3 Na3GTP, and 7 phosphocreatine, pH 7.3 with CsOH, osmolarity 280 mOsm/kg. For experiments where drugs were added to the internal solution and synaptic responses were measured, cells were left for at least 15 min after achieving the whole-cell configuration to allow diffusion to synapses. Signals were recorded using a Multiclamp 700A (Molecular Devices). Responses were filtered at 4–8 kHz and digitized at 10 kHz (Instrutech; ITC-16). All data were acquired, stored, and analyzed on a Macintosh using Axograph. Only cells with a membrane potential greater than −55 mV were included in this study. Access resistance was 5–15 MΩ and was monitored throughout the experiment. Drugs were applied by diluting to the correct concentration in the superfusate.
For afferent stimulation, a bipolar stimulating electrode was placed on the external capsule. Responses evoked by synaptic stimulation were averages of 5–10 sweeps. Paired-pulse facilitation was measured by giving two shocks at an interval of 60 ms. NMDA-receptor-mediated currents were recorded with a caesium-based internal solution from a holding potential of +40 mV for experiments shown in Figure 1, or with a potassium-based internal solution from −60 mV for experiments shown in Figure 2, while perfused in 10 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) and 100 μm picrotoxin to block AMPA and GABAergic currents, respectively. IPSCs were evoked by local stimulation in the lateral amygdala in the presence of NBQX (10 μm) and AP-5 (30 μm), from a holding potential of −40 mV. For puffing experiments, NMDA (100 μm) and glycine (10 μm) were dissolved in aCSF and were puffed onto the dendrites of lateral amygdala neurons for 3–5 ms using a Picospritzer II (Parker Instrumentation) with glycine (10 μm), tetrodotoxin (TTX; 0.5 μm), and NBQX (10 μm) added to the superfusate. LTP was evoked by giving two sets of 100 stimuli at 30 Hz with a 20 s delay between tetani, in the presence of picrotoxin (100 μm) and CGP55845A (1 μm). When pretreating cells with forskolin or isoprenaline before examining the effect of cytochalasin D, slices were perfused in either forskolin (10 μm) or isoprenaline (10 μm) for at least 15 min before recording from a cell with an internal solution containing cytochalasin D. During LTP induction in the presence of apamin and isoprenaline, care was taken to ensure that the EPSP amplitude was similar to that recorded under control conditions. The effects of drugs on evoked and puffed NMDA-mediated responses were examined at the peak amplitude of the response.
Figure 1.
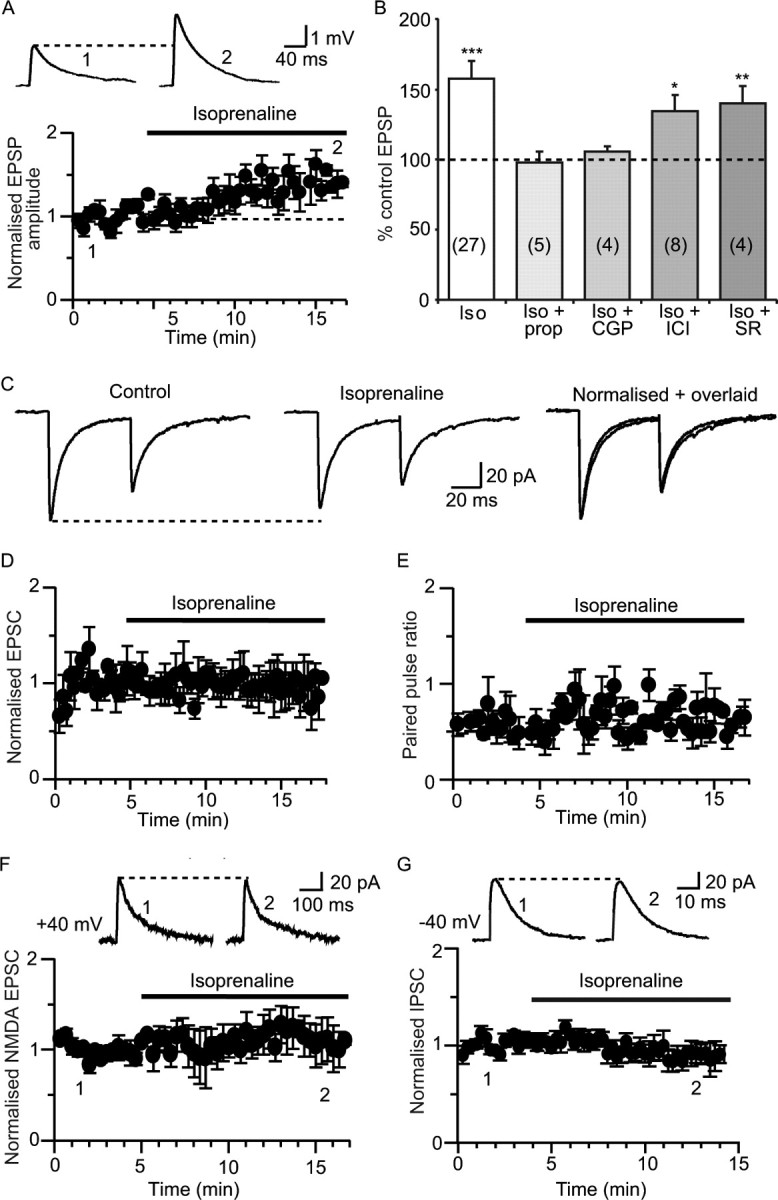
Beta adrenoceptor activation enhances excitatory synaptic transmission. A, Isoprenaline (10 μm) potentiates EPSPs evoked by stimulation of cortical afferents (from 2.9 ± 0.3 mV to 4.5 ± 0.4 mV; n = 23). Representative EPSPs recorded before and after isoprenaline are shown in insets at the time points illustrated on the graph in this and subsequent figures. B, Summary data showing the change in EPSP amplitude in isoprenaline (iso) and blockade of this effect by the nonspecific β adrenoceptor antagonist propranolol (prop; 10 μm) and by the selective β1 adrenoceptor antagonist CGP20712 (CGP; 100 nm) but not by the selective β2 or β3 antagonists, ICI118,551 (ICI; 10 nm) and SR59230A (SR; 100 nm), respectively. Numbers of cells in each measurement are shown in parentheses in the bars in this and subsequent figures. *p < 0.05; **p < 0.01; ***p < 0.001. C, Paired EPSCs recorded at −60 mV in voltage clamp. Isoprenaline has no effect on EPSCs evoked at a 60 ms interval (middle panel; mean amplitude 50.8 ± 13 pA; n = 7) compared with control conditions (left; mean amplitude 53.4 ± 12 pA; n = 7; p > 0.05) and does not change the paired-pulse ratio. Isoprenaline and control traces have been normalized and overlaid on the right panel. Time courses showing the lack of effect of isoprenaline on the EPSC (n = 7), and paired-pulse ratios (n = 7) are shown in D and E, respectively. F, Application of isoprenaline (10 μm) had no effect on NMDA-receptor-mediated EPSC amplitude recorded with a caesium-based internal solution (from 65 ± 17 pA in control to 72 ± 24 pA in isoprenaline; 108 ± 11%; n = 4; p > 0.05). G, Beta adrenergic activation has no effect on IPSCs (IPSCs, measured from a holding potential of −40 mV in the presence of 10 μm NBQX and 30 μm AP-5). Mean IPSC amplitude was 129 ± 26 pA in control and 120 ± 21 pA in isoprenaline (96 ± 9% of control; n = 7; p > 0.05).
Figure 2.

Beta adrenoceptor activation enhances excitatory synaptic transmission through modulation of synaptic SK channels. A, Apamin increases the EPSP from 3.6 ± 0.6 mV to 5.8 ± 1.5 mV (p < 0.05; n = 7). Subsequent application of isoprenaline had no further effect on the EPSP (to 5.4 ± 1.4 mV; n = 7; p > 0.05). B, Potentiation of the EPSP by isoprenaline (middle, from 2.3 ± 0.3 mV in control to 3.5 ± 0.4 mV in isoprenaline) precludes the enhancement by apamin (100 nm; right, mean EPSP amplitude 3.1 ± 0.5 mV; n = 6). C, Postsynaptic infusion of 10 mm BAPTA blocks the enhancement of the EPSP by isoprenaline (mean EPSP amplitude 4.3 ± 0.6 mV in BAPTA vs 4.4 ± 0.7 mV after application of isoprenaline; n = 4), showing that a rise in postsynaptic calcium is required for the actions of isoprenaline. D, Application of the NMDA receptor antagonist d-AP-5 (30 μm) blocks the effect of isoprenaline (from 3.1 ± 0.3 mV in the presence of AP-5 to 3.0 ± 0.6 mV after application of isoprenaline; n = 3). E, Isoprenaline (iso) enhances the NMDA-receptor-mediated EPSC (1 and 2) and precludes enhancement by apamin (ap; 3). Subtraction of the EPSC evoked in isoprenaline from the control EPSC reveals an outward current (4), which has identical kinetics to the pure NMDA-receptor-mediated current (normalized 2 + 4).
Immunohistochemistry.
Animals were perfused transcardially with 2% sodium nitrite solution (in 0.1 m phosphate buffer, pH 7.4) followed by 50 ml of 4% formaldehyde (in 0.1 m phosphate buffer, pH 7.4). Brains were then removed and postfixed overnight in 4% formaldehyde at 4°C, followed by cryoprotection in 20% sucrose (in 0.1 m PBS, pH 7.4) overnight at 4°C. Serial, coronal sections (50 μm) through the amygdala were cut using a freezing microtome. All sections were washed four times (10 min each) in 0.1% PBS before being washed for 30 min in 3% H2O2/PBS solution. After further washes in PBS (5 × 5 min), sections were blocked in a 3% bovine serum albumin/PBS solution. Sections were then incubated for 48 h (at 4°C) in a rabbit anti-β1-adrenoceptor primary antibody (1:5000; Abcam) prepared in a 1% bovine serum albumin (BSA)/PBS solution. However, some sections were not exposed to the primary antibody and formed a secondary only control. All sections were then washed in PBS (3 × 5 min) before being incubated to 2 h at room temperature in a biotinylated donkey anti-rabbit secondary antibody (1:300; Jackson ImmunoResearch). Sections were washed again in PBS (3 × 5 min) and then incubated for 2 h at room temperature in a solution of avidin-biotin-horseradish-peroxidase complex (6 μl A and B/ml of PBS) (Vector Elite Kit). The sections were washed in PBS (3 × 10 min), then in sodium acetate buffer (pH 6.0; 1 × 5 min), and then washed for 15 min in a 2% NiSO4 solution (in sodium acetate buffer, pH 6.0) containing 2 mg/ml d-glucose, 0.4 mg/ml NH4Cl, and 0.025% 3,3-diaminobenzidine. This was followed by another incubation in the same solution but with the addition of glucose oxidase (0.2 μl/ml). The development was observed under a microscope and, at a suitable time point, sections were washed in sodium acetate buffer to stop the reaction. After further PBS washes (3 × 10 min), all sections were mounted onto Superfrost Plus glass slides, dehydrated in ethanol, cleared in xylene, and coverslipped using Depex mounting medium. Sections were visualized and images acquired using Zeiss Axiovision 4.5 software.
Transfection of organotypic cultures and immunostaining.
Amygdala organotypic cultures were prepared according to the standard interface method (Stoppini et al., 1991). Coronal brain slices containing basolateral amygdala were prepared from postnatal day 7–9 Wistar rats under the same conditions as for electrophysiological recordings. Brain slices (300 μm) were cultured on a porous membrane (Millicell inserts; Millipore) using a culture medium containing 50% OptiMEM (Invitrogen), 25% heat inactivated horse serum, 25% HBSS (Sigma) supplemented with d-glucose (6.5 g/L) and penicillin/streptomycin (Invitrogen). The cultures were maintained at 37°C in a 5% CO2 atmosphere.
Gold particles (10 mg, 1.6 μm; Bio-Rad) were mixed with 100 μl 0.05 m spermidine, 25 μg of a green fluorescent protein plasmid pCS2Venus or pEGFPN1 (Clontech), and 25 μg of pJPA5 rSK2 myc3 (kindly provided by J. Adelman, Vollum Institute, Portland, OR), a construct expressing rat SK2 with an extracellular triple-myc epitope tag between S3 and S4. The DNA was precipitated by the addition of 100 μl 5% CaCl2, the particles were washed in 100% ethanol, and resuspended in 3 ml of 100% ethanol containing 0.05 mg/ml polyvinylpyrrolidine. The mix was then used to coat Tefzel tubing on a tube-loading station (Bio-Rad) as per the manufacturer's instructions. Slices were shot <2 h after being cut, using a Helios Gene Gun (Bio-Rad) with a modified barrel ∼3 cm from the slice through a 70 μm nylon mesh (Falcon) at a pressure of 80 PSI.
Organotypic cultures were treated ∼60 h after preparation with media containing 50 μm forskolin [diluted in dimethylsulfoxide (DMSO), 0.05%] or DMSO (0.05%) for 15 min or 30 min at 37°C. Cultures were then washed and incubated with an anti-myc mouse monoclonal antibody (1:1000; Cell Signaling Technology) for 30 min at room temperature, washed in PBS, and fixed with 4% paraformaldehyde/4% sucrose in PBS for 30 min at room temperature. For the 30 min time point, forskolin was also present during incubation with the antibody. The cultures were then incubated in PBS containing 5% normal goat serum/1% BSA for 30 min at room temperature to block nonspecific binding and then in blocking buffer containing Alexa Fluor 568 goat anti-mouse IgG (1:700; Invitrogen) for 45 min at room temperature. The washed slices were mounted using Prolong Gold antifade reagent (Invitrogen) and cells in the lateral amygdala were analyzed on a Zeiss LSM 510 confocal microscope. Fluorescent images were made from green fluorescent neurons within the located basolateral amygdala. Fluorescence Z-stacks were obtained from thin spiny dendrites <100 μm from green fluorescent neurons within the basolateral amygdala. Green (enhanced green fluorescent protein or Venus) and red (Alexa 568) fluorescence images were acquired sequentially using 488 nm excitation (emission, band pass 500–560 nm) and 543 nm excitation (emission, long pass 560), respectively. Z-stacks were deconvolved using the expectation-maximization algorithm for maximum likelihood deconvolution (Conchello, 1998) in the X Windows version of Computational Optical Sectioning Microscopy (www.omrfcosm.omrf.org), low-pass filtered, and flattened into a two-dimensional image. Counts of Alexa-568 puncta were obtained from several secondary and tertiary dendritic sections between 12 and 30 μm in length on each neuron. Up to four segments were sampled and averaged to represent the neuron. Image acquisition, processing, and counts were performed blind to the treatment condition.
Drugs and chemicals.
TTX and apamin were obtained from Alomone Labs. BAPTA, NMDA, glycine, forskolin, dideoxyforskolin, cytochalasin D, latrunculin B, 8-bromo-adenosine 3′, 5′-cyclic monophosphate (8-bromo-cAMP), Rp-cyclic 3′, 5′-hydrogen phosphorothioate adenosine triethylammonium (Rp-cAMPs), and picrotoxin were obtained from Sigma. NBQX, AP-5, propranolol, isoprenaline, CGP20712, ICI118,551, SR59230A, and CGP55845A were obtained from Tocris. Protein kinase A inhibitor 14–22 Amide (PKI) was obtained from Merck. Myristoyl trimethyl ammonium bromide (MiTMAB) and BisT were a gift from Dr. Phil Robinson (Children's Medical Research Institute, Sydney, New South Wales, Australia).
Student's t tests were used for statistical comparisons between groups. Results are expressed a mean ± SEM.
Results
Whole-cell recordings were made from principal neurons in the lateral amygdala, and synaptic responses were evoked by stimulating cortical afferents in the external capsule (Mahanty and Sah, 1999).
Activation of β1 adrenoceptors enhances the EPSP
At resting membrane potentials, stimulation of cortical afferents evokes an EPSP that is mediated by AMPA and NMDA receptors (Faber et al., 2005). Application of the β adrenoceptor agonist isoprenaline (10 μm) enhanced the EPSP (157 ± 13% of control; p < 0.001; n = 27) (Fig. 1 A,B). This effect of isoprenaline was reversible, with the EPSP returning to control values within 10 min of washout (to 100 ± 8% of control values; n = 7; p > 0.05; data not shown) and was blocked by the β adrenoceptor antagonist propranolol (10 μm; 98 ± 8% of control; n = 5; p > 0.05) (Fig. 1 B). Potentiation of the EPSP by isoprenaline was mediated by activation of β1 adrenoceptors because enhancement was blocked in the presence of the selective β1 antagonist CGP20712 (10 nm; to 106 ± 4% of control; n = 4; p > 0.05) (Fig. 1 B). In contrast, the selective β2 antagonist ICI118,5551 (10 nm), or the selective β3 antagonist SR59230A (100 nm) had no effect on the actions of isoprenaline (143 ± 12% of control in ICI118,551; n = 8; p < 0.05 and 140 ± 12% of control in SR59230A; n = 4; p < 0.05) (Fig. 1 B). Consistent with this result, β1 adrenoceptors are clearly present in the lateral nucleus of the amygdala (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
To determine the locus of change that underlies the actions of isoprenaline, we first tested its actions on the EPSC recorded under voltage clamp to limit activation of postsynaptic voltage dependent currents. At a holding potential of −60 mV, the EPSC evoked by stimulation of cortical afferents was unaffected by isoprenaline (94 ± 10%; n = 7; p > 0.05) (Fig. 1 C,D). Application of isoprenaline did not change the paired- pulse ratio (103 ± 10% of control; n = 7; p > 0.05) (Fig. 1 C,E) showing that isoprenaline does not change transmitter release probability (Zucker, 1989) and suggesting a postsynaptic site of action. Under these recording conditions, the EPSC is primarily mediated by AMPA receptors, with minimal contribution of NMDA receptors (Mahanty and Sah, 1999). To test isoprenaline's effects on the NMDA-receptor-mediated component, cells were voltage clamped at +40 mV using a caesium-based internal solution. However, isoprenaline had no effect on the NMDA-receptor-mediated EPSC (108 ± 10%; n = 4; p > 0.05) (Fig. 1 F). These results show that isoprenaline, acting at β adrenoceptors, enhances excitatory synaptic transmission through a postsynaptic mechanism that does not involve a change in transmitter release or modulation of ionotropic glutamate receptor-mediated currents. Moreover, the increase in EPSP amplitude is not attributable to a reduction in disynaptic inhibition (Lang and Paré, 1997) as isoprenaline had no effect on IPSCs evoked by local stimulation in the lateral amygdala in the presence of 10 μm NBQX and 30 μm d-AP-5 (96 ± 9%; n = 7; p > 0.05) (Fig. 1 G).
Beta adrenoceptors modulate postsynaptic SK channels
Excitatory synapses on principal neurons in the lateral amygdala express postsynaptic SK channels that are activated by calcium influx through NMDA receptors and act to shunt the postsynaptic membrane, reducing the amplitude of the evoked EPSP (Faber et al., 2005). Thus, as shown previously (Faber et al. 2005), blockade of SK channels with the selective toxin apamin enhanced synaptic transmission. Apamin (100 nm) increased the amplitude of the EPSP to 155 ± 13% of control (p < 0.01; n = 6) (Fig. 2 A). In the presence of apamin, isoprenaline had no further effect on the EPSP (Fig. 2 A) with the EPSP remaining at 145 ± 16% of control (n = 7), not significantly different from the enhancement produced by apamin (p > 0.05). Conversely, when the EPSP was enhanced by isoprenaline (to 157 ± 13% of control; p < 0.01; n = 6) addition of apamin (100 nm) had no further significant effect (Fig. 2 B) (to 138 ± 17% of control; n = 6; p > 0.05). These results show that the actions of isoprenaline and apamin are mutually exclusive, suggesting that β adrenoceptors enhance the EPSP by acting on SK channels. Consistent with this, the effect of isoprenaline was also blocked when activation of postsynaptic SK channels was inhibited by loading neurons with the calcium chelator BATPA (10 mm; 102 ± 6% of control; n = 4; p > 0.05) (Fig. 2 C) (Faber et al., 2005) confirming a postsynaptic site of action of isoprenaline. Furthermore, blocking the calcium source for SK channel activation, the NMDA receptor (Faber et al., 2005), with d-AP-5 (30 μm), also prevented enhancement of the EPSP by isoprenaline (94 ± 11%; n = 3; p > 0.05) (Fig. 2 D). Together, these data suggest that β adrenoceptor activation augments synaptic transmission by modulating SK channels.
As a final test of this hypothesis, we examined the action of isoprenaline on the NMDA-receptor-mediated EPSC recorded at holding potentials of −60 mV, with extracellular Mg2+ reduced to 0.1 mm and in the presence of NBQX (10 μm) and picrotoxin (100 μm) to block AMPA and GABA receptor-mediated currents. Under these conditions, application of isoprenaline enhanced the EPSC by 167 ± 18% (n = 7; p < 0.05) (Fig. 2 E), and subsequent application of apamin had no significant effect on the EPSC (to 172 ± 23%; n = 4; p > 0.05) (Fig. 2 E). Analysis of the isoprenaline-sensitive current obtained by digital subtraction revealed that the kinetics of this current is similar to that of the NMDA-receptor-mediated current (Fig. 2 E). This result is consistent with our previous demonstration that the SK channel-mediated current at these synapses tracks NMDA receptor activation (Faber et al., 2005) and supports the notion that that β adrenoceptors modulate SK channels. Thus, as with application of apamin (Faber et al., 2005; Ngo-Anh et al., 2005), reducing the SK channel-mediated shunt by applying isoprenaline enhances the excitatory synaptic transmission.
Beta adrenoceptors modulate SK channels via protein kinase A
Beta adrenoceptors are G-protein-coupled receptors that activate adenylyl cyclase, leading to the generation of cAMP and activation of PKA (Gilman, 1987). To test the downstream mechanism of action of isoprenaline, we first applied the adenylyl cyclase activator forskolin (10 μm). Forskolin enhanced the EPSP to 181 ± 21% of control (n = 18; p < 0.005) (Fig. 3 A,C), similar to the increase seen with isoprenaline and apamin. Subsequent application of either isoprenaline (to 201 ± 42% of the control EPSP; n = 6; p > 0.05) (Fig. 3 A,C) or apamin (to 194 ± 58% of the control EPSP; n = 6; p > 0.05) (Fig. 3 B,C) had no further significant effect on the EPSP. In contrast, dideoxyforskolin (10 μm), an inactive isomer of forskolin, had no significant effect on the EPSP (119 ± 20% of control; n = 6; p > 0.05) and did not prevent potentiation of the EPSP by isoprenaline (to 186 ± 32% of control; n = 6; p < 0.05; data not shown).
Figure 3.
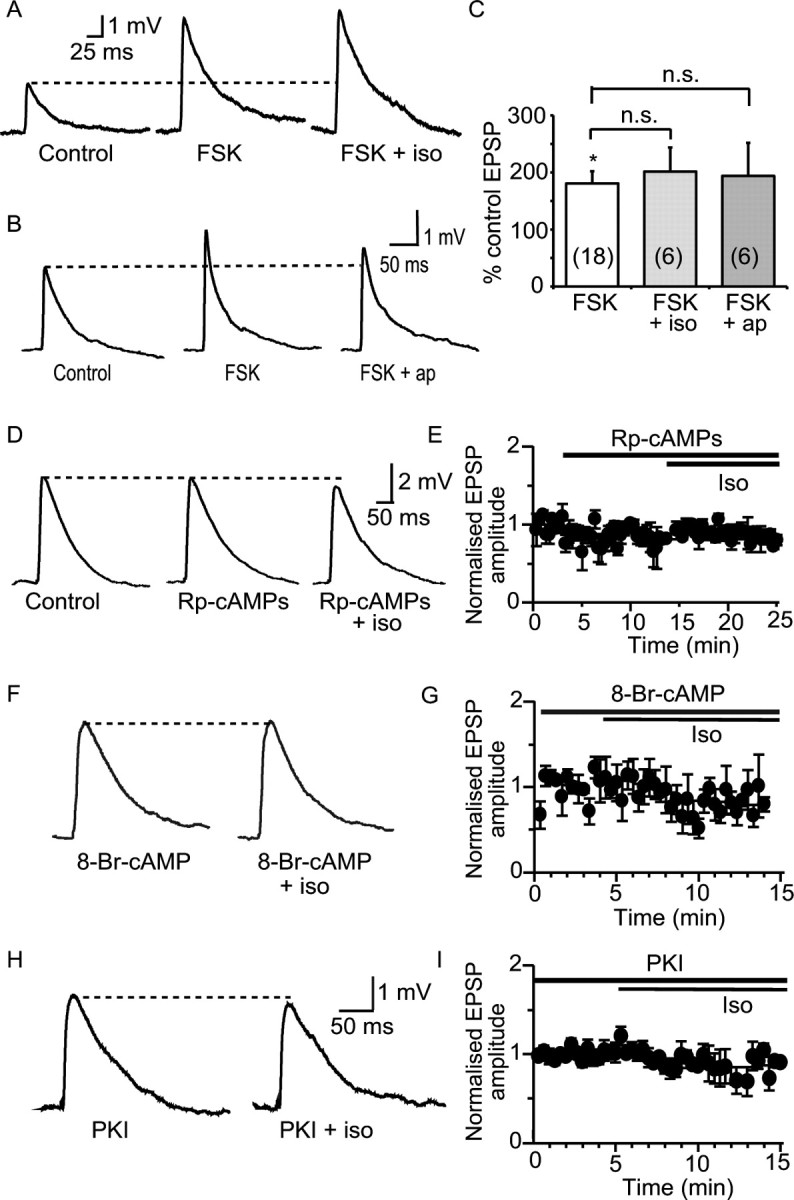
Enhancement of synaptic transmission by β adrenoceptors is mediated through activation of PKA. A, Activation of adenylyl cyclase by forskolin (FSK, 10 μm) augments the EPSP (by 180 ± 20% from 3.7 ± 0.5 mV to 6.3 ± 0.7 mV; n = 18) and precludes the effect of isoprenaline (iso; to 201 ± 42% of control; mean EPSP amplitude in isoprenaline 6.9 ± 0.8 mV; n = 6; p > 0.05). B, Synaptic responses showing that forskolin precludes the augmentation of the EPSP by apamin (to 193 ± 58%; mean EPSP amplitude in apamin 6.3 ± 0.6 mV; n = 6; p > 0.05). C, Summary data showing mean EPSP amplitude after application of forskolin and lack of effect of subsequent treatment with isoprenaline or apamin (ap). D, Inhibition of adenylyl cyclase with Rp-cAMPs (10 μm) has no effect on the EPSP (from 4.3 ± 0.9 mV to 3.8 ± 1.2 mV; n = 4; p > 0.05) but blocks the enhancement of the EPSP by isoprenaline (to 3.3 ± 0.9 mV; p > 0.05). E, Graph showing the mean time course of Rp-cAMPs and isoprenaline on the synaptic responses (n = 3). F, Intracellular perfusion with 8-bromo-cAMP (8-Br-cAMP, 100 μm) precludes the effect of isoprenaline (from 3.3 ± 0.9 mV in 8-bromo-cAMP to 2.9 ± 1 mV in isoprenaline; p > 0.05). G, Time course of effects shown in F for four cells. H, Loading the postsynaptic neuron with the PKA inhibitor PKI blocks the effect of isoprenaline (from 5.2 ± 0.9 mV in the presence of PKI to 4.3 ± 1 mV in the presence of isoprenaline; p > 0.05). Time course for four cells is shown in I. *p < 0.05. n.s., Not significant.
Application of the membrane permeant adenylyl cyclase inhibitor Rp-cAMPs (10 μm) had no effect on the EPSP when applied alone (81 ± 13% of control; p > 0.05; n = 4) (Fig. 3 D,E) but blocked the enhancement by isoprenaline (to 71 ± 7%; p > 0.05; n = 4) showing that activation of adenylyl cyclase is required for the effects of isoprenaline. Loading the postsynaptic neuron with the synthetic cAMP analog, 8-bromo-cAMP (100 μm) also inhibited the effect of isoprenaline on the EPSP (to 80 ± 14% of control; n = 4; p > 0.05) (Fig. 3 F,G), presumably because the increase in cAMP occluded the effect of isoprenaline. Finally, to test if activation of PKA is required for the actions of isoprenaline, we inhibited PKA by loading the specific inhibitor PKI (1 μm) into the postsynaptic neuron. PKI blocked the enhancement of the EPSP by isoprenaline (to 82 ± 8% of control; n = 4; p > 0.05) (Fig 3 H, I). Together, these findings show that isoprenaline acting at β adrenoceptors triggers activation of adenylyl cyclase, leading to an increase in cAMP and activation of PKA, which is necessary and sufficient to cause a postsynaptic enhancement of the EPSP. This effect of isoprenaline requires calcium influx via NMDA receptors (Fig. 2). Because activation of PKA occluded the action of apamin on synaptic transmission, but had no effect on NMDA-receptor-mediated transmission (Fig. 1), these results suggest that activation of PKA uncouples SK channels from postsynaptic NMDA receptors.
To directly examine the postsynaptic effects of forskolin, we tested the effect of forskolin on postsynaptic currents evoked by exogenous application of NMDA. Neurons were voltage clamped at −50 mV and NMDA receptors activated by puffing NMDA (100 μm) onto the dendritic tree (Faber et al., 2005). Forskolin enhanced the current evoked by NMDA to 157 ± 17% of control (n = 4; p < 0.05) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), and subsequent application of apamin had no effect on the NMDA-evoked currents (to 164 ± 13% of control; n = 4; p > 0.05). Conversely, apamin increased the NMDA-evoked currents to 171 ± 13% of control (n = 5; p < 0.05) (Faber et al., 2005) and precluded any subsequent augmentation of the evoked current by forskolin (to 150 ± 24% of control, n = 5) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results confirm that activation of PKA enhances postsynaptic NMDA-evoked currents in lateral amygdala principal neurons, via modulation of SK channels. However, although SK channels have phosphorylation sites for PKA (Ren et al., 2006), these channels are not known to change their biophysical properties in response to phosphorylation. What then explains the lack of effect of apamin after activation of PKA?
SK channels at synapses undergo recycling
At glutamatergic synapses, postsynaptic ionotropic glutamate receptors are anchored to the cytoskeleton and recycled in and out of the postsynaptic membrane both constitutively and during synaptic plasticity (Malinow and Malenka, 2002). Among the plethora of proteins that are present in the postsynaptic density (Sheng and Hoogenraad, 2007), these synapses also express SK channels that are activated by calcium influx via NMDA receptors (Faber et al., 2005; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007; Brosh et al., 2007). When expressed in COS7 cells, phosphorylation of SK channels can reduce their expression levels on the plasmalemma (Ren et al., 2006). Moreover, recent studies have shown that at hippocampal synapses, activation of PKA after tetanic stimulation leads to endocytosis of synaptic SK channels (Lin et al., 2008). We therefore tested whether synaptic SK channels in lateral amygdala principal neurons are also regulated.
At many synapses, postsynaptic ionotropic receptors are anchored at the postsynaptic membrane, and disruption of the cytoskeleton leads to removal of some of these receptors (Zhou et al., 2001; Dillon and Goda, 2005). Thus, we first tested if SK channels may also be similarly anchored. To disrupt the cytoskeleton, we tested two actin depolymerisers, cytochalasin D and latrunculin B (Cooper, 1987; Spector et al., 1989). Loading neurons with cytochalasin D (10 μm) caused the EPSP amplitude to increase over the first 15 min after obtaining the whole-cell configuration (to 135 ± 12% of control; n = 16; p < 0.05) (Fig. 4 A,F). Once the EPSP had stabilized, subsequent application of apamin had no further effect on the EPSP (to 107 ± 17% of the control EPSP; n = 8; p > 0.05) (Fig. 4 B,F). Similarly, loading neurons with latrunculin B (20 μm) also enhanced the EPSP (to 130 ± 8% of control; n = 4; p < 0.05) (Fig. 4 F), and subsequent application of apamin failed to enhance the synaptic response (to 119 ± 22% of the control EPSP; n = 4; p > 0.05). These data suggest that, as for AMPA receptors at hippocampal synapses (Zhou et al., 2001), in pyramidal neurons of the lateral amygdala, SK channels are anchored at the synapse by actin filaments, and their depolymerization leads to removal of postsynaptic SK channels.
Figure 4.

Beta adrenoceptor activation enhances synaptic transmission by regulating SK channel trafficking. Disruption of actin polymerization enhances the EPSP and removes SK channels from the synapse, because loading cells with cytochalasin D causes a run up of the EPSP over the 15 min period after obtaining the whole-cell configuration (A; from 2.8 ± 0.3 mV immediately after break in to 3.4 ± 0.3 mV 15 min later; n = 6). B, C, After perfusion of neurons with cytochalasin D (shown at t = 0 and normalized to the enhancement in cytochalasin D in B and C), neither apamin (B; to 3.4 ± 0.6 mV; n = 5) nor isoprenaline (C; from 2.7 ± 0.2 mV in cytochalasin D to 2.8 ± 0.3 mV after application of isoprenaline; n = 5) have any further effect on the EPSP. D, Pretreatment with isoprenaline blocks the enhancement of the EPSP by cytochalasin D (from 3.5 ± 0.5 mV to 2.7 ± 0.3 mV; n = 6). E, Cytochalasin D (added to the internal solution at 10 μm) has no effect on the NMDA-receptor-mediated EPSC (from 123 ± 14 pA immediately after break in to 113 ± 13 pA, 25 min later; n = 4; p > 0.05). F, Summary graphs showing the effects of actin depolymerizing agents. Cytochalasin D, Cyto D; isoprenaline, iso; forskolin, FSK; latrunculin B, lat B; apamin; ap; n.s., not significant. *p < 0.05.
Activation of PKA by β adrenoceptors blocks the action of apamin (Fig. 3). We propose that synaptic SK channels undergo constitutive recycling from the postsynaptic membrane and β adrenoceptor activation leads to phosphorylation of these channels, trapping the channels in a cytosolic compartment (Ren et al., 2006), blocking their delivery to the postsynaptic membrane, and thus reducing the number of synaptic SK channels. Consistent with this proposal, enhancement of the EPSP by isoprenaline was blocked in neurons loaded with cytochalasin D (to 124 ± 14% of control; n = 6; p > 0.05) (Fig. 4 C,F). Conversely, pretreatment of slices with isoprenaline (n = 6) or forskolin (n = 5) blocked the enhancement of the EPSP by cytochalasin D (Fig. 4 D,F), supporting our proposal that activation of PKA depletes synaptic SK channels, thereby precluding the run up of the EPSP caused by cytochalasin D. In fact, under these conditions, the EPSP was significantly depressed (to 80 ± 5% of control in the presence of isoprenaline and cytochalasin D; n = 6; p < 0.05, and to 81 ± 5% of control in the presence of forskolin and cytochalasin D; n = 5; p < 0.05) (Fig. 4 F), which is likely to be attributable to a reduction in synaptic AMPA receptors caused by actin depolymerization (Kim and Lisman, 1999; Zhou et al., 2001). Enhancement of the EPSP and blockade of the effect of apamin by cytochalasin D and latrunculin B could be attributable to a reduction in postsynaptic NMDA receptors, because a reduction in calcium influx through NMDA receptors would inhibit SK channel activation and thus may augment synaptic transmission (Faber et al., 2005). However, as reported previously for hippocampal neurons (Kim and Lisman, 1999; Zhou et al., 2001), NMDA-receptor-mediated synaptic currents recorded with a caesium internal (thus removing the contribution of SK channels) from a holding potential of +40 mV were not affected by loading neurons with cytochalasin D (to 93 ± 7% of control; n = 4; p > 0.05) (Fig. 4 E) or latrunculin B (to 115 ± 14% of control; n = 4; p > 0.05; data not shown) showing that the actions of actin depolymerization are not attributable to effects on postsynaptic NMDA receptors.
To directly test whether postsynaptic SK channels are removed by activation of PKA, we expressed SK2 channels tagged with an extracellular myc epitope (Ngo-Anh et al., 2005) in lateral amygdala neurons in organotypic brain slices (Stoppini et al., 1991). As shown previously for hippocampal neurons in culture (Ngo-Anh et al., 2005), in principal neurons in the lateral amygdala, we observed punctate SK channel staining on dendritic spines throughout the dendritic tree at a density of 7–9 puncta/10 μm (Fig. 5). This is similar to the average density of spines on basolateral amygdala secondary and tertiary dendritic branches (Power and Sah, 2007) suggesting that almost all spines express SK channels. Application of forskolin for 15 min significantly reduced the number of SK2 puncta from 8.6 ± 0.6 (n = 8) to 6.4 ± 0.7 puncta/10 μm (p < 0.05; n = 12) (Fig. 5 B). A longer application of forskolin (30 min) reduced SK channel puncta from 7.5 ± 0.7 (n = 14) to 3.9 ± 0.5 puncta/10 μm (p < 0.001; n = 22) (Fig. 5 B). Because the myc epitope is attached on an extracellular site, only channels present on the plasmalemma are seen with this antibody, demonstrating that activation of PKA reduces surface expression of SK channels at excitatory synapses in the lateral amygdala.
Figure 5.
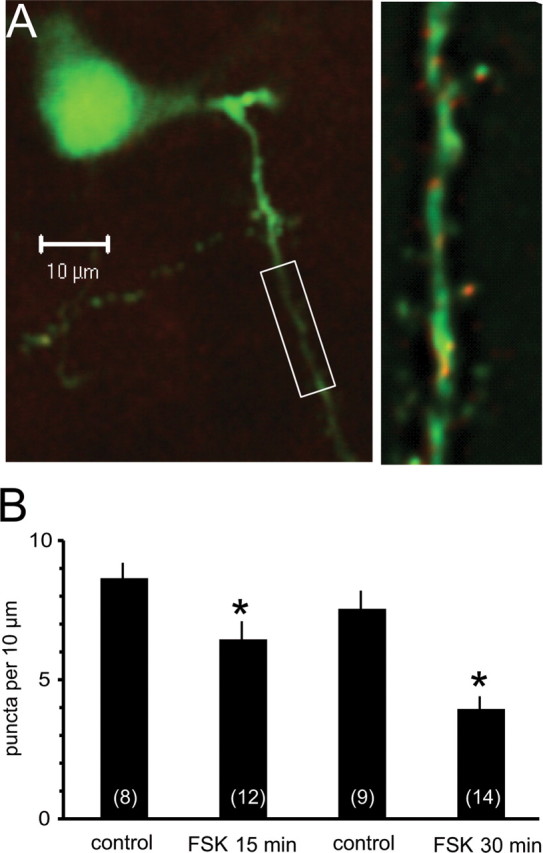
Activation of protein kinase A reduces surface expression of SK channels. Organotypic coronal brain slices were prepared and transfected using a gene gun. A, Principal neuron transfected with SK2 channels with primary and secondary dendrites as shown by venus fluorescence (green). SK2 channels visualized using anti-myc are shown in red and appear as puncta on the dendritic tree. The boxed section of the secondary dendrite is shown on a slightly enlarged view in the right panel. SK2 channel puncta were counted on primary and secondary dendrites and quantified as number of quanta per 10 μm of dendrite. B, Histogram shows mean data for the control group and after treatment with forskolin (FSK) for either 15 min or 30 min. Numbers of cells are shown in the bars. *p < 0.05.
For other synaptic receptors that are modulated by trafficking, such as AMPA (Man et al., 2000) and GABA receptors (Kittler and Moss, 2003), receptor internalization is achieved by endocytosis via clathrin-coated pits, a process that requires the GTPase dynamin (Liu and Robinson, 1995; Schmid et al., 1998). To investigate whether a similar mechanism underlies internalization of SK channels, we loaded principal neurons with two blockers of dynamin, BisT, or MiTMAB (Hill et al., 2004, 2005). Simultaneous dual recordings were made where one cell was loaded with BisT (10 μm) and the other with a control internal solution. Bath application of isoprenaline enhanced the EPSP in control cells (to 122 ± 9% of control; n = 7; p < 0.05) but not in cells loaded with BisT (to 106 ± 10% of control; n = 11; p > 0.05) (Fig. 6 A,C). Similarly, MiTMAB (100 μm) blocked the enhancement of the EPSP by isoprenaline. In fact, EPSP amplitude was slightly reduced by isoprenaline in the presence of MiTMAB (to 89 ± 4% of control; p < 0.05; n = 8) (Fig. 6 B,C).
Figure 6.

Disruption of endocytosis prevents the enhancement of synaptic transmission by β adrenoceptor activation. A, The dynamin inhibitor BisT blocks the effect of isoprenaline. Dual recordings were made where one cell was loaded with BisT and the other with control internal solution. Isoprenaline (Iso) enhances the EPSP recorded in the control cell (bottom; from 3.3 ± 0.6 mV to 4.0 ± 0.8 mV; n = 7) but not in the cell loaded with BisT (top; from 2.7 ± 0.2 mV to 2.8 ± 0.3 mV; n = 11). Example traces are shown on the right. B, MiTMAB (100 μm), applied either by intracellular loading of neurons or by presoaking slices for at least 1 h, blocked the effect of isoprenaline on the EPSP (from 4.0 ± 0.6 mV in MiTMAB to 3.6 ± 0.6 mV in isoprenaline; n = 8). C, Summary graphs showing the effects of dynamin inhibitors, respectively. *p < 0.05, **p < 0.01, ***p < 0.001.
Functional role of SK channel modulation
Activation of postsynaptic SK channels at excitatory synapses limits LTP, and their blockade enhances LTP (Stackman et al., 2002; Faber et al., 2005). We therefore next examined whether isoprenaline enhances synaptic plasticity through modulation of SK channels in the lateral amygdala. Under control conditions (Faber et al., 2005), tetanic stimulation caused long-term potentiation of the EPSP slope to 127 ± 12% of control values (n = 13) (Fig. 7 A). In the presence of isoprenaline, however, LTP was enhanced to 173 ± 17% of control values (n = 7) (Fig. 7 B), significantly greater than seen in control conditions (p < 0.05) (Fig. 7 C). Propranolol blocked the enhancement of LTP by isoprenaline. The EPSP slope recorded 30 min after tetanic stimulation was 128 ± 12% of control in the presence of both propranolol and isoprenaline (n = 6) (Fig. 7 C), which was not significantly different to that recorded under control conditions (p > 0.05), but was significantly less than seen in the presence of isoprenaline alone (p < 0.05). If isoprenaline enhances LTP through a reduction in the SK channel-mediated depression of the EPSP, no further enhancement of LTP should occur when apamin is applied in the presence of isoprenaline. This was found to be the case: LTP elicited in the presence of isoprenaline and apamin was 174 ± 11% (n = 6) (Fig. 7 D) (p > 0.05 compared with in the presence of isoprenaline alone).
Figure 7.

Beta adrenoceptor activation enhances long-term potentiation through modulation of SK channels. A, Under control conditions, tetanic stimulation (shown at time 0 in this and subsequent graphs) causes LTP of the EPSP. Example traces are shown in the bottom at the time points indicated in the graph. B, In the presence of isoprenaline (10 μm), the amount of potentiation after tetanic stimulation is enhanced. C, Graphs showing mean data for LTP evoked in control conditions (open circles), in the presence of isoprenaline (iso; filled circles), and in the presence of isoprenaline and propranolol (prop; 10 μm; filled diamonds). Note that in the presence of propranolol and isoprenaline, LTP was similar to that seen under control conditions. D, Graphs showing mean data for LTP evoked in the presence of isoprenaline alone (filled circles) compared with in the presence of isoprenaline and apamin (ap; 100 nm; filled triangles). No further enhancement of LTP was seen when apamin was applied with isoprenaline compared with in the presence of isoprenaline alone.
Discussion
The synapse is a dynamic structure in which postsynaptic neurotransmitter receptors undergo constant regulation both during basal synaptic transmission and synaptic plasticity (Kneussel, 2002; Malinow and Malenka, 2002). Regulation of the number of postsynaptic receptors is a key mechanism that underlies the changes in synaptic strength seen during several forms of long-term synaptic plasticity. SK channels are widely expressed throughout the CNS (Stocker and Pedarzani, 2000; Sailer et al., 2002) and until recently were thought only to be involved in controlling the firing properties of neurons (Sah and Faber, 2002). However, it is now clear that these channels are also present at glutamatergic synapses where they are activated by calcium influx via NMDA receptors (Faber et al., 2005; Ngo-Anh et al., 2005). Activation of these potassium channels effectively shunts the postsynaptic membrane and reduces the amplitude of excitatory synaptic potentials (Faber et al., 2005; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007). Thus, blockade of SK channels with specific inhibitors, such as apamin, enhances synaptic transmission and likely underlies the effects of apamin on learning (Deschaux et al., 1997; Stackman et al., 2002). In this study, we have shown that in lateral amygdala pyramidal neurons postsynaptic SK channels require the actin cytoskeleton for their stability, being endocytosed constitutively by a dynamin-dependent mechanism, with new channels transported back to the synapse via the actin cytoskeleton. β1 adrenoceptors trigger activation of the adenylyl cyclase-cAMP-PKA pathway, which interferes with this recycling, effectively removing postsynaptic SK channels and thereby enhancing excitatory synaptic transmission. Functionally, the larger amplitude EPSP in the presence of isoprenaline leads to enhanced synaptic plasticity at these synapses. Thus, regulation of SK channels at excitatory synapses provides a new mechanism for controlling synaptic efficacy.
Three lines of evidence support our proposal that synaptic SK channels undergo constitutively recycling and that activation of β adrenoceptors reduces the number of SK channels, thereby potentiating synaptic transmission. First, recycling of SK channels requires endocytosis to remove channels from the postsynaptic density. Consistent with this, the actions of β adrenoceptor activation are inhibited when endocytosis is blocked by inhibitors of dynamin. Second, it is well established that anchoring of postsynaptic receptors is essential for their maintenance at the synapse (Zhou et al., 2001; Dillon and Goda, 2005). Consistent with our proposal that SK channels are also anchored at the postsynaptic density, disruption of the actin cytoskeleton led to removal of SK channels from the synapse and inhibited any further actions of β adrenoceptor activation. Thirdly, activation of PKA significantly reduced the surface expression of SK channels in amygdala principal neurons.
Three isoforms of SK channels are known to be present in neurons: SK1, SK2, and SK3. Although we have not directly identified the type of channels modulated by PKA in lateral amygdala neurons, these are likely to be SK2 channels for the following reasons. First, SK2 channels are expressed in the lateral amygdala (Stocker and Pedarzani, 2000; Sailer et al., 2002). Second, it is the SK2 subunit that is phosphorylated by PKA (Ren et al., 2006) and its overexpression, which would reduce EPSP amplitude in amygdala pyramidal neurons (Faber et al., 2005), depressed rats' performances in the amygdala-dependent behavioral paradigm, cued fear conditioning (Hammond et al., 2006). Thirdly, when expressed in amygdala neurons in organotypic slices, these channels were transported to spines, and activation of PKA significantly reduced their expression levels, confirming that activation of β adrenoceptors removes postsynaptic SK2 channels. Finally, at synapses made by Schaffer Collaterals on hippocampal pyramidal neurons, activation of protein kinase A during induction of long-term potentiation leads to internalization of synaptic SK2 channels and contributes to LTP (Lin et al., 2008).
It has previously been shown that activation of β adrenoceptors in the basal nucleus of the amygdala augments excitatory synaptic transmission at inputs arising from the endopiriform nucleus by enhancing release probability (Huang et al., 1996). However, the effects of isoprenaline we have described at cortical inputs in the lateral nucleus are entirely postsynaptic as they are blocked by postsynaptic infusion of caesium, by buffering postsynaptic calcium with the chelator BAPTA, and by blocking the adenylate cyclase-PKA pathway by postsynaptic loading of inhibitors and activators. Activation of PKA has been reported to enhance calcium influx through NMDA receptors (Raman et al., 1996; Skeberdis et al., 2006). However, increased calcium influx would be expected to enhance SK channel activity, which would be expected to reduce EPSP amplitude. Moreover, consistent with previous results (Skeberdis et al., 2006), activation of PKA had no effect on the amplitude or kinetics of the NMDA-receptor- mediated synaptic current (Fig. 1). The fact that the actions of isoprenaline on excitatory transmission were blocked by pretreatment with apamin, or by postsynaptic infusion of caesium and BAPTA, shows that in the lateral amygdala the effects of isoprenaline on synaptic transmission are mediated entirely by modulation of SK channels.
The laying down of memory traces is thought to result from LTP at excitatory synapses, and enhancement of LTP by β adrenoceptor activation has previously been described for neurons in both the amygdala (Huang et al., 1996; Tully et al., 2007) and in the hippocampus (Sah and Bekkers, 1996; Thomas et al., 1996; Lin et al., 2003; Hu et al., 2007). These effects have been proposed to result from modulation of synaptic integration through blockade of the slow afterhyperpolarization (Sah and Bekkers, 1996; Thomas et al., 1996), modulation of NMDA receptors (Thomas et al., 1996), and by suppression of tonic inhibition (Tully et al., 2007). In the latter study, β adrenergic receptor activation decreased excitability of interneurons, leading to reduced feedforward inhibition after stimulation of thalamic inputs (Tully et al., 2007). This mechanism may also contribute to synaptic plasticity at cortical inputs to principal neurons. However, in the current study, LTP experiments were performed with inhibition blocked, suggesting that under our experimental conditions isoprenaline's actions were entirely mediated by modulation of SK channels. More recently, phosphorylation of GluR1 subunits by β adrenoceptor activation in the hippocampus was shown to lower the threshold for LTP induction (Hu et al., 2007). Our results provide a cellular mechanism that explains the actions of β adrenoceptor activation in enhancing LTP at cortical inputs to the amygdala. Thus, activation of β adrenoceptors leads to a reduction in SK channel expression at excitatory synapses, leading to greater depolarization during repetitive activity. As a consequence, there is a significant increase in postsynaptic calcium attributable to greater alleviation of the Mg2+ block, and synaptic plasticity is enhanced.
The lateral amygdala comprises part of the basolateral complex, a region that is critically involved in the formation and storage of emotional memories (Cahill and McGaugh, 1998; Sah et al., 2003). Noradrenaline is released into the amygdala during increased arousal states and during emotional learning paradigms, enhancing the formation and consolidation of emotionally salient memories (Cahill and McGaugh, 1998; McGaugh, 2000). In addition, glucocorticoids have been shown to facilitate memory consolidation through an action in the amygdala (Roozendaal et al., 1999, 2008). Intra-amygdala infusions of propranolol block both the normally enhanced formation of emotionally salient memories compared with emotionally neutral memories in humans (Cahill et al., 1994) as well as the effects of systemically administered glucocorticoids (Roozendaal et al., 1999), whereas intra-amygdala infusions of β adrenoceptor agonists enhance memory formation (Liang et al., 1986; McGaugh, 2004). Thus, it is clear that the memory enhancing actions of emotional stimuli are mediated by both β adrenoceptor activation and glucocorticoids in the basolateral amygdala, and both appear to act at least in part through modulation of SK channels (this study) (Duvarci and Paré, 2007). Behavioral experiments have shown that, using infusions of blockers of neuromodulators and stress hormones at varying posttraining time points into the basolateral amygdala, the window of enhancement of memory formation by emotional arousal lasts for several hours (Packard et al., 1994; Ferry et al., 1999; Hatfield and McGaugh, 1999). Consistent with this, firing rates of basolateral amygdala neurons following emotional arousal are elevated for up to several hours after emotional arousal (Pelletier et al., 2005), increasing activation of and storage in downstream targets (Cahill and McGaugh, 1998). A small proportion of neurons increase their firing immediately, whereas in the majority of neurons, this is a slowly developing phenomenon, peaking at ∼30 min and remaining elevated for up to several hours (Pelletier et al., 2005). Because levels of noradrenaline are elevated in the amygdala for several hours after emotional arousal (McIntyre et al., 2002), and noradrenaline can rapidly enhance the firing of lateral amygdala neurons by suppressing the slow afterhyperpolarization (Faber and Sah, 2002), in addition to facilitating Hebbian synaptic plasticity by removing SK channels from the synapse (within 15 min) (this study), this suggests that these dual actions of noradrenaline may contribute to the prolonged increased firing of basolateral amygdala neurons after emotional arousal. Thus, our findings add to the growing number of mechanisms that underlie the facilitating actions of noradrenaline on the formation of emotional memories.
Footnotes
This work was supported by grants from the National Health and Medical Research Council of Australia and the Australian Research Council. We thank Phil Robinson for supplying MiTMAB and BisT, John Adelman for supplying the myc-tagged plasmids, Prof. Seong-Seng Tan for the Venus plasmid, and Lachlan Rash for related experiments.
References
- Anderson AK, Wais PE, Gabrieli JD. Emotion enhances remembrance of neutral events past. Proc Natl Acad Sci U S A. 2006;103:1599–1604. doi: 10.1073/pnas.0506308103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Brosh I, Rosenblum K, Barkai E. Learning-induced modulation of SK channels-mediated effect on synaptic transmission. Eur J Neurosci. 2007;26:3253–3260. doi: 10.1111/j.1460-9568.2007.05936.x. [DOI] [PubMed] [Google Scholar]
- Cahill L, McGaugh JL. Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci. 1998;21:294–299. doi: 10.1016/s0166-2236(97)01214-9. [DOI] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh JL. Beta-adrenergic activation and memory for emotional events. Nature. 1994;371:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- Conchello JA. Superresolution and convergence properties of the expectation-maximization algorithm for maximum-likelihood deconvolution of incoherent images. J Opt Soc Am A Opt Image Sci Vis. 1998;15:2609–2619. doi: 10.1364/josaa.15.002609. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Deschaux O, Bizot JC, Goyffon M. Apamin improves learning in an object recognition task in rats. Neurosci Lett. 1997;222:159–162. doi: 10.1016/s0304-3940(97)13367-5. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Paré D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- Ferry B, Roozendaal B, McGaugh JL. Role of norepinephrine in mediating stress hormone regulation of long-term memory storage: a critical involvement of the amygdala. Biol Psychiatry. 1999;46:1140–1152. doi: 10.1016/s0006-3223(99)00157-2. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield T, McGaugh JL. Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol Learn Mem. 1999;71:232–239. doi: 10.1006/nlme.1998.3875. [DOI] [PubMed] [Google Scholar]
- Hill T, Odell LR, Edwards JK, Graham ME, McGeachie AB, Rusak J, Quan A, Abagyan R, Scott JL, Robinson PJ, McCluskey A. Small molecule inhibitors of dynamin I GTPase activity: development of dimeric tyrphostins. J Med Chem. 2005;48:7781–7788. doi: 10.1021/jm040208l. [DOI] [PubMed] [Google Scholar]
- Hill TA, Odell LR, Quan A, Abagyan R, Ferguson G, Robinson PJ, McCluskey A. Long chain amines and long chain ammonium salts as novel inhibitors of dynamin GTPase activity. Bioorg Med Chem Lett. 2004;14:3275–3278. doi: 10.1016/j.bmcl.2004.03.096. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissière S, Lüthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol. 2003;13:341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Kneussel M. Dynamic regulation of GABA(A) receptors at synaptic sites. Brain Res Brain Res Rev. 2002;39:74–83. doi: 10.1016/s0165-0173(02)00159-5. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Paré D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. é. [DOI] [PubMed] [Google Scholar]
- LeDoux J. The emotional brain, fear, and the amygdala. Cell Mol Neurobiol. 2003;23:727–738. doi: 10.1023/A:1025048802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KC, Juler RG, McGaugh JL. Modulating effects of posttraining epinephrine on memory: involvement of the amygdala noradrenergic system. Brain Res. 1986;368:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–4181. doi: 10.1523/JNEUROSCI.23-10-04173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Robinson PJ. Dynamin and endocytosis. Endocr Rev. 1995;16:590–607. doi: 10.1210/edrv-16-5-590. [DOI] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci. 1999;11:1217–1222. doi: 10.1046/j.1460-9568.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Amygdala: role in modulation of memory storage. In: Aggleton JP, editor. The Amygdala: a functional analysis. New York: Oxford UP; 2000. pp. 391–424. [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Hatfield T, McGaugh JL. Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats. Eur J Neurosci. 2002;16:1223–1226. doi: 10.1046/j.1460-9568.2002.02188.x. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in the mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- Packard MG, Cahill L, McGaugh JL. Amygdala modulation of hippocampal-dependent and caudate nucleus-dependent memory processes. Proc Natl Acad Sci U S A. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Pelletier JG, Likhtik E, Filali M, Paré D. Lasting increases in basolateral amygdala activity after emotional arousal: implications for facilitated consolidation of emotional memories. Learn Mem. 2005;12:96–102. doi: 10.1101/lm.88605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Distribution of IP3-mediated calcium responses and their role in nuclear signalling in rat basolateral amygdala neurons. J Physiol. 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Ren Y, Barnwell LF, Alexander JC, Lubin FD, Adelman JP, Pfaffinger PJ, Schrader LA, Anderson AE. Regulation of surface localization of the small conductance Ca2+-activated potassium channel, SK2, through direct phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2006;281:11769–11779. doi: 10.1074/jbc.M513125200. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci U S A. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Barsegyan A, Lee S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Prog Brain Res. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow-afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of LTP. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Sailer CA, Hu H, Kaufmann WA, Trieb M, Schwarzer C, Storm JF, Knaus HG. Regional differences in distribution and functional expression of small-conductance Ca2+-activated K+ channels in rat brain. J Neurosci. 2002;22:9698–9707. doi: 10.1523/JNEUROSCI.22-22-09698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SL, McNiven MA, De Camilli P. Dynamin and its partners: a progress report. Curr Opin Cell Biol. 1998;10:504–512. doi: 10.1016/s0955-0674(98)80066-5. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annual review of biochemistry. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins–novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Pedarzani P. Differential distributions of three Ca2+-activated K+ channel subunits, SK1, SK2 and SK3 in teh adult rat centrl nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]