

Abstract
In this issue of ckj, Tabibzadeh et al. report one of the largest series of patients with MYH9 mutations and kidney disease. The cardinal manifestation of MYH9-related disease is thrombocytopenia with giant platelets. The population frequency of pathogenic MYH9 mutations may be at least 1 in 20 000. The literature abounds in misdiagnosed cases treated for idiopathic thrombocytopenic purpura with immune suppressants and even splenectomy. Additional manifestations include neurosensorial deafness and proteinuric and hematuric progressive kidney disease (at some point, it was called Alport syndrome with macrothrombocytopenia), leucocyte inclusions, cataracts and liver enzyme abnormalities, resulting in different names for different manifestation combinations (MATINS, May–Hegglin anomaly, Fechtner, Epstein and Sebastian syndromes, and deafness AD 17). The penetrance and severity of kidney disease are very variable, which may obscure the autosomal dominant inheritance. A correct diagnosis will both preclude unnecessary and potentially dangerous therapeutic interventions and allow genetic counselling and adequate treatment. Morphological erythrocyte, granulocyte and platelet abnormalities may allow the future development of high-throughput screening techniques adapted to clinical peripheral blood flow cytometers.
Keywords: angiotensin, deafness, inherited kidney disease, MYH9, nephritis, thrombopenia
INTRODUCTION
In this issue of ckj, Tabibzadeh et al. report one of the largest series of patients with MYH9 mutations and kidney disease [1]. This offers the opportunity to emphasize some key concepts to lower the suspicion threshold and improve outcomes, complementing excellent recent reviews [2–4].
WHAT IS MYH9-RELATED DISEASE?
MYH9-related disease (MYH9-RD) refers to clinical manifestations dependent on MYH9 genetic variants. Before the description of the underlying genetic cause in 2000, different syndromes had been described encompassing combinations of potential disease manifestations [5, 6] (Figure 1). OMIM terms the condition MATINS (MAcroThrombocytopenia and granulocyte Inclusions with or without Nephropathy or Sensorineural hearing loss), emphasizing the nearly constant presence of low numbers of large platelets with variable penetrance of other manifestations [7]. Certain genetic variants (e.g. R705H) are associated mainly with neurosensorial deafness caused by cochleosaccular degeneration [2]. The combination of nephropathy and sensorineural hearing loss led to the term ‘Alport syndrome with macrothrombocytopenia’, no longer in use, as Alport syndrome is caused by mutations in COL4A3, COL4A4 or COL4A5 genes.
FIGURE 1.
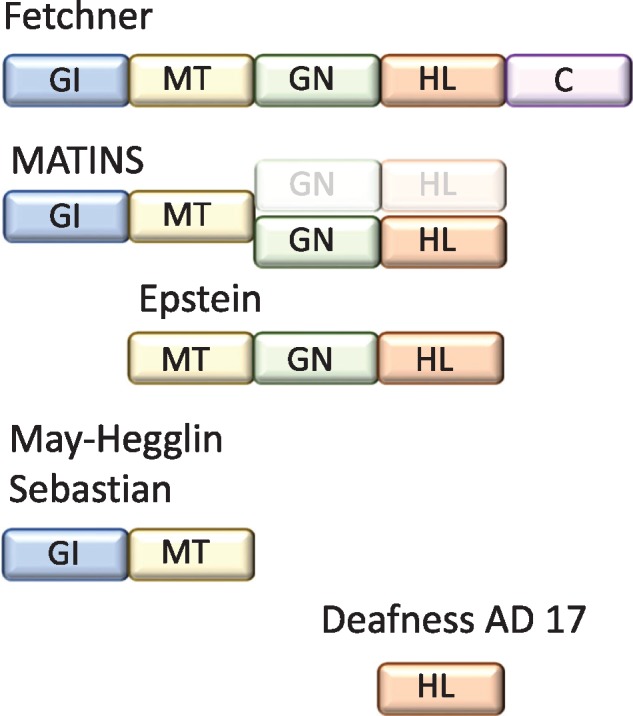
Clinical spectrum of MYH9-related disease. MATINS: MAcroThrombocytopenia and granulocyte Inclusions with or without Nephropathy or Sensorineural hearing loss. Fechtner syndrome: macrothrombocytopenia, granulocyte inclusions, nephropathy, sensorineural hearing loss, cataracts. Epstein syndrome: macrothrombocytopenia, granulocyte inclusions, nephropathy, sensorineural hearing loss. Sebastian syndrome and May–Hegglin anomaly: macrothrombocytopenia and granulocyte inclusions. Granulocyte inclusions visible in May-Grünwald-Giemsa-stained blood smears (Döhle bodies) in the May–Hegglin anomaly differ ultrastructurally from those in Sebastian syndrome, but both stain with anti-heavy chain of non-muscle myosin IIA antibodies. Deafness AD 17: Deafness Autosomal Dominant 17. GI: granulocyte inclusions; MT: congenital macrothrombocytopenia (more than 40% of platelets are >3.9 µM in diameter); GN: glomerular nephropathy; HL: hearing loss, C: cataracts.
HOW FREQUENT IS MYH9-RD?
The frequency of MYH9-RD is unknown. In the EXAC database, six loss-of-function genetic variants (stop codon, frameshift or splice acceptor/donor) were present in around 120 000 alleles, for an allele frequency of around 1 in 20 000 [8]. This is very similar to the estimated frequency in the Greifswald region of Germany; five unrelated families in a population of around 250 000 habitants [9]. If we estimate at least two affected members per family, this yields a frequency of around 1 in 25 000. Since the disease is underdiagnosed and missense variants may also result in disease, we conclude that the frequency of MYH9-RD is at least 1 in 20 000–25 000. The rate of de novo mutations is relatively high at >30% [3].
HOW IS MYH9-RD INHERITED?
MYH9-RD is autosomal dominant. This differs from the classical form of X-linked Alport syndrome, caused by mutations in COL4A5, but it is similar to the autosomal dominant nature of the probably most common form of Alport syndrome [10]. However, the incomplete penetrance and high variability at age of diagnosis of kidney disease or end-stage renal disease (ESRD) may obscure the real inheritance pattern of the kidney condition. As an example, Tabibzadeh et al. report one family with members reaching ESRD at ages ranging from 24 to 78 years [1]. An analysis of the family incidence of more constant features of the disease, such as macrothrombocytopenia, may be more informative. When macrothrombocytopenia is associated to kidney disease, the diagnosis of MYH9-RD should be suspected in the first place, and lab reports should be obtained from family members.
WHAT ARE THE KIDNEY FEATURES OF MYH9-RD?
Tabibzadeh et al. provide an overview of the spectrum of kidney disease [1]. At Nephrology referral, median age was 30 years (range 14–76) and estimated glomerular filtration rate 66 mL/min/1.73 m2, but around 20% were already in ESRD and an additional 50% progressed to ESRD. While the prior term ‘Alport syndrome with macrothrombocytopenia’ and the use of the term ‘nephritis’ in OMIM [7] may evoke the presence of hematuria, this was only observed at presentation or during follow-up in around 50% of patients. By contrast, pathological proteinuria was present in 85% at presentation, may reach nephrotic range and may even lead to nephrotic syndrome. This fits well with the expanding phenotypic spectrum of many classical inherited kidney diseases [11]. While albuminuria was not reported, it would be expected to be more sensitive than proteinuria. Thus, the disease should be considered in any proteinuric kidney disease at any age, especially when accompanied by thrombocytopenia. In this regard, one patient had a renal biopsy diagnosis of focal segmental glomerulosclerosis (FSGS), as previously reported for MYH9-related nephropathy [1, 12]. The most common alternative histological diagnosis was mesangial proliferation without immune deposits [1]. Electron microscopy may additionally show glomerular basement membrane thickening and splitting, and focal foot process effacement [1], pathological findings reminiscent of Alport syndrome, being the main reason why it was considered the same disease for a long time. The focal nature of foot process effacement is a finding associated with secondary FSGS and would argue against treatment with steroids [13]. Kidney outcomes were very variable, with age at ESRD ranging from 15 to 78 years, expanding the upper age range of prior reports [1]. Rapid progression was evident in 4/7 (60%) patients over the age of 40 years, as well as in a 9-year-old child with normal renal function that reached ESRD 6 years later, suggesting that the disease may be accelerated by triggers (Figure 2). Triggers (sepsis and post-partum haemorrhage) were identified in two patients. This possibility of late onset accelerated progression argues for nephrological assessment and monitoring of all affected family members, independently from baseline renal function tests.
FIGURE 2.

Pathogenic working hypothesis with therapeutic implications: two pathways to MYH9-related ESRD. Hypothesis based on the high heterogeneity of kidney disease within families with the same mutation, the clinical evidence of rapidly progressive disease at very different age ranges (from childhood to adulthood to the elderly) suggestive of the existence of triggers and preclinical evidence supporting the existence of triggers such as angiotensin II hyperactivity, together with evidence that a diabetic milieu, angiotensin II or other stressors, decrease MYH9 expression. The direct therapeutic implication is that identification and treatment of triggers may slow disease progression as suggested by certain patients on RAS blockade.
WHAT OTHER CLINICAL MANIFESTATIONS MAY BE EXPECTED IN MYH9-RD?
Figure 1 summarizes additional potential clinical manifestations. Despite the presence of these abnormalities, the diagnosis is not easy and may take decades, either because the abnormalities are missed, the dots are not connected or they are attributed to other conditions, the most common being idiopathic thrombocytopenic purpura (ITP), as clearly illustrated by Tabibzadeh et al. [1] and Furlano et al. [4]. The combined misdiagnosis of ITP and Alport syndrome has even been reported [14]. The degree of thrombocytopenia is very variable (in Tabibzadeh report, platelet numbers ranged from 4000 to 350 000/µL) and thrombocytopenia may be intermittent, i.e. platelet numbers may be normal. Large-sized platelets may be missed because they may not be interpreted as platelets by autoanalysers. The bleeding diathesis is usually mild to moderate, but may lead to significant bleeding, precipitating acute kidney injury, as reported by Tabibzadeh et al. [1]. The high variability in the severity and penetrance of other manifestations is also problematic. Hearing loss may be incapacitating, and may also be late-onset, down-sloping and asymptomatic, and only uncovered by specific studies [15]. Cataracts may be congenital [16], develop later in life or never develop. Granulocyte inclusions will only be found if blood smears are studied. Unexplained abnormal liver enzyme levels are not associated with progressive liver disease but may provide clues for the diagnosis.
The phenotypic spectrum may further expand. There are isolated reports of acute myeloid leukaemia and myelodysplastic syndrome that may be more difficult to diagnose due to the abnormal baseline bone marrow [17]. Extramedullary hematopoiesis may be confounded for tumours [18]. Defective neutrophil migration has been described but the consequences are poorly understood [19]. Additionally, MYH9 is considered a tumour suppressor, and whether the incidence or severity of malignancy differs in patients with germline mutations requires further studies [2]. Congenital haemangioma [20], the MYH9-associated elastin aggregation (MALTA) syndrome characterized by sweat duct proliferations and irregular elastin aggregations [21], and non-syndromic orofacial cleft in Taiwanese patients [22] have also been associated to MYH9 mutations, but not necessarily with MATINS.
WHEN SHOULD NEPHROLOGISTS SUSPECT MYH9-RD?
For nephrologists, MYH9-RD should be part of the differential diagnosis of proteinuric nephropathies at any age, especially if patients present any of the red flags depicted in Table 1 or a histological diagnosis of FSGS or mesangial proliferation without immune deposits. FSGS should always be considered a name in need of a surname: what is the driver of podocyte loss? The diagnosis should also always be considered in patients diagnosed with ITP refered to nephrology.
Table 1.
Red flags for MYH9-RD in the kidney disease patient
| Family history of ITP, bleeding, kidney disease or deafness |
| ITP AND glomerular nephropathy |
| Macrothrombocytopenia AND Döhle body-like granulocyte inclusions |
| Glomerular nephropathy AND sensorineural deafness AND cataracts |
| ‘Alport syndrome-like’ AND thrombocytopenia |
| Steroid-resistant FSGS AND thrombocytopenia |
| Steroid-resistant FSGS AND deafness |
| Early onset cataracts |
| Frequent haematomas |
| Spontaneous haemorrhages AND/OR excessive bleeding provoked by haemostatic challenges as trauma or surgery (post-partum, road accident, anti-platelet agents) |
| Unexplained liver enzyme abnormalities AND kidney disease |
DO ALL DISEASE-CAUSING MYH9 VARIANTS CAUSE KIDNEY DISEASE?
The answer appears to be no, although further work is needed in relation to genotype–phenotype correlations. MYH9 mutations are the most common cause of inherited thrombocytopenia, but, as shown in Figure 1, may not be associated with nephropathy. Motor domain mutations are associated with more severe kidney disease, but kidney disease may also be observed in tail mutations [2] (Figure 3), and even mutations corresponding to the non-helical tailpiece may be associated with kidney disease [1].
FIGURE 3.

Non-muscle myosin heavy chain IIA structure and sites of mutations. The MYH9 gene encodes the heavy chain of non-muscle myosin IIA. (A) Primary structure of the heavy chain of non-muscle myosin IIA. Some of the most frequent mutations are indicated. In Italy, 68% of families carried mutations leading to changes in amino acids 702 (motor domain) or 1424, 1841 and 1933 (tail domain). Motor domain mutations and D1424H are associated with the highest risk of kidney disease [2]. Some mutations are colour-coded in traffic light style as per the risk of kidney disease, red being the highest. (B) Quaternary structure of non-muscle myosin IIA: it is a hexamer composed two heavy chains, two regulatory light chains, whose phosphorylation regulates protein function, and two essential light chains. Key phosphorylation sites on the heavy chain are indicated.
WHAT IS THE PATHOGENESIS?
The MYH9 gene encodes the heavy chain of non-muscle myosin IIA, which is involved in cytokinesis, cell adhesion and cytoskeleton maintenance [2]. Megakaryocytes are normal but proplatelet branching is affected and defective megakaryocyte migration may contribute to ectopic platelet release. Bleeding may be facilitated by the low number of platelets and by defective clot formation. Clot contraction drives the translocation of procoagulant platelets to thrombus surface, but procoagulant platelets failed to translocate and remained inside the thrombi in blood derived from MYH9-deficient mice [23]. In the kidney, podocytes, mesangial cells and tubular cells express non-muscle myosin IIA. While germline knock-in point mutations reproducing human genotypes were associated with diverse manifestations of the disease, including albuminuria and glomerulosclerosis [24], not all authors found that podocyte-specific deficiency was associated to spontaneous glomerular disease in mice, although increased sensitivity to some podocyte stressors was found, which is consistent with human data suggestive of potential ‘triggers’ for chronic kidney disease (CKD) progression (Figure 2) [24–26]. Germline MYH9E1841K/E1841K mice were more sensitive to diverse triggers: albuminuria increased mildly in response to high salt; severe albuminuria and FSGS developed in angiotensin II-induced hypertension; and early mortality followed renal mass reduction [27]. SLIT2 activation of its receptor ROBO2 may be another stressor downregulating MYH9 expression and sensitizing to hypertension-induced podocyte detachment [28]. Both MYH9 mutant and MYH9-deficient cultured podocytes displayed abnormal podocyte cytoskeletal structure, increased motility and mechanical function [27, 29]. Discrepancies regarding the impact of podocyte-specific MYH9 deletion in vivo may also point to the contribution of additional cell types. Thus, beyond podocyte injury, which does take place but may be primary or secondary, defective glomerular haemodynamics or other factors due to disturbances in endothelial, mesangial or tubular cell function may also contribute to the pathogenesis, given the human evidence of focal rather than diffuse foot process effacement. In this regard, MYH9 has also been localized to mesangial, endothelial and proximal, and distal tubular cells in diverse studies [30, 31].
IS THERE A ROLE FOR MYH9 IN ACQUIRED KIDNEY DISEASE?
MYH9 expression was decreased in glomeruli from diabetic patients and animals [32]. Additionally, angiotensin II decreased MYH9 in cultured podocytes via NOX4-mediated oxidative stress and TRPC6 activation, and either angiotensin II or MYH9 knockdown resulted in podocyte actin cytoskeleton reorganization, reduced cell adhesion and increased albumin permeability [32]. These findings suggest a role for MYH9 loss in acquired kidney disease and may explain cases of accelerated CKD progression in patients with MYH9-RD, likely triggered by activation of the renin-angiotensin system (RAS) or other modulators of MYH9 expression. In this regard, the MYH9 rs2032487 single nucleotide polymorphism (SNP) (but not APOL1 variants) has been associated with advanced CKD related to Type 2 diabetes mellitus in the Canary Islands, the Spanish region with the highest prevalence of diabetic kidney disease [33], and the rs3752462 SNP to an increased risk of diabetic kidney disease in Chinese Han individuals [34], reproducing prior SNP associations in other non-African populations (reviewed in Pecci et al. [2]).
THE ASSOCIATION THAT WAS NOT
In 2008, MYH9 was described as a major-effect risk gene for FSGS, underlying an increased risk of ESRD for HIV-associated nephropathy and hypertension-attributed nephropathy in African Americans [35]. This association was later disproved, and the increased risk is now known to be conferred by APOL1 gene variants [36].
HOW IS MYH9-RD MANAGED?
Treatment is symptomatic (Figure 4) [2–4]. However, an a etiologic diagnosis may be useful in driving therapy, thus avoiding immune suppression for misdiagnosed ITP and facilitating the use of thrombopoietin receptor activators to increase platelet production: both eltrombopag and romiplostim have been successfully used, the former in a Phase 3 clinical trial. RAS blockade has been suggested as therapy for MYH9-related nephropathy and successful case reports have been published referring to a decrease in proteinuria associated with stable renal function or mildly progressive disease [1, 37], but treatment failures have also been reported [38]. The reason for the heterogenous response is unclear. Some genetic variants may be too severe to respond to nephroprotective strategies, while RAS blockade may protect from triggers, such as RAS activation, which may increase the severity of the disease by further downregulation of MYH9 expression in some milder genetic variants (Figure 2) [32].
FIGURE 4.

Key MYH9-RD manifestations and therapy.
WHAT NEEDS TO BE LEARNT?
Much remains to be learnt about this rare disease. From the kidney point of view, what are the factors driving the heterogeneity in kidney outcomes even within the same family? Could exome studies provide clues to the highly variable penetrance of kidney disease? Do relatively frequent genetic variants in nephropathy-associated genes (Type IV collagen genes?) contribute to increase severity? What are the triggers for accelerated loss of kidney function? Could these triggers be treatable? When should RAS blockade be started in affected family members? What are cost-effective methods to screen for the disease, given the underdiagnosis, late diagnoses and evidence that it is frequently treated with ineffective and potentially dangerous approaches due to misdiagnosis? In this regard, recent characterization of subclinical red blood cell abnormalities as well as an improved understanding of the flow cytometry characteristics of platelets may one day allow screening for the disease in routine blood cell studies. Abnormalities that could be useful in screening for MYH9-RD are the presence of elongated red blood cells, immunostaining for granulocyte inclusions, assessment of mean platelet diameter, or further standardization of mean platelet volume assessments or cut-off points [39–42]. Patients thus identified should be monitored for early features of kidney involvement (albuminuria). Increased awareness among nephrologists together with the widespread use of massive parallel sequencing will allow the diagnosis of MYH9-RD in patients presenting with a wide range of phenotypes mostly far from the full-blown syndrome.
FUNDING
Sources of support: FIS/Fondos FEDER (PI15/00298, CP14/00133, PI16/02057, PI16/01900, PI18/00362, PI18/01366 ISCIII-RETIC REDinREN RD016/0009), Sociedad Española de Nefrología, FRIAT, Comunidad de Madrid en Biomedicina B2017/BMD-3686 CIFRA2-CM. Salary support: ISCIII Rio Hortega to M.V.P.-G.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Tabibzadeh N, Fleury D, Labatut D. et al. MYH9-related disorders display heterogeneous kidney involvement and outcome. Clin Kidney J 2019; 12: 494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pecci A, Ma X, Savoia A. et al. MYH9: structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018; 664: 152–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balduini A, Raslova H, Di Buduo CA. et al. Clinic, pathogenic mechanisms and drug testing of two inherited thrombocytopenias, ANKRD26-related thrombocytopenia and MYH9-related diseases. Eur J Med Genet 2018; 61: 715–722 [DOI] [PubMed] [Google Scholar]
- 4. Furlano M, Arlandis R, Venegas MDP. et al. MYH9 associated nephropathy. Nefrologia 2019; 39: 133–140 [DOI] [PubMed] [Google Scholar]
- 5. Kelley MJ, Jawien W, Ortel TL. et al. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet 2000; 26: 106–108 [DOI] [PubMed] [Google Scholar]
- 6. Seri M, Cusano R, Gangarossa S. et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet 2000; 26: 103–105 [DOI] [PubMed] [Google Scholar]
- 7.Online Mendelian Inheritance in Man [Internet]. https://www.omim.org/entry/603622?search=myh9&highlight=myh9 (7 July 2019, date last accessed)
- 8. Lek M, Karczewski KJ, Minikel EV. et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature 2016; 536: 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Althaus K, Greinacher A.. MYH9-related platelet disorders. Semin Thromb Hemost 2009; 35: 189–203 [DOI] [PubMed] [Google Scholar]
- 10. Bullich G, Domingo-Gallego A, Vargas I. et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int 2018; 94: 363–371 [DOI] [PubMed] [Google Scholar]
- 11. Ars E, Torra R.. Rare diseases, rare presentations: recognizing atypical inherited kidney disease phenotypes in the age of genomics. Clin Kidney J 2017; 10: 586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lepori N, Zand L, Sethi S. et al. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J 2018; 11: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sethi S, Zand L, Nasr SH. et al. Focal and segmental glomerulosclerosis: clinical and kidney biopsy correlations. Clin Kidney J 2014; 7: 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vassallo D, Erekosima I, Kanigicherla D. et al. Myosin heavy chain-9-related disorders (MYH9-RD): a case report. Clin Kidney J 2013; 6: 516–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song MH, Jung J, Rim JH. et al. Genetic inheritance of late-onset, down-sloping hearing loss and its implications for auditory rehabilitation. Ear and Hearing 2019; doi:10.1097/AUD.0000000000000734 [DOI] [PubMed] [Google Scholar]
- 16. Aoki T, Kunishima S, Yamashita Y. et al. Macrothrombocytopenia with congenital bilateral cataracts: a phenotype of MYH9 disorder with exon 24 indel mutations. J Pediatr Hematol Oncol 2018; 40: 76–78 [DOI] [PubMed] [Google Scholar]
- 17. Tsang HC, Bussel JB, Mathew S. et al. Bone marrow morphology and disease progression in congenital thrombocytopenia: a detailed clinicopathologic and genetic study of eight cases. Mod Pathol 2017; 30: 486–498 [DOI] [PubMed] [Google Scholar]
- 18. Zaninetti C, Boveri E, Melazzini F.. Massive mediastinal enlargement due to extramedullary haematopoiesis in a patient with MYH9-related thrombocytopenia. Br J Haematol 2017; 178: 10. [DOI] [PubMed] [Google Scholar]
- 19. Zehrer A, Pick R, Salvermoser M. et al. A fundamental role of MYH9 for neutrophil migration in innate immunity. J Immunol 2018; 201: 1748–1764 [DOI] [PubMed] [Google Scholar]
- 20. Fomchenko EI, Duran D, Jin SC. et al. De novo MYH9 mutation in congenital scalp hemangioma. Cold Spring Harb Mol Case Stud 2018; 4: a002998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fewings E, Ziemer M, Hörtnagel K. et al. (21 May 2019) MALTA (MYH9 associated elastin aggregation) syndrome: germline variants in MYH9 cause rare sweat duct proliferations and irregular elastin aggregations. J Invest Dermatol; doi:10.1016/j.jid.2019.03.1151 [DOI] [PubMed] [Google Scholar]
- 22. Peng HH, Chang NC, Chen KT. et al. Nonsynonymous variants in MYH9 and ABCA4 are the most frequent risk loci associated with nonsyndromic orofacial cleft in Taiwanese population. BMC Med Genet 2016; 17: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nechipurenko DY, Receveur N, Yakimenko AO. et al. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler Thromb Vasc Biol 2019; 39: 37–47 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y, Conti MA, Malide D. et al. Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood 2012; 119: 238–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnstone DB, Ikizler O, Zhang J. et al. Background strain and the differential susceptibility of podocyte-specific deletion of MYH9 on murine models of experimental glomerulosclerosis and HIV nephropathy. PLoS One 2013; 8: e67839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnstone DB, Zhang J, George B. et al. Podocyte-specific deletion of Myh9 encoding nonmuscle myosin heavy chain 2A predisposes mice to glomerulopathy. Mol Cell Biol 2011; 31: 2162–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cechova S, Dong F, Chan F. et al. E1841K mutation augments proteinuria and podocyte injury and migration. J Am Soc Nephrol 2018; 29: 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fan X, Yang H, Kumar S. et al. SLIT2/ROBO2 signaling pathway inhibits nonmuscle myosin IIA activity and destabilizes kidney podocyte adhesion. JCI Insight 2016; 1: e86934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bondzie PA, Chen HA, Cao MZ. et al. Non-muscle myosin-IIA is critical for podocyte f-actin organization, contractility, and attenuation of cell motility. Cytoskeleton (Hoboken) 2016; 73: 377–395 [DOI] [PubMed] [Google Scholar]
- 30. Arrondel C, Vodovar N, Knebelmann B. et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol 2002; 13: 65–74 [DOI] [PubMed] [Google Scholar]
- 31. Otterpohl KL, Hart RG, Evans C. et al. Nonmuscle myosin 2 proteins encoded by Myh9, Myh10, and Myh14 are uniquely distributed in the tubular segments of murine kidney. Physiol Rep 2017; 5: e13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang JS, Lee SJ, Lee JH. et al. Angiotensin II-mediated MYH9 downregulation causes structural and functional podocyte injury in diabetic kidney disease. Sci Rep 2019; 9: 7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boronat M, Tugores A, Saavedra P. et al. (4 April 2019) Association between polymorphism rs2032487 in the non-muscle myosin heavy chain IIA gene (MHY9) and chronic kidney disease secondary to type 2 diabetes mellitus in a population of the Canary Islands. Endocrinol Diabetes Nutr; doi:10.1016/j.endinu.2019.01.006 [DOI] [PubMed] [Google Scholar]
- 34. Zhao H, Ma L, Yan M. et al. Association between MYH9 and APOL1 gene polymorphisms and the risk of diabetic kidney disease in patients with Type 2 diabetes in a Chinese Han population. J Diabetes Res 2018; 2018: 5068578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kopp JB, Smith MW, Nelson GW. et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 2008; 40: 1175–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tzur S, Rosset S, Shemer R. et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010; 128: 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka M, Miki S, Saita H. et al. Renin-angiotensin system blockade therapy for early renal involvement in MYH9-related disease with an E1841K mutation: a case report. Intern Med 2019; doi:10.2169/internalmedicine.2997-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hashimoto J, Hamasaki Y, Takahashi Y. et al. Management of patients with severe Epstein syndrome: review of four patients who received living-donor renal transplantation. Nephrology (Carlton) 2019; 24: 450–455 [DOI] [PubMed] [Google Scholar]
- 39. Fixter K, Rabbolini DJ, Valecha B. et al. Mean platelet diameter measurements to classify inherited thrombocytopenias. Int J Lab Hem 2018; 40: 187–195 [DOI] [PubMed] [Google Scholar]
- 40. Rabbolini DJ, Chun Y, Latimer M. et al. Diagnosis and treatment of MYH9-RD in an Australasian cohort with thrombocytopenia. Platelets 2017; 1–8 [DOI] [PubMed] [Google Scholar]
- 41. Greinacher A, Pecci A, Kunishima S. et al. Diagnosis of inherited platelet disorders on a blood smear: a tool to facilitate worldwide diagnosis of platelet disorders. J Thromb Haemost 2017; 15: 1511–1521 [DOI] [PubMed] [Google Scholar]
- 42. Smith AS, Pal K, Nowak RB. et al. MYH9-related disease mutations cause abnormal red blood cell morphology through increased myosin-actin binding at the membrane. Am J Hematol 2019; 94: 667–677 [DOI] [PMC free article] [PubMed] [Google Scholar]