

Abstract
Consumption of alcohol (ethanol) during pregnancy can lead to developmental defects in the offspring, the most devastating being the constellation of symptoms collectively referred to as fetal alcohol syndrome (FAS). In the brain, a hallmark of FAS is abnormal cerebral cortical morphology consistent with insult during corticogenesis. Here, we report that exposure to a relatively low level of ethanol in utero (average maternal and fetal blood alcohol level of 25 mg/dl) promotes premature tangential migration into the cortical anlage of primordial GABAergic interneurons, including those originating in the medial ganglionic eminence (MGE). This ethanol-induced effect was evident in vivo at embryonic day 14.5 (E14.5) in GAD67 knock-in and BAC-Lhx6 embryos, as well as in vitro in isotypic telencephalic slice cocultures obtained from E14.5 embryos exposed to ethanol in utero. Analysis of heterotypic cocultures indicated that both cell-intrinsic and -extrinsic factors contribute to the aberrant migratory profile of MGE-derived cells. In this light, we provide evidence for an interaction between ethanol exposure in utero and the embryonic GABAergic system. Exposure to ethanol in utero elevated the ambient level of GABA and increased the sensitivity to GABA of MGE-derived cells. Our results uncovered for the first time an effect of ethanol consumption during pregnancy on the embryonic development of GABAergic cortical interneurons. We propose that ethanol exerts its effect on the tangential migration of GABAergic interneurons extrinsically by modulating extracellular levels of GABA and intrinsically by altering GABAA receptor function.
Keywords: medial ganglionic eminence, fetal alcohol syndrome, ambient GABA, GABAA receptor, GFP+/MGE, telencephalic slice culture
Introduction
Examination of postmortem brains of fetal alcohol syndrome (FAS) patients reveals defects in gross brain morphology attributed to disrupted developmental processes, notably cell growth, differentiation and migration. Abnormal development of the neocortex has been implicated in the pathoetiology of neurobehavioral abnormalities, such as deficits in learning and memory (for review, see Berger-Sweeney and Hohmann, 1997; Copp and Harding, 1999; Gleeson and Walsh, 2000).
Prenatal ethanol exposure initially increases the number of cells in the marginal zone and cortical plate that is followed by enhanced cell death. This sequence of events has been postulated to yield a net diminution of cortical cells and a thinner cortex at later stages of development (Miller 1986, 1996). Other studies attribute decreased cortical cell number to defective cellular migration, including that of the GABAergic cortical interneurons (Moore et al., 1998; Bailey et al., 2004), the majority of which originate in the medial ganglionic eminence (MGE) and migrate tangentially into the cortical anlage (Anderson et al., 1997, 1999, 2001; Tamamaki et al., 1997; Lavdas et al., 1999; Jimenez et al., 2002). Indeed, the GABAergic system plays an important role from the earliest stages of corticogenesis, influencing key developmental processes such as proliferation, neuronal differentiation, and migration (Behar et al., 1994, 1996, 2000; LoTurco et al., 1995; Maric et al., 2001; Ben-Yaakov and Golan, 2003; Manent et al., 2005; Cuzon et al., 2006; Heck et al., 2006; Heng et al., 2007) (for review, see Owens and Kriegstein, 2002).
The GABAergic system is a key target of ethanol action in the brain (for review, see Weiner and Valenzuela, 2006). Ethanol has been shown to influence GABAergic synaptic transmission presynaptically by increasing the release of GABA (Roberto et al., 2003; Carta et al., 2004; Hayward et al., 2004) (for review, see Siggins et al., 2005) and postsynaptically by enhancing GABAA receptor function in a cell type- and subunit composition-dependent manner (Aguayo, 1990; Nishio and Narahashi, 1990; Wafford et al., 1990, 1991; Nakahiro et al., 1991; Sapp and Yeh, 1998; Tatebayashi et al., 1998; Mori et al., 2000). Chronic ethanol exposure has also been shown to alter expression of certain GABAA receptor subunits at synaptic and extrasynaptic sites (Devaud et al., 1995, 1997; Grobin et al., 2000a,b; Papadeas et al., 2001).
The above considerations, together, led us to hypothesize that exposure to ethanol during fetal development may affect the tangential migration of primordial GABAergic interneurons from the MGE to the developing neocortex. The disposition of tangentially migrating MGE-derived cells was analyzed in vivo using the GAD67-GFP (Δneo) and bacterial artificial chromosome (BAC)-Lhx6-GFP transgenic mouse lines (Gong et al., 2003; Tamamaki et al., 2003). Experiments in vitro examined tangential migration in a telencephalic slice coculture system (Cuzon et al., 2006). We found that ethanol exposure in utero enhanced tangential migration, notably that of GABAergic cells derived from the MGE. Whole-cell patch-clamp recording of MGE-derived cells revealed that ethanol exposure in utero elevated ambient GABA and increased sensitivity to GABA. Thus, both cell-extrinsic and -intrinsic mechanisms contribute to the ethanol-induced effect on tangential migration.
Materials and Methods
All procedures involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Dartmouth Medical School Institutional Animal Care and Use Committee. Mice of similar age (8–10 weeks) and weight (21.0 ± 0.71 g) were housed in rooms with controlled lighting (10:00 A.M. light/10:00 P.M. dark) and given mouse chow and water ad libitum until alcohol treatment began. In vivo experiments used two transgenic mouse lines. One of these transgenic mouse lines was created by the National Institute of Neurological Disorders and Stroke GENSAT BAC Transgenics Project (Gong et al., 2003) using a BAC vector in which the expression of enhanced green fluorescent protein (eGFP) is under the control of the entire transcription unit and associated regulatory domain for the Lhx6 gene. Because cells arising from the MGE uniquely express Lhx6 (Lavdas et al., 1999), the BAC-Lhx6 mouse facilitated analysis of the migration of MGE-derived cells in real time. The other transgenic mouse line was the GAD67-GFP (Δneo), referred to hereafter as GAD67-GFP knock-in, that expresses eGFP under the regulation of the endogenous GAD67 promoter (Tamamaki et al., 2003). Because GAD67 encodes a key enzyme in the biosynthesis of GABA and is specifically expressed in GABAergic cells, the GAD67-GFP knock-in mouse allowed for the analysis of all primordial GABAergic cells including those from the MGE (Tanaka et al., 2006). In vitro experiments involving slice cocultures used transgenic mice expressing GFP under the control of a β-actin/cytomegalovirus (CMV) promoter/enhancer, referred to hereafter as GFP β-actin mice (Okabe et al., 1997). The GAD67-knock-in and the GFP β-actin mice are of C57BL/6 background. The BAC-Lhx6 mouse line, originally obtained from GENSAT of Swiss-Webster background, was backcrossed into a C57BL/6 background and all mice used were of the F5 generation or later.
Chronic treatment with ethanol.
After detection of vaginal plug [embryonic day 0.5 (E0.5)], pregnant dams were individually housed and assigned to one of three groups: ethanol fed, pair fed (PF), or ad libitum. Mice were maintained on a liquid diet (Research Diets, New Brunswick, NJ) supplemented with ethanol [1 or 2% (w/v); ethanol fed] or an isocaloric control diet containing maltose and dextrins (PF and ad libitum). Dams in the ethanol-fed and ad libitum groups were given their respective liquid diets ad libitum, whereas dams in the PF group received similar amounts of food consumed on the previous day by their ethanol-fed counterparts. All mice received water ad libitum. Fresh liquid food was replenished daily in graduated feeding tubes between 3:00 and 5:00 P.M., when the amount of liquid food consumed and the weight of the dam were also measured. Mice were maintained on their respective diets until being killed by CO2 asphyxiation (E14.5).
BAL.
Timed pregnant dams were bled via the tail vein on the morning before being killed during the dark cycle between 3:00–5:00 A.M., when the blood alcohol level (BAL) tends to be highest (Miller, 1992). Trunk blood from pregnant dams and blood/amniotic fluid from embryos were also collected at the time of killing (8:00 A.M.). The alcohol content was assayed using an Analox Instruments (Lunenburg, MA) GM7 series analyzer. Ethanol is oxidized by the enzyme alcohol oxidase, in the presence of molecular oxygen, to acetaldehyde and hydrogen peroxide. A Clark-type amperometric oxygen electrode monitored the rate of oxygen consumption, which is directly proportional to alcohol concentration.
Preparation of isotypic and heterotypic slice cocultures.
Telencephalic slice cocultures were prepared from E14.5 embryos obtained from the mating of GFP transgenic mice under the control of β-actin/CMV/enhancer sequence and wild-type C57BL/6J mice as described previously by Cuzon et al. (2006). Briefly, 250 μm coronal slices from the anterior half of the cerebral hemisphere were obtained using a vibroslicer (World Precision Instruments, Sarasota, FL). Only slices in which the medial and lateral ganglionic eminences were clearly demarcated by the ganglionic sulcus were used. The MGE from GFP-expressing embryos were microdissected, minced, and a small piece was transplanted onto the MGE of a slice obtained from a wild-type embryo. Isotypic slice cocultures were those in which the GFP-expressing MGE (GFP+/MGE) explants and wild-type host slice were derived from the same treatment group. Heterotypic slice cocultures consisted of explants and host slice pairings from different treatment groups. The cocultures were maintained for up to 2 d in vitro (DIV) in a humidified 5% CO2 atmosphere at 37°C in medium containing DMEM/F12 (1:1; Invitrogen, Grand Island, NY), d-glucose (6 mg/ml), 10% fetal calf serum (Hyclone, Logan, UT), l-glutamine (2 mm; Sigma, St. Louis, MO), and penicillin/streptomycin (Cellgro, Herndon, VA). In some experiments, bicuculline methiodide (20 μm; Sigma), an antibody raised against GABA (rabbit anti-GABA; 1 μg/ml or 1:1000; Sigma), or ethanol (25 mg/dl) was added to the culture medium.
Immunohistochemistry.
Brains isolated from E14.5 embryos were immerse-fixed in either 4% paraformaldehyde (PFA) or in 0.1% glutaraldehyde in 4% PFA, cryoprotected in 30% sucrose, sectioned on a cryostat (20 μm) along the coronal plane and mounted directly onto gelatin-coated slides. Sections stained with an antibody against bromodeoxyuridine (BrdU) were preincubated in 2N HCl for 30 min at 37°C. Sections were blocked in 10% normal goat serum/0.01% Triton X-100 in PBS. Dilutions of primary antibodies were as follows: mouse anti-GFP at 1:1000 (Invitrogen) and rabbit anti-GABA at 1:1000 (Sigma). Secondary antibodies used were Alexa-Fluor 488, Alexa Fluor 568, and Alexa Fluor 555 (Invitrogen) diluted 1:800 in PBS. Sections were counterstained with 4,6,diamidino-2-phenylindole (DAPI) (Sigma) and coverslipped in FluroSave Reagent (Calbiochem, La Jolla, CA).
Slice cocultures were routinely fixed and immunostained with antibody against GFP as detailed previously (Cuzon et al., 2006). For both cryosections and slice cocultures, the cortex beyond the corticostriate juncture was divided into 100 μm consecutive cortical bins and the number of migrating GFP+/MGE cells in each cortical bin was normalized to the number of GFP+/MGE cells located before the corticostriate juncture (CSJ) to derive at a “crossing index” (Cuzon et al., 2006). Crossing index was compared between different treatment groups within cortical bins using one-way ANOVA and the Holm–Sidak method. Data were presented as mean ± SEM.
Time-lapse video microscopy.
The migration of individual GFP+/MGE cells was monitored using time-lapse videomicroscopy (Cuzon et al., 2006). Embryonic slice cocultures (1–2 DIV), were transferred to a custom-made mini-incubator on the stage of an upright microscope (BX61WI; Olympus, Center Valley, PA) equipped with a spinning disc confocal disc scanning unit (Olympus), maintained in a humidified atmosphere saturated with 95% oxygen/5% carbon dioxide at 37°C. GFP+/MGE cells were visualized within slice cocultures using a xenon light source. A set of images was collected at 5 min intervals at a series of depths through the thickness of the slice (z-series) to compensate for cells that migrated out of a given plane of focus. Each set of z-series images was flattened to produce a single image that was used to determine the rate of migration of GFP+/MGE cells before the CSJ and in the neocortex. The rate of migration of GFP+/MGE cells was compared between the control and ethanol-exposed treatment groups using Student's t test. Data were presented as the mean rate ± SEM.
Electrophysiology.
Embryonic slices were transferred to a recording chamber, stabilized by an overlying platinum ring strung with plastic string mesh, and maintained at 32–34°C on a heated stage fit onto an upright microscope (BX51WI; Olympus, Melville, NY). Slices were perfused at a rate of 0.5 ml/min with oxygenated artificial CSF (aCSF) containing (in mm) 124 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 0.01 glycine, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The slices were not perfused in the set of experiments assessing ambient GABA to minimize wash-out of endogenous GABA, and a constant gentle stream of 95% oxygen/5% CO2 was applied over the surface of the static aCSF.
GFP+/MGE cells were identified under fluorescence illumination and under Hoffman modulation optics (Modulation Optics, Greenvale, NY) using a 40× water-immersion objective (3 mm working distance; Olympus). Real-time images were captured using an analog video camera attached to a video frame grabber (Integral Technologies, Indianapolis, IN) and displayed on a computer monitor, which aided the navigation and placement of the drug and recording pipettes. Patch pipettes were pulled from glass capillaries (1.5 mm outer diameter; 0.86 mm inner diameter) (Sutter Instruments, Novato, CA) to a resistance of 8–10 MΩ. Recording pipettes were filled with an internal solution composed of (in mm) 140 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, supplemented with Mg2+ ATP. Recordings were made using an AxoPatch 200A amplifier (Molecular Devices, Foster City, CA). Membrane currents of MGE-derived cells were monitored at a holding potential of −65 mV under whole-cell patch-clamp conditions, and filtered at 5 kHz, digitized using Clampex v9.0 and analyzed with Clampfit v9.0 (Molecular Devices). Statistical analysis was performed using SigmaStat 3.0 (SPSS, Chicago, IL). Mean peak current amplitude of drug-evoked currents was analyzed using Student's t test. Data were reported as mean ± SEM.
GABA (0.1–500 μm) and bicuculline methiodide (20 μm) were dissolved in external solution, stored as frozen stock and diluted to working concentrations with aCSF immediately before each recording session. The drug solutions were loaded into separate barrels of an 8-barrel drug pipette assembly and applied using regulated pulses of pressure (≤3 psi; Picospritzer; General Valve, Fairfield, NJ) within 10 μm of the soma of the cell under study. The timing and duration of the pressure pulses were controlled by a digital timing unit (Pulse train 1831; World Precision Instruments). One of the barrels of the multibarrel assembly was routinely filled with aCSF, which was applied between drug applications to clear drugs from the vicinity of the cell. This also served as vehicle control and to control for mechanical artifacts attributable to bulk flow.
Results
In this study, we analyzed the effect of in utero ethanol exposure on the migration of MGE-derived cells both in vitro (Cuzon et al., 2006) and in vivo. The experiments were complemented by the use of the GAD67-GFP knock-in mouse line (Tamamaki et al., 2003), which facilitated analysis of putative GABAergic neurons in general, and the BAC-Lhx6 mouse line (Gong et al., 2003), which permitted analysis of those derived from the MGE.
Pregnant dams were initially fed a liquid diet containing 1% ethanol (w/v) in an attempt to establish an effective, physiologically relevant level of gestational ethanol consumption. Treatment with 1% ethanol yielded inconsistent BAL, undetectable in 5 of 12 cases and 12.7 ± 1.1 mg/dl when detectable (7 of 12 cases). In addition, the 1% ethanol-treated groups were indistinguishable from both pair-fed and ad libitum control groups when parameters used to assess tangential migration were examined (e.g., crossing index and distribution within the developing neocortex) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Therefore, unless noted otherwise, further results reported in the present study were derived from experiments in which pregnant dams were fed liquid diet containing 2% ethanol. Because there was no significant difference in crossing index between pair-fed and ad libitum control groups; the data derived from these two groups were pooled and referred to hereafter as “control.”
Consumption of 2% ethanol during pregnancy did not affect litter size (control, 7.10 ± 2.02; ethanol-treatment, 7.50 ± 2.07; Student's t test; p = 0.69), in contrast to treatment with higher ethanol concentrations, which have been reported to decrease litter size and body and brain weight (Sanchis et al., 1986). The size of the head and brain was notably greater in ethanol-treated embryos (Fig. 1 A,B), as were body weight and brain weight (Fig. 1 C). However the ratio of brain weight to body weight was unaltered (Fig. 1 C).
Figure 1.

The mouse model of low-level chronic ethanol consumption used to examine the effect of in utero ethanol exposure on corticogenesis increases the size of embryos. A, B, Images of control (A1) and 2% ethanol-exposed (A2) embryos and brains (B) at E14.5. C, The body and brain weight were significantly increased with ethanol exposure (Student's t test; *p < 0.05) without altering the brain to body weight ratio (Student's t test; p = 0.96) as measured in the BAC-Lhx6 embryos (control, n = 8; ethanol, n = 9). Data are expressed as mean ± SEM. Asterisks denote a statistical difference compared with control. D, BAL was not significantly different between dams and embryos at time of killing within treatment groups (Student's t test; GAD67, n = 3, p = 0.86; BAC-Lhx6, n = 10, p = 0.44; GFP β-act, n = 7, p = 0.70) or between treatment groups (GAD67, 28.94 ± 1.97 mg/dl; BAC-Lhx6, 25.03 ± 1.59 mg/dl; GFP β-act, 24.95 ± 1.36 mg/dl; ANOVA, p = 0.42). Data are expressed as mean ± SEM. GAD67, GAD67-GFP transgenic mice; GFP β-act, GFP under the control of a β-actin/CMV promoter/enhancer sequence.
Blood alcohol level of pregnant dams measured between 5 and 7 h into the dark cycle before being killing (E13.5) and in trunk blood collected at the time of killing did not differ (data not shown). In addition, as illustrated in Figure 1 D, BAL did no differ between dams and embryos at time of killing nor between dams of the transgenic mouse lines used in this study.
Number and density of GABAergic cells in the neocortex at E14.5
We first determined whether ethanol exposure altered the number and/or distribution of GABA-immunopositive cells within the embryonic neocortex. The density of GABA-immunopositive cells in E14.5 cryosections obtained from brains of control (Fig. 2 A1) and ethanol-treated (Fig. 2 A2) embryos was calculated in terms of location within (1) cortical bins that reflected distance from the CSJ and (2) the embryonic cortical layers. As summarized in Figure 2, B and C, such analyses revealed significant increases in GABA-immunopositive cells in the ethanol-treated group in both distance from the CSJ (Fig. 2 B) and throughout the marginal zone–cortical plate (MZ–CP), intermediate (IZ), and subventricular–ventricular zones (SVZ–VZ) (Fig. 2 C).
Figure 2.

Ethanol consumption during pregnancy increases the number of GABA-immunoreactive cells in the neocortex. A, Representative GABA-immunoreactive E14.5 telencephalic sections obtained from control (n = 10) and ethanol exposed in utero (n = 7) wild-type littermates of BAC-Lhx6 litters. Note that immunostaining for GABA is clearly evident in the cells in the marginal, intermediate, and subventricular zones of the neocortex. Scale bar, 100 μm. B, C, Ethanol exposure increased the density of GABA-immunopositive cells throughout the extent of the neocortex as seen in cortical bins (B) and in layer density (C). Asterisks denote a significant difference from control (Student's t test; p < 0.01).
A comparison of the disposition of putative GABAergic cells within the neocortical anlage of E14.5 GAD67-GFP knock-in embryos with and without exposure to ethanol in utero corroborated the above findings (Fig. 3 A,B). With ethanol exposure, the number of GAD67-GFP cells increased significantly in each cortical layer (Fig. 3 C) without affecting the ratio of total GAD67-GFP cells between the cortical layers (Fig. 3 D). These results suggest that the increase in the number of putative GABAergic cells resulting from ethanol exposure in utero is not specific for any given layer of the embryonic neocortex.
Figure 3.
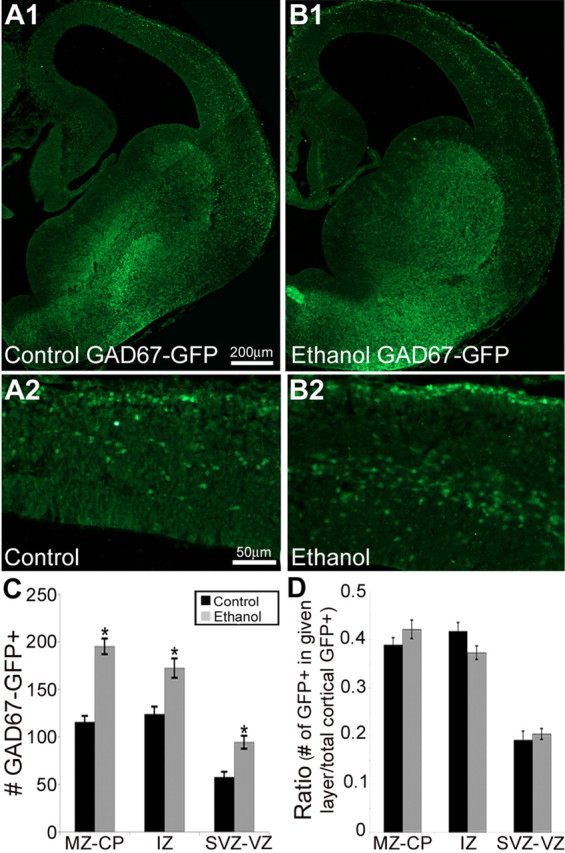
A, B, Representative sections from E14.5 GAD67-GFP knock-in control (A; n = 22) and ethanol-treated (B; n = 18) embryos. A2 and B2 are enlargements of the neocortex in A1 and B1, respectively. C, Exposure to ethanol in utero increased the number of GAD67-GFP cells throughout the cortical layers (Student's t test; MZ–CP, p < 0.001; IZ, p = 0.003; SVZ–VZ, p < 0.001). D, However, the ratio between the cortical layers is unaltered (Student's t test; MZ–CP, p = 0.26; IZ, p = 0.12; SVZ–VZ, p = 0.92). Data are expressed as mean density ± SEM. Asterisks denote a significant difference from control (Student's t test, p < 0.005).
Given the observed overall increase in GABAergic cells, we then assessed whether ethanol exposure in utero increased cell proliferation and/or migration. To assess cell proliferation, pregnant dams at E14.5 were injected intraperitoneally with BrdU 45 min before being killed. Cryosections from fixed brains were immunoprocessed using an antibody against BrdU and counterstained with DAPI (Fig. 4 A,B). Ethanol exposure decreased the BrdU labeling index in all proliferating zones, including the MGE, suggesting decreased proliferative activity at the time of BrdU injection (control: 0.40 ± 0.051; ethanol: 0.27 ± 0.055; Student's t test; p < 0.05). Because, at E14.5, the majority of migrating primordial GABAergic cortical interneurons in the rodent arise from the MGE (Anderson et al., 1997, 1999, 2001; Tamamaki et al., 1997; Lavdas et al., 1999; Jimenez et al., 2002) the decreased BrdU labeling index in the region likely reflects the proliferative status of this subset of MGE-derived cells.
Figure 4.

Exposure to ethanol in utero increases cell differentiation. A, B, Representative 20-μm-thick cryosections of E14.5 brains stained with an antibody against BrdU obtained from control (A) and ethanol-exposed (B) wild-type embryos. Scale bar, 100 μm. C, Individual cells were monitored in still images taken every 5 min for 12 h in isotypic slice cocultures in which an MGE explant from a GFP β-actin embryo was transplanted onto the MGE of a wild-type littermate. Arrowheads show an example of migrating MGE-derived cells. D, Cells monitored in slices from in utero ethanol exposed embryos (n = 142; before CSJ, 37.52 ± 1.56 μm/h; after CSJ, 37.65 ± 1.08 μm/hr) migrated faster than those from control slices (n = 288; before CSJ, 16.52 ± 0.57 μm/h; after CSJ, 16.58 ± 0.77 μm/hr) both before and after the CSJ (ethanol, n = 142). Data are expressed as mean ± SEM. Asterisks indicate a significant difference from control (Student's t test; p < 0.005).
To assess the rate of cell migration from the MGE into the neocortex, individual GFP+/MGE cells proximal and distal to the CSJ were tracked in isotypic slice cocultures by real-time videomicroscopy and the distance traveled over time calculated (Fig. 4 C). The GFP+/MGE cells from ethanol-treated slice cocultures migrated significantly faster than those of their control counterparts (Fig. 4 D). Notably, there was no significant difference in the migration rate before and after the CSJ in either the control or ethanol-treated groups (Fig. 4 D) (Student's t test; control, p = 0.96; ethanol treated, p = 0.95). Thus, exposure to ethanol in utero increased the rate of migration of MGE-derived cells consistently throughout their migratory path.
Tangential migration of MGE-derived cells
We asked whether MGE-derived cells contributed to the increase in GABAergic neurons in the embryonic neocortex observed after ethanol exposure in utero. Counts of GFP+/MGE cells in cryosections from brains of E14.5 BAC-Lhx6 embryos (Fig. 5 A) revealed that exposure to ethanol in utero increased the number of MGE-derived cells in the SVZ–VZ, IZ, and CP–MZ (Fig. 5 B) without altering the percentage of total GFP+/MGE cells within each cortical layer (Fig. 5 C). The increased number of GFP+/MGE cells accounted for the increase in crossing index as illustrated in Figure 5 D. The observation that the distribution of GFP+/MGE cells within cortical layers is unaltered suggests that the tangential migratory routes are not affected by in utero ethanol exposure.
Figure 5.

Exposure to ethanol in utero increases the number of MGE-derived cells in the neocortex without altering the ratio in Bac-Lhx6 mice. A–D, Representative cryosections from E14.5 Bac-Lhx6 embryos obtained from control (A1; n = 17) or ethanol-treated (A2; n = 23) embryos. GFP+/MGE cells were counted within the cortical layers (B, C) and within 100-μm-wide cortical bins to reflect the distance from the CSJ (D) of cryosections obtained from control (black bars) and ethanol-treated (gray bars) embryos. Ethanol exposure significantly increased the number of GFP-immunopositive cells throughout the extent of the neocortex (B, D; Student's t test; p < 0.01). However, the ratio of GFP-positive cells was not altered (C; Student's t test; VZ–SVZ, p = 0.89; IZ, p = 0.35; CP–MZ, p = 0.41). Data are expressed as mean ± SEM. Asterisks denote a significant difference from control.
Using NeuN-immunoreactivity as a marker for neuronal differentiation (Fig. 6 A,B), ethanol exposure in utero resulted in an increase in the ratio of GFP+/MGE cells that were NeuN-immunopositive, concomitant with a decrease in the ratio of NeuN-immunonegative GFP+/MGE cells in the neocortex of BAC-Lhx6 E14.5 embryos (Fig. 6 C). This is consistent with the notion that ethanol exposure in utero drives MGE-derived cells toward differentiation.
Figure 6.

Exposure to ethanol in utero increases the differentiation of MGE-derived cells in BAC-Lhx6 mice. A, B, Cryosections of control (n = 8) and ethanol-treated embryos (n = 9) immunostained for NeuN (A1, B1) and GFP (A2, B2). A3 and B3 are overlays of control and ethanol-treated sections, respectively. Scale bar, 100 μm. Arrows are examples of double-labeled cells. C, The ratio of GFP-immunopositive cells that were colabeled with NeuN was increased with ethanol treatment (control, 0.28 ± 0.04; ethanol, 0.43 ± 0.03; Student's t test, p < 0.05), whereas the ratio of cell GFP-immunopositive-only cells were decreased with in utero ethanol exposure (control, 0.72 ± 0.04; ethanol, 0.57 ± 0.03; Student's t test, p < 0.05). Data are expressed as mean ± SEM. Asterisks indicate a significant difference from control.
Extrinsic and intrinsic mechanisms
The interplay between environmentally derived (extrinsic) and cell-autonomous (intrinsic) factors have been implicated in a host of regulatory processes critical for neuronal differentiation and maturation (for review, see Komuro and Rakic, 1998; Nguyen et al., 2001; Ben-Ari, 2002; Flames and Marin, 2005; Heng et al., 2007). In this light, we determined crossing index in isotypic and heterotypic slice cocultures, the latter consisting of (1) GFP+/MGE explants from in utero ethanol-exposed embryos placed onto control slices or (2) control GFP+/MGE explants placed onto slices from in utero ethanol-exposed embryos (Fig. 7 A). Figure 7 B illustrates that, within the first 300 μm neocortical segment distal to the CSJ, the crossing indices in isotypic slice cocultures from in utero ethanol-treated embryos and both types of heterotypic slice cocultures were significantly higher than those in control slice cocultures. These observations are in line with the notion that ethanol affects both cell-intrinsic and -extrinsic factors that lead to increased cortical entry of MGE-derived cells. Our previous report that ambient GABA, via activation of GABAA receptors, facilitates tangential migration (Cuzon et al., 2006), led us to test the hypothesis that ethanol interacts in utero with the embryonic GABAergic system to manifest its intrinsic and extrinsic influences on migration from the MGE into the developing neocortex.
Figure 7.

Both cell-intrinsic and -extrinsic factors contribute to the effect of increased tangential migration of MGE-derived cells seen after exposure to ethanol in utero. A, Table depicting the heterotypic slice cocultures obtained from GFP β-actin embryos used in this experiment. B, Crossing indices of slice cocultures obtained from control, ethanol, and mixed cocultures. Data are expressed as mean ± SEM. Asterisks indicate a significant difference (ANOVA, p < 0.05).
Extrinsic mechanisms: ambient GABA
Isotypic slice cocultures were maintained in the absence or presence of ethanol, ethanol plus bicuculline (20 μm), or ethanol plus an antibody raised against GABA (1:1000). The concentration of ethanol (25 mg/dl) was adjusted to simulate the BAL in the amniotic fluid of ethanol-treated embryos (Fig. 1 D). Ethanol exposure increased the crossing index in the slice cocultures, an effect that was prevented by bicuculline to block GABAA receptor activation, or an antibody to sequester ambient GABA (Fig. 8). Addition of 2 μm GABA, mimicking the ambient level measured in ethanol-exposed slices (Fig. 9 G), increased the number of MGE-derived cells in the neocortex to a level similar to that measured in the ethanol-exposed treatment group. This suggests that an interaction between ethanol and ambient GABA in the extracellular milieu may underlie an extrinsic mechanism that regulates tangential migration of MGE-derived cells.
Figure 8.

The GABAergic system contributes extrinsic mechanisms that are altered with ethanol exposure in utero. Crossing index of isotypic slice cocultures obtained from GFP β-actin embryos demonstrates that blocking the GABAA receptor with bicuculline (Bicu) or sequestering ambient GABA with a GABA-specific antibody blocks the effect of ethanol. Addition of 2 μm GABA, similar to the ambient level of GABA observed in ethanol-exposed embryos increases the crossing index to levels that are indistinguishable from that of ethanol-treated slice cocultures. Data are expressed as mean ± SEM. Asterisks denote a significant difference (***different from control, ethanol + Bicu, ethanol + GABA; **different from control and ethanol + Bicu only; ANOVA and Holm–Sidak Method, p < 0.05). EtOH, Ethanol.
Figure 9.

Prenatal ethanol exposure increases ambient GABA. A–D, Application of 20 μm bicuculline (Bicu) to GFP+/MGE cells recorded from the MGE (A, C) and the IZ (B, D) of control and in utero ethanol-exposed acute BAC-Lhx6 slices, respectively. E, Mean bicuculline-induced outward current from cells recorded from the neocortex (cortex; 14.87 ± 2.02 pA; n = 16) of control and the MGE (n = 22) and neocortex (n = 21) of in utero ethanol-exposed slices (MGE, 31.55 ± 4.5 pA; neocortex, 35.70 ± 2.35 pA; ANOVA, *p < 0.05). F, Example of a GABA concentration–response curve (black circles and regression line) obtained from a GFP+/MGE cell in the neocortex of an in utero ethanol-exposed slice and the bicuculline-induced current (gray circle) used to calculate the concentration of ambient GABA. G, Calculations based on bicuculline-induced outward current recorded from cells in the cortex of control (0.43 ± 0.11 μm) and the MGE (2.47 ± 0.24 μm) and neocortex (2.73 ± 0.57 μm) of in utero ethanol-exposed slices reveal a significant difference in ambient GABA concentration (ANOVA, *p < 0.01).
Ethanol exposure in utero elevated the level of ambient GABA that tonically activated a GABAA receptor-mediated current in MGE-derived cells. As reported previously (Cuzon et al., 2006), this tonic current could be revealed in individual tangentially migrating cells by blocking GABAA receptor activation with focal application of a saturating concentration of bicuculline. In the present study, the same concentration of bicuculline (20 μm) uncovered significantly larger outward currents in GFP+/MGE cells recorded in acute slices from E14.5 BAC-Lhx6 embryos in the in utero exposed group than control (Fig. 9). The bicuculline-induced current monitored in each cell was fit to the GABA concentration-response curve derived from the same cell to estimate the concentration of ambient GABA (Fig. 9 F). As summarized in Figure 9 G, the concentration of ambient GABA in the MGE–CSJ region and the IZ of the neocortex of in utero ethanol-exposed embryos reached 2.60 ± 0.40 μm, which was significantly higher than that estimated to be present in the MGE–CSJ region and IZ of the neocortex of control embryos (0.43 ± 0.11 μm) in this and a previous study (Cuzon et al., 2006). Importantly, a tonic current was detected in GFP+/MGE cells located in the MGE–CSJ area of acute slices from E14.5 ethanol-exposed embryos that was not evident in the MGE–CSJ area of control, ethanol-naive embryos (Fig. 9 F) (Cuzon et al., 2006). This finding, although consistent with the demonstration of an elevated ambient GABA, prompted an additional series of experiments to examine the possibility that in utero ethanol exposure altered the properties of GABAA receptors, including those expressed in cells in the MGE.
Intrinsic mechanisms: receptivity to GABA
We first monitored the peak amplitude of whole-cell current responses to a set concentration of GABA in GFP+/MGE cells located in the MGE–CSJ region and IZ of the neocortex of acute slices from control and ethanol-exposed E14.5 BAC-Lhx6 embryos. Whole-cell membrane capacitance in tangentially migrating GFP+/MGE cells recorded in the MGE–CSJ area (control, 0.141 ± 0.02 pF; ethanol, 0.166 ± 0.03 pF) and the IZ of the neocortex (control, 0.154 ± 0.04 pF; ethanol, 0.145 ± 0.02 pF) were not significantly different (data not shown). In control slices, focal application of GABA (100 μm) elicited current responses in the IZ of the neocortex (148.85 ± 19.66 pA) that were much more robust than those monitored in the MGE–CSJ region of the same slice (25.25 ± 4.16 pA) (Fig. 10 A). We previously postulated that this reflects MGE-derived cells maturing and acquiring increased responsiveness to GABA as they migrate from the MGE to the neocortex (Cuzon et al., 2006). However, in slices from embryos exposed to ethanol in utero, the same concentration of GABA elicited robust current responses even in GFP+/MGE cells in the MGE–CSJ region (156.20 ± 10.72 pA). These GABA-activated current responses were significantly larger than monitored in control slices (Student's t test, p < 0.001) and were comparable in peak amplitude to those recorded in the IZ of either control (148.85 ± 19.66 pA) or ethanol-exposed slices (138.06 ± 12.64 pA).
Figure 10.
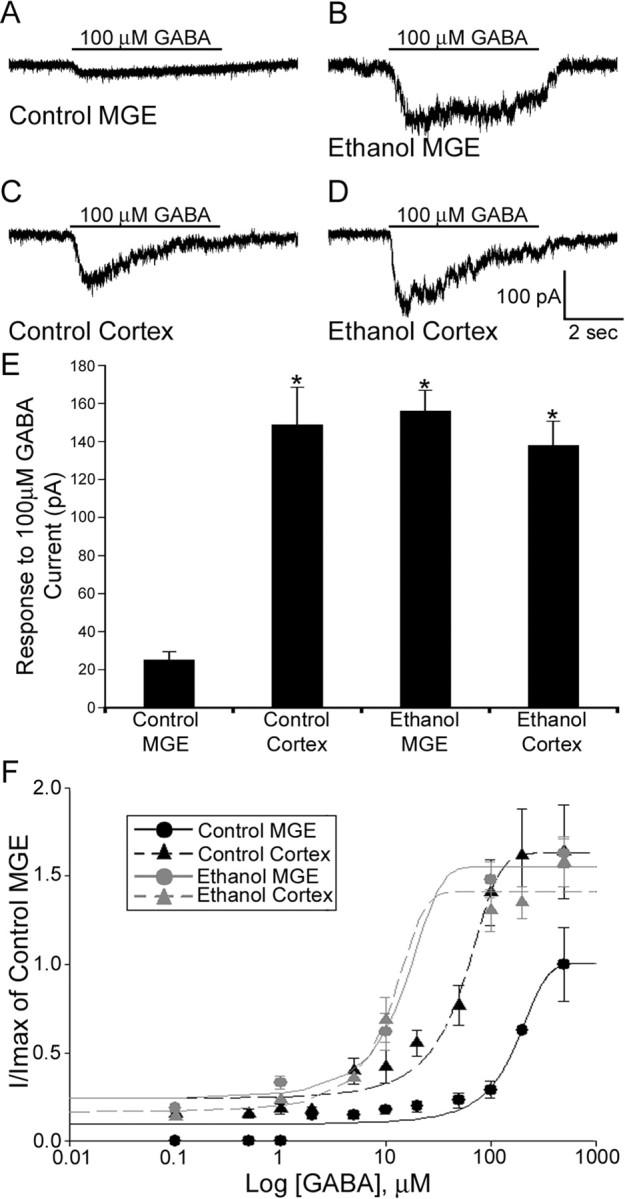
GFP+/MGE cells recorded in slices obtained from BAC-Lhx6 in utero ethanol-exposed embryos are more sensitive to GABA than those recorded within control slices. A–D, Responses to application of 100 μm GABA to GFP+/MGE cells recorded from the MGE (A, B) and neocortex (C, D) of control and in utero ethanol exposed acute BAC-Lhx6 slices, respectively. E, Average response of GFP+/MGE cells recorded within the MGE and the IZ of the neocortex in slices from control and in utero ethanol-exposed E14.5 embryos (control MGE, 25.25 ± 4.16 pA; control cortex, 148.85 ± 19.66 pA; ethanol MGE, 156.20 ± 10.72 pA; ethanol cortex, 138.06 ± 12.64 pA; ANOVA, p < 0.001). Data are expressed as mean amplitude ± SEM. Asterisks denote a significant difference from control MGE (p < 0.001). F, GABA concentration–response curves for GFP+/MGE cells recorded in the MGE and IZ of the neocortex of acute slices obtained from control and in utero ethanol-treated E14.5 BAC-Lhx6 embryos normalized to the max amplitude of cells recorded in the MGE of control slices.
Concentration-response profiles were then determined with incremental concentrations of GABA (0.1–500 μm) for GFP+ cells in the MGE–CSJ and IZ of acute BAC-Lhx6 slices from E14.5 control and in utero ethanol-exposed embryos. In Figure 10 B, the peak current amplitude monitored in each GFP+ cell was normalized to that obtained from GFP+/MGE cells in the MGE region of control slices and plotted semilogarithmically as a function of GABA concentration. The apparent affinity (EC50) to GABA was significantly different for cells recorded in the MGE of control (160.05 ± 0.09 μm) versus in utero ethanol-exposed embryonic slices (13.87 ± 0.08 μm), as were those determined for cells in the IZ (control, 49.32 ± 0.06 μm; in utero ethanol exposure, 11.54 ± 0.04 μm). Overall, in GFP+/MGE cells in the MGE, in utero ethanol exposure shifted the EC50 leftward and increased the apparent efficacy to approximate that estimated in the IZ. These results, together, suggest that in utero exposure to ethanol results in upregulated expression of GABAA receptors of either the same or different isoforms, or both. Thus, aside from extrinsically modifying the ambient level of GABA, in utero ethanol exposure intrinsically altered GABAA receptor function.
Discussion
The basic premise underlying this study is that cortical malformations associated with ethanol exposure in utero manifest themselves early by compromising the developmental profile of neuronal populations during corticogenesis. In this light, we tested the hypothesis that exposure to ethanol in utero disrupts the embryonic development of GABAergic cortical interneurons. Clearly, the most severe outcome of excessive gestational ethanol consumption is FAS. However, we report here that chronic consumption of a relatively low level of ethanol (2% w/v), resulting in BAL (25 mg/dl, corresponding to 5.4 mm ethanol) three times lower than that considered as being intoxicated in the USA (80 mg/dl, corresponding to 17 mm ethanol), can affect the development of GABAergic cortical interneurons. The major findings are (1) that exposure to moderate levels of ethanol in utero increases the number of embryonic cortical GABAergic neurons, and (2) that this can be accounted for by enhanced tangential migration and cortical entry of MGE-derived cells. We propose that ethanol exposure in utero invokes both cell-intrinsic and -extrinsic mechanisms, specifically, by interacting with the GABAergic system in changing the ambient level of GABA (extrinsic) as well as GABAA receptor properties (intrinsic).
Ethanol exposure in utero increases GABAergic cells in the embryonic neocortex
At E14.5, MGE-derived cells contribute prominently to the tangentially migrating wave of the subpopulation of cortex-bound GABAergic interneurons that postnatally express somatostatin- and parvalbumin-immunoreactivity (Xu et al., 2004). Other subpopulations arise from the lateral and caudal ganglionic eminences (Anderson et al., 2001; Nery et al., 2002, 2003; Xu et al., 2004; Butt et al., 2005). Although our study focused on migration from the MGE under conditions of in utero ethanol exposure, the results also demonstrated a clear-cut increase in the number and density of GABA-immunopositive cells in wild-type and GFP-immunopositive cells in GAD67-GFP knock-in and BAC-Lhx6 transgenic mice. Thus, the ethanol-induced effect uncovered here likely applies to cells of other extracortical origins destined to become GABAergic cortical interneurons.
In utero ethanol exposure decreased cell proliferation in the MGE, pushed MGE-derived cells toward differentiation, and increased their rate of migration. These factors contribute to the increase in GABAergic cells in the embryonic neocortex. In utero ethanol exposure elevated ambient GABA and promoted functional expression of GABAA receptors displaying a higher apparent affinity for GABA. Because GABA is depolarizing at embryonic and early postnatal ages (Owens et al., 1996; Howard et al., 2007), an enhanced GABA-induced depolarization would favor suppressed cellular proliferation, enhanced differentiation, and altered migration (Behar et al., 1994, 1996, 2000; LoTurco et al., 1995; Cancedda et al., 2007).
The increase in the number and density of MGE-derived cells in the embryonic neocortex after in utero ethanol exposure was not accompanied by abnormal cellular distribution or cortical patterning. This suggests that ethanol exposure in utero does not interfere with cues that regulate cortical lamination nor is it selective in altering the development of subpopulations of GABAergic cortical interneurons. In fact, earlier studies demonstrated that normal cortical patterning prevails in the adult and postnatal rat cortex exposed to ethanol in utero (Miller 1986, 1993). However, the same studies by Miller and colleagues reported an overall loss of cortical neurons postnatally. A recent report also indicated decreased number of GABAergic cells in the adult guinea pig and rat cortices (Moore et al., 1998; Bailey et al., 2004), an effect that could be attributed to ethanol inducing apoptosis (Bhave and Hoffman, 1997; Cartwright et al., 1998; Cheema et al., 2000; Dikranian et al., 2005). The discrepant results could be reconciled by hypothesizing cell death occurring in the immature cortex later than E14.5, including early postnatal ages, that is exacerbated by gestational ethanol. If so, then an accelerated ushering of supernumerary GABAergic interneurons into the cortical anlage may lead to their premature demise, resulting in a net decrease when examined at postnatal ages. Alternatively, GABAergic interneurons that are improperly integrated into the synaptic circuitry may be subject to death and elimination. These hypotheses await experimental testing in future studies.
Ethanol exposure in utero enhances tangential migration by invoking extrinsic and intrinsic GABAergic mechanisms
The results of the present study indicate that ethanol exposure in utero influences tangential migration by (1) raising the ambient level of GABA, which we defined as being an extrinsic factor, and (2) altering the receptivity to GABA of migrating GABAergic interneurons, which we considered to be intrinsic. Two lines of evidence prompted us to test the hypothesis that ethanol interacts in utero with the embryonic GABAergic system. First, GABAergic transmission has long been implicated in ethanol action in the brain (for review, see Hanchar et al., 2004; Paul, 2006; Mody et al., 2007; Santhakumar et al., 2007); evidence is quite compelling that ethanol targets presynaptic, postsynaptic, and extrasynaptic GABAergic mechanisms to exert its effects that range from acute intoxication to longer-term development of tolerance and dependence (Aguayo, 1990; Nishio and Narahashi, 1990; Devaud et al., 1995, 1997; Tatebayashi et al., 1998; Grobin et al., 2000a,b; Roberto et al., 2003; Hayward et al., 2004). Second, GABA affects a variety of tropic and trophic processes during development that is seemingly unrelated to its role in mediating synaptic transmission and regulating neuronal excitability in the adult brain (for review, see Ben-Ari, 2002; Owens and Kriegstein, 2002; Heng et al., 2007); we and others have demonstrated that GABA, by way of GABAA receptors, regulates migration of embryonic cortical neurons, including that of GABAergic interneurons (Behar et al., 1994, 1996, 2000; Cuzon et al., 2006).
Ethanol enhances tangential migration by elevating ambient GABA in utero
In many brain regions examined to date, ambient levels of GABA and/or glutamate have been demonstrated and implicated in maintaining a baseline level of activity that regulates neuronal excitability and circuit function (Ge et al., 2006; Kirmse and Kirischuk, 2006; Herman and Jahr, 2007; LeMeur et al., 2007). With specific regard to GABA, the prevailing thought is that synaptic spillover results in the ambient extracellular accumulation of GABA, which tonically activates high-affinity, slowly desensitizing extra-synaptic GABAA receptor isoforms with cell-type specific pharmacological properties (Nusser and Mody, 2002; Fritschy and Brunig, 2003; Caraiscos et al., 2004). In the rodent embryonic neocortex, despite the fact that few, if any, functional GABAergic synapses or synaptic specializations are present on the tangentially migrating, immature GABAergic interneurons, ambient GABA is present at submicromolar concentrations (∼0.5 μm) that tonically activate bicuculline-sensitive GABAA receptors in MGE-derived cells and sustains tangential migration (Cuzon et al., 2006).
In the MGE and neocortex of embryos exposed to ethanol in utero, the ambient level of GABA was elevated and reached micromolar range (∼2.5 μm). A priori, an elevated ambient GABA could become manifest as the result of enhanced GABA release (Roberto et al., 2003; Hayward et al., 2004), increase in GABA-releasing subpopulation of cells, or both. It is noteworthy that the elevated ambient GABA tonically activated MGE-derived cells to a greater extent, and that GABA in this concentration range enhanced tangential migration to the same degree as exposure to 2% ethanol in utero.
In utero ethanol exposure augments GABA responsiveness of MGE-derived cells
The expression of GABAA receptor subunits is dynamic during the developmental stages of migration, differentiation, and synaptogenesis (Laurie et al., 1992; Poulter et al., 1992, 1993; Fritschy et al., 1994). For example, as development progresses, the expression of the α1 subunit increases, and α1 subunit-containing GABAA receptor isoforms exhibiting fast deactivation and desensitization kinetics become predominant at mature synapses (Fritschy et al., 1994; Gingrich et al., 1995; Bianchi et al., 2002). Activation of the GABAA receptor itself has been implicated in accelerating this progression (Poulter et al., 1997).
In the present study, the electrophysiological data suggest that prenatal ethanol exposure promoted the expression and/or activation of higher affinity GABAA receptors in MGE-derived cells, and that this may contribute to enhanced migration in utero. A caveat here is that bicuculline did not completely arrest in utero ethanol-enhanced tangential migration. It is likely that activation of GABAA receptors is not the only underlying mechanism, considering the pleiotropic effects of ethanol and the myriad regulators of cell cycle, migration, and cell type specification that ethanol may influence in utero (for review, see Marin and Rubenstein, 2001; Guillemot, 2007; Nakajima, 2007). In any event, because subunit combinations dictate GABAA receptor properties (for review, see Verdoorn et al., 1990; Lüddens and Wisden, 1991; Rudolph et al., 2001), and because certain GABAA receptor isoforms are selectively sensitive to ethanol (for review, see Aguayo et al., 2002; Boehm et al., 2004; Hanchar et al., 2004; Paul, 2006; Lovinger and Homanics, 2007; Santhakumar et al., 2007), it would be important to determine which GABAA receptors subunits are differentially expressed with prenatal ethanol exposure. Profiling of GABAA receptor subunit mRNAs (α1–5, β1–3, γ1–3, δ) in E14.5 brains of control and ethanol-exposed embryos suggests differential expression of GABAA receptor subunit cadres in both MGE and neocortex (our unpublished data). This preliminary finding is being extended in ongoing studies to analyze differential expression of GABAA receptors specifically in MGE-derived cells with and without ethanol exposure.
Footnotes
This work was supported by Public Health Service Grants RO1 MH069826 (H.H.Y.) and F31 AA014698 (V.C.C). We thank Drs. Donald Bartlett and James Leiter for their critical reading of this manuscript.
References
- Aguayo LG. Ethanol potentiates the GABAA-activated Cl− current in mouse hippocampal and cortical neurons. Eur J Pharmacol. 1990;187:127–130. doi: 10.1016/0014-2999(90)90349-b. [DOI] [PubMed] [Google Scholar]
- Aguayo LG, Peoples RW, Yeh HH, Yevenes GE. GABAA receptors as molecular sites of ethanol action. Direct or indirect actions? Curr Top Med Chem. 2002;2:869–885. doi: 10.2174/1568026023393426. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisestat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Anderson S, Mione M, Yun K, Rubenstein JL. Differential origins of neocortical projection and local circuit neurons: role for Dlx genes in neocortical interneuronogenesis. Cereb Cortex. 1999;9:646–654. doi: 10.1093/cercor/9.6.646. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Brien JF, Reynolds JN. Chronic prenatal ethanol exposure alters the proportion of GABAergic neurons in layers II/III of the adult guinea pig somatosensory cortex. Neurotoxicol Teratol. 2004;26:59–63. doi: 10.1016/j.ntt.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Colton CA, Somogyi R, Olah Z, Lehel C, Barker JL. GABA-induced chemokinesis and NGF-induced chemotaxis of embryonic spinal cord neurons. J Neurosci. 1994;14:29–58. doi: 10.1523/JNEUROSCI.14-01-00029.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL. GABA stimulates chemotaxis and chemokinesis of embryonic neurons via calcium-independent mechanisms. J Neurosci. 1996;16:1808–1818. doi: 10.1523/JNEUROSCI.16-05-01808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Scott CA, Greene CL, Barker JL. GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb Cortex. 2000;10:899–909. doi: 10.1093/cercor/10.9.899. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature versus nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Ben-Yaakov G, Golan H. Cell proliferation in response to GABA in the postnatal hippocampal slice cultures. Int J Dev Neurosci. 2003;21:153–157. doi: 10.1016/s0736-5748(03)00027-3. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, Hohmann CF. Behavioral consequences of abnormal cortical development: insights into developmental disabilities. Behav Brain Res. 1997;86:121–142. doi: 10.1016/s0166-4328(96)02251-6. [DOI] [PubMed] [Google Scholar]
- Bhave SV, Hoffman PL. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J Neurochem. 1997;68:578–586. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Haas KF, Macdonald RL. α1 and α6 subunits specify distinct desensitization, deactivation and neurosteroid modulation of GABAA receptors containing the δ subunit. Neuropharmacol. 2002;43:492–502. doi: 10.1016/s0028-3908(02)00163-6. [DOI] [PubMed] [Google Scholar]
- Boehm SL, Ponomarev I, Jennings AW, Whiting PJ, Rosahl TW, Garrett EM, Blendov YA, Harris RA. γ-aminobutyric acid A receptor subunit mutant mice: new perspectives on alcohol actions. Biochem Pharmacol. 2004;68:1581–1602. doi: 10.1016/j.bcp.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo . J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos V, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Roshal TW, Wafford KA, Macdonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc Natl Acad Sci USA. 2004;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol enhances GABA-ergic transmission to cerebellar granule cells via an increase in golgi cell excitability. J Neurosci. 2004;24:3746–3751. doi: 10.1523/JNEUROSCI.0067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright MM, Tessmer LL, Smith SM. Ethanol-induced neural crest apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol Clin Exp Res. 1998;22:142–149. [PubMed] [Google Scholar]
- Cheema ZF, West JR, Miranda RC. Ethanol induces FAS/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol Clin Exp Res. 2000;24:535–543. [PubMed] [Google Scholar]
- Copp AJ, Harding BN. Neuronal migration disorders in humans and in mouse models-an overview. Epilepsy Res. 1999;36:133–141. doi: 10.1016/s0920-1211(99)00047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon VC, Yeh PW, Cheng Q, Yeh HH. Ambient GABA promotes cortical entry of tangentially migrating cells derived from the medial ganglionic eminence. Cereb Cortex. 2006;16:1377–1388. doi: 10.1093/cercor/bhj084. [DOI] [PubMed] [Google Scholar]
- Devaud LL, Smith FD, Grayson DR, Morrow AL. Chronic ethanol consumption differentially alters the expression of γ-aminobutyric acidA receptor subunit mRNAs in rat cerebral cortex: competitive, quantitative reverse transcriptase-polymerase chain reaction analysis. Mol Pharmacol. 1995;48:861–868. [PubMed] [Google Scholar]
- Devaud LL, Fritschy JM, Sieghart W, Morrow AL. Bidirectional alterations of GABAA receptor subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. J Neurochem. 1997;69:126–130. doi: 10.1046/j.1471-4159.1997.69010126.x. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney J. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev Brain Res. 2005;155:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Flames N, Marin O. Developmental mechanisms underlying the generation of cortical interneuron diversity. Neuron. 2005;46:377–381. doi: 10.1016/j.neuron.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Brunig I. Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther. 2003;98:299–323. doi: 10.1016/s0163-7258(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci. 1994;14:5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Goh ELK, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;493:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich KJ, Roberts WA, Kass RS. Dependence of the GABAA receptor gating kinetics on the α-subunit isoform: implications for structure-function relations and synaptic transmission. J Physiol (Lond) 1995;489:529–543. doi: 10.1113/jphysiol.1995.sp021070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Scahmbra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Fritschy JM, Morrow AL. Chronic ethanol administration alters immunoreactivity for GABAA receptor subunits in rat cortex in a region-specific manner. Alcohol Clin Exp Res. 2000a;24:1137–1144. [PubMed] [Google Scholar]
- Grobin AC, Papadeas ST, Morrow AL. Regional variations in the effects of chronic ethanol administration on GABAA receptor expression: potential mechanisms. Neurochem Inter. 2000b;37:453–461. doi: 10.1016/s0197-0186(00)00058-9. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hanchar HJ, Wallner M, Olsen RW. Alcohol effects on γ-aminobutyric acid type A receptors: are extrasynaptic receptors the answer? Life Sci. 2004;76:1–8. doi: 10.1016/j.lfs.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Hayward ML, Martin AE, Brien JF, Dringenberg HC, Olmstead MC, Reynolds JN. Chronic prenatal ethanol exposure impairs conditioned responding and enhances GABA release in the hippocampus of the adult guinea pig. J Pharmacol Exp Therap. 2004;308:644–650. doi: 10.1124/jpet.103.059261. [DOI] [PubMed] [Google Scholar]
- Heck N, Kilb W, Reiprich P, Kubota H, Furukawa T, Fukuda A, Luhmann HJ. GABAA receptors regulate neocortical neuronal migration in vitro and in vivo. Cereb Cortex. 2006;17:138–148. doi: 10.1093/cercor/bhj135. [DOI] [PubMed] [Google Scholar]
- Heng JI, Moonen G, Nguyen L. Neurotransmitters regulate cell migration in the telencephalon. Eur J Neurosci. 2007;26:537–546. doi: 10.1111/j.1460-9568.2007.05694.x. [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concetration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MZ, Burger RM, Rubel EW. A developmental switch to GABAergic inhibition dependent on increases in Kv1-type K+ currents. J Neurosci. 2007;27:2112–2123. doi: 10.1523/JNEUROSCI.5266-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez D, Lopez-Mascaraque LM, Valverde F, De Carlos JA. Tangential migration in neocortical development. Dev Biol. 2002;244:155–169. doi: 10.1006/dbio.2002.0586. [DOI] [PubMed] [Google Scholar]
- Kirmse K, Kirischuk S. Ambient GABA constraints the strength of GABAergic synaspses at Cajal-Retzius cells in the developing visual cortex. J Neurosci. 2006;26:4216–4227. doi: 10.1523/JNEUROSCI.0589-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J Neurobiol. 1998;37:110–130. [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMeur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol (Lond) 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath JS, Davis MBE, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Homanics GE. Tonic for what ails us? High-affinity GABAA receptors and alcohol. Alcohol. 2007;41:139–143. doi: 10.1016/j.alcohol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüddens H, Wisden W. Function and pharmacology of multiple GABAA receptor subunits. Trends Pharmacol Sci. 1991;12:49–51. doi: 10.1016/0165-6147(91)90495-e. [DOI] [PubMed] [Google Scholar]
- Manent J-B, Demarque M, Jorquera I, Pellegrino C, Ben-Ari Y, Aniksztejn L, Represa A. A noncanonical release of GABA and glutamate modulates neuronal migration. J Neurosci. 2005;25:4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric D, Liu Q-Y, Maric I, Chaudry S, Chang Y-H, Smith SV, Sieghart W, Fritschy J-M, Barker JL. GABA expression dominates neuronal lineage progression in the embryonic rat neocortex and facilitates neurite outgrowth via GABAA autoreceptor/Cl− channels. J Neurosci. 2001;21:2343–2360. doi: 10.1523/JNEUROSCI.21-07-02343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science. 1986;233:1308–1311. doi: 10.1126/science.3749878. [DOI] [PubMed] [Google Scholar]
- Miller MW. Circadian rhythm of cell proliferation in the telencephalic ventricular zone: effect of in utero exposure to ethanol. Brain Res. 1992;595:17–24. doi: 10.1016/0006-8993(92)91447-m. [DOI] [PubMed] [Google Scholar]
- Miller MW. Migration of cortical neurons is altered by gestational exposure to ethanol. Alcohol Clin Exp Res. 1993;17:304–314. doi: 10.1111/j.1530-0277.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Limited ethanol exposure selectively alters the proliferation of precursor cells in the cerebral cortex. Alcohol Clin Exp Res. 1996;20:139–143. doi: 10.1111/j.1530-0277.1996.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Mody I, Glykys J, Wei W. A new meaning for “Gin and Tonic”: tonic inhibition as the target for ethanol action in the brain. Alcohol. 2007;41:145–153. doi: 10.1016/j.alcohol.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DB, Quintero MA, Ruygrok AC, Walker DW, Heaton MB. Prenatal ethanol exposure reduces parvalbumin-immunoreactive GABAergic neuronal number in the adult rat cingulate cortex. Neurosci Lett. 1998;249:25–28. doi: 10.1016/s0304-3940(98)00378-4. [DOI] [PubMed] [Google Scholar]
- Mori T, Aistrup GL, Nishikawa K, Marszalec W, Yeh JZ, Narahashi T. Basis of variable sensitivities of GABAA receptors to ethanol. Alcohol Clin Exp Res. 2000;24:965–971. [PubMed] [Google Scholar]
- Nakahiro M, Arakawa O, Narahashi T. Modulation of γ-aminobutyric acid receptor-channel complex by alcohols. J Pharmacol Exp Ther. 1991;259:235–240. [PubMed] [Google Scholar]
- Nakajima K. Control of tangential/non-radial migration of neurons in the developing cerebral cortex. Neurochem Int. 2007;51:121–131. doi: 10.1016/j.neuint.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Nery S, Corbin JG, Fishell G. Dlx2 progenitor migration in wildtype and Nkx2.1 mutant telencephalon. Cereb Cortex. 2003;13:895–903. doi: 10.1093/cercor/13.9.895. [DOI] [PubMed] [Google Scholar]
- Nguyen L, Rigo JM, Rocher V, Belachew S, Malgrange B, Rogister B, Leprince P, Moonen G. Neurotransmitters as early signals for central nervous system development. Cell Tissue Res. 2001;35:187–202. doi: 10.1007/s004410000343. [DOI] [PubMed] [Google Scholar]
- Nishio M, Narahashi T. Ethanol enhancement of GABA-activated chloride current in rat dorsal root ganglion neurons. Brain Res. 1990;518:283–286. doi: 10.1016/0006-8993(90)90982-h. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominani K, Nakanishi T, Nishimune Y. “Green mice” as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MBE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens F, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Papadeas S, Grobin AC, Morrow AL. Chronic ethanol consumption differentially alters GABAA receptor alpha1 and alpha4 subunit peptide expression and GABAA receptor-mediated 36Cl(−) uptake in mesocorticolimbic regions of rat brain. Alcohol Clin Exp Res. 2001;25:1270–1275. [PubMed] [Google Scholar]
- Paul SM. Alcohol-sensitive GABA receptors and alcohol antagonists. Proc Natl Acad Sci USA. 2006;103:8307–8308. doi: 10.1073/pnas.0602862103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter MO, Barker JL, O'Carroll AM, Lolait SJ, Mahan LC. Differential and transient expression of GABAA receptor alpha-subunit mRNAs in the developing rat CNS. J Neurosci. 1992;12:2888–2900. doi: 10.1523/JNEUROSCI.12-08-02888.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter MO, Barker JL, O'Carroll AM, Lolait SJ, Mahan LC. Co-existent expression of GABAA receptor beta 2, beta 3, and gamma 2 subunit messenger RNAs during embryogenesis and early postnatal development of the rat central nervous system. Neuroscience. 1993;53:1019–1033. doi: 10.1016/0306-4522(93)90486-y. [DOI] [PubMed] [Google Scholar]
- Poulter MO, Ohannesian L, Larmet Y, Feltz P. Evidence that GABAA receptor subunit mRNA expression during development is regulated by GABAA receptor stimulation. J Neurochem. 1997;68:631–639. doi: 10.1046/j.1471-4159.1997.68020631.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and post synaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA. 2003;100:2053–2058. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestini F, Möhler H. GABAA receptor subtypes: dissecting their pharmacological functions. Trends Pharmacol Sci. 2001;22:188–194. doi: 10.1016/s0165-6147(00)01646-1. [DOI] [PubMed] [Google Scholar]
- Sanchis R, Sancho-Tello M, Guerri C. The effects of chronic alcohol consumption on pregnant rats and their offspring. Alcohol Alcohol. 1986;21:295–305. [PubMed] [Google Scholar]
- Santhakumar V, Wallner M, Otis TS. Ethanol acts directly on extrasynaptic subtypes of GABAA receptors to increase tonic inhibition. Alcohol. 2007;41:211–221. doi: 10.1016/j.alcohol.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp DW, Yeh HH. Ethanol-GABAA receptor interactions: a comparison between cell lines and cerebellar purkinjie cells. J Pharmacol Exp Ther. 1998;284:768–776. [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Fujimori KE, Takauji R. Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J Neurosci. 1997;17:8313–8323. doi: 10.1523/JNEUROSCI.17-21-08313.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J-I, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tanaka DH, Maekawa K, Yanagawa Y, Obata K, Murakami F. Multidirectional and multizonal migration of GABAergic interneurons in the developing cerebral cortex. Development. 2006;133:2167–2176. doi: 10.1242/dev.02382. [DOI] [PubMed] [Google Scholar]
- Tatebayashi H, Motomura H, Narahashi T. Alcohol modulation of single GABA(A) receptor-channel kinetics. NeuroReport. 1998;9:1769–1775. doi: 10.1097/00001756-199806010-00018. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Draguhn A, Ymer S, Seeburg PH, Sakmann B. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron. 1990;4:919–928. doi: 10.1016/0896-6273(90)90145-6. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Burnette DM, Dunwiddie TV, Harris RA. Genetic differences in the ethanol sensitivity of GABAA receptors expressed in Xenopus oocytes. Science. 1990;249:291–293. doi: 10.1126/science.1695761. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Burnett DM, Liedenheimer NJ, Burt DR, Wang JB, Kofuji P, Dunwiddie TV, Harris RA, Sikela JM. Ethanol sensitivity of the GABAA receptor expressed in Xenopus oocytes requires 8 amino acids contained in the γ2L subunit. Neuron. 1991;7:27–33. doi: 10.1016/0896-6273(91)90071-7. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]