

Abstract
Axon growth is critical to the establishment of neuronal connectivity. The E3 ubiquitin ligase Cdh1-anaphase-promoting complex (Cdh1-APC) and its substrate the transcriptional modulator SnoN form a cell-intrinsic pathway that orchestrates axonal morphogenesis in the mammalian brain. How the Cdh1-APC/SnoN pathway is controlled in the nervous system remained unknown. Here, we report that the TGFβ-regulated signaling protein Smad2 plays a key role in regulating the Cdh1-APC/SnoN pathway in neurons. We find that Smad2 is expressed in primary granule neurons of the developing rat cerebellar cortex. The Smad signaling pathway is basally activated in neurons. Endogenous Smad2 is phosphorylated, localized in the nucleus, and forms a physical complex with endogenous SnoN in granule neurons. Inhibition of Smad signaling by several distinct approaches, including genetic knock-down of Smad2, stimulates axonal growth. Biochemical evidence and genetic epistasis analyses reveal that Smad2 acts upstream of SnoN in a shared pathway with Cdh1-APC in the control of axonal growth. Remarkably, Smad2 knock-down also overrides the ability of adult rat myelin to inhibit axonal growth. Collectively, our findings define a novel function for Smad2 in regulation of the Cdh1-APC/SnoN cell-intrinsic pathway of axonal morphogenesis, and suggest that inhibition of Smad signaling may hold therapeutic potential in stimulating axonal growth after injury in the CNS.
Keywords: cerebellum, axon growth, Smad2, SnoN, Cdh1-APC, myelin, SB431542
Introduction
Axon growth and guidance are essential events in the developing nervous system that ensure the proper connectivity of neurons. Extrinsic cues, including guidance factors and morphogens, coordinate the growth and navigation of axons through the complex environment of the nervous system (Tessier-Lavigne and Goodman, 1996; Dickson, 2002; Charron and Tessier-Lavigne, 2005). Neurons also harbor cell-intrinsic programs that govern axonal growth and patterning (Chen et al., 1997; Konishi et al., 2004; Stegmuller et al., 2006). In particular, growing evidence supports an important role for transcription factors and the ubiquitin proteasome system in axonal development (Campbell and Holt, 2001; Watts et al., 2003; Goldberg, 2004; Konishi et al., 2004; Stegmuller and Bonni, 2005; Kuo et al., 2006; Polleux et al., 2007).
The E3 ubiquitin ligase Cdh1-anaphase-promoting complex (Cdh1-APC) has emerged as a key regulator of axonal morphogenesis in the mammalian brain (Konishi et al., 2004). The transcriptional regulator SnoN is a critical substrate of Cdh1-APC in neurons (Stegmuller et al., 2006). Gain- and loss-of-function analyses indicate that SnoN enhances axonal growth. Cdh1-APC stimulates the ubiquitination and subsequent degradation of SnoN in neurons. Genetic epistasis experiments reveal that Cdh1-APC and SnoN function in a shared pathway whereby Cdh1-APC limits axonal growth by inhibiting SnoN. Thus, Cdh1-APC and SnoN comprise a cell-intrinsic pathway that orchestrates axonal growth by regulating transcription. Identification of the transcriptional protein Id2 as another crucial substrate of neuronal Cdh1-APC in the control of axon growth further strengthens the view that neuronal Cdh1-APC operates in the nucleus as a critical regulator of axonal morphogenesis (Lasorella et al., 2006; Kim and Bonni, 2007).
A major question that remains to be addressed is how the Cdh1-APC cell-intrinsic pathway of axonal development is regulated in neurons. An important clue is provided by the finding that SnoN acts as a substrate of neuronal Cdh1-APC (Stegmuller et al., 2006). SnoN function is intimately linked to TGFβ-Smad signaling in dividing cells (Sun et al., 1999; Liu et al., 2001). TGFβ ligand binding induces the heterodimeric association of TGFβ type I and II serine/threonine receptor kinases (Shi and Massague, 2003; ten Dijke and Hill, 2004). Activated TGFβ type I receptor recruits and phosphorylates Smad2 and Smad3, which in turn associate with the protein Smad4 (Shi and Massague, 2003; ten Dijke and Hill, 2004). Complexed Smads translocate to the nucleus, where they regulate transcription of TGFβ-responsive genes (Attisano and Wrana, 2000). SnoN associates with Smad2 and Smad3 and thereby modulates TGFβ-dependent transcription (Stroschein et al., 1999; He et al., 2003; Sarker et al., 2005; Hsu et al., 2006). Conversely, TGFβ signaling regulates SnoN activity in a Smad2/3-dependent manner (Bonni et al., 2001; Stroschein et al., 2001; Wan et al., 2001). According to these studies, TGFβ-activated Smad2 and Smad3 act as scaffolding molecules to recruit SnoN to Cdh1-APC, leading to the ubiquitination and subsequent degradation of SnoN. These data support the model that Smad2 and Smad3 act upstream of SnoN in TGFβ signaling. The intimate relationship of SnoN with Smad signaling raises the intriguing question of whether and how Smad signaling might regulate the Cdh1-APC/SnoN cell-intrinsic pathway of axonal morphogenesis in neurons.
In this study, we report a critical role for Smad signaling in regulating the function of the Cdh1-APC/SnoN pathway in axonal growth. Smad2 is expressed and associates with SnoN in granule neurons of the cerebellar cortex. Inhibition of endogenous Smad signaling in primary granule neurons by several distinct approaches, including genetic knock-down of Smad2 by RNAi, stimulates axonal growth, suggesting that Smad signaling inhibits axonal growth. We also find that Smad2 acts together with Cdh1-APC in a shared pathway upstream of SnoN in the control of axonal growth. Strikingly, Smad2 knock-down also overrides myelin inhibition of axonal growth. These findings implicate the Smad pathway in axon growth control and suggest that this pathway holds potential for therapeutic approaches to enhance the intrinsic ability of neurons to extend axons after injury and disease.
Materials and Methods
Plasmids and reagents.
The U6/smad2 and pcDNA3/Smad2 plasmids were kindly provided by Daniel Bernard (McGill University, Montreal, Quebec, Canada) (Bernard, 2004). To generate the pcDNA3/Smad2-Rescue expression plasmid, silent mutations in Smad2 cDNA were introduced using QuikChange site directed mutagenesis kit (Stratagene, La Jolla, CA). The U6/cdh1 and U6/snon plasmids have been described previously (Stegmuller et al., 2006). The pCMV5/Smad6 and pCMV5/Smad7 expression plasmids were a kind gift from Shirin Bonni (University of Calgary, Calgary, Alberta, Canada). SB431542 and SB505124 were purchased from Sigma (St. Louis, MO).
Cerebellar granule neuron culture and transfections.
Granule neurons were isolated from postnatal Long–Evans rat cerebellum [postnatal day 6 (P6)] as described previously (Konishi et al., 2002). Neurons were plated on polyornithine-coated glass coverslips and kept in basal medium Eagle (Invitrogen, Carlsbad, CA) supplemented with 10 μg/ml insulin and 2 mm glutamine, penicillin, and streptomycin, with 10% calf serum and 2 mm glutamine, penicillin, and streptomycin, or with 10% calf serum (Hyclone Laboratories, Logan, UT), 25 mm KCl, and 2 mm glutamine, penicillin, and streptomycin (referred to as full media). Neurons were treated with the mitosis inhibitor cytosine-β-d-arabinofuranoside (10 μm) when kept in serum-supplemented media to inhibit proliferation of non-neuronal cells. Neurons were transfected 8 h after plating using the calcium-phosphate method with indicated plasmids together with a GFP expression plasmid to visualize transfected neurons. To rule out the possibility that the effects of RNAi or protein expression on axonal length were caused by any effect of these manipulations on cell survival, the anti-apoptotic protein Bcl-xL was coexpressed in all our experiments. The expression of Bcl-xL itself has little or no effects on axonal length (Konishi et al., 2004). Neurons were fixed in paraformaldehyde after 3 d in vitro and subjected to immunocytochemistry using a GFP antibody (Invitrogen).
Axon growth assay and morphometry.
Axonal growth and morphometry was done as described previously (Konishi et al., 2004; Stegmuller et al., 2006). Briefly, images of transfected GFP-positive neurons were captured in a blinded manner using a Nikon (Tokyo, Japan) Eclipse TE2000 epifluorescence microscope. Axonal growth was analyzed by measuring the length of axons using SPOT software.
Western blot and subcellular fractionation analyses.
Granule neurons were harvested at indicated days in vitro (DIV), and lysates were analyzed by SDS-PAGE followed by Western blotting using the Smad2/3 (BD, Franklin Lakes, NJ), Smad2 (Abcam, Cambridge, MA), phosphoS465/467-Smad2 (EMD Biosciences, San Diego, CA), SnoN (Santa Cruz Biotechnology, Santa Cruz, CA), or 14-3-3β antibody (Santa Cruz Biotechnology). For subcellular fractionation, granule neurons were scraped into detergent-free buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, protease inhibitors) and mechanically disrupted using a 2 ml Dounce. Nuclei were spun down [500 × g, 4°C, Eppendorf (Hamburg, Germany) table centrifuge], and supernatant was collected as the cytoplasmic fraction. Nuclei were subjected to one wash in 0.1% NP40 supplemented buffer A. Nuclei were then lysed in buffer B (20 mm HEPES, pH 7.9, 400 mm NaCl, 1 mm EDTA, 1 mm EGTA, protease inhibitors) and pelleted (maximum speed, 4°C, Eppendorf table centrifuge). Supernatant was collected as the nuclear fraction.
In vivo electroporation. In vivo electroporation was performed as described previously (Stegmuller et al., 2006).
Isolation of myelin.
Myelin was prepared as described previously (Norton and Poduslo, 1973). Briefly, brains from Long–Evans adult rats were homogenized in 10.5% sucrose and subjected to a series of 10.5%/30% sucrose gradient ultracentrifugation steps alternating with nongradient ultracentrifugations. Purified myelin was resuspended in water, and integrity of myelin proteins was determined by SDS-PAGE followed by Coomassie staining. Glass coverslips, precoated with polyornithine, were coated overnight at 4°C with 13.3 μg/ml myelin diluted in PBS.
Results
To investigate the mechanistic basis of SnoN function in axonal growth in the mammalian brain, we characterized the role of SnoN-interacting proteins in neurons. In dividing cells, SnoN associates with the transcription factors Smad2 and Smad3 and thus plays a critical role in the TGFβ-Smad signaling pathway (Sun et al., 1999; Liu et al., 2001). We therefore asked whether the Smad proteins play a role in the regulation of axonal growth.
We first characterized the expression of Smad2 and Smad3 in granule neurons of the developing rat cerebellar cortex. Immunoblotting with a widely used antibody that recognizes both Smad2 and Smad3 revealed expression of Smad2/3 as a doublet in cerebellar granule neurons (Fig. 1A). Smad2/3 was also found to be expressed in cerebral cortical and hippocampal neurons, suggesting that these proteins are widely expressed in the brain (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Smad2 has a slightly slower mobility than Smad3 on SDS-PAGE (De Bosscher et al., 2004; Seay et al., 2005). We confirmed the identity of the lower mobility Smad2/3-immunoreactive band as Smad2 using RNAi specifically targeting Smad2 (see Fig. 5C, lanes 1, 3). When Smad2 and Smad3 were allowed to separate further by SDS-PAGE, we found that Smad2 is expressed at higher levels than Smad3 in granule neurons (Fig. 1B).
Figure 1.

Smad2/3 is expressed in the cerebellum and interacts with SnoN in neurons. A, Cerebellar granule neurons were lysed at the indicated DIV after plating and immunoblotted using an antibody that recognizes Smad2 and Smad3 and an antibody to 14-3-3, the latter to serve as loading control. B, Cerebellar granule neuron (CGN) lysates were separated by SDS-PAGE for an extended period of time and subjected to immunoblotting using the Smad2/3 antibody to distinguish Smad2 and Smad3. C, Cultured granule neurons (DIV2) were subjected to immunofluorescence analysis using the Smad2/3 antibody (top) and the DNA dye bisbenzimide (bottom). D, Granule neurons were subjected to subcellular fractionation. Nuclear (N) and cytoplasmic (C) fractions were immunoblotted using the Smad2/3, SnoN, and 14-3-3 antibodies. The asterisk indicates nonspecific band. A large portion of Smad2/3 is present in the nucleus. E, F, Granule neuron lysates were subjected to immunoprecipitation (IP) using the hemagglutinin control antibody and the SnoN antibody followed by immunoblotting with the Smad2/3 antibody (E) or reciprocal coimmunoprecipitation (F). Double asterisks indicate heavy chain IgGs. Endogenous SnoN and Smad2/3 form a physical complex in neurons.
Figure 5.
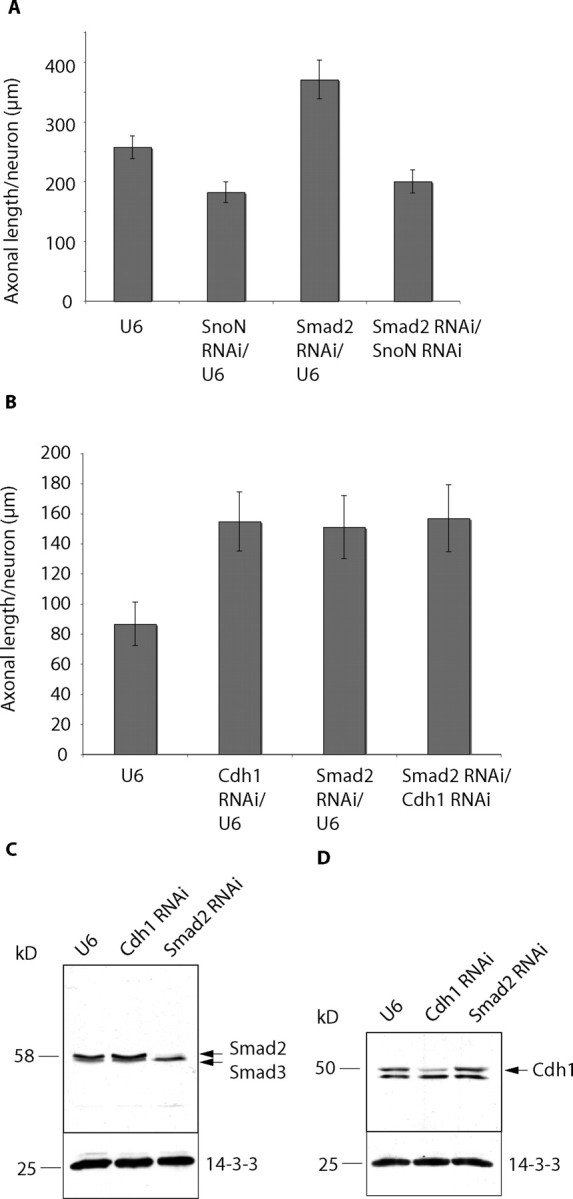
Smad2 acts in the Cdh1-APC/SnoN pathway of axonal morphogenesis. A, Neurons transfected with the control U6, U6/smad2, or U6/snon RNAi plasmid or both U6/smad2 and U6/snon plasmids were maintained in media supplemented with 10% calf serum and membrane-depolarizing concentrations of KCl and analyzed as in Figure 3B. Although Smad2 RNAi increased and SnoN RNA; reduced axonal length, simultaneous knock-down of Smad2 and SnoN reduced axonal growth (ANOVA, p = 0.0633). A total of 438 neurons were measured. B, Neurons transfected with the control U6, U6/cdh1, or U6/smad2 RNAi plasmid or both U6/cdh1 and U6/smad2 plasmids were placed in media supplemented with insulin and analyzed as in Figure 3B. Knock-down of Cdh1 and Smad2 individually increased axonal length compared with control neurons (ANOVA, p < 0.001), but no additive effect was detected after simultaneous knock-down of Cdh1 and Smad2. A total of 429 neurons were measured. C, D, Granule neurons were transfected using an electroporation-based nucleofection method with the U6 control, U6/cdh1 RNAi, or U6/smad2 RNAi plasmid. Lysates were analyzed 5 d later by immunoblotting using the Smad2/3 (C), Cdh1 (D), or 14-3-3 antibody. 14-3-3 served as loading control. The Smad2 RNAi and Cdh1 RNAi plasmids led to specific knock-down of endogenous Smad2 and Cdh1, respectively, in granule neurons.
To determine whether Smad2 acts in concert with SnoN to regulate axonal growth and whether that is linked to the function of SnoN as a key target of Cdh1-APC in the control of axonal growth, we first tested whether SnoN interacts with Smad2 in neurons. Both immunofluorescence and fractionation studies showed that a large portion of Smad2/3 is localized in the nucleus, where SnoN resides (Fig. 1C,D). In coimmunoprecipitation analyses, in which we immunoprecipitated SnoN followed by Smad2/3 immunoblotting, endogenous Smad2 and Smad3 associated with endogenous SnoN (Fig. 1E). The reciprocal approach of immunoprecipitating Smad2/3 followed by SnoN immunoblotting confirmed the interaction (Fig. 1F). Together, these results suggest that endogenous SnoN forms a physical complex with endogenous Smad2 and Smad3 in neurons.
Because Smad2 was the predominantly expressed SnoN-associated protein in granule neurons, we focused our investigations on the role of Smad2 in neurons. Using an antibody that specifically recognizes Smad2 but not Smad3, we characterized the expression of Smad2 in the developing cerebellar cortex. We found high expression of Smad2 in granule neurons of the internal granule layer at P6 and P8, a decrease in expression at P13, and very low levels at P30 (Fig. 2). Smad2 expression was also found in the external granular layer and in Purkinje cells. Thus, Smad2 is expressed in the first two postnatal weeks, overlapping with SnoN expression and during the peak period of granule neuron axonal morphogenesis in the cerebellar cortex (Stegmuller et al., 2006).
Figure 2.
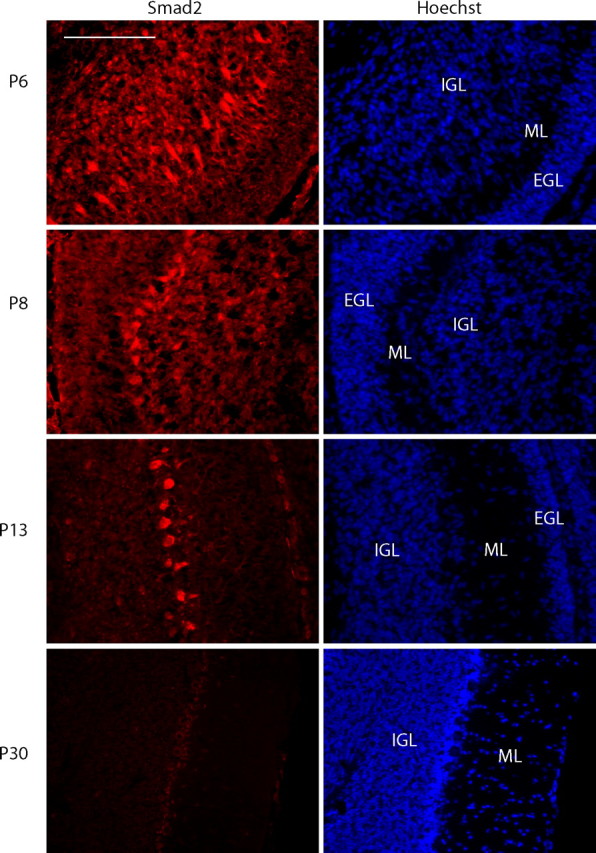
Smad2 expression in the developing cerebellar cortex. Sagittal cerebellar sections from P6, P8, P13, and P30 were subjected to immunohistochemistry using an antibody that specifically recognizes Smad2. Scale bar, 100 μm. EGL, External granular layer; ML, molecular layer; IGL, internal granular layer. Smad2 is expressed in the postnatal cerebellum.
We next assessed Smad2 function in the regulation of axonal growth. We used a plasmid-based method of RNAi to acutely knock-down Smad2 in cerebellar granule neurons (Gaudilliere et al., 2002). Primary granule neurons were transfected with the U6 or U6/smad2 plasmid together with a GFP expression plasmid, and 3 d after transfection, axonal length of transfected GFP-positive neurons was measured. We found that axonal length was robustly increased in Smad2 knock-down neurons compared with control U6-transfected neurons (Fig. 3A). Quantitation of these results revealed that Smad2 RNAi increased axonal length by nearly 50% (Fig. 3B). Total axonal length was also significantly increased after Smad2 RNAi (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). To further characterize the role of Smad2 in axonal morphogenesis, we asked whether Smad2 RNAi leads to a difference in axonal branching. We counted axonal protrusions (>15 μm), determined the number of protrusions per axon, and normalized to either axonal or total axonal length to calculate the axonal branching index. We did not find a difference in axonal branching index between control U6 transfected neurons and Smad2 RNAi neurons (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Collectively, these results suggest that Smad2 specifically inhibits the growth of the major axonal fiber in granule neurons.
Figure 3.

Smad2 suppresses axonal growth in cerebellar granule neurons. A, Granule neurons were transfected 8 h after plating with the Smad2 RNAi or control U6 plasmid together with the GFP expression plasmid and maintained in media supplemented with insulin. Three days after transfection, neurons were fixed and subjected to immunocytochemistry using an antibody to GFP. Images of control U6-transfected and U6/smad2-transfected neurons are shown, with asterisks indicating cell bodies and arrows indicating axons. Scale bar, 100 μm. Smad2 knock-down neurons had longer axons than control U6-transfected neurons. B, Axonal length was measured in GFP-positive transfected neurons using SPOT software. Axonal length was significantly longer in Smad2 knock-down neurons compared with control U6-transfected neurons (t test, p < 0.0001). A total of 268 neurons were measured. C, 293T cells were transfected with the Smad2 RNAi plasmid or control U6 plasmid together with Smad2-WT or Smad2-Rescue. Lysates were immunoblotted using the Smad2/3 and 14-3-3 antibodies, the latter to serve as loading control. Smad2 RNAi led to the knock-down of Smad2-WT, but failed to induce knock-down of Smad2-Rescue. D, Granule neurons transfected with the Smad2 RNAi or control U6 plasmid together with pcDNA3 vector, Smad2-WT, or Smad2-Rescue expression plasmid were analyzed as in Figure 2B. Whereas expression of Smad2-WT did not affect Smad2 RNAi-mediated increase in axonal length, expression of Smad2-Rescue reversed the Smad2 RNAi phenotype (ANOVA, p < 0.0001). A total of 297 neurons were measured.
To determine whether the Smad2 RNAi-induced phenotype is attributable to specific knock-down of Smad2 and not to off-target effects of RNAi, we performed a rescue experiment. We designed a Smad2 rescue (Smad2-Rescue) expression construct that harbors multiple silent mutations in the targeting region. Whereas Smad2 RNAi induced the efficient knock-down of Smad2 encoded by wild-type cDNA (Smad2-WT), Smad2 RNAi failed to induce knock-down of Smad2-Rescue (Fig. 3C). We next determined the ability of Smad2-Rescue to reverse Smad2-mediated enhancement of axonal growth. We transfected granule neurons with the control U6 or the Smad2 RNAi plasmid together with Smad2 WT or Smad2-Rescue expression plasmid. Expression of Smad2-WT had little effect on the ability of Smad2 RNAi to increase axonal length (Fig. 3D). In contrast, expression of Smad2-Rescue significantly reduced axonal length in the background of Smad2 RNAi. These findings indicate that Smad2 RNAi enhancement of axonal growth is the result of specific knock-down of Smad2 in neurons.
In other experiments, we found that Smad2 RNAi had little or no effect on survival of granule neurons (data not shown). In addition, both control and Smad2 knock-down neurons expressed markers of differentiated postmitotic granule neurons, including the neuron-specific class III β-tubulin (data not shown), MEF2 (myocyte enhancer factor 2), and the dendrite-specific protein MAP2 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), suggesting that Smad2 is not required for general aspects of granule neuron differentiation.
To investigate the role of Smad2 in the developing cerebellar cortex in vivo, we performed an in vivo electroporation assay to acutely knock down Smad2 in the postnatal rat cerebellum. We electroporated P3 rat pups with the control plasmid (U6-cmvGFP) or an RNAi plasmid encoding Smad2 hairpin RNAs and GFP bicistronically (U6/smad2-cmvGFP) (Fig. 4A) and analyzed coronal cerebellar sections at P8. We were unable to determine whether Smad2 knock-down stimulates axonal growth in vivo, because measurement of axonal length is not technically feasible in the cerebellar cortex. We therefore assessed the effect of Smad2 knock-down on the patterning of granule neuron parallel fibers. We found that Smad2 knock-down had little or no effect on parallel fiber patterning (Fig. 4B). These data support the view that Smad2 specifically controls the growth but not patterning of axons.
Figure 4.
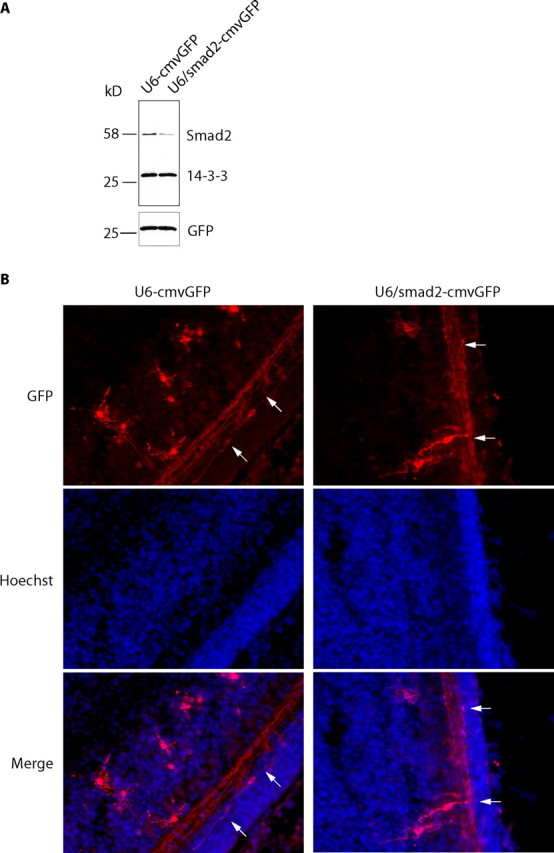
In vivo Smad2 knock-down does not appear to affect parallel fiber patterning in the cerebellar cortex. A, Lysates of 293T cells transfected with the control U6-cmvGFP plasmid and the U6/smad2-cmvGFP RNAi plasmid were immunoblotted with the Smad2, 14-3-3, or GFP antibody. A bicistronic plasmid encoding a Smad2 hairpin RNA and GFP efficiently knocks down endogenous Smad2. B, The control U6-cmvGFP plasmid or U6/smad2-cmvGFP RNAi plasmid together with a Bcl-Xl expression plasmid were injected into the cerebella of P3 rat pups. Pups were subjected to electroporation and killed 5 d later. Coronal cerebellar sections were analyzed by immunohistochemistry using a GFP antibody. Arrows indicate parallel fibers. No difference was observed in parallel fiber patterning.
The interaction of Smad2 with SnoN plays a dual role in TGFβ signaling, serving to modulate Smad2-dependent transcription or alternatively to allow the Smad2-recruited ubiquitin ligase Cdh1-APC to target SnoN for ubiquitination and consequent degradation (Stroschein et al., 1999, 2001; Wan et al., 2001; He et al., 2003; Sarker et al., 2005; Hsu et al., 2006). Because of the dual function of the Smad2/SnoN interaction, we characterized the functional relationship of Smad2 and SnoN in neurons using epistasis analyses. We compared the effect of the simultaneous knock-down of both SnoN and Smad2 to the effect of knock-down of SnoN or Smad2 alone on axonal length in granule neurons. We found that whereas Smad2 RNAi enhanced and SnoN RNAi reduced axonal length, simultaneous knock-down of Smad2 and SnoN led to the appearance of short axons, resembling the SnoN knock-down phenotype (Fig. 5A). Thus, SnoN knock-down suppressed the ability of Smad2 RNAi to stimulate axonal growth. These results suggest that Smad2 acts upstream of SnoN in the control of axonal morphogenesis in neurons.
Because the ubiquitin ligase Cdh1-APC also acts upstream of SnoN in neurons and because of Smad2's function in recruiting Cdh1-APC to its substrate SnoN (Stroschein et al., 2001; Wan et al., 2001; Stegmuller et al., 2006), we next determined the role of Smad2 in the Cdh1-APC pathway of axonal growth. We measured the effect of Smad2 RNAi or Cdh1 RNAi alone or together on axonal length in granule neurons. Axonal length was increased in Cdh1 knock-down as well as Smad2 knock-down neurons (Fig. 5B). Simultaneous knock-down of both Cdh1 and Smad2 in granule neurons did not result in additive effects of axonal length when compared with individual knock-down of Cdh1 and Smad2 (Fig. 5B). In control experiments, we asked whether knock-down of Smad2 or Cdh1 affect Cdh1 and Smad2 expression, respectively. We used a nucleofection method to transfect granule neurons with the control U6, Smad2 RNAi, or Cdh1 RNAi plasmid and analyzed lysates of transfected neurons by immunoblotting. Endogenous Smad2 was specifically reduced in Smad2 RNAi neurons but not in control neurons or Cdh1 RNAi neurons (Fig. 5C). Endogenous Cdh1 expression was specifically reduced in Cdh1 RNAi neurons, but not in control or Smad2 RNAi neurons (Fig. 5D). These data indicate that knock-down of Cdh1 and Smad2 does not reduce Smad2 and Cdh1 expression, respectively. Together, our results suggest that Cdh1 and Smad2 operate in a shared pathway, whereby Smad2 and Cdh1-APC both act upstream of SnoN in neurons to regulate axonal growth.
Identification of a novel function for Smad2 as an inhibitor of axon growth raised the question of the role of the TGFβ-regulated Smad signaling pathway in the control of axonal development. We first monitored Smad2 activation in granule neurons after exposure to TGFβ1. In cycling cells, TGFβ ligand binding triggers the phosphorylation of Smad2 at the key regulatory sites serines 465 and 467, leading to the consequent turnover of SnoN (Stroschein et al., 2001; Wan et al., 2001). Surprisingly, we found that Smad2 was basally phosphorylated at serines 465 and 467 in neurons, and exposure of neurons to TGFβ1 did not further stimulate the Smad2 phosphorylation (data not shown). Consistent with these results, in subcellular fractionation analyses, Smad2 phosphorylated at serines 465 and 467 was localized in the nucleus (Fig. 6A). These results suggest that Smad signaling is constitutively active in neurons. Corroborating our findings are results in transgenic mice engineered to express a TGFβ-induced Smad-responsive luciferase reporter demonstrating that high basal activity of TGFβ signaling is present in the brain (Luo et al., 2006).
Figure 6.
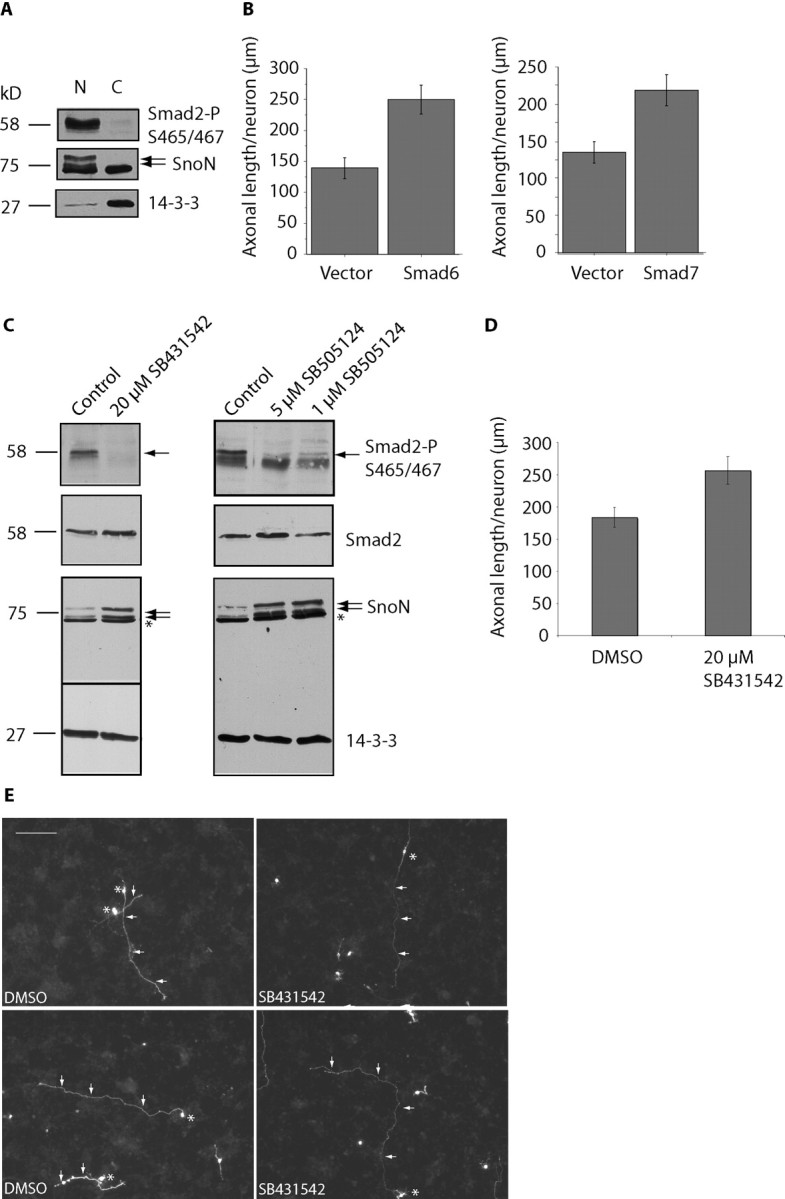
TGFβ-Smad signaling suppresses axonal growth. A, Granule neurons were subjected to subcellular fractionation and analyzed with immunoblotting using an antibody that recognizes Smad2 specifically when phosphorylated at serines 465 and 467 as well as the SnoN and 14-3-3 antibodies. Phosphorylated Smad2 is localized in the nucleus in granule neurons. B, Neurons transfected with the control pCMV5 vector, Smad6, or Smad7 expression plasmids were placed in media supplemented with 10% calf serum and analyzed as in Figure 3B. Expression of Smad6 or Smad7 significantly increased axonal length (t test, p < 0.001). A total of 247 and 305 neurons were measured, respectively. C, Lysates of granule neurons exposed to SB431542 and SB505124 at the indicated concentrations for 48 h were immunoblotted with the SnoN, phosphoS465/467-Smad2, Smad2/3, and 14-3-3 antibodies. Exposure of neurons to small molecule inhibitors of TGFβ receptors reduced Smad2 phosphorylation and increased SnoN levels in neurons. Asterisks indicate nonspecific band. D, Granule neurons transfected with the GFP expression plasmid at DIV0 and placed in media supplemented with 10% calf serum. Neurons were treated with SB431542 or its vehicle (DMSO) starting at DIV1 for 48 h and analyzed as in Figure 3B. Treatment with SB431542 significantly increased axonal length (t test, p < 0.01). A total of 170 neurons were measured. E, Images of control vehicle- and SB431542-treated neurons are shown, with asterisks indicating cell bodies and arrows indicating axons. Scale bar, 100 μm. Neurons that were treated with SB431542 have longer axons than vehicle-treated neurons.
Having identified a high basal level of Smad2 activation in neurons, we next determined the effect of manipulation of endogenous TGFβ-Smad signaling in neurons. The proteins Smad6 and Smad7 inhibit the Smad signaling pathway primarily at the level of TGFβ receptors (Hayashi et al., 1997; Imamura et al., 1997). We tested the effect of Smad6 or Smad7 on axonal growth in granule neurons. We found that expression of Smad6 or Smad7 in neurons robustly increased axonal length (Fig. 6B). These results suggest that inhibition of Smad signaling stimulates axonal growth.
We also characterized the biochemical and biological response of neurons to small molecule inhibitors of TGFβ receptors, SB431542 and SB505124 (Inman et al., 2002; DaCosta Byfield et al., 2004). Exposure of granule neurons to both small molecule inhibitors led to loss of Smad2 phosphorylation at serines 465 and 467 (Fig. 6C). These results suggest that small molecule inhibitors of TGFβ receptors inactivate Smad signaling in neurons. Together with the reduction in Smad2 phosphorylation, SnoN levels were significantly increased in neurons after exposure to SB431542 or SB505124 (Fig. 6C). These results suggest that activation of Smad2 signaling promotes SnoN turnover, consistent with the model that activated Smad2 recruits SnoN to Cdh1-APC, leading to the subsequent degradation of SnoN (Stroschein et al., 2001).
To test the idea that TGFβ-Smad signaling inhibits axonal growth, we measured axon length in neurons exposed to the small molecule inhibitor SB431542. Neurons were transfected 8 h after plating and treated for 48 h with 20 μm of the inhibitor or the vehicle (DMSO). We found that axonal length was significantly increased after exposure to SB431542 (Fig. 6D,E). Together, our findings suggest that constitutive TGFβ-Smad signaling suppresses axon growth in neurons.
The identification of a novel function for the Smad pathway in the control of axonal growth in granule neurons of the developing rat cerebellum led us next to consider the possibility that this pathway might also contribute to the intrinsic inability of neurons to grow axons under conditions that axons encounter after injury in the nervous system. Myelin proteins are thought to play an important role in the inhibition of axonal growth in the injured CNS (Caroni and Schwab, 1988; Filbin, 2003). Inhibition of Cdh1-APC overrides the ability of myelin to suppress axonal growth (Konishi et al., 2004). Because we found in this study that the TGFβ-Smad2 pathway acts together with Cdh1-APC to inhibit axonal growth, we asked whether Smad2 knock-down might also stimulate axon growth after myelin inhibition. We plated granule neurons on myelin or control substrate and transfected these neurons with the Smad2 RNAi or control U6 plasmid together with the GFP expression plasmid. As expected, myelin significantly inhibited axonal growth (Fig. 7A,B). Strikingly, Smad2 knock-down robustly increased axonal length compared with control-transfected neurons on myelin (Fig. 7A,B). These results suggest that inhibition of Smad2 overcomes myelin inhibition of axonal growth. Collectively, our data support the conclusion that Smad signaling engages the Cdh1-APC/SnoN pathway to limit the intrinsic ability of neurons to extend axons. By regulating the Cdh1-APC/SnoN pathway, Smad signaling may play an important role in the failure of CNS neurons to regenerate. Thus, inhibition of Smad signaling may offer novel approaches in the treatment of CNS injury and disease.
Figure 7.

Smad2 knock-down overrides myelin inhibition of axonal growth. A, Images of neurons plated on polyornithine- or myelin-coated coverslips (13.3 μg/ml) and transfected with the control U6 or U6/smad2 RNAi plasmid were analyzed at DIV3 as in Figure 3A. Asterisks and arrows indicate cell bodies and axons, respectively. Scale bar, 100 μm. B, Quantitation of neurons in A analyzed as in Figure 3B. Axonal length is significantly reduced in control U6-transfected neurons on myelin compared with polyornithine substrate (ANOVA, p < 0.001). Axonal length is significantly increased in Smad2 knock-down neurons compared with control U6-transfected neurons on myelin (ANOVA, p < 0.0005). A total 130 of neurons were measured.
Discussion
In this study, we have uncovered a novel function for the Smad signaling pathway in control of axonal morphogenesis. Genetic knock-down of Smad2 by RNAi in primary neurons stimulated axonal growth. Likewise, expression of the inhibitory Smad protein Smad6 or Smad7 significantly increased axonal length. We also found that Smad signaling influences the growth of axons by regulating the Cdh1-APC/SnoN cell-intrinsic pathway of axonal morphogenesis. Endogenous SnoN associates with endogenous Smad2 in neurons. Epistasis analyses revealed that Smad2 acts together with the ubiquitin ligase Cdh1-APC upstream of the transcriptional modulator SnoN to control axonal growth. Exposure of neurons to small molecule inhibitors of TGFβ receptors inactivated Smad2 phosphorylation and concomitantly increased the levels of SnoN protein. Consistent with these results, the small molecule inhibitors stimulated the growth of axons in granule neurons. Importantly, we also found that genetic knock-down of Smad2 overrides the ability of adult rat brain myelin to suppress axonal growth. Collectively, our findings suggest that the TGFβ-Smad signaling pathway plays a critical role in regulating axonal development and may harbor promising targets for drug discovery aimed at enhancing recovery in the nervous system after injury or disease.
Identification of a novel function for the Smad proteins in the control of axonal growth in mammalian CNS neurons bears several significant ramifications. This finding extends our understanding of the Cdh1-APC/SnoN pathway as a pivotal cell-intrinsic regulator of axonal morphogenesis. Both biochemical and genetic lines of evidence in our study support the conclusion that in neurons Smad2 acts together with Cdh1-APC upstream of SnoN in the control of axonal growth. These findings suggest that TGFβ-Smad signaling plays a key role in regulating the Cdh1-APC/SnoN cell-intrinsic pathway in neurons.
Our results support the model that Smad2 facilitates the ability of Cdh1-APC to regulate SnoN turnover in neurons, suggesting that Smad2 may act upstream of SnoN in the control of axonal growth. Our findings raise the intriguing possibility that SnoN might regulate the function of other transcription factors in neurons that remain to be identified. It will be important to explore this question in future investigations. Clues may emerge from identification of the program of gene expression that is controlled by SnoN in neurons.
Aside from SnoN, the SCF scaffold protein Skp2 and the signaling protein HEF1 have been identified as substrates of Cdh1-APC that are targeted for degradation by the proteasome in response to TGFβ signaling in dividing cells (Bashir et al., 2004; Nourry et al., 2004; Wei et al., 2004; Liu et al., 2007). Like SnoN, the degradation of HEF1 and Skp2 is enhanced in the presence of activated Smad3 (Bashir et al., 2004; Nourry et al., 2004; Wei et al., 2004; Liu et al., 2007). Collectively, these observations raise the interesting question of whether Skp2 and HEF1 also function downstream of Smad signaling and the Cdh1-APC/SnoN pathway in controlling axonal growth in CNS neurons.
Consistent with the role of Smad signaling in regulating the Cdh1-APC/SnoN pathway in neurons, we found that inhibition of Smad signaling endows neurons with the ability to grow axons on the substrate myelin. Because myelin is thought to play an important role in inhibiting axonal regeneration in the injured CNS (Caroni and Schwab, 1988; Filbin, 2003), modulation of the TGFβ-Smad2 and Cdh1-APC/SnoN pathway may have implications in brain injury. Interestingly, components of this pathway that inhibit axonal growth, including Cdh1 and Smad2/3, are expressed in the adult brain (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (Gieffers et al., 1999). In contrast, the axon growth-promoting protein SnoN is dramatically downregulated in the adult brain, which correlates with the inability of the CNS to regenerate (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). It remains to be determined whether Cdh1-APC activity leads to increased SnoN turnover in the adult brain. Together, our observations suggest that components of TGFβ-Smad signaling as well as the Cdh1-APC/SnoN pathway represent potential targets in the development of drugs that should stimulate axonal regeneration after injury and disease in the CNS.
TGFβ signaling contributes to CNS development, and its role is increasingly appreciated in neurological diseases (Buisson et al., 2003; Gomes et al., 2005; Tesseur and Wyss-Coray, 2006). TGFβs are thought to have prosurvival effects in distinct populations of neurons (Unsicker and Krieglstein, 2002; Brionne et al., 2003). The TGFβ related bone morphogenetic proteins (BMPs) also contribute to CNS development. In the cerebellum, BMPs control the early stages of granule neuron development. In particular, BMPs control the number, proliferation, and differentiation of granule neuron precursors (Angley et al., 2003; Rios et al., 2004; Krizhanovsky and Ben-Arie, 2006; Qin et al., 2006; Machold et al., 2007).
Mounting evidence implicates an important neuroprotective role of TGFβs in Alzheimer's disease and ischemia (Buisson et al., 2003; Tesseur and Wyss-Coray, 2006). Thus, TGFβs may have beneficial impact on neurons after brain injury. Among the TGFβ superfamily, TGFβ1, TGFβ2, and their receptors are upregulated at the site of injury (McTigue et al., 2000; Lagord et al., 2002). Our study suggests that TGFβ signaling engages an intrinsic mechanism of axon growth inhibition. Thus, in the setting of injury, although TGFβ signaling may have prosurvival effects on neurons, it might simultaneously counteract axon regeneration by suppressing intrinsic programs of axonal growth. It will be important in future studies to identify the particular TGFβ ligand and cognate receptor that operate in an autocrine or paracrine manner to inhibit axonal growth in neurons. In addition, elucidation of the TGFβ-regulated mechanisms that protect neurons as well as those that inhibit axonal growth will be important in devising strategies that both protect neurons and stimulate axonal regeneration in the CNS.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS051255 (A.B.), the Harvard Medical School-Merck Sponsored Research Program, the Christopher Reeve Foundation, a Charles A. King Trust (Bank of America, Cotrustee) Postdoctoral Fellowship (J.S.), and NIH Training Grant GM077226 (M.A.H.). We thank Shirin Bonni for helpful discussions and critical reading of this manuscript.
References
- Angley C, Kumar M, Dinsio KJ, Hall AK, Siegel RE. Signaling by bone morphogenetic proteins and Smad1 modulates the postnatal differentiation of cerebellar cells. J Neurosci. 2003;23:260–268. doi: 10.1523/JNEUROSCI.23-01-00260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone beta subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, Wrana JL. TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587–595. doi: 10.1038/35078562. [DOI] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Buisson A, Lesne S, Docagne F, Ali C, Nicole O, MacKenzie ET, Vivien D. Transforming growth factor-beta and ischemic brain injury. Cell Mol Neurobiol. 2003;23:539–550. doi: 10.1023/A:1025072013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- Charron F, Tessier-Lavigne M. Novel brain wiring functions for classical morphogens: a role as graded positional cues in axon guidance. Development. 2005;132:2251–2262. doi: 10.1242/dev.01830. [DOI] [PubMed] [Google Scholar]
- Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- DaCosta Byfield S, Major C, Laping NJ, Roberts AB. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2004;65:744–752. doi: 10.1124/mol.65.3.744. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Hill CS, Nicolas FJ. Molecular and functional consequences of Smad4 C-terminal missense mutations in colorectal tumour cells. Biochem J. 2004;379:209–216. doi: 10.1042/BJ20031886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci USA. 1999;96:11317–11322. doi: 10.1073/pnas.96.20.11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL. Intrinsic neuronal regulation of axon and dendrite growth. Curr Opin Neurobiol. 2004;14:551–557. doi: 10.1016/j.conb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Gomes FC, Sousa Vde O, Romao L. Emerging roles for TGF-beta1 in nervous system development. Int J Dev Neurosci. 2005;23:413–424. doi: 10.1016/j.ijdevneu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Jr, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- He J, Tegen SB, Krawitz AR, Martin GS, Luo K. The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J Biol Chem. 2003;278:30540–30547. doi: 10.1074/jbc.M304016200. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Sarker KP, Pot I, Chan A, Netherton SJ, Bonni S. Sumoylated SnoN represses transcription in a promoter-specific manner. J Biol Chem. 2006;281:33008–33018. doi: 10.1074/jbc.M604380200. [DOI] [PubMed] [Google Scholar]
- Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Kim AH, Bonni A. Thinking within the D box: initial identification of Cdh1-APC substrates in the nervous system. Mol Cell Neurosci. 2007;34:281–287. doi: 10.1016/j.mcn.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Ben-Arie N. A novel role for the choroid plexus in BMP-mediated inhibition of differentiation of cerebellar neural progenitors. Mech Dev. 2006;123:67–75. doi: 10.1016/j.mod.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–290. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Lagord C, Berry M, Logan A. Expression of TGFbeta2 but not TGFbeta1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol Cell Neurosci. 2002;20:69–92. doi: 10.1006/mcne.2002.1121. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Stegmuller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, de la Torre-Ubieta L, Pagano M, Bonni A, Iavarone A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442:471–474. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- Liu W, Wu G, Li W, Lobur D, Wan Y. Cdh1-anaphase-promoting complex targets Skp2 for destruction in transforming growth factor beta-induced growth inhibition. Mol Cell Biol. 2007;27:2967–2979. doi: 10.1128/MCB.01830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sun Y, Weinberg RA, Lodish HF. Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev. 2001;12:1–8. doi: 10.1016/s1359-6101(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Luo J, Lin AH, Masliah E, Wyss-Coray T. Bioluminescence imaging of Smad signaling in living mice shows correlation with excitotoxic neurodegeneration. Proc Natl Acad Sci USA. 2006;103:18326–18331. doi: 10.1073/pnas.0605077103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold RP, Kittell DJ, Fishell GJ. Antagonism between Notch and bone morphogenetic protein receptor signaling regulates neurogenesis in the cerebellar rhombic lip. Neural Develop. 2007;2:5. doi: 10.1186/1749-8104-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Popovich PG, Morgan TE, Stokes BT. Localization of transforming growth factor-beta1 and receptor mRNA after experimental spinal cord injury. Exp Neurol. 2000;163:220–230. doi: 10.1006/exnr.2000.7372. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Nourry C, Maksumova L, Pang M, Liu X, Wang T. Direct interaction between Smad3, APC10, CDH1 and HEF1 in proteasomal degradation of HEF1. BMC Cell Biol. 2004;5:20. doi: 10.1186/1471-2121-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Ince-Dunn G, Ghosh A. Transcriptional regulation of vertebrate axon guidance and synapse formation. Nat Rev Neurosci. 2007;8:331–340. doi: 10.1038/nrn2118. [DOI] [PubMed] [Google Scholar]
- Qin L, Wine-Lee L, Ahn KJ, Crenshaw EB., III Genetic analyses demonstrate that bone morphogenetic protein signaling is required for embryonic cerebellar development. J Neurosci. 2006;26:1896–1905. doi: 10.1523/JNEUROSCI.3202-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios I, Alvarez-Rodriguez R, Marti E, Pons S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development. 2004;131:3159–3168. doi: 10.1242/dev.01188. [DOI] [PubMed] [Google Scholar]
- Sarker KP, Wilson SM, Bonni S. SnoN is a cell type-specific mediator of transforming growth factor-beta responses. J Biol Chem. 2005;280:13037–13046. doi: 10.1074/jbc.M409367200. [DOI] [PubMed] [Google Scholar]
- Seay U, Sedding D, Krick S, Hecker M, Seeger W, Eickelberg O. Transforming growth factor-beta-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J Pharmacol Exp Ther. 2005;315:1005–1012. doi: 10.1124/jpet.105.091249. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stegmuller J, Bonni A. Moving past proliferation: new roles for Cdh1-APC in postmitotic neurons. Trends Neurosci. 2005;28:596–601. doi: 10.1016/j.tins.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Stegmuller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science. 1999;286:771–774. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- Stroschein SL, Bonni S, Wrana JL, Luo K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001;15:2822–2836. doi: 10.1101/gad.912901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Liu X, Ng-Eaton E, Lodish HF, Weinberg RA. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor beta signaling. Proc Natl Acad Sci USA. 1999;96:12442–12447. doi: 10.1073/pnas.96.22.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Wyss-Coray T. A role for TGF-beta signaling in neurodegeneration: evidence from genetically engineered models. Curr Alzheimer Res. 2006;3:505–513. doi: 10.2174/156720506779025297. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Krieglstein K. TGF-betas and their roles in the regulation of neuron survival. Adv Exp Med Biol. 2002;513:353–374. doi: 10.1007/978-1-4615-0123-7_13. [DOI] [PubMed] [Google Scholar]
- Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-beta signaling by targeting SnoN for destruction. Mol Cell. 2001;8:1027–1039. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]