

Abstract
Ca2+ influx through NMDA receptors (NMDARs) leads to channel inactivation, which limits Ca2+ entry and protects against excitotoxicity. Extensive functional data suggests that this Ca2+-dependent inactivation (CDI) requires both calmodulin (CaM) binding to the C0 cassette of the NR1 subunit's C terminus (CT) and regulation by α-actinin-2, but a molecular understanding of CDI has been elusive. Here we used a number of methods to analyze the molecular nature of the interaction among CaM, α-actinin-2, and the NR1 CT. We found that a single CaM binds to two NR1 CTs in a Ca2+-dependent manner and promotes their reversible “dimerization.” Expressed NMDARs containing NR1 concatamers in which the NR1 C termini are “uncoupled” display markedly reduced CDI. In contrast to current models, α-actinin-2 does not bind to the NR1 CT. We propose a new model for CDI in which the noncanonical Ca2+/CaM-dependent dimerization of the two NR1 subunits inactivates the channel by propagating a conformational change from the short NR1 CT to the nearby channel pore.
Keywords: calcium, calmodulin, channel, glutamate, inactivation, NMDA receptors
Introduction
NMDA receptors (NMDARs) are key targets of glutamate, the major excitatory neurotransmitter in brain, and play a prominent role in neuronal development, synaptic plasticity, and the complex neuronal circuits that underlie learning and memory. These postsynaptic Ca2+-permeable, glutamate-gated, and voltage-dependent ion channels are heterotetramers containing two NR1 subunits and two NR2 subunits. The different members of the glutamate-binding NR2 subunits display specific agonist- and antagonist-binding properties and therefore provide molecular diversity (Waxman and Lynch, 2005). The NR1 subunits, which bind the coagonist glycine, add to the molecular diversity through alternative splicing in both their N and C termini. Three exons (C0, C1, and C2) encode the NR1 intracellular C terminus (CT) (see Fig. 1A). All NR1 subunits contain the invariant C0; the C1 and C2 cassettes are alternatively spliced in a developmentally regulated manner (Zukin and Bennett, 1995).
Figure 1.

CaM interacts with NR1 CT in Ca2+-dependent manner. A, Schematic of NR1 subunit and demarcation of the C0, C1, and C2 cassettes in the CT. Amino acid 880 (see text) is also identified. B, Coomassie-stained gel showing purification of 6xHis-tagged NR1 CT (amino acids 838–938) coexpressed with CaM and CaM mutants. Immunoblots with CaM and 6xHis antibodies are shown to confirm protein identification. C, Immunoblot showing CaM purification with a GST-NR1 CT construct in the presence of Ca2+ (1 mm) but not EGTA (5 mm). A Coomassie-stained gel shows that the GST fusion proteins were equally loaded. D, Anti-CaM immunoblot after GST pull down of GST-NR1 (amino acids 838–938) with added CaM in the presence of Ca2+ (1 mm) or EGTA (5 mm). For B–D, lane numbers are in the boxes for reference in the text.
Ca2+-dependent inactivation (CDI) of NMDARs provides important feedback inhibition of Ca2+ influx, preventing excessive Ca2+ entry that can lead to neurodegeneration and excitotoxicity (Coyle and Puttfarcken, 1993). Extensive functional data have clearly defined a role in CDI for calmodulin (CaM) (Ehlers et al., 1996; Rycroft and Gibb, 2004), the NR1 C0 cassette (Zhang et al., 1998), and the actin-binding protein α-actinin-2 (Krupp et al., 1999; Rycroft and Gibb, 2004). Addition of CaM to patches from hippocampal neurons reduced NMDAR single-channel mean open time and open probability; in contrast, addition of α-actinin decreased single-channel shut time and increased open probability (Rycroft and Gibb, 2004). Nevertheless, a precise molecular understanding of CDI is lacking. In heterologous expression systems, the NR1 C0 cassette is necessary for CDI (Zhang et al., 1998; Krupp et al., 1999). Two putative Ca2+/CaM-binding sites have been identified within the NR1 CT, an essential binding site that resides in C0 (Ehlers et al., 1996) and a higher-affinity binding site in C1 that is functionally redundant (Zhang et al., 1998; Krupp et al., 1999). α-Actinin-2 has been reported to bind, via its central rod domain, to the same site as CaM in the NR1 C0 domain (Krupp et al., 1999; Leonard et al., 2002). Ca2+/CaM binding to the NR1 CT is proposed to displace α-actinin-2, thereby leading to channel inactivation (Wyszynski et al., 1997; Zhang et al., 1998; Krupp et al., 1999; Leonard et al., 2002). Recently it was suggested that Ca2+-free CaM (apoCaM) is prebound to the NR1 C0 domain via the CaM C-terminal lobe (Akyol et al., 2004). If apoCaM is prebound to C0, however, α-actinin-2 binding to NR1 would be precluded and the displacement model inconsistent.
Here we used precise biochemical methods to show that a single CaM binds two NR1 CT in a noncanonical manner and drives a Ca2+-dependent dimerization of the NR1 CTs. In contrast to a recent report (Akyol et al., 2004), we found that apoCaM did not interact with NR1. Moreover, we demonstrated that α-actinin-2 did not interact with the NR1. Thus, our data suggest a major revision to the model of CDI and how Ca2+ influences NMDAR signaling events.
Materials and Methods
Molecular biology.
NR1 CT (amino acids 838–938), NR1 C0 (amino acids 834–863), NR1 C0–C1 (amino acids 834–900), NR1 (amino acids 834–880), α-actinin-2 rod domain (amino acids 274–746), α-actinin-2 rod domain + CT (amino acids 274–894), and NR2B CT (amino acids 1120–1482) were all subcloned into the first multiple cloning site (MCS 1) in pETDuet-1 (Invitrogen, Carlsbad, CA), which contains the sequence for an N-terminal 6xHis tag. CaM, CaM12, CaM34, CaM1234, α-actinin-2 rod domain (amino acids 274–746), α-actinin-2 rod domain + CT (amino acids 274–894), and myc-tagged NR1 CT were subcloned into the second MCS in pETDuet-1, in various combinations with the constructs described above. Additionally, the NR1 CT (amino acids 838–938) was cloned into pGEX4T-1 (GE Healthcare, Piscataway, NJ). We also used CaM, CaM12, and CaM1234 cloned into pSGC02, as described previously (Kim et al., 2004). Rat NR1 and NR2B templates were provided by Dr. D. S. Bredt (University of California, San Francisco, San Francisco, CA). The NR1 concatamers and the associated NR1 fourth transmembrane domain (TM4), previously described, were provided by S. Schorge (University College London, London, UK) (Schorge and Colquhoun, 2003). NR2A, in which the 5′-untranslated region has been removed to increase expression (Wood et al., 1996) and with a YFP-tagged to the C terminus, was provided by A. VanDongen (Duke University, Durham, NC).
Protein expression and copurification.
Proteins were expressed in BL-21 (DE3) cells after induction with 1 mm isopropyl-1-thio-β-d-galactopyranoside for 48–72 h at 16°C. Cells were harvested and resuspended in 500 mm NaCl, 20 mm Tris-HCl, glycerol 25% (v/v), 5 mm imidazole plus 1 μm CaCl2 (for lysates containing 6xHis-tagged proteins) or 100 μm CaCl2 (for lysates containing GST-fusion proteins), pH 7.5, supplemented with EDTA-free protease inhibitor mixture (Roche, Indianapolis, IN). Cell extracts were prepared by passage through a French pressure cell and then centrifuged at 100,000 × g for 90 min.
Supernatants of 6xHis-tagged proteins or complexes containing 6xHis-tagged proteins were applied to Talon metal affinity resin (Clontech, Mountain View, CA). The column was then washed in buffer supplemented with 30 mm imidazole, and proteins were eluted with buffer supplemented with 250 mm imidazole and 10 μm CaCl2.
Supernatants of GST-tagged protein complexes were applied to glutathione Sepharose 4B (GE Healthcare). The column was then washed in lysis buffer, and proteins were eluted in lysis buffer supplemented with 10 mm glutathione, pH 7.5. CaM was purified as described previously (Singla et al., 2001).
Gel filtration.
Gel filtration was performed on a Superdex 200 HR 10/30 column on an AKTA FPLC (GE Healthcare) in 500 mm NaCl, 20 mm Tris-HCl, pH 7.5, supplemented with EGTA-buffered 10 μm free CaCl2 calculated using WEBMAXC (Patton et al., 2004) or 5 mm EGTA. The column was calibrated with molecular mass standards (GE Healthcare), indicated by arrowheads from left to right, respectively, in Figure 2B that included bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), and Ribonuclease A (13.7 kDa).
Figure 2.
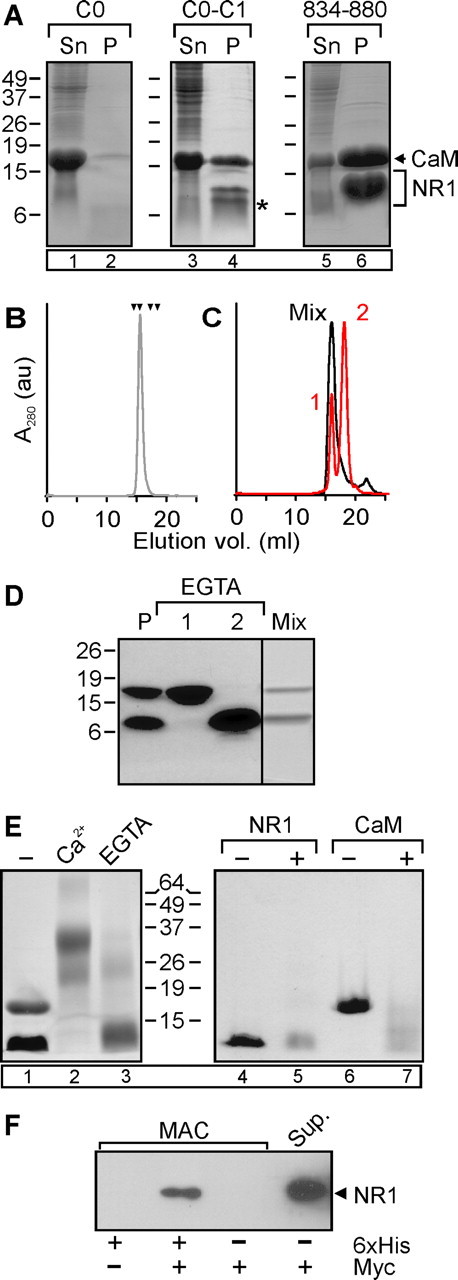
Ca2+/CaM drives “dimerization” of the NR1 CT. A, Coomassie-stained gel of bacterial lysate supernatants (Sn) and metal-affinity purified material (P) of the NR1 C0, NR1 C0–C1, NR1 834–880, or entire NR1 CT, C0-C2 (indicated by bracket) coexpressed with CaM (indicated by arrow). The asterisk denotes the presumed proteolyzed fragment (see text). B, Gel filtration analysis of the purified NR1 834–880/CaM complex. Arrowheads represent, from left to right, standards: bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa). C, Gel filtration of the material after preequilibration in 5 mm EGTA (orange) and after the material from the two peaks (labeled “1” and “2”) was remixed in Ca2+ (gray). D, Coomassie-stained gel of the purified complex (P), material from gel filtration peaks 1 and 2, and material from the peak eluted after the material was remixed in Ca2+. E, Coomassie-stained gel of NR1 834–880/CaM complex after cross-linking with DSG in the presence of Ca2+ (10 μm) or EGTA (5 mm). F, Anti-myc immunoblot after metal affinity purification (MAC) of CaM coexpressed with 6xHis-tagged NR1 CT and myc-tagged NR1 CT, as indicated. For A and E, lane numbers are in the boxes for reference in the text.
Chemical cross-linking.
Cross-linking was conducted with 2.5 mm disuccinimidyl glutarate (DSG; Pierce Endogen, Rockford, IL) in DMSO added to the purified proteins (20–50 μm), after their dialysis into 50 mm sodium phosphate buffer, pH 7.5, with 500 mm NaCl, for 5–30 min at 25°C. The reaction was stopped by adding Tris-HCl, pH 8.0, to a final concentration of 0.1 m.
GST pull down.
GST fusion proteins were purified with glutathione Sepharose 4B (GE Healthcare) according to the manufacturer's recommendations and retained on the matrix. CaM was added and incubated overnight at 4°C in 150 mm NaCl, 50 mm Tris, 0.1% Triton, pH 7.4, supplemented either with 1 mm CaCl2 or 5 mm EGTA. The beads were washed extensively, and the bound proteins were eluted with SDS-PAGE sample buffer.
Multiangle light scattering.
The molecular mass of the complex of CaM with NR1 and individual components were determined using size exclusion chromatography coupled with multiangle light scattering. Proteins (100 μl, 10 mg/ml) were injected onto a Superdex S75 10/300 GL column (GE Healthcare) equilibrated with 20 mm Tris.HCl, pH 7.5, 300 mm NaCl, and 5 mm CaCl2, and eluted at 0.5 ml/min. The eluted protein was monitored by an 18-angle light-scattering system (DAWN HELEOS; Wyatt Technology, Santa Barbara, CA) at 658 nm and a refractive index detector (Optilab rEX; Wyatt Technology). Molar mass and polydispersity of the protein were obtained by data analysis using ASTRA (Wyatt Technology).
Immunoblots.
Proteins were separated by 10–18% SDS-PAGE under reduced condition and transferred to nitrocellulose membranes. The following primary antibodies were used: monoclonal anti-CaM (Millipore, Billerica, MA), monoclonal anti-Hisx6 (Covance, Princeton, NJ), monoclonal anti-myc (Santa Cruz Biotechnology, Santa Cruz, CA). Proteins were detected by enhanced chemiluminescence (Pierce Endogen).
Electrophysiology.
HEK 293 cells were transfected with YFP-NR2A and the indicated NR1 construct by calcium phosphate precipitation. Whole-cell currents were measured 24–48 h after transfection. Cells were held at −70 mV, and currents were elicited by application of 100 μm glutamate/100 μm glycine in solution containing the following (in mm): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 10 glucose, 10 HEPES, and 0.01 EDTA, pH 7.3. The intracellular solution contained the following (in mm): 180 N-methyl-d-glucamine, 40 CsCl2, 2 Na2-ATP, 2 MgCl2, 10 HEPES, and 10 EGTA, pH 7.2.
CDI was calculated as the ratio of the sustained current (Isus) to the peak current (Ipeak) during the application of glutamate. Statistical comparisons were done using a two-tailed Student's t test.
Results
CaM binds to the NR1 CT in a Ca2+-dependent manner
To determine the Ca2+ dependence of CaM interaction with the NR1 CT, we tested whether CaM or CaM1234, a Ca2+-insensitive CaM that harbors an Asp→Ala mutation in each of the four Ca2+-binding sites, copurified with a 6xHis-tagged NR1 CT (amino acids 838–938, containing C0, C1, and C2 domains) (Fig. 1A) after coexpression in Escherichia coli. CaM and CaM1234 were expressed at similar levels (Fig. 1B). However, in the presence of 10 μm Ca2+, only CaM copurified with the NR1 CT (Fig. 1B, lane 1, middle gel). Moreover, coexpression of CaM1234 (compared with CaM) significantly decreased the NR1 CT yield in the bacterial cell lysate and in the subsequent purified material, suggesting that expression of the NR1 CT required Ca2+/CaM (Fig. 1B, lane 4, bottom gel). The absence of CaM1234 copurification contrasts with a recent report, suggesting that the NR1 CT can bind Ca2+-free CaM (Akyol et al., 2004). Because CaM1234 is a mutant that may not have properties identical to apoCaM, we directly examined whether wild-type (WT) CaM could interact with NR1 in the absence of Ca2+. Because metal-affinity purification of 6xHis-tagged proteins is compromised by Ca2+ chelators such as EGTA, we instead assayed for the interaction between CaM and GST-NR1 CT (amino acids 838–938). Lysate prepared from bacteria coexpressing GST-NR1 CT and CaM was equally divided and each fraction purified over glutathione agarose, one in 1 mm Ca2+ and the other in 5 mm EGTA. As shown in Figure 1C, CaM copurified only in the presence of Ca2+. Moreover, when we added purified CaM to GST-NR1 CT prebound to glutathione agarose, CaM was retained in the presence of 1 mm Ca2+ but not in 5 mm EGTA (Fig. 1D, lanes 3, 5). No CaM binding was observed to GST control (lanes 1 and 2). These data suggest that CaM interaction with NR1 is Ca2+ dependent, as originally proposed (Ehlers et al., 1996).
To determine whether calcification of the N lobe or C lobe of CaM was required for interaction with the NR1 CT, we coexpressed CaM12, in which the N lobe is rendered Ca2+ insensitive (by Asp→Ala mutations) or CaM34 (C-lobe mutant) with the 6xHis-tagged NR1 CT. Both were expressed at comparable levels to CaM. However, only CaM12 copurified with the NR1 CT (Fig. 1B). Similar to the result with CaM1234, coexpression of CaM34 reduced the NR1 CT yield. Thus, Ca2+ binding to the C lobe, but not the N lobe, is required for interaction between CaM and the NR1 CT.
The minimal CaM interaction site in the NR1 CT contains amino acids 834–880
We used the coexpression strategy to map the Ca2+/CaM-binding site(s) within the NR1 CT. The two potential CaM-binding sites (one in C0 and another in C1) have only been evaluated as isolated peptides and not within the context of the entire CT. Because functional data suggested that only the C0 domain is necessary for CDI (Zhang et al., 1998), we first tested the 6xHis-tagged NR1 C0 domain (amino acids 834–863), but were unable to generate significant amounts of the purified complex (Fig. 2A, lane 2). This suggested that additional determinants in the NR1 CT were necessary. We next tested a CT containing C0–C1 (amino acids 834–900). This construct yielded significant levels of NR1 expression and supported CaM copurification, yet when the material was analyzed by SDS-PAGE we consistently observed an additional, faster migrating band (Fig. 2A, asterisk, lane 4). Mass spectrometry predicted that this faster-migrating band represented 6xHis-tagged NR1 with amino acids 834–880 prompting us to test the sufficiency of this shorter domain for complex formation with CaM. As shown in Figure 2A (lane 6), CaM copurified with this NR1 fragment.
In 10 μm Ca2+, this purified material eluted from a gel filtration column as a single peak that contained both the NR1 fragment and CaM (Fig. 2B). Multiangle light scattering analysis predicted a molecular weight (MW) of 29.0 ± 0.7 kDa with 1% fitting error and a polydispersity of 1.001 (N = 5), consistent with a 2:1 stoichiometry between the NR1 CT and CaM. Complex formation was Ca2+ dependent. When the purified material was preequilibrated in EGTA before gel filtration, we observed two clearly separable peaks; the first peak contained only CaM, and the later eluting peak contained only the NR1 CT (Fig. 2C,D). Light scattering on the NR1 peak predicted a MW of 7.9 ± 0.1 kDa with 8% fitting error and a polydispersity of 1.001 (N = 3), suggesting that the isolated NR1 molecules were monomers. After remixing the separated fractions in the presence of 10 μm Ca2+, the majority of the material eluted again as a single peak that contained both CaM and the NR1 CT, and at an elution volume equivalent to the original complex (Fig. 2C,D). These data suggested that a single CaM drives reversible “dimerization” of two NR1 CTs dependent on Ca2+.
The 2:1 stoichiometry suggests a noncanonical mode of interaction between CaM and the NR1 CT. To confirm this stoichiometry, we performed irreversible chemical cross-linking of the purified complex with DSG followed by SDS-PAGE. The product migrated with a predicted MW of ∼31 kDa (Fig. 2E, lane 2). In control experiments, neither CaM nor the NR1 CT, isolated as in Figure 2, C and D, yielded any material that migrated near 31 kDa (Fig. 2E, lanes 5, 7).
This Ca2+/CaM-dependent dimerization was not an artifact of the NR1 CT truncated at amino acids 880, because we were able to confirm that more than one NR1 CT was present within a purified complex generated with the entire NR1 CT (amino acids 838–938). We coexpressed a myc-tagged NR1 CT along with the 6xHis-tagged NR1 CT and CaM and purified the complex by metal-affinity chromatography. Myc-tagged NR1 CT was efficiently copurified with the 6xHis-tagged NR1 CT (Fig. 2F). Negative-control experiments showed that copurification of the myc-tagged NR1 CT was dependent on the presence of the 6xHis-tagged NR1 CT.
The NR1 CT does not bind α-actinin-2
To assess the role of α-actinin-2 in the CaM-dependent dimerization of the NR1 CT, we first attempted to demonstrate α-actinin-2 interaction with the NR1 CT using our coexpression assays. We coexpressed the 6xHis-tagged NR1 CT (amino acids 838–938) with the α-actinin-2 rod domain and assayed whether α-actinin-2 copurified on metal-affinity chromatography. The α-actinin-2 rod domain was expressed to high levels (a prominent band corresponding to the α-actinin-2 rod domain was apparent even in the bacterial lysate), but none copurified with the NR1 CT (Fig. 3A). We also found that a longer α-actinin-2 construct containing both the rod domain and the α-actinin CT (a domain with homology to CaM) did not copurify with the NR1 CT. These negative results are not likely caused by the presence of Ca2+ in the buffers during purification (α-actinin-2 has been postulated to interact with the NR1 CT in low Ca2+), because neither the α-actinin-2 nor the NR1 CT bind Ca2+. In addition, identical experiments performed without added Ca2+ yielded similar results (data not shown). Moreover, under the same conditions, we were able to demonstrate that the α-actinin-2 domain was able to copurify with the NR2B CT (Wyszynski et al., 1997). Confirmation that this copurified band was α-actinin-2 came by showing that it migrated similar to purified 6xHis-tagged α-actinin-2 rod domain + CT (Fig. 3D, right lane). Thus, CaM but not α-actinin-2 appears to be a binding partner for the NR1 CT.
Figure 3.
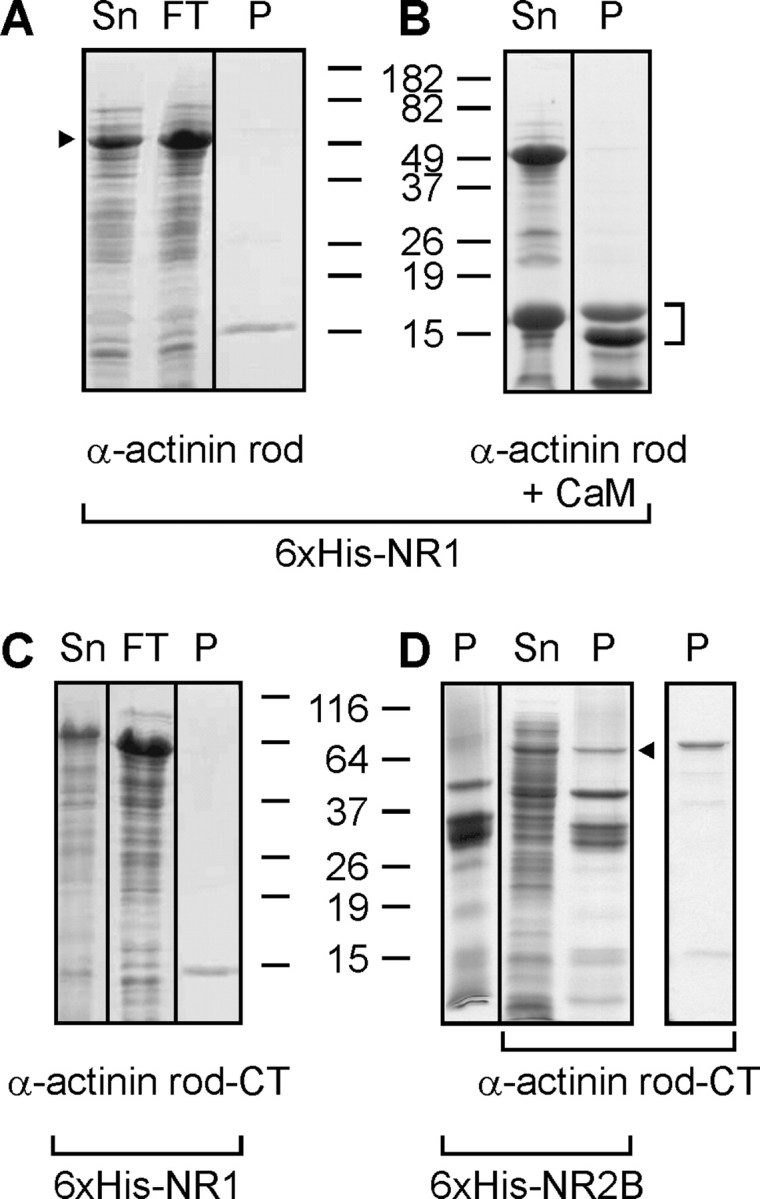
The NR1 CT does not bind α-actinin-2. A, B, Coomassie-stained gels showing supernatant (Sn), column flow-through (FT), and metal-affinity purified material (P) of 6xHis-tagged NR1 CT coexpressed with the α-actinin-2 rod domain (indicated by arrowhead to left of A) without (A) or with (B) CaM. The NR1 or NR1/CT complex is indicated by a bracket to right of B. C, Same as in A, but with α-actinin-2 rod domain plus α-actinin-2 CT. D, Same as in C, but with 6xHis-tagged NR2B CT (amino acids 1120–1482). Arrow denotes successful copurification of the α-actinin-2 rod plus CT domains with NR2B. On right is shown a metal-affinity purification of 6xHis-tagged α-actinin-2 rod plus CT domains.
Uncoupling the NR1 CTs blocks CDI
We propose a new model for NMDAR CDI in which Ca2+/CaM binding to the NR1 CTs induces a Ca2+/CaM-dependent dimerization of the two NR1 CTs within a NMDAR (Fig. 4A); we predict that this propagates a conformational change to the channel pore that leads to channel closure. After cellular Ca2+ returns to basal levels, CaM should dissociate and the two NR1 CTs separate (Fig. 2D), allowing recovery from inactivation. To test this hypothesis, we examined CDI (Isus/Ipeak) in currents from NMDARs in which one of the NR1 CTs can be uncoupled or eliminated from the rest of the receptor. This strategy used NR1 concatamers (Schorge and Colquhoun, 2003) in which the first component of the concatamer contained a NR1 subunit truncated in the extracellular loop before the fourth transmembrane segment (thereby removing the fourth transmembrane domain and the CT). This “NR1t” subunit was then fused to the N terminus of a second NR1 subunit, forming an NR1t→NR1 concatamer (Fig. 4B). The fourth transmembrane domain and CT removed from the first component of the concatamer could be expressed as a separate polypeptide (“TM4”). These constructs (or WT NR1) were then expressed in HEK 293 cells along with NR2A, which supports CDI (Krupp et al., 1996), and CDI was quantified as the ratio of the sustained current to the peak current (Isus/Ipeak) during glutamate activation. When nonconcatamerized WT NR1 subunits were expressed, Isus/Ipeak was 51 ± 5% (Fig. 4B,C). The observed inactivation was clearly Ca2+ dependent, because removal of Ca2+ from the extracellular solution almost eliminated inactivation (Fig. 4B,C). With the concatamers, we then eliminated one of the NR1 CTs (using NR1t→NR1) or uncoupled one of the NR1 CTs from the rest of the NMDAR receptor (by expressing TM4 with the NR1t→NR1 concatamer). In both cases, CDI was greatly attenuated (Fig. 4B,C). There was no significant difference between NR1t→NR1 and NR1t→NR1 + TM4 (p = 0.08). Although the currents obtained from the concatamers were smaller than from nonconcatamerized WT NR1, as previously observed (Schorge and Colquhoun, 2003), the current amplitudes from all sets overlapped, and there was no correlation between absolute current amplitude and CDI within any set (data not shown).
Figure 4.

A, Model for CDI of NMDARs by Ca2+/CaM-driven dimerization of the NR1 CT. Schematic of the NMDAR, with focus on the two NR1 subunits. In the presence of Ca2+, Ca2+/CaM drives dimerization of the CTs in the two NR1 subunits, leading to channel closure and inactivation. B, Exemplar current traces and schematics of the WT NR1 with and without extracellular Ca2+ (top); the NR1t→NR1 concatamer (middle); and the NR1t→NR1 concatamer plus TM4, the separate fourth transmembrane domain, and the C terminus (bottom). Calibration: 5 s and 500 pA for WT/+Ca2+; 20 pA for WT/−Ca2+; 50 pA for NR1t→NR1; and 100 pA for NR1t→NR1+TM4. C, Summary data for CDI (Isus/Ipeak) showing that removal of Ca2+ or decoupling of the two NR1 C termini abolishes CDI. The number of cells tested is shown for each condition.
Discussion
Overall, these data are consistent with the previous reports that have defined roles for the NR1 CT and for CaM in CDI of NMDARs, but present a new molecular hypothesis for the mechanics of CDI. Attenuation of CDI when the two NR1 CTs are uncoupled, as shown in Figure 4B,C, supports the “dimerization model” presented in Figure 4A. In certain respects, the Ca2+/CaM-driven dimerization model for NMDAR inactivation presented here is similar to that proposed for the Ca2+/CaM activation of SK K+ channels (Schumacher et al., 2001), albeit with a noteworthy difference: two CaM molecules interact with two SK CTs in a twofold symmetrical arrangement to drive Ca2+-dependent dimerization of two SK CTs, whereas we find an asymmetric 2:1 minimal stoichiometry between CaM and the NR1 CT. This disparity may reflect the homotetrameric nature of SK channels compared with the NMDARs containing both NR1 and NR2 subunits.
The asymmetric stoichiometry between CaM and the NR1 CTs highlights that the nature of the interaction between CaM and the NR1 CT appears unusual and falls into a category of noncanonical CaM interactions that have recently been defined. These interactions differ from the well characterized mode in which the Ca2+-saturated lobes of CaM wrap around a single α-helical target in the binding protein (Hoeflich and Ikura, 2002).
The demonstration that α-actinin-2 does not bind to the NR1 CT also fosters a significant revision of those models that postulated that Ca2+/CaM drives CDI by displacing α-actinin-2 from the NR1 CT (Zhang et al., 1998; Krupp et al., 1999). Functional data obtained in hippocampal neurons (Rycroft and Gibb, 2004) and in heterologous expression systems (Krupp et al., 1999; Michailidis et al., 2007) clearly support a role for α-actinin-2 in the regulation of NMDAR gating, but biochemical data demonstrating a direct interaction between α-actinin-2 and the NR1 CT are less established. NR1 CT binding to α-actinin-2 was originally observed in a yeast two-hybrid screen using the NR1 CT as a bait (Wyszynski et al., 1997), but immunoprecipitation experiments from rat brain with either an anti-NR1 antibody or anti-α-actinin 2 antibody showed that most of the NR1 was not associated with α-actinin-2 (Dunah et al., 2000). Additionally the failure of surface plasmon resonance experiments (Krupp et al., 1999) to detect saturation binding between α-actinin-2 and a NR1 C0 peptide even with 500 nm α-actinin-2 (when CaM binding saturated at <70 nm under identical conditions) suggests that binding affinity between α-actinin-2 and NR1 is very low and that the reported Kd from the derived binding isotherm is an underestimate. Although a recent study suggests that α-actinin-1 has a higher affinity for the NR1 CT than α-actinin-2 (Merrill et al., 2007), the functional experiments demonstrating α-actinin-mediated effects on NMDAR currents used α-actinin-2 isoform; it seems unlikely, then, that a direct interaction of α-actinin-1 with NR1 rather than α-actinin-2 could explain CDI. Thus, we predict that the role for α-actinin-2 in CDI of NMDARs does not include competition with CaM for an NR1-binding site.
Our finding that only Ca2+/CaM but not apoCaM binds to the NR1 CT also contrasts with recent studies that suggested that apoCaM can interact with the NR1 CT (Akyol et al., 2004; Merrill et al., 2007). Rather, our data are consistent with the original report describing a Ca2+/CaM interaction with the NR1 CT (Ehlers et al., 1996). The reasons for the discrepancies are not obvious, but high levels of apoCaM (∼50 μm) were required to detect interaction with NR1. We propose that apoCaM is bound to proteins other than NR1 before Ca2+ rises to induce CDI.
Footnotes
This work was supported by grants to G.S.P. from the National Institutes of Health (HL 71165) and the American Heart Association (0740030N). G.S.P. was a recipient of the Irma T. Hirschl Career Scientist Award and the Esther Aboodi Assistant Professor of Medicine (Columbia University, New York, NY), which supported part of this work. We thank Becky Klein (University of Washington, Seattle, WA) for the suggestion to use the concatamers and S. Schorge (University College London, London, UK) for graciously sharing them.
References
- Akyol Z, Bartos JA, Merrill MA, Faga LA, Jaren OR, Shea MA, Hell JW. Apo-calmodulin binds with its c-terminal domain to the N-methyl-d-aspartate receptor NR1 C0 region. J Biol Chem. 2004;279:2166–2175. doi: 10.1074/jbc.M302542200. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Wyszynski M, Martin DM, Sheng M, Standaert DG. alpha-actinin-2 in rat striatum: localization and interaction with NMDA glutamate receptor subunits. Brain Res Mol Brain Res. 2000;79:77–87. doi: 10.1016/s0169-328x(00)00102-9. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. Calcium-dependent inactivation of recombinant N-methyl-d-aspartate receptors is NR2 subunit specific. Mol Pharmacol. 1996;50:1680–1688. [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Bayer K-U, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW. Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and α-actinin. J Biol Chem. 2002;277:48441–48448. doi: 10.1074/jbc.M205164200. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Malik Z, Akyol Z, Bartos JA, Leonard AS, Hudmon A, Shea MA, Hell JW. Displacement of alpha-actinin from the NMDA receptor NR1 C0 domain by Ca2+/calmodulin promotes CaMKII binding. Biochemistry. 2007;46:8485–8497. doi: 10.1021/bi0623025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidis IE, Helton TD, Petrou VI, Mirshahi T, Ehlers MD, Logothetis DE. Phosphatidylinositol-4,5-bisphosphate regulates NMDA receptor activity through α-actinin. J Neurosci. 2007;27:5523–5532. doi: 10.1523/JNEUROSCI.4378-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Regulation of single NMDA receptor channel activity by alpha-actinin and calmodulin in rat hippocampal granule cells. J Physiol (Lond) 2004;557:795–808. doi: 10.1113/jphysiol.2003.059212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorge S, Colquhoun D. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J Neurosci. 2003;23:1151–1158. doi: 10.1523/JNEUROSCI.23-04-01151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- Singla SI, Hudmon A, Goldberg JM, Smith JL, Schulman H. Molecular characterization of calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:29353–29360. doi: 10.1074/jbc.M101744200. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-d-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Wood MW, VanDongen HMA, VanDongen AMJ. The 5′-untranslated region of the N-methyl-d-aspartate receptor NR2A subunit controls efficiency of translation. J Biol Chem. 1996;271:8115–8120. doi: 10.1074/jbc.271.14.8115. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]
- Zukin RS, Bennett MV. Alternatively spliced isoforms of the NMDARI receptor subunit. Trends Neurosci. 1995;18:306–313. doi: 10.1016/0166-2236(95)93920-s. [DOI] [PubMed] [Google Scholar]