

Abstract
Metastatic tumors and malignant gliomas make up the majority of cancers in the brain. They are invariably fatal and there is currently no cure. From in vitro comparisons of a number of viruses, we selected one that appeared the best in selectively killing glioblastoma cells. This replication-competent virus, the glioma-adapted vesicular stomatis virus strain VSVrp30a, was used for in vivo tests with the underlying view that infection of tumor cells will lead to an increase in the number of viruses subsequently released to kill additional tumor cells. Intravenous injection of VSVrp30a expressing a green fluorescent protein reporter, rapidly targeted and destroyed multiple types of human and mouse tumors implanted in the mouse brain, including glioblastoma and mammary tumors. When tumors were implanted both in the brain and peripherally, emulating systemic cancer metastasis, tumors inside and outside the brain were simultaneously infected. Intranasal inoculation, leading to olfactory nerve transport of the virus into the brain, selectively infected and killed olfactory bulb tumors. Neither control cortical wounds nor transplanted normal mouse or human cells were targeted, indicating viral tumor selectivity. Control viruses, including pseudorabies, adeno-associated, or replication-deficient VSV, did not infect the brain tumor. Confocal laser time-lapse imaging through a cranial window showed that intravenous VSV infects the tumor at multiple sites and kills migrating tumor cells. Disrupted tumor vasculature, suggested by dye leakage, may be the port of entry for intravenously delivered VSV. Quantitative PCR analysis of how VSVrp30a selectively infected tumor cells suggested multiple mechanisms, including cell surface binding and internalization.
Keywords: brain cancer, glioma, metastasis, vesicular stomatitis virus, oncolytic virus, fluorescence microscopy
Introduction
Cancer in the brain is one of the deadliest diseases. A common brain cancer is glioblastoma, which is a primary cancer of the brain; tumor cells can migrate from the body of a tumor into multiple brain systems. Metastases, which migrate into the brain from cancer originating outside the CNS, can colonize multiple sites within the brain. Each year >200,000 people in the United States are diagnosed with a brain tumor (Central Brain Tumor Registry of the United States, 2005). The malignant disease usually starts with subtle changes in cerebral function, becomes manifest with seizures or raised intracranial pressure, progresses to loss of motor and mental function, and finally to death. There is no cure or effective treatment, and the median survival is measured in months despite modern therapy (Posner and Chernik, 1978; DeAngelis, 2001; Ohgaki and Kleihues, 2005; Stupp et al., 2005). Failure of conventional treatment modalities such as surgery, radiation and chemotherapy results from multifocality of disease, infiltrative growth within otherwise normal brain tissue, impermeability of the CNS to therapeutic agents, substantial tumor genotypic variability, persistence of cancer-initiating cells, and rapid genetic evolution of resistant clones. Cancer is a systemic disease, and multiple metastatic lesions or large infiltrative gliomas require an efficient delivery method to target dispersed tumor cells.
A unique approach to combating brain cancer is the use of viruses (Aghi and Rabkin, 2005). Nonreplicating viruses are used to deliver toxic or therapeutic genes. Such viral vectors have so far been only partially effective because only a small percentage of tumor cells are infected (Puumalainen et al., 1998; Rainov and Ren, 2003). An alternate strategy is the use of replicating viruses that target, infect, produce progeny virus to infect more tumor cells, and kill infected cells. Some viruses that have been engineered to kill dividing tumor cells, such as recombinant herpes simplex or adenovirus, have shown promise, but these viruses have not been able to disseminate to all regions of the tumor (Khuri et al., 2000; Pulkkanen and Yla-Herttuala, 2005). Other viruses, including reovirus, measles, or pox viruses, show merit for infecting tumor cells (Parato et al., 2005). In a previous series of in vitro experiments on nine different viruses with reported intrinsic oncolytic potential against gliomas, a rhabdovirus, vesicular stomatis virus VSVrp30a, outperformed other candidates (Wollmann et al., 2005). It was also superior in infecting glioma xenografts in a subcutaneous glioma model. VSV also has potent oncolytic activity against a broad spectrum of systemic cancer cells tested in vitro (Stojdl et al., 2003). To clearly analyze viral infection and spread in brain tumors, we used red fluorescent protein (RFP)-expressing cancer cells and a recombinant VSV that expresses green fluorescent protein (GFP). A battery of genetically distinct human cancer cell lines and several mouse xenograft models allowed us to study different aspects of VSV oncolysis. A single intravenous dose of VSV simultaneously and selectively targeted multifocal brain tumors and subcutaneous flank tumors. Time-lapse confocal laser imaging of VSV oncolysis in the live mouse brain suggested that infection spread within the tumor, probably enhanced by a local leaky blood–brain barrier (BBB). Onco-selectivity of VSV can be attributed in part to its high sensitivity to interferon antiviral defense, which allows normal cells to attenuate infection but appears to be defective in the majority of tumors tested (Stojdl et al., 2000, 2003). In addition, we show with quantitative reverse transcription (RT)-PCR experiments that multiple additional mechanisms, including more efficient virus binding and internalization, enhance preferential targeting of tumor cells by VSVrp30a.
Materials and Methods
Cell culture.
U87MG human high-grade glioma cells and 4T1 mouse mammary carcinoma cells were obtained from American Type Culture Collection (Rockville, MD). A549 and Calu-1 human lung carcinomas and T-47D, MCF-7, and BT474 human breast carcinoma cells were provided by the Yale Cancer Center (Yale University, New Haven, CT). U118MG glioma cells were kindly provided by Dr. R. Matthews (Yale University). Primary human glioblastoma cultures were established from tissue derived from two patients undergoing resective surgery. Nontumor primary human astrocytes were prepared as explant cultures from human temporal lobectomy material, removed for intractable epilepsy, solely for the benefit of the patient, and the use was approved by the Yale University Human Investigation Committee. Astrocyte identity was confirmed by positive immunofluorescence for human glial fibrillary acidic protein. Cells were maintained in Minimal Essential Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 1% sodium pyruvate, 100 μm nonessential amino acids, and 25 mm HEPES buffer. For VSV susceptibility experiments, 2 × 105 cells were cultured in six-well plates and infected with VSVrp30a at a multiplicity of infection (MOI) of 1. The total number of viable and infected cells was counted at 12 h intervals in 15 high-magnification microscopic fields.
Transfection.
For stable transfection, RFP [tetrameric phosphorylated Discosoma red (pDsRed1)–N1 and monomeric pDsRed-monomer–C1], cyan fluorescent protein [phosphorylated cytomegalovirus (pCMV)–CFP], and blue fluorescent protein (pCMV–BFP) plasmids were used. pCMV–CFP and pCMV–BFP were generated by exchanging the RFP expression cassette from pDsRed monomer–C1 with CFP or BFP cassettes from pRSET/CFP (Invitrogen). Cells were transfected using Lipofectamine 2000 reagent (Invitrogen), enriched, and maintained with G418 (Sigma, St. Louis, MO). rU87 and rU118 cells were sorted for brightest fluorescence on a FACSvantage SE fluorescence-activated cell sorter (BD Biosciences, San Jose, CA). Growth characteristics were assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Invitrogen) assay according to the instructions of the manufacturer. Optical density was read at 570 nm using a Dynatech (Alexandria, VA) MR500 ELISA plate reader and corrected from background control.
Mouse procedures.
Animal experiments and postoperative care were performed in accordance with institutional guidelines of the Yale University Animal Care and Use Committee. Immunodeficient homozygous CB17-SCID (CB17SC-M) and NCr-Nude mice (NCr-Foxn1nu) at an age of 4–5 weeks were obtained from Taconic Farms (Germantown, NY). Animals were stereotactically grafted with 3 × 105 tumor cells bilaterally into the striatum (2 mm lateral, 0.4 mm rostral to bregma at 3 mm depth) or cortex or with 1 × 105 cells unilaterally in the left olfactory bulb (0.75 mm lateral, 4.5 mm rostral to bregma at 1.5 mm depth). For cranial window experiments, a teardrop-shaped skin flap (with an anteroposterior axis along the parasagittal plane crossing the midpupillary line) was removed, and a 5-mm-wide round craniectomy was drilled under an operating microscope (Zeiss, Oberkochen, Germany). rU87 glioma cells, 5 × 104, were injected along a trajectory parallel to the cortex immediately below the pia mater. The craniotomy was covered with a round glass coverslip of 8 mm diameter and glued to the bone margins using cyanoacrylate. Animals were followed with daily measurements of weight, food and water consumption, and overall health and activity, and the cranial window animals were given parenteral antibiotics. During in vivo brain imaging, mice were lightly anesthetized to reduce movement, and the head was maintained in a horizontal position during image acquisition.
Animals were killed with a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde. For determination of blood–brain barrier integrity, 2% Evans Blue (Sigma) in 100 μl of sterile saline was injected intravenously 1 h before the animals were killed.
Viruses.
Ten days after tumor grafting, 100 μl of medium containing virus (107 plaque forming units (PFU) of VSVrp30a, VSV–GFP, VSV–ΔG–GFP, or recombinant pseudorabies virus (PRV)–GFP or 1012 virions adeno-associated virus serotype 2 (AAV)–GFP] was injected through the tail vein. VSV–GFP and VSV–ΔG–GFP were generously provided by Dr. J. K. Rose (Yale University). PRV–GFP strain 152 was kindly provided by Dr. L. W. Enquist (Princeton University, Princeton, NJ) (Smith et al., 2000) and AAV–GFP was kindly provided by Dr. K. R. Clark (Ohio State University, Columbus, OH). Details on the generation and characteristics of the tumor-adapted VSV–GFP isolate VSVrp30a were described previously (Wollmann et al., 2005). VSV, PRV, and AAV were all shown to infect glioma cells previously (Wollmann et al., 2005).
Immunocytochemistry.
Cryosections were mounted and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). For cleaved caspase 3 immunocytochemistry, 20 μm sections were incubated with a 1:150 dilution of polyclonal rabbit cleaved caspase 3 antiserum (Cell Signaling Technology, Danvers, MA), followed by detection with a 1:200 dilution of biotinylated anti-rabbit antibody (Invitrogen) and ABC reagents detected with diaminobenzidine and hydrogen peroxide (Vector Laboratories, Burlingame, CA). For von Willebrand factor staining, a 1:500 dilution of polyclonal rabbit antibody (Abcam, Cambridge, MA) was used following the same procedure outlined above.
Imaging.
Histological sections were studied either on an Olympus Optical (Tokyo, Japan) IX71 fluorescent microscope fitted with a SPOT-RT camera (Diagnostic Instruments, Sterling Heights, MI) or on an Olympus Optical Fluo-300 laser confocal microscope using Fluoview 300 software version 4.3. Corresponding phase-contrast, green, red, and blue fluorescence photomicrographs were fused and corrected for color and contrast using Adobe Photoshop 7 software (Adobe Systems, San Jose, CA). Several high-resolution pictures were used for montage of whole coronal sections.
Tumor area calculations and cell and nuclear counting were done using NIH ImageJ software. Animals with cranial windows were imaged daily or more frequently under an Olympus Optical SZXZB12 stereo microscope and Olympus Optical Fluo-300 laser confocal microscope.
Virus binding and entry experiments.
Binding, entry, and uncoating of VSVrp30a were compared in rU87 glioma cells and normal human astrocytes using quantitative RT-PCR. To assess membrane-bound fraction, cells were incubated at 4°C with VSVrp30a for 20 min and washed five times before RNA extraction. To quantify intracellular virus, cells were trypsinized for 10 min and washed five times in PBS. VSV was pooled in the endosomal compartment by incubating for 90 min in the presence of 5 mm ammonium chloride (Sigma). Ammonium chloride blocks acidification of the endosome and therefore blocks viral uncoating into the cytoplasm. These experiments were done in triplicate using an MOI of 10.
Real-Time PCR for viral GFP.
RNA was extracted from cell lysates using RNeasy kit (Qiagen, Valencia, CA), and 1 μg of total RNA was reverse transcribed by random hexamer priming using the Super Script III reverse transcriptase kit (Invitrogen). This was followed by quantitative PCR using TaqMan gene expression assays (Applied Biosystems, Foster City, CA) for GFP and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences for GFP were as follows: sense, 5′-GAGCGCACCATCTTCTTCAAG-3′; antisense, 5′-TGTCGCCCTCGAACTTCAG-3′.
Results
Tumor cells stably transfected with red fluorescent protein gene reliably form tumors
To choose the optimum nontoxic fluorescent protein that would allow simultaneous imaging with GFP encoding virus, we tested several different fluorescent proteins, including cyan (pCMV–CFP), blue (pCMV–BFP), and red (tetrameric pDsRed1–N1 and monomeric pDsRed-monomer–C1). BFP was subject to quick photobleaching, and CFP was not practical for use simultaneously with GFP-expressing VSV because of overlap in their fluorescent spectrum. Transfection of U87 high-grade glioma cells with tetrameric RFP resulted in toxicity in vitro. Stable transfection of U87MG or U118MG glioma cells with a plasmid encoding the monomeric DsRed protein resulted in bright red fluorescence and did not alter growth characteristics as demonstrated by MTT assay. Over a 24 h period, absorbance measurements increased by 89.8 ± 33.2% in rU87 cells and by 103.8 ± 32.5% in U87MG cells (statistically nonsignificant p = 0.75, n = 6, Student's t test), indicating that the red glioma cells were replicating at their usual rate. After selection in G418 and fluorescence-activated cell sorting, all tumor cells expressed red fluorescent protein (Fig. 1 A).
Figure 1.
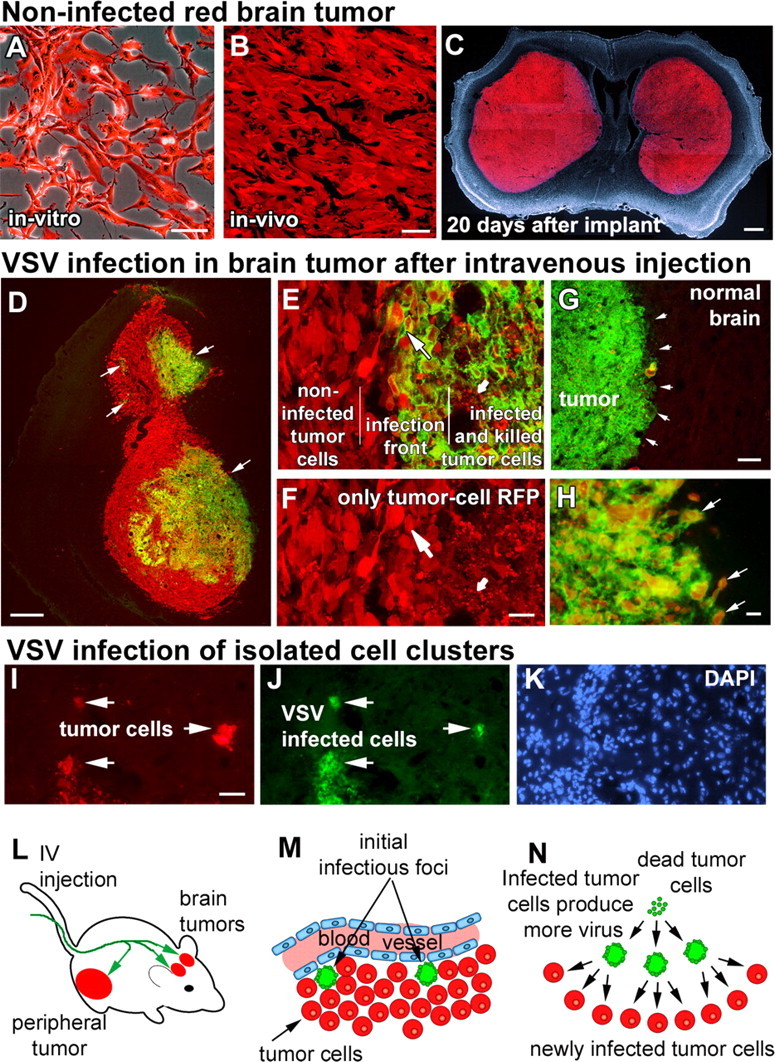
Intravenously injected VSVrp30a infects intracranial xenografts at multiple sites and spreads within the tumor to infect the tumor mass and infiltrating tumor cells. Stably RFP-transfected and fluorescence-activated cell-sorted U87MG human high-grade glioma cells grew in vitro without any signs of cytotoxicity (A) and formed brain tumors with no signs of spontaneous regression (B), eventually resulting in the animal's demise (C). When tumor-bearing animals were given a single intravenous injection of VSVrp30a, multiple independent foci (arrows in D) of VSV infection were found inside the tumor mass. Tumor cells in infected areas demonstrated marked cytopathy, and most were lysed (small arrow in E, F). Viral progeny released from these initially infected cells spread to neighboring tumor cells (large arrow in E, F) creating distinct areas of cells in different phases of VSV infection. In addition to infecting the whole tumor, VSVrp30a also infected individual infiltrative tumor cells close to the tumor mass (arrows in H) as well as in isolated clusters remote from the main tumor bulk (I–K). Despite widespread infection within the tumor mass (green fluorescent area in G marked with arrows), no infection was noted in the surrounding brain parenchyma. L–N schematize the general outline of VSV oncolysis in the SCID mouse brain tumor model: After intravenous injection (L), VSVrp30a infection starts at few scattered foci (M) around brain tumor vasculature and spreads to infect the whole tumor (N). Scale bars: A, B, 50 μm; C, 500 μm; D, 300 μm; E, F, H–K, 20 μm; G, 100 μm.
Bilateral brain injections of either rU87 or rU118 cells in SCID or nude mice yielded tumors in 100% (90 of 90) of injections. Ten days after injection, noninfected rU87 xenografts in SCID mice had a mean calculated volume of 7.13 ± 5.76 mm3 (n = 4; calculated as ellipsoid volume: 4/3 π x y z). Horizontal and vertical radii x and y represent half of the horizontal and vertical diameters in the coronal section that harbored the largest tumor cross-section area. The anteroposterior radius z represents half the tumor craniocaudal length, calculated by multiplying section thickness with the number of sections where the tumor is present. Tumor xenografts grew predominantly by expansion (Fig. 1 C), although in a number of cases we found tumor cells separated from the main tumor body and invading the surrounding parenchyma. Tumors grew with no spontaneous regression or hemorrhage until they reached a lethal size (Fig. 1 C). For humane reasons, mice were killed at the onset of symptoms before tumor-induced death.
Intravenously injected VSVrp30a targets multifocal brain tumors with high selectivity
In a previous study, we showed that, in animals bearing two subcutaneous tumors, intratumoral VSV injection into one tumor resulted in selective infection also in the second tumor on the contralateral side of the body. This strongly suggested vascular viral dissemination and led us to the hypothesis that VSV could be delivered to brain tumors through the intravascular route. However, entry into brain tumors is more complex than elsewhere in the body because the blood–brain barrier can be a major obstacle, even for relatively small-sized chemical or biological agents (Pardridge, 2002). We tested whether intravenously administered VSV could target intracranial tumor xenografts. In mice that were implanted with bilateral brain rU87 xenografts (n = 9 animals, 18 tumors), intravenous injection of 107 PFU of VSVrp30a through the tail vein consistently resulted in infection of the intracranial tumors. A single injection was sufficient to infect gliomas. After an injection of 107 PFU, VSVrp30a infection was found in different regions inside the tumor, suggesting multiple sites of virus seeding (Fig. 1 D). Infection inside the tumor started with a time lag after intravenous delivery but was found in all tumors analyzed after 24 h.
VSVrp30a replicates and spreads within the tumor mass
VSV is a replication-competent virus and spreads within the tumor. Early in the course of infection in the non-infected areas, red tumor cells with intact cellular outlines were seen. Within the VSV-infected area, infected and dead tumor cells and debris (Fig. 1 E,F, small arrows) were surrounded by VSV-infected cells with membranous GFP staining (Fig. 1 E,F, large arrows). The presence of newly infected cells at the boundary between killed and non-infected cells suggests that VSV spreads within the tumor. Analyzing animals at different time points within the course of VSV infection and using serial histological sections through the tumor, we found that the virus had spread from initially infected foci to encompass the whole tumor (Fig. 1 G). In addition to the widespread infection and tumor cell killing inside the main tumor mass, tumor cells that were migrating into the normal brain at the tumor margin were also infected, with remarkable sparing of neighboring normal brain cells in immediate contact with the tumor (Fig. 1 H). Although most tumor xenografts grew by expansion, some isolated cell clusters of tumor cells remote from the main tumor bulk were observed. Of note, these distant cell conglomerates were also targeted by VSV (Fig. 1 I–K). The course of VSV oncolysis in the brain is schematized in Figure 1 L–N.
To document viral spread within the tumor mass, we killed animals 24, 48, and 72 h post-inoculation (HPI) and demonstrated a continuously increasing area of infection in the tumor mass, resulting in tumor cell death, as indicated by the loss of cellular integrity and the appearance of small granular debris after viral infection (Fig. 2). Six tumors were used for analysis of VSV and four for VSVrp30a infection at each time point. In animals killed early in the course of infection, we documented areas of infected and killed tumor cells surrounded by other cells that appeared live at the time of death but with early signs of infection and expression of viral reporter gene (Fig. 2). Intact cellular outlines and homogeneous intracellular DsRed signal were used to distinguish healthy cells from dead or dying cells. In addition, DAPI counterstain showed nuclear breakdown commonly found in dead cells, and areas of dead cells showed small granular debris in the place of intact cells (see additional details below and Fig. 4). The mean infected area in coronal brain sections with the largest tumor diameter increased from ∼50 to 60% at 1 d post-inoculation (DPI) to what appeared to be infection of almost the entire tumor by 3 DPI (Fig. 2). As shown in the fractionated bar graph in Figure 2 A, the proportion of viable tumor cells showing normal cellular and nuclear morphology decreased simultaneously. Finally, the spread of infection was documented in real time using the cranial window animal model as described below.
Figure 2.
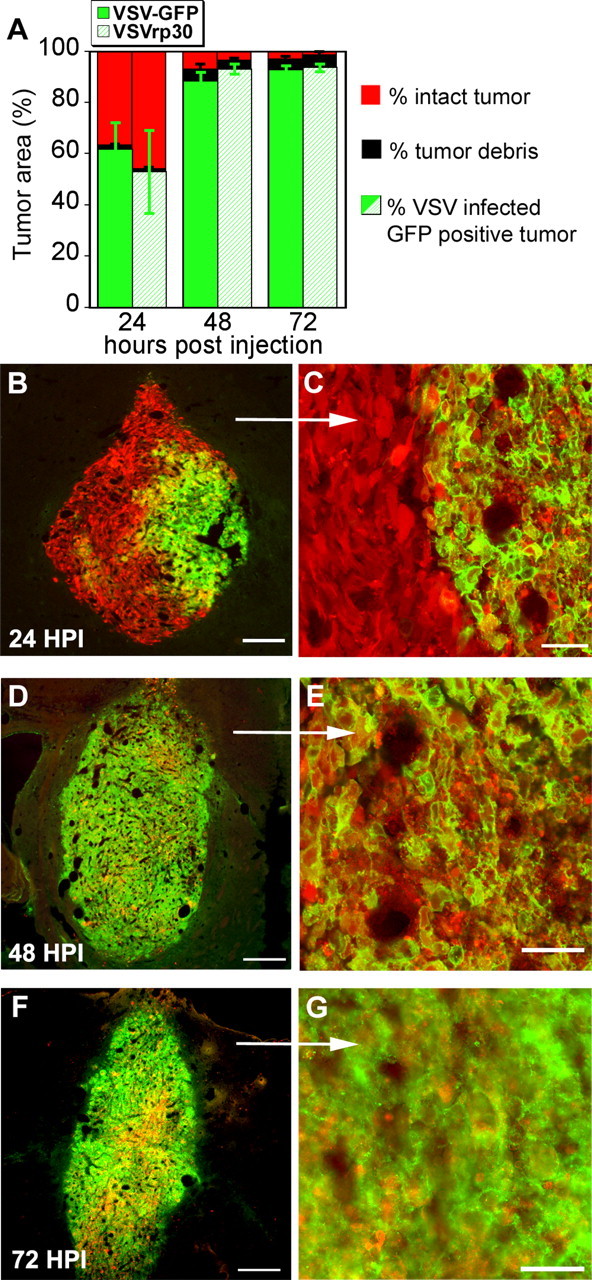
The whole tumor mass is infected within 72 h. The spread of VSVrp30a infection encompasses the whole tumor mass within a few days. A shows the time progression of the VSV oncolysis. n = 6 for VSV; n = 4 for VSVrp30a at each time point. The infected area increases and the number of viable cells decreases. Black indicates the area of tumor lysis with little GFP detection because of GFP diffusion out of dead cells. VSVrp30a has a slightly higher rate of spread within the tumor than VSV–GFP (not statistically significant) at 2 and 3 d after inoculation. B–G show three representative rU87 xenografts analyzed 24 (B, C), 48 (D, E), and 72 (F, G) h after infection respectively. Scale bars: B, 100 μm; C, E, G, 50 μm; D, F, 300 μm.
Figure 4.
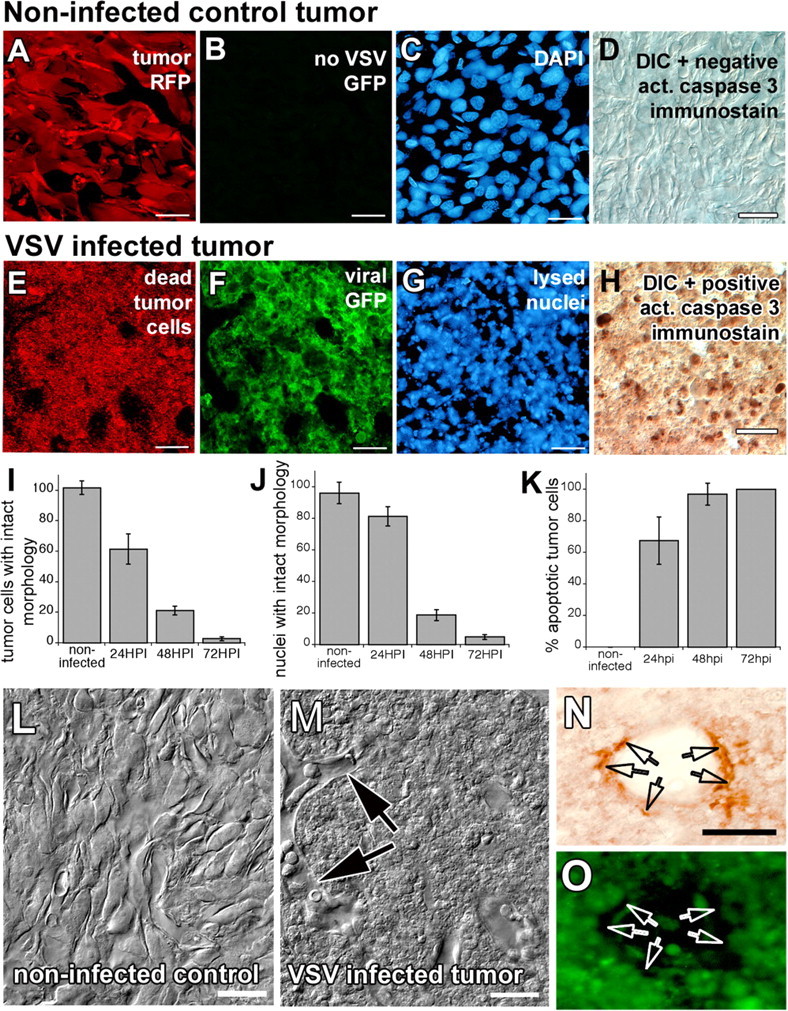
VSV infection of brain tumor xenografts results in widespread tumor cell death. Slice preparations from uninfected (A–D) and infected (E–H) rU87 xenografts in SCID mice. Uninfected tumor xenografts have normal cellular morphology as outlined by cytoplasmic RFP transgene expression (A), have no viral green fluorescence (B), have normal appearing nuclei (C), and do not stain with the apoptosis marker cleaved caspase 3 (D). VSV-infected tumor cells, in contrast, express viral GFP (F), lose cellular integrity (E), undergo chromatolysis (G), and stain positively for the apoptotic marker (H) at 48 HPI. Graphs (I–K) indicate the increase in viral tumor cell killing during the first 3 d after virus injection. Laser confocal DIC imaging shows uninfected (L) and infected (M) glioma xenografts. Note that, despite widespread cellular debris inside the VSV-infected tumor xenograft, vessel architecture is spared and endothelial cells appear morphologically healthy (arrows). Noninfected cells constituting the vessel wall stained positive for endothelial marker von Willebrand factor (arrows in N, O). Scale bar, 50 μm.
In vitro, VSVrp30a was more effective at selective destruction of gliomas than the parental VSV–GFP. To determine whether this difference persisted in vivo, we compared the relative abilities of the two VSVs to infect gliomas in the mouse brain using the same concentration of virus inoculum. Both VSVrp30a (n = 9 animals, 18 tumors) and VSV–GFP (n = 9 animals, 18 tumors) successfully targeted the brain tumors after tail vein inoculation VSV–GFP. A high tumor selectivity of VSV infection was observed during the time period of our study. In animals implanted with multiple tumors, all brain tumors were infected simultaneously (Fig. 3). VSV infection and resultant oncolysis were observed in all tumor masses regardless of the size and location. Small and large tumors were infected at different localizations in the brain whether striatal, cortical, subventricular, or within the olfactory bulb. Tumors in the brain and periphery were also simultaneously infected (see below). In animals killed within the first 72 HPI, when the entire tumor mass was infected by VSVrp30a, we did not observe GFP expression in the brain parenchyma surrounding the tumor, choroid plexus, or in the leptomeninges. After this period of high tumor-specific infection (72–96 HPI in our model), when the whole tumor bulk showed marked cytopathy, we did observe select periventricular groups of infected neurons and ependymal cells in immunodeficient mice.
Figure 3.
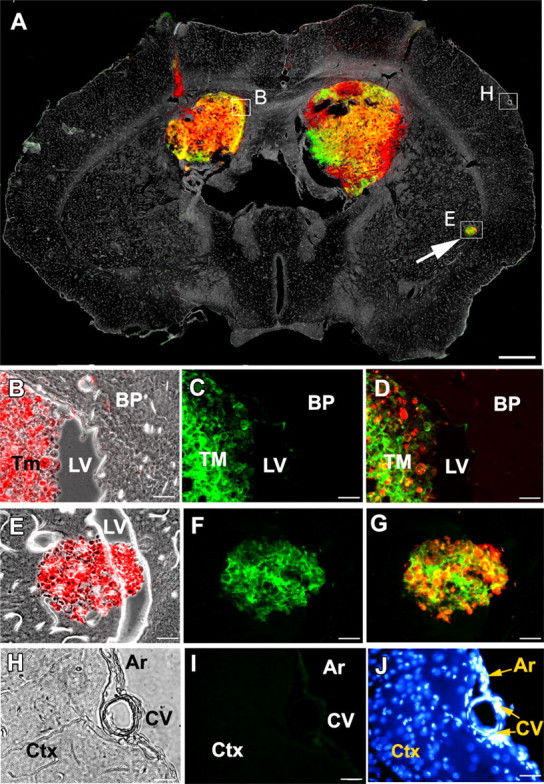
VSVrp30a targets multiple separate xenografts in the brain. A single intravenous injection of VSVrp30a was given to an SCID animal bearing bilateral striatal red fluorescent rU87 grafts and a third incidental tumor mass that has seeded through the CSF to the lateral ventricle. A is a montage of fluorescent and phase-contrast images from a representative coronal brain slice prepared 48 HPI. A shows colocalization of viral-encoded GFP and tumor cell-expressed RFP, simultaneously in all tumor masses. B–D were taken from the ventricular border of the right xenograft as marked on A. E–G are from the incidental tumor mass at the ventricular wall (arrow). H–J are from brain parenchyma, distant from the tumor graft. H shows phase-contrast image, and I shows the absence of viral fluorescence. J shows healthy nuclei of cells in the brain parenchyma (BP), arachnoid (Ar), and cortical blood-vessels (CV). Similarly, no infection was observed outside the tumor in the slice preparation at 72 HPI. Ctx, Cortex; LV, lateral ventricle; Tm, TM, tumor. Scale bars: A, 500 μm; B–J, 20 μm.
VSVrp30a infection causes tumor cell death
VSV infection results in death of tumor cells in vitro. Similarly, VSVrp30a induced widespread tumor cell death in the brain in vivo. Such a fast and efficient elimination of the tumor is important because remaining resistant cells have the potential of repopulating the tumor mass, which is commonly observed in gliomas after conventional treatment. Widespread cell death was detected within the tumor mass. Cell death was indicated by loss of cellular outlines on confocal and light microscopy. Differential interference contrast (DIC) microscopy showed loss of cellular outlines and diffusion of intracellular tumor cell DsRed with fluorescent imaging and nuclear fragmentation assessed with DAPI staining (Fig. 4). Infected tumors stained positively for the cellular apoptosis execution protein activated caspase 3 (Fig. 4 H). There was a statistically significant decrease in the number of cells with intact cellular (p < 0.0001, ANOVA test) and nuclear morphology (p < 0.0001, ANOVA test) and a statistically significant increase in the number of tumor cells immunostained positive for activated caspase 3 (p < 0.0001, ANOVA test) after infection (Fig. 4 I–K). Infected tumor cells underwent lysis several hours after expression of viral proteins. An important observation was that the mouse neovasculature and its cellular constituents, including endothelial cells, were not infected, as shown by the intact vessel architecture in DIC images (Fig. 4 M) and the positive staining for the endothelial marker von Willebrand factor of these non-infected cells (Fig. 4 N,O). After widespread tumor cell lysis, both viral GFP and tumor DsRed intensity decreased in the tumor mass, which can be attributed to loss of cellular integrity and resultant diffusion of intracellular content.
Oncolytic potential of VSVrp30a generalizes to human glioma and mammary carcinoma
VSVrp30a was adapted to human gliomas by passaging it on U87MG cells for many generations. Therefore, to test the hypothesis that the in vivo oncolytic potential of VSVrp30a was not restricted to the cell line on which it was developed, we also used two other types of tumor implanted into the mouse brain. One of the cell types was U118MG, a cell line derived from high-grade human glioma but with different genetic anomalies than found in U87MG. The two glioma types have been shown to differ from each other in their p53 status, c-myc, and epidermal growth factor receptor and PDGF (platelet-derived growth factor) receptor overexpression; they share p14, p16 deletion, and low expression of multidrug resistance gene. U118 cells were stably transfected and sorted using the same protocol we used for U87MG cells and will be called rU118 herein. SCID mice bearing rU118 human glioblastoma xenografts (n = 12 tumors) were killed 48 and 72 h (six tumors each) after a single intravenous injection of VSVrp30a; widespread selective infection, and apoptotic cell death was noted (Fig. 5 A–C). There was no significant statistical difference in VSV induction of apoptosis between rU118 tumors and rU87 tumors 72 HPI (p = 0.1855, Student's t test), suggesting that the virus targeted both types of glioma cells.
Figure 5.
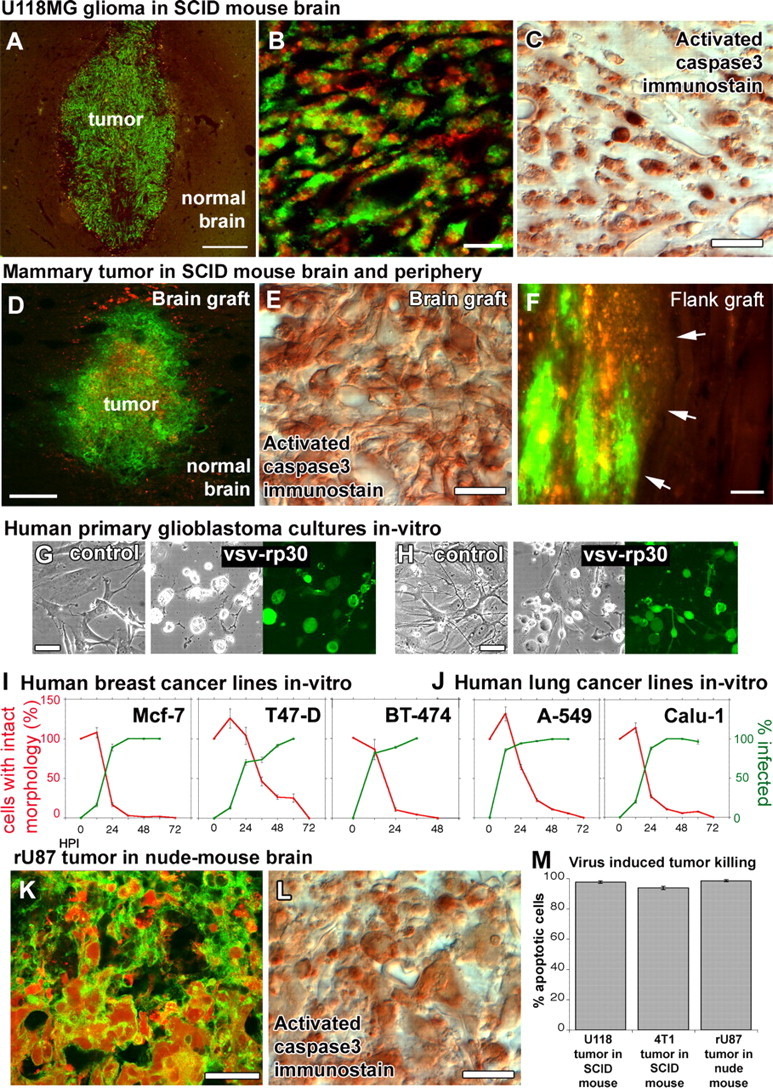
Brain and mammary cancers were targeted in different xenograft models of the brain and the periphery. To exclude the possibility that VSVrp30a tumor targeting might be specific to the rU87 glioma cell line in the SCID mouse model, we tested the virus for its oncolytic activity on different cancer cell lines in vivo and in vitro. Xenografts of rU118 high-grade glioma cells were targeted with VSVrp30a, and the virus spread throughout the whole tumor mass and induced apoptosis within 72 h (A–C). 4T1 mouse mammary cancer cells in the brain (D, E) and in the flank (F) were also effectively and selectively targeted and killed in SCID mice after a single intravenous injection of VSVrp30a (arrows indicate tumor–normal tissue border). Tissue from patients undergoing glioblastoma surgery was cultured and completely infected by VSVrp30a within 36 h (G, H). Three other human breast carcinoma (I) and two human lung carcinoma cell lines (J) were tested in vitro, and all were killed within 48–72 h after the addition of VSVrp30a at a multiplicity of infection of 1. T-cell-deficient nude mice received the same rU87 glioma xenograft and were subjected to the identical virus injection protocol as in the T- and B-cell-deficient SCID mouse model. Comparable with SCID mice, glioma xenografts in the nude mouse brain were efficiently targeted and killed with comparable kinetics (K–M). At 3 d after virus injection, virus-induced apoptosis was observed in the majority of tumor cells. Scale bars: A, D, 300 μm; B, 50 μm; C, 20 μm; E, 30 μm; F, 100 μm; G, H, 30 μm; K, L, 20 μm.
We also tested a mouse mammary carcinoma, the 4T1 cell. 4T1 cells were transiently transfected with the monomeric DsRed gene and injected into the brain and periphery 48 h after transfection. We used 4T1 mouse mammary carcinoma cells to create an animal model with simultaneous peripheral and multiple brain tumors to mimic the setting of a disseminated systemic cancer. VSVrp30a infected all cranial (Fig. 5 D,E) and subcutaneous flank grafts (Fig. 5 F) in SCID mice (n = 2 mice) after a single intravenous injection.
VSVrp30a kills a wide variety of human cancer cells that can cause brain tumors
To test the tumor cell tropism, we tested VSVrp30a on five additional human carcinoma lines. Our choice was guided by the fact that lung and breast cancers are the most common sources for brain metastasis. All human cancer lines tested, including A549, Calu-1 lung carcinoma, and T-47D, MCF7, and BT-474 breast carcinoma, were infected and killed by VSVrp30a at an MOI of 1. All cells exhibited viral green fluorescence by 60 h after infection, and all cells, dividing and nondividing, in the culture dish were killed by 48–72 h after infection (Fig. 5 I,J). In addition, multiple cultures were established from tissue derived from resected glioblastoma surgery. These primary high-grade gliomas were grown in culture and infected with VSVrp30a at an MOI of 1. Complete infection and presence of cytopathic effects were observed within 36 h after virus application (Fig. 5 G,H), indicating that the oncolytic capacity of VSV seen on established glioma and other cancer cell lines could translate to tumor cells directly derived from human cancer patients.
IV delivery of VSV efficiently targets and lyses brain xenografts in nude mice
SCID mice are deficient in both humoral and cellular immune defense. Nude mice, conversely, have an immune defect primarily affecting T-cells. To test whether the less compromised immune status would interfere with tumor targeting, we tested striatal rU87 glioma xenografts in NCr-Nude mice (n = 6 tumors). Tumor xenografts were infected and destroyed with similar efficiency and kinetics in the nude mouse as earlier in the SCID mouse (Fig. 5 K–L). At 72 h after virus inoculation, there was no significant difference in dying cells revealed by activated caspase 3 immunostaining between rU87 tumors in SCID animals, rU118 tumors in SCID animals, and rU87 tumors in nude mice (p = 0.085, ANOVA test). All brain tumors were similarly targeted by the virus.
Intranasal VSV targets olfactory bulb tumors
After intranasal inoculation, VSV can enter the CNS. Nasal mucosal infection results in infection of olfactory neurons, and VSV can gain access to the CNS through the olfactory nerve, which projects to the glomeruli in the olfactory bulb (van den Pol et al., 2002). We tested the hypothesis that VSV could enter the brain along the olfactory nerve and target brain tumors in the olfactory bulb. rU87 cells were stereotactically grafted into one of the two olfactory bulbs in SCID mice. Four days after tumor implantation, animals were inoculated in each nostril with 25 μl containing 2.5 × 107 PFU of VSVrp30a. VSV was transported to the periphery of both olfactory bulbs after intranasal inoculation and infected the olfactory nerve and the tumor but did not show much infection in deeper parts of the control contralateral bulb lacking tumor (Fig. 6 F). SCID mice with VSV in the olfactory bulb survived VSV infection for 2 weeks. A long survival in this tumor model allowed us to demonstrate that few tumor cells could be found in the bulb after the viral challenge. In animals killed 3 DPI (four tumors) or 5 DPI (six tumors), VSV infection was located within the tumor but not in the surrounding parenchyma or in the intact contralateral olfactory bulb (Fig. 6). In nine additional mice killed 7, 10, 15, 16, or 17 d after virus inoculation, we found cellular debris indicative of a viral infection but did not find surviving tumor cells, suggesting that the virus was successful in eliminating the tumor.
Figure 6.
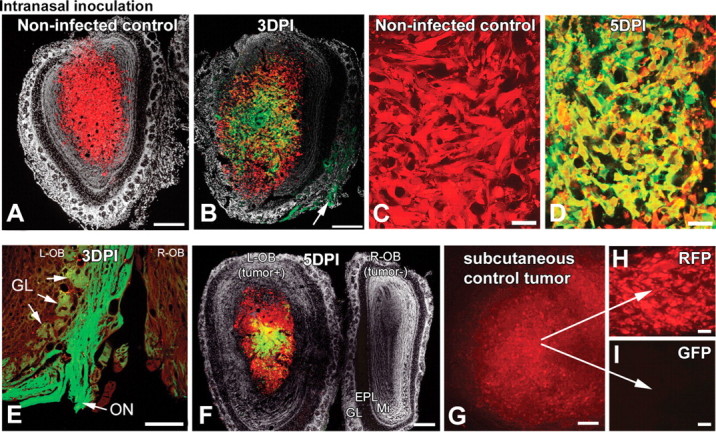
Gliomas within the olfactory bulb are infected after intranasal inoculation. Intranasally given VSV can enter the brain through the olfactory nerve. When rU87 glioma cells were unilaterally implanted into the olfactory bulbs of SCID mice, tumor xenografts grew similar to the striatal xenografts (A, C). Four days after implantation, animals were challenged with intranasal VSVrp30a and the virus was documented to infect the olfactory xenografts (B, D, F). VSV replication was documented in axons within the olfactory nerve (E, and arrow in B) and in some but not all of the olfactory glomeruli, which indicates the port of VSV entry (B, E). Virus can enter both tumor-containing and control bulbs through both olfactory nerves, but only tumor-bearing side shows infection in the central part (F). Simultaneously grown subcutaneous tumor grafts (G) did not show infection after intranasal VSVrp30a application (I), indicating that virus travels through the olfactory route rather than systemically. Stereomicroscopic picture of an intact subcutaneous flank tumor (G) and section of the same tumor (H). L-OB, Left olfactory bulb; R-OB, right olfactory bulb; EPL, external plexiform layer; GL, glomeruli; Mi, mitral cell layer; ON, olfactory nerve. Scale bars: A, B, G, 300 μm; E, F, 200 μm; C, D, H, I, 50 μm.
Although we found evidence of infected olfactory nerve fibers that could serve as entry to the CNS, there is still the possibility that VSV entered the blood circulation through intranasal application and might reach the olfactory bulb via the circulatory system. To address this question, we placed peripheral sentinel tumors into the flank of three animals simultaneously with olfactory tumor grafting. Seven days later, mice received 2.5 × 107 PFU VSVrp30a into each nostril. At 3 d after inoculation, both olfactory bulb and peripheral tumors were analyzed. Interestingly, no viral infection was found at the remote sentinel tumor (Fig. 6 G–I), whereas strong GFP expression was evident at the nasal mucosa, olfactory nerve and bulb, and at the olfactory tumor graft, suggesting a direct path of VSVrp30a through the olfactory nerve route.
Real-time microscopy of VSVrp30a oncolysis in cortical glioma xenografts
To study the kinetics of VSV oncolysis, we also used a time-lapse in situ human brain tumor imaging model. rU87 cells were implanted subpially into the SCID mouse cortex (n = 3) under a glass coverslip permanently mounted on a parasagittal craniectomy (Fig. 7 A,B). By imaging tumor cells in the living animal brain using fluorescent and laser confocal microscopy, we could detect VSV infection as early as 16 HPI after tail vein injection of 107 PFU of VSVrp30a (Fig. 7 C) and follow viral infection over several days in the live brain. Tumor growth and infection were observed every 4–8 h over a course of 4 d. VSV infection spread through the whole tumor bulk within 96 h (Fig. 7 C); although multiple points of infection were seen, the majority of tumor cells appeared to be infected by a locally spreading VSV infection. Laser confocal microscopy gave us a cellular or even subcellular resolution, allowing us to see not only single cells but also their processes. Using this technique, we found that VSV infected single infiltrating red tumor cells but not the surrounding normal brain tissue (Fig. 7 D,E). Vascular patency and circulation appeared normal in tumor vessels within the infected area even at advanced stages of tumor cell lysis; in live real-time imaging, erythrocytes could be seen moving through the uninfected walls of the blood vessels within the tumor even when all surrounding tumor cells were infected and dying.
Figure 7.
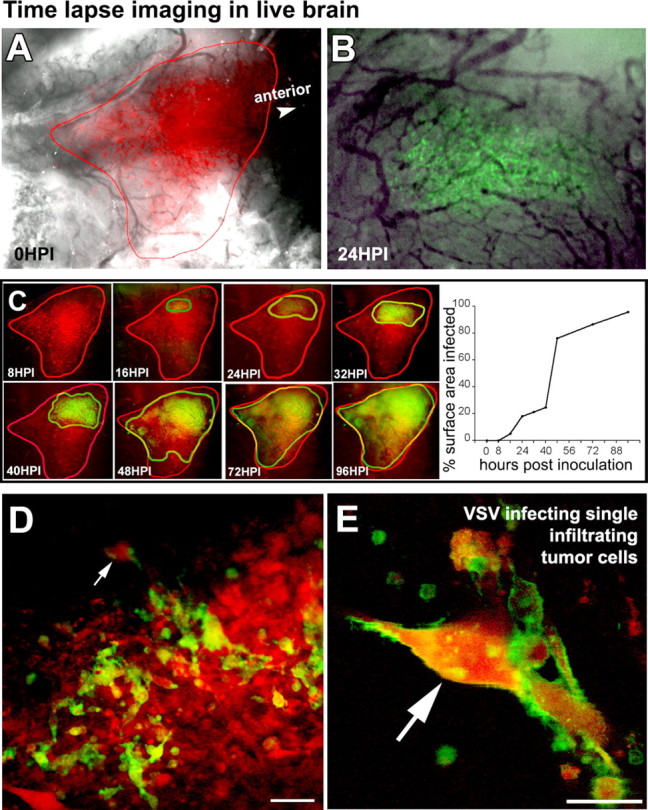
Time-lapse confocal imaging of VSV targeting of glioma in live mouse brain. Kinetics of viral oncolysis were analyzed using rU87 cells implanted subpially in SCID mouse cortex under a cranial glass coverslip. A shows the localization of the tumor cells in the living animal brain. Fluorescence microscopy during 96 h after an intravenous injection of VSVrp30a showed that the viral GFP expression became apparent 16 h after inoculation (B). The infection spread within the tumor and progressed to encompass the whole tumor mass (C). In situ laser confocal microscopy revealed that single infiltrative tumor cells were targeted after such an intravenous viral delivery. Note that single tumor cells infiltrating the cortex (arrow) express viral membranous GFP (D, E). Scale bars: D, 50 μm; E, 10 μm.
Mechanism of VSV tumor targeting
The blood–brain barrier may be leaky in some types of tumors. In normal circumstances, VSV does not enter the CNS through the hematogenous route. Therefore, to explore what makes tumor xenografts permeable to VSV, we tested blood–brain barrier integrity in the xenograft neovasculature. Strong Evans Blue staining was detected within bilateral striatal tumors in SCID (n = 4 tumors) and nude mice (n = 2 tumors) 1 h after intravenous injection of the dye. On all sections, the Evans Blue stained area completely overlapped with the red tumor mass, indicating a uniform permeability of the tumor to dye (Fig. 8 A,B). Control experiments were performed to address the effect of needle injury alone on Evans Blue leakage. Evans Blue leakage was observed only immediately after the injury. Ten days after injury, the same time given for tumor formation, no dye leakage could be observed (Fig. 8 C). All brains showed dye leakage in circumventricular areas in which the blood–brain barrier is normally permissive for dye infiltration.
Figure 8.
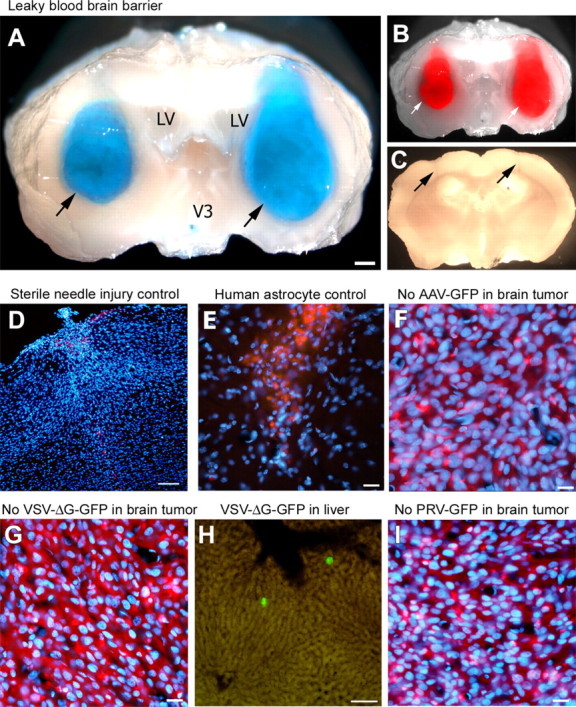
VSV selectively enters brain tumors through a leaky blood–brain barrier but does not infect control wounds or normal brain cell implants. After intravenous injection, Evans Blue leaked into tumor xenografts in which it colocalized (arrows) with tumoral RFP fluorescence, indicating an incompetent blood–brain barrier within the entire tumor (A, B). No Evans Blue leakage was observed in areas with needle injury only (see arrows) (C). Nonspecific brain damage inflicted by a sterile needle injury (D) or human astrocyte transplants (E) in the same brain region used for tumor grafting was not enough to allow VSVrp30a entry after intravenous injection of the same dose of VSVrp30a as in experimental animals. Tumor targeting in the brain required replication-competent VSV, and replication-deficient VSV (VSVdeltaG-GFP) did not infect rU87 xenografts in the mouse brain, but numerous infected cells were documented in the liver (G, H). No infection in the brain tumor, liver, or spleen was detected after intravenous injection of pseudorabies virus (PRV–GFP) or adeno-associated virus (AAV–GFP), both of which are known to infect glioma cells (F, I). LV, Lateral ventricle. Scale bars: A, 300 μm; D, H, 100 μm; E, G, 50 μm; F, I, 20 μm.
Sterile needle injury or human astrocyte transplants do not get infected
To test the hypothesis that tumor targeting of VSV was not a consequence of nonspecific parenchymal brain damage at the injection site, bilateral sterile stab wounds were done using the same (Hamilton point style 2) needle at the same striatal coordinates (n = 4 wounds). When the same amount of intravenous virus was injected 10 d after this needle injury, we did not detect any viral GFP within the brain (Fig. 8 D). In parallel, we addressed the question of whether the virus targets human cells, or transplanted cells in general, rather than tumor cells specifically. When human (n = 4 injections) or mouse (n = 4 injections) normal astrocytes were transplanted into the same coordinates used for tumor cells, no viral GFP expression was detected in the brain 72 h after intravenous virus injection (Fig. 8 E). These data support the view that VSVrp30a does not enter the brain because of the injury of cell transplantation, nor does it target normal human or mouse cells transplanted into the brain. Infection of glioma xenografts appears specific for replication-competent VSV. To test whether replication was essential for effective tumor targeting, we tested a recombinant replication deficient VSV (VSV–ΔG–GFP) for its capacity to infect intracranial xenografts. This virus can infect cells but, because of the deletion of the VSV-G gene, cannot produce infectious progeny, resulting in only a single cycle of infection. Intravenous injection of 107 PFUs of VSV–ΔG–GFP to animals with bilateral striatal tumors (n = 2) did not result in tumor infection (Fig. 8 G). As a positive control, GFP-expressing scattered cells were detected in the liver after intravenous injection of VSV–ΔG–GFP (Fig. 8 H). To exclude the possibility that targeting of intracranial tumor xenografts by VSV was not the result of nonspecific viral entry, we used two other viruses that were previously shown to infect or lyse glioma cells in vitro. Intravenous injection of a replication-incompetent adeno-associated virus type 2 (AAV–GFP; n = 2; 3 DPI) or a replication-competent pseudorabies virus (PRV–GFP; n = 2; 2 DPI) did not result in infection of intracranial rU87 xenografts (Fig. 8 F,I). All three of these viruses were shown previously to infect tumor cells in vitro, and this was reconfirmed in vitro for the stocks used in this study.
Mechanism of VSV tumor selectivity
Our data are consistent with the view that VSV selectively targets glioma in the brain, suggesting that some mechanism underlies the relative selectivity of the virus to the tumor. A correlation exists between VSV infection and various defects in the interferon system in tumor cells (Stojdl et al., 2000; Balachandran et al., 2001). We previously compared a series of five different brain tumor cell lines and normal human control astrocytes and demonstrated an interferon-dependent selectivity of VSV for glial tumor cells over their nontumor counterpart (Wollmann et al., 2007). However, antiviral defense may not exclusively account for the selectivity, and several additional mechanisms have been proposed (Wagner and Rose, 1996; Balachandran and Barber, 2004; Barber, 2005; Lyles and Rupprecht, 2007). No specific receptor has been identified for VSV; after binding, the virion undergoes clathrin-mediated endocytosis, and, after acidification in the endosome, the uncoated virus enters the cytoplasm. To test the hypothesis that VSVrp30a binds and enters tumor cells with a higher efficiency, we compared viral binding and entry in rU87 glioma cells and primary normal adult human astrocytes in vitro using quantitative PCR (Fig. 9). Viral quantity was normalized against cellular GAPDH. In separate quantitative PCR experiments, we confirmed that GAPDH was expressed at comparable quantities in normal astrocytes and glioma cells (results not shown). To test for the quantity of VSV that binds to cell surface, we incubated the cells with virus at 4°C. After 20 min of incubation, we washed the cells five times with buffered medium before RNA extraction. Compared with human astrocytes, the quantity of cell-bound VSVrp30a at 20 min after infection appeared to be slightly (1.8 times) greater for rU87 glioma cells (statistically significant; p = 0.005, n = 6, Student's t test) (Fig. 9 A). When we quantified surface-bound plus intracellular VSVrp30a after a 30 min incubation at an MOI of 10, we again found a somewhat greater viral quantity in rU87 cells (p > 0.05, n = 6, Student's t test) (Fig. 9 B). We then compared how efficiently the cells internalized VSV. For this purpose, we incubated the cells with VSVrp30a at an MOI of 10 for 30 min and incubated them for another 1 h after changing the medium three times to eliminate unbound virus. At 90 min after infection, cells were washed five times with PBS and trypsinized, and intracellular VSV was quantified. Finally, to measure only the endosomal fraction, the same experiment was duplicated in the presence of ammonium chloride, which inhibits viral uncoating from the endosomes. At 90 min after infection, we found 2.6 times more virus in the endosomes and 3.6 times more virus intracellularly in rU87 cells compared with normal astrocytes (both statistically significant; p = 0.001, n = 6, and p = 0.003, n = 6, respectively, Student's t test) (Fig. 9 C,D). These data suggest that VSV binding to the plasma membrane and virus internalization appear to be greater in glioma than in normal astrocytes. These factors may contribute to the greater level of VSV infection found in glioma.
Figure 9.

VSV binding and entry is more efficient in rU87 glioma cells. Quantification of VSV by RT-PCR at different intervals in distinct cellular compartments indicated that VSVrp30a binds and enters the rU87 human glioma cell line more efficiently than its normal cellular counterpart. Cell-surface-bound VSV after 20 min of incubation with the virus was significantly greater in rU87 cells compared with normal human astrocytes (A). When surface-bound plus internalized virus was quantified at 30 min, there was a trend toward more virus in rU87 but the difference was not significant (B). At 90 min after infection, there was significantly more VSV in the endosomal compartment in rU87 cells (C) when compared with astrocytes. Also the total amount of intracellular virus was significantly greater in rU87 cells (D). All experiments were done in triplicate using VSVrp30a at an MOI of 10.
Discussion
Metastatic tumors and glioblastomas make up the majority of cancers in the brain and generally result in death within months; there is currently no cure. This therapeutic failure is mostly attributable to the incomplete elimination of all cancer cells. We show that intravenous inoculation of VSVrp30a resulted in selective infection and destruction of human glioblastoma and mammary cancer cells transplanted into the mouse brain. Systemic delivery of a limited dose of the replication-competent VSVrp30a resulted in selective infection of multifocal tumors and in production of viral progeny that spread throughout the tumor within 72 h. Such a local self-amplification is not found with other conventional cytotoxic treatments such as chemotherapy or radiation. Different types of cancer cells were targeted, with sparing of normal brain cells, underlining VSV as a potential therapeutic against multiple types of cancer in brain.
Tumor targeting by systemic delivery
Gliomas and brain metastases are different in their tumor biology, and growth pattern and kinetics. The mode of action of viral therapeutics differs fundamentally from conventional methods such as radiation or chemotherapy. The unique property of VSV that separates it from many other conventional treatment technologies is that VSV shows oncolytic activity against a broad spectrum of cancer types, including both systemic cancers and primary cancers of the brain, despite substantial genotypic variability. We tested VSV as a potential oncolytic candidate that can be intravenously administered to treat multiple types of brain cancer regardless of the kind of tumor or the status of systemic disease.
Our study shows that a single intravenous injection of VSVrp30a effectively targets multifocal or infiltrative tumors simultaneously in the brain and periphery. By 3 d after inoculation, tumors generally were completely or almost completely infected, and tumor cells were dying or dead. In contrast to VSV, several other oncolytic viruses (adenovirus, herpes simplex) have been applied by systemic injections and were able to infect only a small part of the tumor (Rainov and Kramm, 2001; Lyons et al., 2006; Liu and Kirn, 2007). In our experiments, after intravenous injection, selective viral infection was found in all tumors and tumor types, and in both SCID and nude mice on different genetic backgrounds, indicating a high efficiency of VSV-tumor targeting. A notable finding was the demonstration of simultaneous oncolytic infection in both subcutaneous flank and brain tumors in the same animal after a single systemic virus injection. In addition to demonstrating the efficiency of a systemic VSV-based therapy, this finding emphasized the potential of VSV to infect widely metastatic cancers. Given the oncolytic activity of VSV against a range of cancer cells with diverse genetic defects (Stojdl et al., 2003; Lichty et al., 2004; Barber, 2005), an intravenous injection of VSV can potentially be used to target simultaneously both brain and peripheral metastases of a systemic cancer.
Direct intratumoral injection may be useful for some tumors. To date, such intratumoral injections of viruses have resulted in a very limited viral spread around the injection site (Pulkkanen and Yla-Herttuala, 2005). The ability of VSV to infect multiple independent central and peripheral sites and spread in them is substantive, making it possible to treat disseminated disease and even target minute tumor colonies that are otherwise difficult to detect. Xenograft models are limited in that human gliomas often grow expansively rather than infiltratively in mice. Nonetheless, infiltrating cells were observed within the host brain, and these were targeted by VSV.
Olfactory nerve route
We also tested a novel approach of targeting brain tumors through neural circuits. VSV can enter the brain through the olfactory nerve (Reiss et al., 1998; van den Pol et al., 2002). Inoculation of the olfactory mucosa led to complete infection of the tumor via the olfactory nerve; after infection, tumor cells were absent from the olfactory bulb. Relatively little infection was found outside the tumor in the tumor- bearing bulb or in the contralateral normal bulb. This finding indicates that neural routes of VSV dissemination in brain can be exploited for targeting tumors in specific regions and facilitating of distant tumors or infiltrative tumor cells with minimal infection in normal brain parenchyma.
Detection of recombinant virus and tumor
To localize the virus, we used a green fluorescent reporter gene coupled to the VSV-G gene in the position of the fifth VSV gene. This would shift the viral L-gene to the sixth position, resulting in attenuated L-protein synthesis and a slight reduction in replication (Dalton and Rose, 2001), an advantage when considering treatment of the brain. Live microscopic imaging of the brain allowed us for the first time to follow the time course with single-cell resolution, from before inoculation to a point when VSV had spread throughout the tumor. In experiments in which we imaged VSV oncolysis through a glass window above the brain using time-lapse confocal laser microscopy, green viral infection of red cortical tumors spread throughout the tumor, mostly by local expansion of the area of infected cells. Blood vessels appeared mostly undamaged even late in tumor infection. Furthermore, vessel cells that stained positive for the endothelial marker von Willebrand factor appeared to be spared from infection, favoring the hypothesis that virus entry is facilitated by vessel leakage. With live recording, blood cells could be seen moving through patent vessels throughout the tumor. In control experiments, VSV did not enter the normal brain through the vasculature, nor did it enter the brain away from the tumor in mice bearing tumors, raising the question as to how VSV entered the brain. We tested the hypothesis that a local disruption of the BBB existed using intravenous dye injections and found dye entered the brain only in areas of tumors but not at ∼10-d-old control needle injuries, indicating an incompetent BBB. This leakage was selective for the tumor vasculature because additional experiments showed that simple focal brain damage or implantation of nonmalignant cells into the brain did not result in VSVrp30a infection in the CNS. The ability to cross the BBB does not generalize to all viruses because we found no evidence that pseudorabies virus or AAV infected the tumor. Even a leaky BBB prevented adenovirus and herpes from entering brain tumors (Rainov and Kramm, 2001). Furthermore, a replication-deficient VSV (Miller et al., 2004; Okuma et al., 2006) that can replicate only in the first cell infected without subsequent release of infectious virions showed no tumor invasion; that the replication-incompetent virus can be detected was shown by the presence of a few solitary infected, GFP-expressing liver cells and by in vitro experiments in which the virus showed robust infection and reporter gene expression in cultured glioma. This suggests that ongoing viral replication is needed to initiate tumor infection. VSV entering the tumor may therefore arise, at least in part, from infectious daughter virions released by infected peripheral cells.
Infiltrating tumor cells that migrate away from the main tumor body into normal brain escape local treatments and are an important cause of tumor recurrence. Confocal imaging in both live and fixed sections demonstrated that these cells are selectively targeted by VSVrp30a, most likely a result of virus progeny diffusing from the main tumor bulk. Despite the heavy infection of the tumor and infiltrative tumor cells, we did not observe infection in neighboring normal astrocytes or neurons in immediate contact with the tumor, indicating the high tumor specificity of the infection. In this study, VSV rapidly infected and killed two types of glioma and one type of mammary cancer in vivo. It also killed five systemic carcinoma cell lines (lung and breast cancer) and cells from two primary glioblastoma in vitro, indicating the potential for VSV to kill a diverse array of cancers that may arise or metastasize in the brain. In addition to its diverse tropism, VSV, unlike some other viruses, is not dependent on specific genetic tumor defects such as the p53 status, RAS or myc oncogene activity, cell cycle status, or hypoxic environment of the tumor cell (Balachandran et al., 2001; Connor et al., 2004; Wollmann et al., 2005).
Mechanisms of viral selectivity for cancer cells
We found a remarkably high degree of selective infection of the brain tumors with relatively little infection of the surrounding normal brain; the ratio of infected tumor cells to infected normal brain cells was 10,000:1, or better. A large fraction of various tumors display defects in their interferon system (Stojdl et al., 2003). VSV is highly sensitive to interferon-mediated antiviral defense mechanisms with which normal cells effectively control VSV infection and spread (Trottier et al., 2005). We and others demonstrated previously VSV mutants and recombinants that are enhanced in their susceptibility to interferon have shown promise in targeting cancers (Stojdl et al., 2003; Ahmed et al., 2004; Ebert et al., 2005). VSVdeltaM51, which has an enhanced interferon susceptibility, has also been shown to target gliomas systemically and prolong survival (Lun et al., 2006). We took an alternative strategy and isolated VSVrp30a by raising VSV for many generations on gliomas under selective pressure (Wollmann et al., 2005). VSVrp30a showed enhanced binding and internalization in glioma cells, mechanisms that could contribute to tumor selectivity.
Because human cancers show more complex genotypic alterations compared with rodent tumors, we used human glioblastoma models in immunodeficient mice (Hahn and Weinberg, 2002). A limiting factor in this study is that such mice are more susceptible to viral infections of any kind. Wild-type mice and humans survive peripheral VSV infections, but immunodeficient mice do not, making it difficult to study long-term results of VSV targeting of human cancers in animals. Despite the vulnerability of immunodeficient mice to VSV, we nevertheless demonstrated a therapeutic time window of complete tumor oncolysis with minimal brain infection outside the tumor. With longer survival times than used in the present study, VSV could potentially infect normal brain cells. Attenuated recombinant VSV strains may provide an additional margin of safety when used in the brain of nonhuman primates (Johnson et al., 2007) and in mice (Flanagan et al., 2003; Clarke et al., 2007) after intracerebral injections or intranasal administration.
Footnotes
This work was supported by National Institutes of Health Grants A1/NS48854 and CA124737 (A.N.v.d.P.). K.Ö. was supported by an Anna Fuller Molecular Oncology Fellowship. We thank Dr. P. K. Ghosh for expertise in generating fluorescent tumor cells, Dr. D. D. Spencer for supplying human brain tissue, Y. Yang and V. Rogulin for excellent technical facilitation, and Dr. J. Rose for helpful suggestions.
References
- Aghi M, Rabkin S. Viral vectors as therapeutic agents for glioblastoma. Curr Opin Mol Ther. 2005;7:419–430. [PubMed] [Google Scholar]
- Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Balachandran S, Barber GN. Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell. 2004;5:51–65. doi: 10.1016/s1535-6108(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Balachandran S, Porosnicu M, Barber GN. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol. 2001;75:3474–3479. doi: 10.1128/JVI.75.7.3474-3479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. VSV-tumor selective replication and protein translation. Oncogene. 2005;24:7710–7719. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- Central Brain Tumor Registry of the United States. Chicago: Central Brain Tumor Registry of the United States; 2005. Statistical report: primary brain tumors in the United States, 1998–2002. [Google Scholar]
- Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J Virol. 2007;81:2056–2064. doi: 10.1128/JVI.01911-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JH, Naczki C, Koumenis C, Lyles DS. Replication and cytopathic effect of oncolytic vesicular stomatitis virus in hypoxic tumor cells in vitro and in vivo. J Virol. 2004;78:8960–8970. doi: 10.1128/JVI.78.17.8960-8970.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton KP, Rose JK. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- DeAngelis L. M. Brain tumors. N Engl J Med. 2001;344:114–123. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- Ebert O, Harbaran S, Shinozaki K, Woo SL. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 2005;12:350–358. doi: 10.1038/sj.cgt.7700794. [DOI] [PubMed] [Google Scholar]
- Flanagan EB, Schoeb TR, Wertz GW. Vesicular stomatitis viruses with rearranged genomes have altered invasiveness and neuropathogenesis in mice. J Virol. 2003;77:5740–5748. doi: 10.1128/JVI.77.10.5740-5748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, Gore M, Ironside J, MacDougall RH, Heise C, et al. a controlled trial of intratumoral ONYX-015, a selectivelyreplicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Liu TC, Kirn D. Systemic efficacy with oncolytic virus therapeutics: clinical proof-of-concept and future directions. Cancer Res. 2007;67:429–432. doi: 10.1158/0008-5472.CAN-06-2871. [DOI] [PubMed] [Google Scholar]
- Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D, Lichty B, Power A, Johnston RN, Hamilton M, et al. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- Lyles DS, Rupprecht CE. Rhabdoviridae. In: Knipe D. M., Howley P. M., editors. Fields Virology. Lippincott: Williams and Wilkins; 2007. pp. 1363–1408. [Google Scholar]
- Lyons M, Onion D, Green NK, Aslan K, Rajaratnam R, Bazan-Peregrino M, Phipps S, Hale S, Mautner V, Seymour LW, Fisher KD. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol Ther. 2006;14:118–128. doi: 10.1016/j.ymthe.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Miller MA, Lavine CL, Klas SD, Pfeffer LM, Whitt MA. Recombinant replication-restricted VSV as an expression vector for murine cytokines. Protein Expr Purif. 2004;33:92–103. doi: 10.1016/j.pep.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- Okuma K, Boritz E, Walker J, Sarkar A, Alexander L, Rose JK. Recombinant vesicular stomatitis viruses encoding simian immunodeficiency virus receptors target infected cells and control infection. Virology. 2006;346:86–97. doi: 10.1016/j.virol.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Drug and gene delivery to the brain: the vascular route. Neuron. 2002;36:555–558. doi: 10.1016/s0896-6273(02)01054-1. [DOI] [PubMed] [Google Scholar]
- Posner JB, Chernik NL. Intracranial metastases from systemic cancer. Adv Neurol. 1978;19:579–592. [PubMed] [Google Scholar]
- Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–598. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- Puumalainen AM, Vapalahti M, Yla-Herttuala S. Gene therapy for malignant glioma patients. Adv Exp Med Biol. 1998;451:505–509. doi: 10.1007/978-1-4615-5357-1_78. [DOI] [PubMed] [Google Scholar]
- Rainov NG, Kramm CM. Vector delivery methods and targeting strategies for gene therapy of brain tumors. Curr Gene Ther. 2001;1:367–383. doi: 10.2174/1566523013348445. [DOI] [PubMed] [Google Scholar]
- Rainov NG, Ren H. Oncolytic viruses for treatment of malignant brain tumours. Acta Neurochir Suppl. 2003;88:113–123. doi: 10.1007/978-3-7091-6090-9_17. [DOI] [PubMed] [Google Scholar]
- Reiss CS, Plakhov IV, Komatsu T. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann N Y Acad Sci. 1998;855:751–761. doi: 10.1111/j.1749-6632.1998.tb10655.x. [DOI] [PubMed] [Google Scholar]
- Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. Pseudorabies virus expressing enhanced green fluorescent protein: A tool for in vitro electrophysiological analysis of transsynaptically labeled neurons in identified central nervous system circuits. Proc Natl Acad Sci USA. 2000;97:9264–9269. doi: 10.1073/pnas.97.16.9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant-temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Trottier MD, Palian BM, Reiss CS. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology. 2005;333:215–225. doi: 10.1016/j.virol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Dalton KP, Rose JK. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J Virol. 2002;76:1309–1327. doi: 10.1128/JVI.76.3.1309-1327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RR, Rose JK. Rhabdoviridae: the viruses and their replication. In: Fields B. N., Knipe D. M., editors. Fields virology. Raven Press: Philadelphia; 1996. [Google Scholar]
- Wollmann G, Tattersall P, van den Pol AN. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79:6005–6022. doi: 10.1128/JVI.79.10.6005-6022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmann G, Robek MD, van den Pol AN. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J Virol. 2007;81:1479–1491. doi: 10.1128/JVI.01861-06. [DOI] [PMC free article] [PubMed] [Google Scholar]