

Abstract
Mutations in the hypoxia-inducible factor angiogenin (ANG) have been identified in Amyotrophic Lateral Sclerosis (ALS) patients, but the potential role of ANG in ALS pathogenesis was undetermined. Here we show that angiogenin promotes motoneuron survival both in vitro and in vivo. Angiogenin protected cultured motoneurons against excitotoxic injury in a PI-3-kinase/Akt kinase-dependent manner, whereas knock-down of angiogenin potentiated excitotoxic motoneuron death. Expression of wild-type ANG protected against endoplasmic reticulum (ER) stress-induced and trophic-factor-withdrawal-induced cell death in vitro, whereas the ALS-associated ANG mutant K40I exerted no protective activity and failed to activate Akt-1. In SOD1G93A mice angiogenin delivery increased lifespan and motoneuron survival, restored the disease-associated decrease in Akt-1 survival signaling, and reversed a pathophysiological increase in ICAM-1 expression. Our data demonstrate that angiogenin is a key factor in the control of motoneuron survival.
Keywords: ALS, SOD1, angiogenin, motoneuron, hypoxia, neuroprotection
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by relatively selective motoneuron loss in the spinal cord, brainstem and motor cortex, for which there is no cure (Cleveland and Rothstein, 2001). Although the specific etiology of ALS is largely unknown, 20% of familial ALS cases carry mutations in the gene encoding Cu/Zn superoxide dismutase-1 (SOD1) (Rosen et al., 1993). Transgenic mice over-expressing human mutant SOD1 develop a disease phenotype and pathology which closely mimics that seen in ALS patients (Gurney et al., 1994).
Recent studies have suggested that defects in hypoxic signaling and/or increased sensitivity of motoneurons to hypoxia may underlie this selective vulnerability (Oosthuyse et al., 2001; Lambrechts et al., 2003; Greenway et al., 2006). This hypothesis was first suggested by the finding that mice with deletions in the hypoxic response element of the vascular endothelial growth factor (VEGF) gene develop adult-onset motoneuron degeneration and paralysis (Oosthuyse et al., 2001). However, mutations in VEGF or genes involved in hypoxic-inducible factor (HIF) signaling have not been identified in ALS patients (Lambrechts et al., 2003).
We have recently reported 7 missense mutations in the hypoxia-inducible gene ANG in 15 individuals, 4 with familial and 11 with apparently “sporadic” ALS (Greenway et al., 2006). ANG codes for angiogenin, a 14.1 kDa protein, which is a member of the pancreatic ribonuclease A (RNaseA) superfamily (Fett et al., 1985). Angiogenin is a potent inducer of neovascularization (Moroianu and Riordan, 1994; Kishimoto et al., 2005; Tsuji et al., 2005). In endothelial and tumor cells, angiogenin can undergo nuclear translocation, and is able to bind to DNA and stimulate rRNA transcription (Moroianu and Riordan, 1994; Tsuji et al., 2005). Five of the reported missense mutations (Q12L, K17E, K17I, R31K, C39W, K40I) affect functionally important residues involved in nuclear import, nuclear localization, or ribonucleolytic activity (Shapiro et al., 1989; Greenway et al., 2006). Subsequent genetic studies have identified further ANG mutations that may interfere with nuclear localization, ribonucleolytic, and angiogenic activity (Crabtree et al., 2007; Wu et al., 2007). Previously, we could detect significant angiogenin expression in motoneurons (Greenway et al., 2006), a finding confirmed in other studies (Crabtree et al., 2007; Wu et al., 2007). We could also provide preliminary evidence for a neuroprotective activity of angiogenin in vitro (Kieran et al., 2005). In this study we examined the biological role of angiogenin and the ALS-associated angiogenin mutant K40I in controlling motoneuron survival, and analyzed the signal transduction pathways involved in this protection. We also show that treatment of SOD1G93A mice with angiogenin, even after disease onset, significantly increased motoneuron survival.
Materials and Methods
Primary motoneuron cultures.
Primary motoneuron cultures were established from E13 C57 mouse embryos (Camu and Henderson, 1992). Spinal cord ventral horns were dissected, the ventral horn tissue was trypsinized in Ham F10 medium (Invitrogen) and dissociated in complete medium containing 0.4% BSA and 0.1 mg/ml DNase1 (Sigma) and seeded at 5 × 104/ml. After 7 d cultures were exposed to either AMPA (50 μm, 24 h, Tocris Cookson) or tunicamycin (500 nm, Qbiogene) and cotreated with human recombinant angiogenin protein (100 ng/ml, R&D Systems), heat-denatured angiogenin, or the PI-3-K inhibitor wortmannin (100 nm, Sigma).
Motoneuron survival in vitro in primary motoneuron cultures was determined by MTT assay as well as direct counts of motoneuron survival, as previously described (Kieran et al., 2005).
Cloning of angiogenin and site-directed mutagenesis.
The full-length cDNA of human ANG was amplified by PCR (sense primer: 5′ GGAGCCTGTGTTGGAAGAGA-3′; antisense primer: 5′-TGAATGTTG-CCACCACTGTT-3′) and inserted into the PCR-Blunt II-TOPO vector (Invitrogen). K40I mutation was inserted in the sequence using QuikChange XL site-directed mutagenesis kit (Stratagene). The ANG fragment was subcloned into pIRES2-DsRed2 vector (Clontech).
Expression and purification of recombinant angiogenin protein.
Two truncated variants of human ANG, encoding the wild-type (WT) or K40I mutant protein, were inserted as 0.4 kb BamHI-EcoRI fragments into the pcDNA4-HisMaxA vector (Invitrogen). Purified plasmids were used to transfect HEK293 cells using Lipofectamine 2000 (Invitrogen).
NSC34 transient transfection and cell survival.
Cells were transfected with pIRES-DsRed2 plasmids using Lipofectamine 2000 and 24 h later exposed to tunicamycin (500 nm) for 24 h. Apoptotic cell death in transfected cells (DsRed2 fluorescence) was assessed after staining of nuclei with Hoechst 33258 (Sigma, 1 μg/ml). Only strongly condensed and/or fragmented nuclei were considered apoptotic.
Western blotting.
Primary antibodies used in this study were Akt-1, phospho-Akt, ERK1/2 and phosho-ERK1/2 (1:1000; Cell Signaling Technology), ICAM-1 (1:500; R&D Systems), β-actin, (1:2500), and β-tubulin (1:2000) (both Sigma).
Transfection with siRNA.
Cells were transfected with predesigned siRNA's to murine ANG1 (siGENOME-ON-TARGETplus, Dharmacon) using SMARTpool reagents (Dharmacon), according to manufacturer's instructions.
Treatment of SOD1G93A mice with ANG.
TgN[SOD1-G93A]1Gur (Jax Laboratories) were injected daily with 1 μg of human recombinant angiogenin i.p. Uptake was studied using fluorescein-labeled angiogenin (EZ-label Fluorescein Protein Labeling, Pierce). For lifespan, body weight and stride length each experimental group (treatment from 50 or 90 d) contained angiogenin-treated (n = 12) and untreated SOD1G93A (n = 15) littermates, as well as untreated WT littermates (n = 15). Probability of survival was determined using Kaplan–Meyer analysis.
Assessment of disease progression in vivo.
Body weight (Kieran et al., 2004), PAGE test (Weydt et al., 2003), and stride length (Gurney et al., 1994) assessments were performed blind, twice weekly. The number of surviving sciatic motoneurons was assessed by counting sciatic motoneurons in each ventral horn on every third section between the L2 and L5 levels of the spinal cord (Kieran et al., 2004).
Results
Angiogenin protects primary motoneurons from stress-induced cell death in vitro
It has been proposed that excitotoxicity involving overactivation of Ca2+ permeable α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors and selective loss of excitatory amino acid transporters (EAATs), may be involved in ALS pathogenesis (Rothstein et al., 1990). To investigate whether angiogenin has a neuroprotective activity against excitotoxic injury, primary ventral horn motoneuron cultures were exposed to the glutamate agonist AMPA, and treated with human recombinant angiogenin protein or vehicle. Direct counts of motoneuron survival in immunolabeled and trypan-blue-stained motoneuron cultures demonstrated that angiogenin had a direct protective effect on motoneurons (Fig. 1A). Endoplasmic reticulum (ER) stress-induced apoptosis has also been implicated in the pathophysiology of ALS (Kikuchi et al., 2006). Tunicamycin, an inhibitor of N-linked glycosylation, was applied to induce ER stress. Angiogenin also protected motoneurons from ER stress-induced neuronal injury (Fig. 1B).
Figure 1.
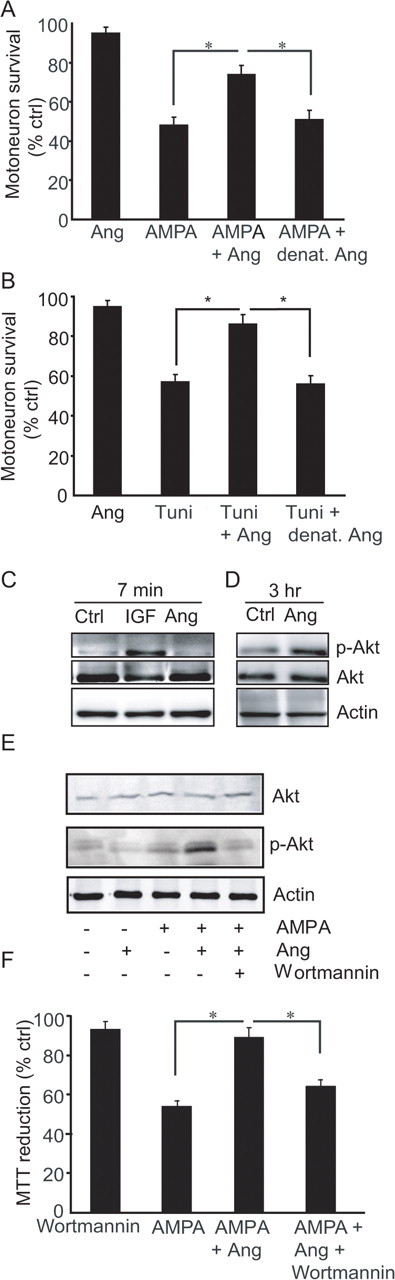
Angiogenin is a neuroprotective factor for cultured motoneurons. A, Direct counts of motoneuron survival in primary ventral horn motoneuron cultures exposed to AMPA (50 μm, 24 h), AMPA and angiogenin (100 ng/ml), angiogenin alone and AMPA and heat denatured angiogenin (*p ≤ 0.05, mean ± SEM, n = 12). B, Direct counts of motoneuron survival in primary motoneuron cultures exposed to tunicamycin (500 nm, 24 h) and/or angiogenin (100 ng/ml, *p ≤ 0.05, mean ± SEM, n = 12). C–E, Western blot analysis of Akt-1 and Ser473-phosphorylated Akt-1 (p-Akt) expression in angiogenin- and IGF-I-treated (C, D), and angiogenin-plus AMPA-treated (E) primary motoneuron cultures. F, Inhibition of the PI-3-K pathway using wortmannin (100 nm, 24 h) reduces the neuroprotective effect of angiogenin in primary motoneuron cultures exposed to AMPA (*p ≤ 0.05, mean ± SEM, n = 3).
In endothelium, angiogenin expression is required for cell signaling of growth factors including basic fibroblast growth factor, epidermal growth factor and VEGF (Kishimoto et al., 2005). Survival signaling through these growth factors involves activation of the PI-3-K/Akt pathway. Western blotting experiments using a Ser435-phospho-specific Akt1 antibody demonstrated an activation of Akt-1 in motoneuron cultures 3 h after treatment with angiogenin, but unlike classical growth factors not immediately after treatment (Fig. 1C,D). We next tested the hypothesis that angiogenin mediated its neuroprotective activity via an increased PI-3-K/Akt signaling. In motoneuron cultures, the neuroprotective effect of angiogenin was abolished by cotreatment with the PI-3-K inhibitor wortmannin, which blocked the potent activation of Akt-1 in AMPA-stressed, angiogenin-treated cultures (Fig. 1E,F).
Expression of wild-type ANG, but not the ALS-associated ANG K40I mutant, protects NSC34 cells against ER stress-induced cell death
NSC34 is a hybrid cell line from embryonic mouse spinal cord and mouse neuroblastoma cells with characteristics of primary motoneurons (Cashman et al., 1992). We compared the effects of WT ANG and the ALS-associated ANG K40I mutation on survival of NSC34 cells. Cultures were transiently transfected with a pIRES-DsRed2 plasmid containing WT ANG or the mutated K40I ANG sequence. Human angiogenin levels in transfected NSC34 cells were assessed by means of Western blotting and quantitative real-time PCR (Fig. 2A). pIRES-DsRed2/ANG-transfected cells showed significantly reduced levels of ER stress-induced apoptosis when compared with empty vector-transfected cells (Fig. 2B). In pIRES-DsRed2/K40I ANG-transfected cells, the apoptosis level was similar to empty vector-transfected cells, demonstrating a loss of neuroprotective activity of the ALS-associated mutant (Fig. 2B).
Figure 2.
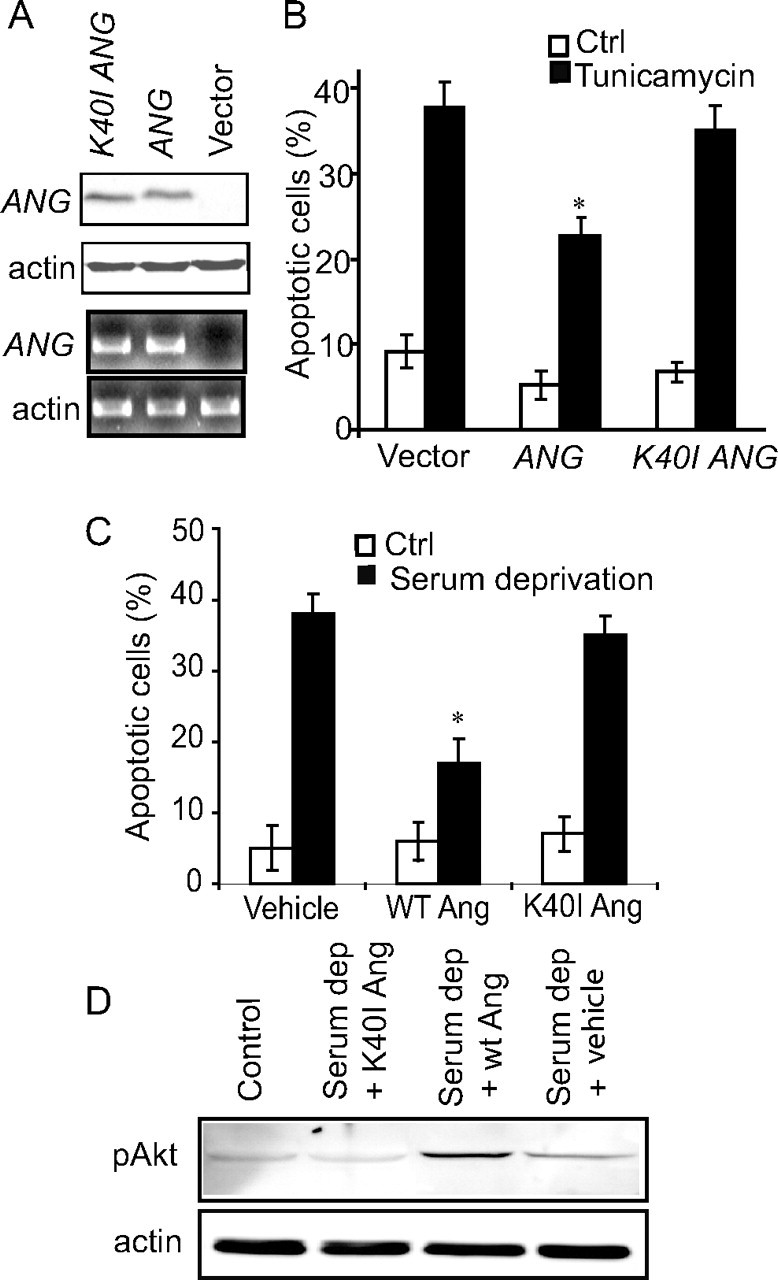
Lack of activity of the ALS-associated ANG mutants and potentiation of stress-induced cell injury by ANG knockdown. A, Western blot (two top panels) and quantitative real-time PCR analysis (two bottom panels) showing human ANG levels in NSC34 cells transiently transfected with pIRES-DsRed2 constructs alone, or containing either ANG, or K40I mutant ANG. B, NSC34 cells transiently transfected with pIRES-DsRed2/ANG are more resistant to tunicamycin-induced ER stress (500 nm, 24 h) than cells transfected with K40I ANG or with empty vector. Apoptotic cell death was assessed after Hoechst 33258 staining. (*p < 0.01 vs treated empty vector, mean ± SEM, n = 7–10). C, Wild-type recombinant angiogenin protein protects NSC34 cells from serum deprivation-induced toxicity (24 h), whereas mutant K40I angiogenin protein shows no protective effect (*p < 0.01, mean ± SEM, n = 7–10). D, Wild-type angiogenin protein, but not K40I mutant angiogenin protein, elevated the levels of pAkt-1 in NSC34 cells during trophic factor withdrawal.
We next compared the effects of recombinant WT angiogenin protein and the ALS-associated K40I mutant protein on Akt-1 survival signaling of NSC34 cells. WT angiogenin protein, but not K40I mutant angiogenin protein protected NSC34 cells against serum deprivation-induced apoptosis (Fig. 2C). WT angiogenin protein, but not K40I mutant angiogenin protein elevated the levels of active, phosphorylated Akt-1 in NSC34 cells during trophic factor withdrawal (Fig. 2D), suggesting that the ALS-associated mutation interferes with survival signaling pathways.
Knock-down of endogenous angiogenin potentiates stress-induced cell death in motoneurons in vitro
We next examined the role of endogenous angiogenin in stress-induced motoneuron death in vitro. Knock-down of murine ANG1, the proposed orthologue of human ANG, was achieved using two siRNA constructs targeting different regions (Fig. 3A). In primary motoneuron cultures transfected with ANG1 siRNA, AMPA-induced excitotoxic injury was significantly increased compared with mock-transfected cells or cells transfected with a scrambled sequence (Fig. 3B), suggesting that endogenous angiogenin is required for maintaining motoneuron survival.
Figure 3.

Knock-down of endogenous angiogenin potentiates stress-induced cell death in motoneurons in vitro. A, Western blot analysis demonstrating efficient knock-down of murine ANG1 in NSC34 cells with siRNAs 48 h after transfection. B, Apoptotic cell death of primary motoneuron cultures transfected with murine ANG1 siRNA and treated with AMPA was assessed after Hoechst 33258 staining. (*p < 0.01 vs nontransfected sister cultures, mean ± SEM, n = 7–10).
Angiogenin treatment, even after symptom onset, increases lifespan and motor function in SOD1G93A mice
In light of the significant neuroprotective effects of angiogenin observed in the in vitro models of motoneuron degeneration, the effect of angiogenin treatment in the SOD1G93A mouse model of ALS (Gurney et al., 1994) was examined. Animals were sex-matched and randomized, and treated systemically with human recombinant angiogenin protein (1 μg, daily) from either 50 d (before symptom onset) or 90 d (after symptom onset) as a more clinically relevant paradigm. In control experiments, uptake of fluorescently labeled angiogenin in spinal cord gray and white matter could be detected 2 h after a single administration (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Survival analysis of SOD1G93A mice showed that treatment with angiogenin from 50 d significantly increased lifespan (Fig. 4A). Of note, a similar protective effect was observed in animals treated from 90 d onwards, i.e., after symptom onset (Fig. 4A). Administration of angiogenin to WT littermates for 75 d had no effect on mouse viability or breeding behavior.
Figure 4.
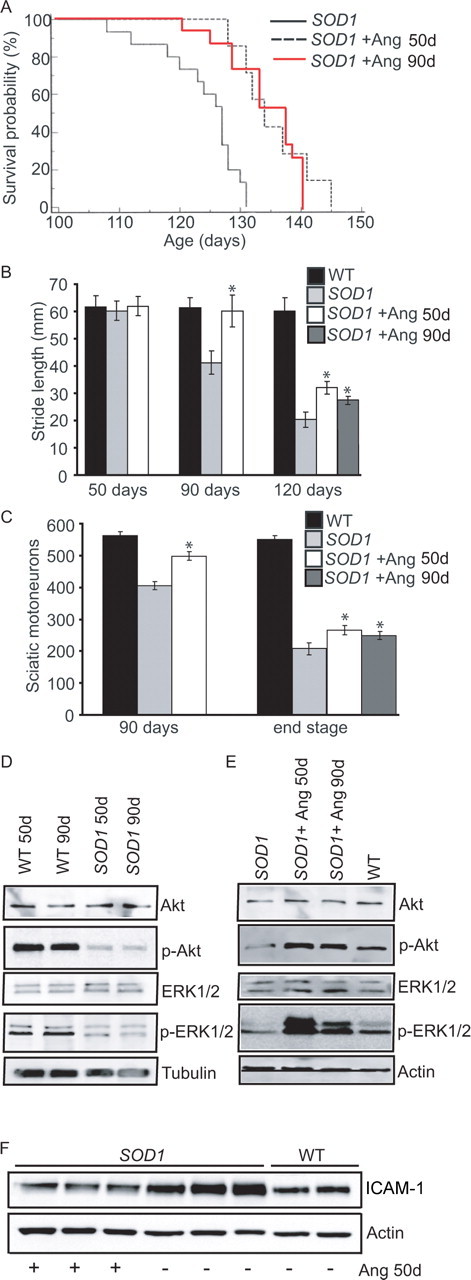
Angiogenin treatment after symptom onset in SOD1G93A mice delays disease progression and extends lifespan. A, Kaplan–Meyer analysis of probability of survival. Treatment from 50 d, angiogenin-treated SOD1 mice survived for 135 d (±1.5, n = 12), whereas vehicle-treated SOD1 mice survived for only 122.8 d (±2.2, n = 15, p < 0.0001). Treatment from 90 d, angiogenin-treated SOD1 mice survived for 132.8 d (±1.8, n = 12), whereas vehicle-treated SOD1 mice survived for only 122.2 d (±2.1, n = 15, p = 0.0005). B, Stride length analysis in the identical treatment paradigms (*p ≤ 0.05, mean ± SEM). C, Quantitative analysis of sciatic motoneuron survival (*p ≤ 0.05, mean ± SEM, n = 6). D, Western blot analysis of Akt-1, p-Akt, ERK1/2 and p-ERK1/2 in spinal cord homogenate of wild-type and untreated SOD1G93A mice. E, Western blot analysis of Akt, p-Akt, ERK1/2 and p-ERK1/2 in spinal cord homogenate from wild-type littermates, untreated SOD1G93A mice, SOD1G93A mice treated with angiogenin from 50 or 90 d. F, Western blot analysis of ICAM-1 in spinal cord homogenate from wild-type littermates, untreated SOD1G93A mice, and SOD1G93A mice treated with angiogenin from 50 d.
Because of the similar effect of 50 and 90 d treatments, a multivariate analysis was performed on the pooled data from both conditions (p value from Bartlett's test for homogeneity = 0.9622). There was no evidence of a difference in survival between both treatments (p = 0.5176), nor evidence of gender bias (p = 0.5076). However, for the pooled data (n = 54), the estimated survival was 11.4 d longer for angiogenin-treated (134.1 d) compared with vehicle-treated mice (122.7 d) and remained highly significant (p < 0.0001).
Motor performance was analyzed by stride length measurements (Gurney et al., 1994), showing that motor weakness was delayed in angiogenin-treated SOD1G93A mice (Fig. 4B). This improvement was accompanied by improved paw grip endurance (PaGE) test performance and a delay in body weight decline in angiogenin-treated SOD1G93A mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To examine the effect of angiogenin treatment on hindlimb muscles, the weights of the tibialis anterior (TA) and extensor digitorum longis (EDL) muscles were recorded during disease progression (supplemental Table 1, available at www.jneurosci.org as supplemental material). In angiogenin-treated SOD1G93A mice the decline in muscle weight reduction was delayed compared with untreated littermates, demonstrating that improvement in motoneuron survival during disease progression delayed hindlimb muscle denervation and atrophy.
Angiogenin treatment protects motoneurons in SOD1G93A mice
To correlate motor performance with histological data, the effect of angiogenin treatment on motoneuron survival in the spinal cord was assessed by counting the number of sciatic motoneurons in the lumbar spinal cord (Kieran et al., 2004). Treatment with angiogenin significantly increased sciatic motoneuron survival in SOD1G93A mice at both 90 d and at the disease end stage (Fig. 4C). This improvement in motoneuron survival correlated with the observed delay in disease progression in angiogenin-treated SOD1G93A mice.
In vitro, angiogenin neuroprotective activity required cell survival signaling through the PI-3-K/Akt pathway. Interestingly, we detected a significant decrease in the presence of active, Ser473-phosphorylated Akt-1 in the anterior horn and motoneurons of SOD1G93A mice compared with WT littermates (Fig. 4D; supplemenral Fig. 3, available at www.jneurosci.org as supplemental material), suggesting that SOD1G93A mice do not appropriately activate this endogenous survival pathway. The levels of active, phophorylated ERK-1 and -2 were also reduced (Fig. 4D). Of note, treatment with angiogenin, either before or after symptom onset, potently restored the disease-associated decrease in Akt-1 and ERK1/2 survival signaling in vivo (Fig. 4E), and reversed the disease-associated increase in ICAM-1, a marker for activated endothelium, that was detectable in SOD1G93A mice (Fig. 4F). Both vehicle- and angiogenin-treated SOD1G93A mice exhibited blood-spinal cord-barrier leakage during disease progression (Zhong et al., 2008) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Discussion
In this study we have shown that ANG has definite neuroprotective activities on motoneurons, and that the protective effect of angiogenin observed in the in vitro and in vivo models of motoneuron degeneration may relate to its potent (re-)activation of cell survival signaling pathways. Our data and other recently published data (Dewil et al., 2007) point to a strong disruption of Akt-1 survival signaling during SOD1G93A-mediated motoneuron degeneration in mice in vivo. Levels of phospho-Akt-1 have also been found to be reduced in motoneurons from sporadic and familial ALS patients (Dewil et al., 2007). Our recently reported genetic evidence for ANG mutations in ALS patients (Greenway et al., 2006), and the data presented here suggest that angiogenin may be required and sufficient to maintain motoneuron survival.
The ability of angiogenin to positively modulate PI-3-kinase/Akt-1 and ERK1/2 signaling (Hetman and Gozdz, 2004) may play an important role in its control of motoneuron survival. Angiogenin is known to positively modulate growth factor signaling (Kishimoto et al., 2005). Recent data suggest that angiogenin activates two distinct signaling pathways (Xu et al., 2002; Tsuji et al., 2005; Kim et al., 2007): Although binding to a 170 kDa receptor has been shown to be required for nuclear translocation, stimulation of rRNA synthesis, and cell proliferation (Moroianu and Riordan, 1994; Kim et al., 2007), angiogenin alternatively can bind to a 42 kDa binding protein identified as α-actin (Hu et al., 1993), a process that has been shown to promote Akt-1 activation (Kim et al., 2007). Our data suggests that the latter signaling pathway is activated by angiogenin in neural cells. However, because the 170 kDa angiogenin receptor has not yet been cloned, a molecular characterization of its involvement in the protective effects reported here cannot be sufficiently addressed experimentally at present. We could also demonstrate that the mutant K40I angiogenin protein failed to activate Akt-1, and that this was associated with a loss of neuroprotective activity. Recent genetic and biochemical studies have shown that multiple ALS-associated ANG mutations including K40I ANG reduce the protein's ribonucleolytic activity (Greenway et al., 2006; Crabtree et al., 2007; Wu et al., 2007). It is therefore tempting to hypothesize that decreased ribonucleolytic activity and Akt-1 activation may be inextricably linked, and that defects in this interaction play an important role in motoneuron degeneration associated with ANG mutations.
Effects of angiogenin on endothelial cells, angiogenesis, and local blood flow may play additional, potentially significant roles in vivo. However, these effects may not necessarily explain the selective vulnerability of motoneurons in familial and “sporadic” ALS associated with ANG mutations. We also found that angiogenin treatment of SOD1G93A mice reversed rather than enhanced the pathophysiological increase in ICAM-1 expression, a marker for activated endothelium. The effects of angiogenin delivery on the lengths of capillary networks and early reductions in local blood flow in SOD1G93A mice may need to be explored in more detail in further studies. However, the high levels of angiogenin expression directly in motoneurons (Greenway et al., 2006), and the critical role of angiogenin in the control of motoneuron survival demonstrated here suggest that the neural activities of angiogenin may play important physiological roles.
Previously, we could demonstrate a modest elevation in serum ANG levels in ALS patients at time of diagnosis (Cronin et al., 2006). Angiogenin concentrations in the human or murine spinal cord, and the pharmacokinetics of angiogenin in humans, or in mice, are currently not known. However, our data indicate accumulation of fluorescein-conjugated angiogenin in the murine spinal cord (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), despite potential inactivation or first pass effects. Potentially more importantly, in the ALS mice blood-spinal cord-barrier leakage (Zhong et al., 2008) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) may provide a direct access of angiogenin to the injured spinal cord.
In conclusion, our data demonstrate that angiogenin is a critical determinant of motoneuron survival, and suggest a critical role for angiogenin in the pathophysiology of ALS.
Footnotes
This work was supported by grants from Science Foundation Ireland to J.H.M.P. (03/RP1/B344), Enterprise Ireland to D.K., O.H., and J.H.M.P. (PC2005/133; PC2007/045), and a Health Research Board Fellowship and studentship to M.J.G. and D.C., respectively. D.K., M.J.G., O.H., and J.H.M.P. are inventors on patents relating to the use of angiogenin for the treatment of CNS diseases. We are grateful to Dr. Patrick Dicker (Department of Epidemiology and Public Health, Royal College of Surgeons in Ireland) for his help with statistical analysis. NSC34 cells (Dr. N. Cashman, University of Toronto, Toronto, Ontario, Canada) were obtained from Prof. P. J. Shaw, University of Sheffield, United Kingdom.
References
- Camu W, Henderson CE. Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. J Neurosci Methods. 1992;44:59–70. doi: 10.1016/0165-0270(92)90114-s. [DOI] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Crabtree B, Thiyagarajan N, Prior SH, Wilson P, Iyer S, Ferns T, Shapiro R, Brew K, Subramanian V, Acharya KR. Characterization of human angiogenin variants implicated in amyotrophic lateral sclerosis. Biochemistry. 2007;46:11810–11818. doi: 10.1021/bi701333h. [DOI] [PubMed] [Google Scholar]
- Cronin S, Greenway MJ, Ennis S, Kieran D, Green A, Prehn JH, Hardiman O. Elevated serum angiogenin levels in ALS. Neurology. 2006;67:1833–1836. doi: 10.1212/01.wnl.0000244466.46020.47. [DOI] [PubMed] [Google Scholar]
- Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W, Van den Bosch L. Vascular endothelial growth factor counteracts the loss of phospho-Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2007;33:499–509. doi: 10.1111/j.1365-2990.2007.00850.x. [DOI] [PubMed] [Google Scholar]
- Fett JW, Strydom DJ, Lobb RR, Alderman EM, Bethune JL, Riordan JF, Vallee BL. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry. 1985;24:5480–5486. doi: 10.1021/bi00341a030. [DOI] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Jr, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- Hu GF, Strydom DJ, Fett JW, Riordan JF, Vallee BL. Actin is a binding protein for angiogenin. Proc Natl Acad Sci U S A. 1993;90:1217–1221. doi: 10.1073/pnas.90.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kieran D, Sebastia J, Greenway MJ, Hardiman O, Prehn JH. The role of angiogenin in motoneuron degeneration. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(Suppl 1):42. [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guégan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Kang DK, Kim HY, Kang SS, Chang SI. Angiogenin-induced protein kinase B/Akt activation is necessary for angiogenesis but is independent of nuclear translocation of angiogenin in HUVE cells. Biochem Biophys Res Commun. 2007;352:509–513. doi: 10.1016/j.bbrc.2006.11.047. [DOI] [PubMed] [Google Scholar]
- Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF. Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene. 2005;24:445–456. doi: 10.1038/sj.onc.1208223. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Moroianu J, Riordan JF. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc Natl Acad Sci U S A. 1994;91:1677–1681. doi: 10.1073/pnas.91.5.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, Mckenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van Den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Shapiro R, Fox EA, Riordan JF. Role of lysines in human angiogenin: chemical modification and site-directed mutagenesis. Biochemistry. 1989;28:1726–1732. doi: 10.1021/bi00430a045. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Sun Y, Kishimoto K, Olson KA, Liu S, Hirukawa S, Hu GF. Angiogenin is translocated to the nucleus of HeLa cells and is involved in ribosomal RNA transcription and cell proliferation. Cancer Res. 2005;65:1352–1360. doi: 10.1158/0008-5472.CAN-04-2058. [DOI] [PubMed] [Google Scholar]
- Weydt P, Hong SY, Kliot M, Möller T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14:1051–1054. doi: 10.1097/01.wnr.0000073685.00308.89. [DOI] [PubMed] [Google Scholar]
- Wu D, Yu W, Kishikawa H, Folkerth RD, Iafrate AJ, Shen Y, Xin W, Sims K, Hu GF. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann Neurol. 2007;62:609–617. doi: 10.1002/ana.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZP, Tsuji T, Riordan JF, Hu GF. The nuclear function of angiogenin in endothelial cells is related to rRNA production. Biochem Biophys Res Commun. 2002;294:287–292. doi: 10.1016/S0006-291X(02)00479-5. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]