

Abstract
Long-term potentiation (LTP) of inputs relaying sensory information from cortical and thalamic neurons to principal neurons in the lateral amygdala (LA) is thought to serve as a cellular mechanism for associative fear learning. Nitric oxide (NO), a messenger molecule widely implicated in synaptic plasticity and behavior, has been shown to enhance LTP in the LA as well as consolidation of associative fear memory. Additional evidence suggests that NO-induced enhancement of LTP and amygdala-dependent learning requires signaling through soluble guanylyl cyclase (sGC) and cGMP-dependent protein kinase (cGK). Mammals possess two genes for cGK: the prkg1 gene gives rise to the cGK type I isoforms, cGKIα and cGKIβ, and the prkg2 gene encodes the cGK type II. Reportedly, both cGKI and cGKII are expressed in the amygdala, and cGKII is involved in controlling anxiety-like behavior. Because selective pharmacological tools for individual cGK isoforms are lacking, we used different knock-out mouse models to examine the function of cGKI and cGKII for LTP in the LA and pavlovian fear conditioning. We found robust expression of the cGKI specifically in the LA with cGKIβ as the prevailing isoform. We further show a marked reduction of LTP at both thalamic and cortical inputs to the LA and a selective impairment of auditory-cued fear memory in cGKI-deficient mutants. In contrast, cGKII null mutants lack these phenotypes. Our data suggest a function of cGKI, likely the β isoform, in the LA, supporting synaptic plasticity and consolidation of fear memory.
Keywords: amygdala, cGMP, PKG, fear conditioning, long-term memory, long-term potentiation
Introduction
Storage of emotionally arousing events critically depends on the lateral nucleus of the amygdala (LA) and is associated with changes of synaptic transmission in its sensory inputs (Sigurdsson et al., 2007). Excitatory neurons of the LA express neuronal nitric oxide synthase (NOS) (Schafe et al., 2005), a source for activity-dependent generation of nitric oxide (NO) known to modulate synaptic plasticity and behavior. In line with this, in vivo data imply increases of NO production during pavlovian fear conditioning that correlate with levels of freezing behavior (Izquierdo et al., 2006). Disrupting NO signaling results in marked suppression of long-term potentiation (LTP) in the LA and long-term auditory-cued fear memory (Schafe et al., 2005). Additional findings suggest that NO mediates its effect by activating soluble guanylyl cyclase, leading to increases in intracellular cGMP (Chien et al., 2003, 2005; Ota et al., 2008). cGMP-dependent protein kinase (cGK) is a major transducer of cGMP signals in neurons (Feil et al., 2005a). A recent study using intra-LA infusion of cGK activators and inhibitors provided compelling evidence for the function of cGK in fear memory consolidation (Ota et al., 2008). Complementary electrophysiological experiments revealed that synaptic plasticity in thalamic inputs is regulated by cGK. Analyses of neuronal functions of cGK have to take into account that mammals possess two genes, prkg1 and prkg2, encoding cGKI and cGKII (Wernet et al., 1989; Uhler, 1993; Jarchau et al., 1994). Additional isoforms, cGKIα and cGKIβ, arise from using alternative promoters and 5′ exons. cGKI and cGKII have both been implied in the regulation of hippocampal synaptic plasticity (Arancio et al., 2001; Kleppisch et al., 2003; Serulle et al., 2007), but their role for associative fear learning and synaptic plasticity in the LA remains unclear because of the lack of selective pharmacological tools. In this study, we used different knock-out models for cGKI and cGKII to elucidate this issue. Data from in situ hybridization and immunohistochemical analyses suggested that neurons of the amygdala express both cGKI and cGKII (El-Husseini et al., 1995, 1999; Werner et al., 2004; Feil et al., 2005b), and activity of cGKII plays a role for anxiety-like behavior (Werner et al., 2004). Hitherto, detailed information regarding expression of the cGKIα and cGKIβ in the amygdala is missing. Here, we demonstrate that the cGKI isoform is expressed most robustly specifically in the LA and identify the cGKIβ as the predominant isoform. We further functionally link cGKI expression in the LA to fear conditioning and synaptic plasticity: cGKI-deficient mutants showed an impairment of auditory-cued fear memory paralleled by a marked reduction of LTP in thalamic and cortical inputs to the LA. In contrast, cGKII-deficient mice showed normal fear conditioning and LTP in the LA under the same experimental conditions. Importantly, cGKI-deficient mutants displayed normal pain threshold and no marked phenotype in open-field, elevated plus maze, and light/dark box tests. Our findings support the view that mechanism(s) mediated by cGK, likely the Iβ isoform, promote persistent changes of synaptic transmission in the LA and consolidation of fear memory.
Materials and Methods
Experimental animals
For electrophysiological experiments, we used brain slices from 4- to 5-week-old apparently healthy cGKI null mutants (cGKI−/−), 6- to 8-week-old cGKIα smooth muscle rescue mice, 6- to 8-week-old cGKII (cGKII−/−), and the corresponding litter-matched controls (Pfeifer et al., 1996; Wegener et al., 2002; Weber et al., 2007). For behavioral analysis, we used 10- to 16-week-old cGKIα smooth muscle rescue mice, cGKII−/−mice, and the corresponding litter-matched controls. cGKIα smooth muscle rescue mice are deficient for endogenous cGKI but express the Iα isoform under the control of the SM22 promotor exclusively in smooth muscle cells, thus representing a total knock-out for cGKI in neuronal tissue. Moreover, adult mice of this genotype are healthy and suitable for behavioral analysis. All mice used were bred on a 129Sv background, and experiments with the different mutant mice were compared with their respective litter-matched controls. The genotype of the experimental animals was unknown to the investigator. For genotyping, the following primers were used: cGKI, 5′-GTCAAGTGACCACTATG-3′, 5′-CCT-GGCTGTGATTTCACTCCA-3′, and 5′-AAATTATAACTTGTCAAATTCTTG-3′; and cGKII, 5′-GGTGAAGTTTAGGTGAACCAAGAA-3′, 5′-GCCTGCTCTTTACTGAAGGCTCT-3′, and 5′-CACCATGTCCT-TGATCTGCT-3′. This study conforms to the German animal protection laws.
Western blot analysis
Control and mutants mice were killed by cervical dislocation, and the brain was removed. Whole brain or tissue of a specific brain region was shock frozen in liquid nitrogen. To isolate amygdala tissue, coronal slices (400 μm thick) containing the amygdala were cut using a microtome HM 650 V (Microm), and a region including the lateral nucleus, the basolateral complex, and the central nucleus was dissected thoroughly under the microscope using a special blade. Frozen tissue was homogenized in extraction buffer (20 mm Tris-HCl, pH 8.3, 0.7% SDS, 1.7% 2-mercaptoethanol, and 0.2 mm PMSF) using a FastPrep-24 (MP Biomedicals). Homogenates were denatured (95°C, 10 min) and centrifuged at 13,000 × g for 5 min. Probes were then precipitated from supernatants and resuspended in an appropriate volume of SDS sample buffer, heated again at 95°C for 10 min, separated by SDS-PAGE, and blotted onto a polyvinylidene difluoride membrane (Immobilin-P; Millipore). To block the unspecific binding sites, the membrane was blocked in 5% milk powder in 1× TRIS-buffered saline with 0.1% Tween 20 for 1 h before incubation with the primary antibody (4°C, overnight). For detection of cGKI, we used a rabbit polyclonal anti-cGKI common antibody (1:200 dilution) (Pfeifer et al., 1998). For detection of the α and β isoform, we used a goat polyclonal anti-cGKIα antibody (sc-10335; 1:500 dilution; Santa Cruz Biotechnology) and a goat polyclonal anti-cGKIβ antibody (sc-10342; 1:500 dilution; Santa Cruz Biotechnology), respectively. cGKII was detected with a rabbit polyclonal anti-cGKII antibody (sc-25430, 1:1000 dilution; Santa Cruz Biotechnology). As a loading control, we used a rabbit polyclonal antibody raised against β-actin (1:50,000 dilution; Abcam). Primary antibodies were detected with either anti-rabbit (Cell Signaling Technology) or anti-goat HRP-conjugated secondary antibodies (Santa Cruz Biotechnology). Finally, the ECL Western blotting analysis system (GE Healthcare) was used for detection of the target proteins.
Immunohistochemistry
Animals kept under deep anesthesia were perfused with 10% phosphate-buffered Formalin. After decapitation, the brain was dissected and postfixed for 4 h in the same fixative solution. Subsequently, the tissue was washed with PBS, dehydrated, and embedded in paraffin. Coronal sections (10 μm thick) containing the amygdala were prepared and used for staining. Before staining, a heat-induced antigen retrieval was performed with sodium citrate buffer (10 mm sodium citrate, pH 6.0, 0.05% Tween 20), and unspecific bindings sites were blocked with 1.5% normal goat serum in PBS for 1 h. The sections were then incubated (4°C, overnight) with a primary antibody raised against an epitope common to both cGKI isoforms (1:50 dilution). For detection of the primary antibody, we used the Vectastain ABC AP kit (Vector Laboratories) based on the avidin–biotin technique. Sections were incubated with biotinylated goat anti-rabbit secondary antibody and the ABC complex, and alkaline phosphatase substrate was used as a chromogene. Finally, sections were mounted in Aquatex (Merck). Sections from cGKIα smooth muscle rescue animals were also stained with the cGKI common antibody and used as a control for unspecific binding of the primary antibody. Hoechst 33324 (5 μg/ml; Fluka) was used for dual labeling of cellular nuclei in a few sections that were then mounted in PermaFluor (Beckman Coulter). Parallel Nissl staining of neighboring sections was used to identify the complex of the amygdala and other brain regions.
Electrophysiological recordings
Slice preparation.
Brain slices (300–400 μm thick) were obtained from experimental animals (see above) deeply anesthetized with ether before decapitation. The whole brain was dissected and placed in ice-cold artificial CSF (aCSF) of the following composition (in mm): 10 glucose, 124 NaCl, 3 KCl, 1.25 KH2PO4, 26 NaHCO3, 2 MgSO4, and 2.5 CaCl2, pH 7.4 (bubbled with 95% O2/5% CO2). Coronal slices containing the amygdala were prepared in the same solution using a microtome HM 650 V (Microm) and retained 1–2 h in a storage chamber for recovery before transferring them into a submersion-type recording chamber perfused with aCSF (10 ml/min) at 30°C.
Synaptic transmission and plasticity.
Field potentials (FPs) were recorded in cortical and thalamic inputs of control mice, apparently healthy cGKI null mutants, cGKIα smooth muscle rescue mice, and cGKII null mutants using an Axoclamp 2B Amplifier (Molecular Devices) and aCSF-filled glass electrodes (3–6 MΩ) placed within the lateral nucleus of the amygdala. FPs were evoked by stimuli (100 μs) delivered through a bipolar concentric tungsten electrode (TM53CCINS; WPI) placed near the external capsule and directly above the central nucleus of the amygdala, respectively. FP amplitude was used to assess synaptic strength. The stimulus intensity was adjusted to produce an FP with ∼50% of the maximal amplitude. The same intensity was used while delivering high-frequency stimulation (HFS) (five trains at 100 Hz for 1 s with 90 s intermission between trains) for induction of LTP. Before HFS was applied, the response to single stimuli had to remain stable for at least 20 min. Stimulation frequency during this baseline period and after the HFS was 0.066 Hz. LTP was estimated as the increase of the FP amplitude 1 h after HFS expressed as the percentage of the baseline FP amplitude. Stimulation and data acquisition were controlled by PULSE software via an ITC-16 computer interface (HEKA). All experiments were performed in the presence of 50 μm picrotoxin (Roth). Whole-cell patch-clamp recordings of EPSCs were obtained from pyramidal neurons in the LA of brain slices from 2- to 3-week-old mice using an EPC-9 amplifier (HEKA). Pyramidal cells were identified visually and by their firing properties. Slices were superfused with aCSF containing picrotoxin (50 μm), and patch electrodes (∼3 MΩ resistance) were filled with a solution containing the following (in mm): 10 glucose, 10 HEPES, 95 K-gluconate, 20 KCl, 10 NaCl, 1 MgCl2, 0.2 EGTA, 1 KATP, 1 Na2GTP, pH 7.2. EPSCs were evoked by stimulating either cortical or thalamic afferents with 100 μs square pulses delivered through a bipolar concentric tungsten electrode (see above). Paired-pulse facilitation (PPF) was assessed as the amplitude ratio of EPSCs (EPSC2/EPSC1) elicited by a pair of stimuli delivered with interpulse intervals of 25 and 50 ms. Holding potential was −70 mV for all voltage-clamp experiments.
Behavior
Male 10- to 16-week-old control, litter-matched cGKIα smooth muscle rescue and cGKII−/− mice (for details, see above, Experimental animals) were used for behavioral testing. All animals were separated to individual plastic cages with ad libitum access to food and water and kept under inverse 12 h light/dark cycle (lights off at 8:00 A.M.) at least 2 weeks before starting behavioral testing. All tests were performed during the dark phase.
Fear conditioning.
Experiments were performed in two contexts: (1) the conditioning chamber with a metal grid shock floor and (2) the test context differing in shape, odor, and illumination (TSE Systems). All chambers were cleaned thoroughly after each trial. For auditory fear conditioning, naive experimental animals were placed into the conditioning chamber and left to explore for 3 min before a tone (9 kHz tone, 80 dB) was presented for 30 s that coterminated with a single electric foot shock (0.7 mA, 2 s). Mice were returned to their home cages 1 min later. To test auditory-cued fear memory at various phases, different cohorts of experimental animals mice were placed into a neutral context (black Plexiglas cylinder cleaned with 1% acetic acid) 2 h, 24 h, and 7 d after conditioning, respectively. In each case, freezing behavior of the corresponding animals in the neutral context was monitored for 3 min without presentation of the tone (control period) and 3 min with presentation of the tone (test period). Contextual fear memory was tested in the afternoon of days 1 and 7 by reexposing the mice to the conditioning context for 3 min without tone presentation. Videotaped sessions were analyzed offline by means of customized software assessing the duration of freezing episodes, which were defined by the absence of all movements except for respiration.
Open field.
Open-field analysis was performed in the conditioning chamber described above (22 × 22 cm) lacking the foot-shock grid. Horizontal (total distance moved) and vertical (exploratory rearings) locomotive activity was assessed using infrared sensor beams and monitored for 10 min in the dark (0 lux). Thigmotaxis was determined as the percentage of time animals spent <5.5 cm away from the side walls.
Elevated plus maze.
The elevated plus maze was made out of gray polyvinyl chloride and consisted of a plus-shaped platform elevated 40 cm above the floor, with two open (30 × 5 cm) and two closed (30 × 5 × 15 cm) arms and a connecting central zone (5 × 5 cm). The open arms were lit by white light of 300 lux, the neutral zone by 115 lux, and the closed arms by 5 lux. The test was performed between 10:00 A.M. and 12:00 A.M. Animals were kept in the test room for 24 h before starting the experiment. They were placed onto the central compartment facing the closed arm. During a 5 min exposure, the following parameters were recorded/calculated by an experienced observer by means of a video/computer system as described by Kromer et al. (2005): (1) the number of full entries into open and closed arms (i.e., the animal entered the corresponding arm with all four paws), (2) percentage of entries into open arms (ratio of open arm entries to sum of entries into all arms), and (3) the relative time on the open arms expressed as a percentage of the total time spent on either arms.
Light/dark box.
Mice were tested under 300 lux in boxes connected by a doorway. The light compartment had nontransparent white Plexiglas walls. The non-illuminated black compartment consisted of black plastic walls. The floor was painted white and black, respectively. Light and dark compartment were connected by a short tunnel (5 × 5 cm). The number of entries to and the time spent in the lit compartment were recorded. Compartments and floor were thoroughly cleaned with soap and water between sessions.
Pain threshold.
Naive animals were placed individually into the conditioning chamber. After 3 min, electric foot shocks (2 s) were applied starting with an intensity of 0.1 mA. The intensity was increased stepwise by 0.1 mA until the animal showed the first signs of pain (jumping and vocalization), and the corresponding value was taken as the pain threshold. The pause between successive stimuli was always 30 s.
Analysis and statistics
Data are expressed as mean ± SEM. For estimation of LTP, the field potential amplitude was normalized to the averaged baseline response before tetanization at time 0. Statistical analyses were performed using ANOVA for repeated measures, followed by Newman–Keuls post hoc test if appropriate, or using Student's t test for two independent means. Significance was accepted if p < 0.05.
Results
The LA is a site with most dense expression of cGKI
To get a more precise picture of cGKI and cGKII expression, we initially performed immunohistochemical and Western blot analyses for the cGKI (including analysis for Iα and Iβ isoforms) and cGKII focusing on regions known to be related to associative fear learning such as the amygdala. Besides cellular layers of the cortex and hippocampus, the amygdala represented the region with most prominent staining by the cGKI common antibody in sections from wild-type mice (Fig. 1A). Within the amygdala complex, expression of cGKI was most abundant in the LA, whereas expression in other nuclei of the amygdala, e.g., the basolateral nucleus, was rather weak (Fig. 1B). Similarly, expression of cGKI in thalamic nuclei was negligible. A more detailed analysis at larger magnifications suggested that cGKI expression in the LA is ample in the neuropil, which likely includes presynaptic structures (Fig. 1B). There was also distinct expression in the cytosol, i.e., the primary localization of cGKI, of postsynaptic neurons as evidenced by ring-shaped staining surrounding their nuclei. These results are compatible with the view that both presynaptic and postsynaptic cGKI-mediated mechanisms can contribute to synaptic plasticity and, ultimately, associative memory. The specificity of the antibody used was confirmed in sections from cGKIα smooth muscle rescue mice that showed no staining (Fig. 1A, bottom), which also demonstrates that the neuronal tissue of cGKIα smooth muscle rescue mice lacks the protein, ensuring that these mice are well suited to study its function in the CNS. Confirming this notion, immunoblots of whole-brain extracts from cGKIα smooth muscle rescue mice yielded only a vague band that was attributable to expression of reconstituted cGKIα in vascular smooth muscle (Fig. 1D). A cGKIβ-specific antibody yielded a robust band in Western blots of probes from the amygdala of wild-type mice (Fig. 1E). In contrast, only faint staining was observed in immunoblots for the amygdala using a cGKIα-specific antibody that yielded a robust band for cerebellar tissue. Collectively, the findings above suggest that the LA is a region with most dense expression of the cGKI protein and that cGKIβ represents the prevailing isoform expressed in this region. Former reports regarding the expression of cGKII in the amygdala were commonly based on detection of mRNA (El-Husseini et al., 1995, 1999; Werner et al., 2004). We confirmed expression of cGKII protein in the amygdala using immunoblot (Fig. 1E). However, immunohistochemical staining (data not shown) suggested that the level of cGKII expression in the amygdala is by far lower than that in thalamic nuclei. However, cGKII protein expressed in the soma of presynaptic neurons in the auditory thalamus may also have a functional impact on synaptic plasticity in the LA (cf. Apergis-Schoute et al., 2005). Noteworthy, cGKI knock-out mice showed no upregulation of the cGKII in the amygdala and vice versa.
Figure 1.

Expression of cGK in brain regions related to associative fear learning. A, Immunohistochemical staining of brain sections from wild-type (top) and cGKIα smooth muscle rescue (bottom) mice using an anti-cGKI common antibody detecting both the α and the β isoform. B, Stainings of the amygdala complex from wild-type mice at larger magnifications. The two bottom panels illustrate dual labeling for cGKI (left) and for cellular nuclei (right) using the anti-cGKI common antibody and Hoechst 33324, respectively. Identical cells are indicated by arrows. C, cGKIα smooth muscle rescue mice represent a cGKI knock-out model in which the α isoform was reconstituted selectively in smooth muscle tissue (in red) on the background of the null mutation, i.e., all other tissues including the brain lack the cGKI protein as confirmed by immunohistochemical and Western blot analyses. D, Immunoblots performed with probes from the entire brain of wild-type mice (WT), cGKI null mutants (I-KO), and cGKIα smooth muscle rescue mice (Res) using the anti-cGKI common antibody (cGKIcom). E, Immunoblots with tissue probes from the amygdala (Amy) and cerebellum (Cer) using an anti-cGKIα (cGKIα), anti-cGKIβ (cGKIβ), and anti-cGKII (cGKII) antibody, respectively. Specificity of the signal obtained with wild-type probes (WT) was proven using additional probes from cGKI (I-KO) and cGKII (II-KO) null mutants, respectively. β-Actin served as a loading control.
LTP in the LA is impaired in cGKI-deficient mice but normal in cGKII null mutants
In the next step, we evaluated the functional importance of cGKI and cGKII for synaptic plasticity in the amygdala by examining LTP in cortical and thalamic inputs to the LA in brain slices from wild-type mice and litter-matched cGKI null mutation (cGKI−/−). To avoid a major impact of the severe gastrointestinal phenotype exhibited by adult cGKI−/− mice, we used only animals that did not yet develop overt symptoms for our electrophysiological analysis. Suggesting a universal function of cGKI for modulation of synaptic transmission in the LA, cGKI−/− mice showed a marked reduction of LTP in both cortical and thalamic inputs compared with control mice (Fig. 2C). Tetanic stimulation of cortical (Fig. 2A) and thalamic (Fig. 2B) afferents, respectively, induced robust LTP in the LA in control mice with an FP amplitude 1 h after tetanus amounting to 125.8 ± 5.8% (cortical, 11 slices, 6 animals) and 129.2 ± 4.9% (thalamic, 13 slices, 7 animals) of its amplitude during baseline. Under the same conditions, the FP amplitudes in cGKI−/− mice 1 h after tetanus were 111.1 ± 3.3% (cortical, 14 slices, 7 animals) and 113.4 ± 3.8% (thalamic, 10 slices, 5 animals) of the baseline amplitude, respectively. Two-way ANOVA for repeated measures revealed a significant interaction genotype × time for both auditory inputs to the LA (cortical, F(12,276) = 1.83, p = 0.0042; thalamic, F(12,252) = 1.83, p = 0.044). The reduction of LTP in cGKI−/− mice reached significance within ∼20 min after the tetanus for the cortical inputs and within 35 min after tetanus for thalamic inputs (p < 0.01, Newman–Keuls post hoc test). To eliminate the possibility that reduced LTP was related to possible alterations in cerebral circulation attributable to the smooth muscle dysfunction of cGKI−/− mice, we additionally examined LTP in so-called cGKIα smooth muscle rescue mice (for details, see Materials and Methods). Results obtained with these mutants confirmed those with cGKI null mutants (Fig. 2D). Again, control mice showed robust LTP in the LA (cortical, 137.0 ± 6.0%, 11 slices, 5 animals; thalamic, 142.1 ± 5.7%, 9 slices, 4 animals). In the corresponding experiments with slices from litter-matched cGKIα smooth muscle rescue mice, potentiation of the FP amplitude was 117.6 ± 4.3% (11 slices, 5 animals) for cortical and 121.8 ± 4.6% (8 slices, 4 animals) for thalamic inputs to the LA. When tested with ANOVA for repeated measures, the reduction of LTP in the LA of cGKIα smooth muscle rescue mice proved significant both for cortical (genotype, F(1,20) = 6.92, p = 0.016; genotype × time, F(12,240) = 3.32, p < 0.0005) and thalamic (genotype, F(1,15) = 5.11, p = 0.039) inputs.
Figure 2.

LTP in the amygdala of transgenic mouse models lacking either cGKI or cGKII. Synaptic transmission was assessed by recording FPs in the lateral nucleus of the amygdala in response to stimulation of cortical (A) and thalamic (B) inputs, respectively. A strong tetanus (5 times for 1 s at 100 Hz with 90 s pause) was applied at time 0. Picrotoxin (50 μm) was present throughout recording. C, Time course of the FP evoked by stimulation of cortical (left) or thalamic (right) inputs recorded in brain slices from 4- to 5-week-old cGKI null mutants (□) and their litter-matched controls (■). D, Time course of the FP evoked by stimulation of cortical (left) or thalamic (right) inputs recorded in brain slices from 6- to 8-week-old cGKIα smooth muscle rescue mice (□) and their litter-matched controls (■). E, Time course of the FP evoked by stimulation of cortical (left) or thalamic (right) inputs recorded in brain slices from 6- to 8-week-old cGKII null mutants (◇) and their litter-matched controls (♦). The insets show representative FP recordings. Calibration: 2 ms, 250 μV. *p < 0.05, **p < 0.01, ***p < 0.005, respectively (ANOVA and Newman–Keuls post hoc test).
In contrast to the cGKI-deficient mouse models, cGKII−/− mice exhibited normal LTP in the LA (Fig. 2E). The FP amplitude 1 h after tetanus amounted to 120.8 ± 2.9% (wild type, 28 slices, 17 animals) versus 118.8 ± 4.2% (cGKII−/−, 24 slices, 11 animals) in cortical and 120.5 ± 2.8% (wild type, 24 slices, 11 animals) versus 119.0 ± 5.1% (cGKII−/−, 12 slices, 5 animals) in thalamic inputs. These findings, in nice agreement with our expression data, point out the cGKI isoform as a key regulator of synaptic plasticity in the LA.
cGKI-deficient mouse models show normal basal synaptic transmission in the LA
Impairment of synaptic plasticity observed in cGKI null mutants may result from a general defect in synaptic transmission. To elucidate this issue, we analyzed characteristics of basal excitatory synaptic transmission in control and cGKI-deficient mice (Fig. 3). Input–output relation (IOR) assessed by the FP amplitude was not significantly altered in cortical and thalamic inputs to the LA of cGKI null mutants compared with control mice. Recordings of EPSCs and EPSPs from single pyramidal cells in the LA of these genotypes did not reveal any overt differences in the basal excitatory synaptic transmission (data not shown). PPF represents another important parameter of synaptic transmission likely primarily related to presynaptic function (Zucker, 1989). Our whole-cell patch-clamp analysis of EPSCs revealed no differences between cGKI null mutants and control mice in PPF for interpulse intervals of 25 and 50 ms (Fig. 3). Current-clamp EPSP recordings further confirmed that PPF was intact in these animals (data not shown). The same analyses of basal synaptic transmission were performed for cGKIα smooth muscle rescue mice, a cGKI-deficient mouse model used in behavioral tests (see below). These mice too showed normal IOR and PPF in cortical and thalamic inputs to the LA (Fig. 3).
Figure 3.
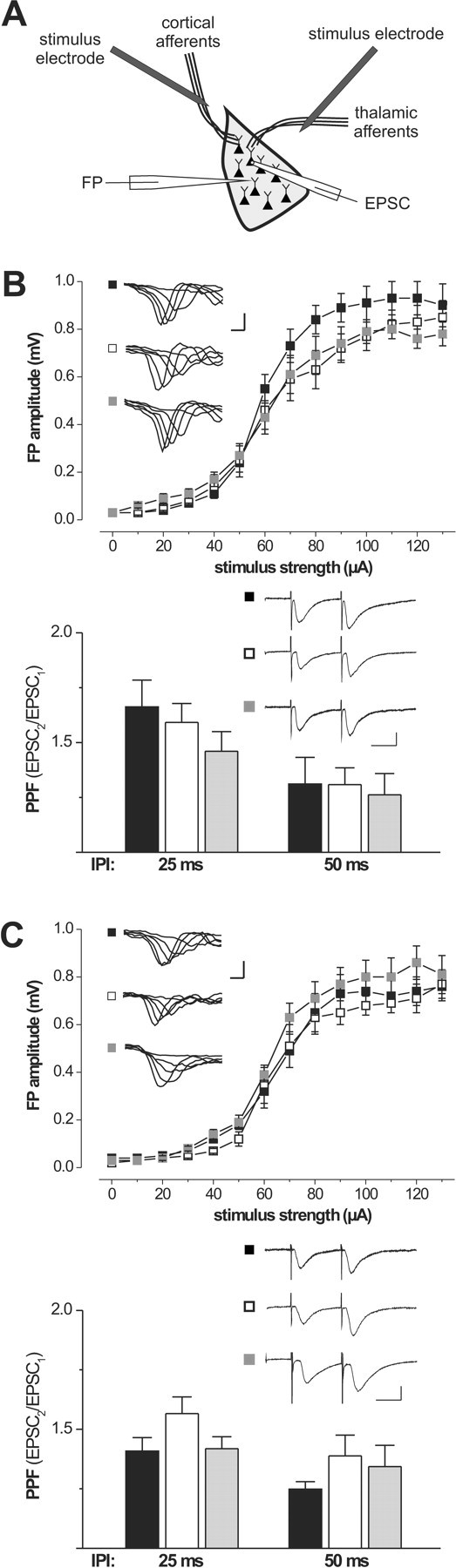
Basal excitatory synaptic transmission in the amygdala of transgenic mouse models lacking the cGKI. A, FPs and EPSCs were recorded in the lateral nucleus of the amygdala in response to stimulation of either cortical or thalamic afferents relaying to pyramidal cells in the LA. B, Input–output relations of FPs and PPF of EPSCs recorded from control mice (black squares and bars; 6 animals, 21 inputs to the LA, and 8 pyramidal cells), cGKI null mutants (white squares and bars; 5 animals, 12 inputs to the LA, and 10 pyramidal cells), and cGKIα smooth muscle rescue mice (gray squares and bars; 5 animals, 11 inputs to the LA, and 11 pyramidal cells) in response to stimulation of cortical inputs. Representative FPs are shown in the corresponding insets; calibration: 1 ms, 500 μV. PPF in cortical inputs of the three genotypes is illustrated by representative EPSCs recorded with an interpulse interval (IPI) of 50 ms. Calibration: 25 ms, 25 pA. C, Input–output relations of FPs and PPF of EPSCs recorded from control mice (black squares and bars; 5 animals, 11 inputs to the LA, and 7 pyramidal cells), cGKI null mutants (white squares and bars; 4 animals, 9 inputs to the LA, and 11 pyramidal cells), and cGKIα smooth muscle rescue mice (gray squares and bars; 4 animals, 13 inputs to the LA, and 11 pyramidal cells) in response to stimulation of thalamic inputs. Representative FPs are shown in the corresponding insets. Calibration: 1 ms, 500 μV. PPF in thalamic inputs of the three genotypes is illustrated by representative EPSCs recorded with an interpulse interval of 50 ms. Calibration: 25 ms, 25 pA.
cGKIα smooth muscle rescue mice behave normally in the open field, elevated plus maze, and light/dark box
Next, we aimed to evaluate whether modulatory function of cGKI for synaptic plasticity in the LA has a functional impact on related behavior. Because cGKI null mutants do not allow to perform behavioral studies, we used the so-called cGKIα smooth muscle rescue mice (Weber et al., 2007). These mice are cGKI-deficient in neuronal tissue (Fig. 1), healthy compared with the cGKI null mutants, and, therefore, well suited for behavioral analysis. To assess possible general behavioral defects in these mice, we initially performed a 10 min analysis of locomotive and exploratory behavior in the open field. Behavior of cGKIα smooth muscle rescue mice in the open field was not different from that of their control littermates (Fig. 4). For example, cGKIα smooth muscle rescue mice showed normal general locomotive activity assessed by the time they were moving (control, 23.1 ± 1.6% of total time, 15 animals; cGKIα smooth muscle rescue, 26.5 ± 1.5% of total time, 11 animals) (Fig. 4A) and the total distance traveled (control, 34.3 ± 4.4 m; cGKIα smooth muscle rescue, 38.7 ± 5.2 m) (Fig. 4B). In addition, we found no difference between the genotypes in the time mice performed vertical movements (rearings) (control, 4.9 ± 0.9% of total time; cGKIα smooth muscle rescue mice, 4.6 ± 1.6% of total time) (Fig. 4D) and time spent near the wall of the box (thigmotaxis) (control, 78.2 ± 4.5% of total time; cGKIα smooth muscle rescue, 77.5 ± 5.9% of total time) (Fig. 4C), suggesting that cGKIα smooth muscle rescue mice have normal exploratory behavior and innate fear, respectively. Importantly, none of the parameters examined differed in any particular time segment of analysis as illustrated in the corresponding time courses. We performed additional tests of anxiety-related behavior in the elevated plus maze and in the light/dark box. Fitting with the open-field data, there was no difference between control and cGKIα smooth muscle rescue mice in the elevated plus maze (Fig. 5): the number of open arm entries were slightly but not significantly decreased in cGKIα smooth muscle rescue (26.5 ± 10.5% of overall entries, 8 animals) compared with control mice (40.3 ± 11.7% of overall entries, 7 animals; p = 0.4). Accordingly, the total number of entries (12.0 ± 2.0 vs 12.0 ± 2.4) and the number of entries to the closed arms (data not shown) were also not significantly different between the two genotypes. Furthermore, no difference between the genotypes was detected in the light/dark box test: the number of entries to and the time spent in the lit compartment amounted to 0.6 ± 0.4 and 4.2 ± 3.9% of total time (control, 7 animals) versus 2.0 ± 0.9 and 11.4 ± 7.9% of total time (cGKIα smooth muscle rescue mice, 8 animals), respectively. Collectively, these data argue against an altered level of innate fear and anxiety in cGKIα smooth muscle rescue mice.
Figure 4.

cGKIα smooth muscle rescue mice show normal locomotive activity. Adult (10- to 16-week-old) control (filled symbols and bars; 15 animals) and cGKIα smooth muscle rescue (open symbols and bars, 11 animals) mice were exposed to an open field for 10 min under 0 lux, and the time course of the following parameters was analyzed in 2 min intervals. A, Percentage of the time animals were moving. B, Distance animals moved during 2 min intervals. C, Percentage of the time spent in quadrants adjacent to the wall of the experimental chamber. D, Percentage of the time spent performing vertical movements (rearings). Insets illustrate the corresponding mean values for the entire 10 min period.
Figure 5.

cGKIα smooth muscle rescue mice show normal anxiety-related behavior in an elevated plus maze. Adult (10- to 16-week-old) control (filled bars; 7 animals) and cGKIα smooth muscle rescue (open bars; 8 animals) mice were placed to the neutral (central) zone of an elevated plus maze facing closed arms and left to explore the elevated plus maze for 5 min. Shown are the entries to the open arm as the percentage of the total number of arm entries (left) and the total number of entries to open and closed arms (right).
Locomotive activity of cGKII−/− mice in the open field appeared to be slightly decreased compared with that of their littermate controls (time active, 33.2 ± 1.2% of total time vs 37.2 ± 0.9% of total time; distance moved, 36.4 ± 1.6 vs 52.5 ± 3.1 m; 10 animals for each genotype; p < 0.05), perhaps attributable to a growth retardation of the mutant mice (cf. Pfeifer et al., 1996). Fitting with data from a previous report (Werner et al., 2004), cGKII−/− mice also showed increased thigmotaxis (control, 37.0 ± 5.6%, 10 mice; cGKII−/−, 56.7 ± 6.6%).
cGKIα smooth muscle rescue mice exhibit a selective impairment of auditory-cued fear memory
To assess the function of cGKI and cGKII for learning behavior related to synaptic plasticity in the LA, we next performed a comparative analysis of pavlovian fear conditioning. Several groups of matched experimental animals were given a single presentation of a tone (30 s) coterminating with a foot shock (2 s), and freezing behavior was tested at just one time point (2 h, 1 d, or 7 d) after conditioning in a neutral context (auditory-cued fear memory) (Fig. 6A–C), followed by a test in the conditioning chamber (contextual fear memory) (Fig. 6D). As expected, three-way ANOVA for repeated measure revealed a significant general effect of the tone presentation (F(1,62) = 415, p < 0.0001), indicating that all genotypes formed an auditory-cued fear memory. In addition, there was a highly significant interaction for genotype × tone (F(1,62) = 8.67, p = 0.004), indicating that the cGKIα smooth muscle rescue mice exhibited weaker tone–shock association. No significance was reached for the genotype × time × tone interaction, likely attributable to the fact that, for each time point, separate cohorts of experimental animals were tested. Therefore, we additionally performed a t test separately for each time point. This test revealed that cGKIα smooth muscle rescue mice exhibited significantly reduced fear memory 1 and 7 d after conditioning but not 2 h after conditioning, even when using the one-tailed version of the test (2 h, t(25) = 0.83, p = 0.209; 1 d, t(17) = 2.4, p = 0.014; 7 d, t(20) = 1.88, p = 0.037) (Fig. 6A–C), supporting the view that cGKIα smooth muscle rescue mice exhibit a selective deficit of long-term memory. These differences were specifically related to the auditory stimulus and not observed in its absence, i.e., during the 3 min before tone presentation. Noteworthy, as illustrated in the corresponding time course of freezing to tone during the 3 min presentation period (Fig. 6A–C, bottom rows), the immediate response of cGKIα smooth muscle rescue mice to the tone was the same as in the control for all time points tested (2 h, 1 d, or 7 d). This argues against the contribution of a sensory deficit (e.g., in hearing) or a general motivational defect to the phenotype observed.
Figure 6.

Auditory-cued and contextual fear memory in 10-to 16-week-old control (filled bars and symbols) and cGKIα smooth muscle rescue (open bars and symbols) mice. Animals were exposed to a single 2 s foot shock coterminating with a 30 s tone. Fear memory was assessed by the percentage of the total time animals spent freezing. A–C, Separate litter-matched batches (9–15 animals for all groups) were tested before conditioning (pre-cond) and either 2 h, 1 d, or 7 d after conditioning in a neutral context (cylinder) for 3 min without (no tone) followed by 3 min with presentation of the conditioned stimulus (tone), respectively. Bottom rows show plots of the time course of freezing analyzed in 30 s intervals before (0–180 s) and during (180–360 s) tone presentation. D, Freezing during reexposure of the experimental animals to the conditioning context (box) 1 and 7 d after conditioning, respectively. E, Pain threshold determined by the minimal intensity of the foot shock causing the behavioral responses vocalizing and jumping. *p < 0.05.
cGKIα smooth muscle rescue and control mice froze to the same extent during reexposure to the conditioning chamber at all times, indicating that there was no difference between genotypes in contextual fear memory. This was statistically confirmed by a three-way ANOVA for repeated measures (genotype, F(1,37) = 0.11, p = 0.74; genotype × context, F(1,37) = 0.83, p = 0.37; genotype × time × context, F(1,37) = 0.05, p = 0.82). Noticeably, all experimental animals showed rather low levels of freezing during reexposure to the conditioning chamber, likely attributable to the genetic background (Sv129). However, all groups showed significantly more freezing in the conditioning chamber than during the 3 min period before tone presentation in the neutral context (F(1,37) = 56.5, p < 0.001), indicating the formation of a contextual fear memory.
Together, the data above reveal that cGKIα smooth muscle rescue mice have normal contextual fear memory but a selective impairment of long-term auditory-cued fear memory. The latter is indicative for disruption of memory consolidation.
We used the same fear conditioning protocols as above to study auditory-cued and contextual fear memory in wild-type and cGKII−/− mice. cGKII−/− mice froze to the same extent as the wild type when reexposed to the tone in a neutral context and during reexposure to the conditioning context without presentation of the tone, respectively (Fig. 7). Thus, in agreement with our findings for synaptic plasticity, cGKII was dispensable for amygdala-related associative fear memory.
Figure 7.
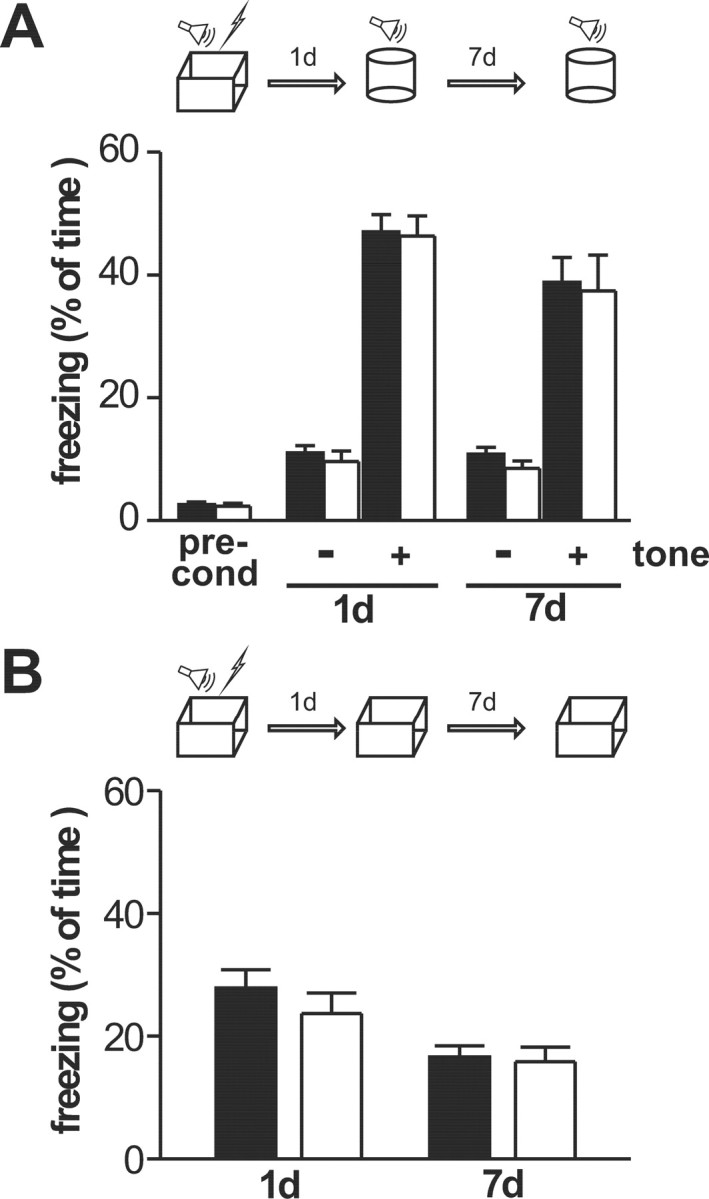
Associative fear memory in 10- to 16-week-old wild-type (filled bars; 12 animals) and cGKII−/− (open bars; 11 animals) mice. Animals were exposed to a single 2 s foot shock coterminating with a 30 s tone. Fear memory was assessed by the percentage of the total time animals spent freezing. A, Freezing before conditioning (pre-cond) and auditory-cued fear memory was tested 1 and 7 d after conditioning in a neutral context (cylinder) for 3 min without (−) followed by 3 min with (+) tone presentation, respectively. B, Contextual fear memory was tested 1 and 7 d after conditioning in the corresponding context (box).
cGKIα smooth muscle rescue mice display a normal pain threshold
Auditory-cued fear memory is believed to rely on potentiation of auditory inputs to the LA by simultaneous strong activity in converging nociceptive inputs that arises from the foot shock (for review, see Sigurdsson et al., 2007). To interpret the phenotype of cGKIα smooth muscle rescue mice in auditory-cued fear memory, it is important to exclude the possibility that nociception is altered in these mutants. Previously, it was reported that cGKI null mutants show normal acute thermal nociception (Tegeder et al., 2004). To assess the role of cGKI in processing pain caused by an electric foot shock, we determined the minimal current intensity that caused vocalizing or jumping (Fig. 6E). cGKIα smooth muscle rescue mice displayed the same threshold intensity as their litter-matched controls for both behavioral responses (control, 0.39 ± 0.03 mA for vocalizing and 0.37 ± 0.03 mA for jumping, 7 animals; cGKIα smooth muscle rescue mice, 0.37 ± 0.02 mA for vocalizing and 0.36 ± 0.02 mA for jumping, 7 animals).
Discussion
The present study demonstrates for the first time a critical function of the cGKI in auditory-cued fear memory and synaptic plasticity thought to underlie behavioral changes occurring during pavlovian fear conditioning. This conclusion is based on the finding that a loss of the cGKI protein in the brain caused a marked reduction of freezing behavior to a conditioned tone and a decrease of LTP in the LA. Slightly divergent from findings of a recent study using pharmacological tools to modulate the activity cGK (Ota et al., 2008), cGKI-deficient mutants exhibited impaired LTP in both thalamic and cortical inputs. Remarkably, a function in fear memory and synaptic plasticity was specifically associated with the cGKI, whereas the cGKII involved in anxiety-related behavior (Werner et al., 2004) was dispensable because cGKII null mutants showed normal fear conditioning and synaptic plasticity.
Fitting well with the function of cGKI for associative fear learning, we identified the lateral nucleus of the amygdala as a hot spot of its expression within the CNS. Moreover, among regions involved in pavlovian fear conditioning, the LA appears to display a maximal expression level of the cGKI protein, whereas relevant thalamic nuclei showed no significant expression (Fig. 1). There are two isoforms of the cGKI, Iα and Iβ, that differ in their N-terminal domain and cGMP sensitivity. For the amygdala, we detected an expression pattern of the two cGKI isoforms reminiscent of that observed in the cortex and olfactory bulb (Feil et al., 2005b) with cGKIβ as the principal isoform. In contrast, the cGKIα protein highly expressed in cerebellar tissue was virtually absent in the amygdala (Fig. 1D).
A wealth of data, including some evidence with regard to the LA (Huang and Kandel, 1998), supports the view that NO supports LTP in the brain acting as a retrograde messenger turning on presynaptic signaling cascades contributing to synaptic plasticity. Accordingly, one would expect a presynaptic localization of cGKI functionally related to auditory-cued fear memory and synaptic plasticity in the LA. Indeed, our immunohistochemical analysis revealed expression of cGKI in the neuropil, likely including presynaptic terminals and axonal structures (Fig. 1B). However, cGKI was also detected in the cytosol of postsynaptic neurons (Fig. 1B), suggesting that it may be involved in presynaptic and postsynaptic mechanisms contributing to synaptic plasticity and, ultimately, associative memory. Noteworthy, loss of cGKI had no effect on IOR and PPF in cortical and thalamic inputs to the LA (Fig. 3), arguing against a critical function of cGKI for presynaptic transmitter release per se.
Because of the lack of selective pharmacologic tools, studies aimed to resolve the role of individual cGK isoforms in NO/cGMP-dependent signaling heavily depend on suitable transgenic mouse models. A previous analysis of conditional neuronal cGKI knock-out mice (cGKhko) generated using the Cre/loxP system and the NEX–Cre deleter mouse revealed no phenotype in spatial learning, auditory-cued, and contextual fear memory (Kleppisch et al., 2003). Recombination in these mice is expected to be highly efficient in neurons of the neocortex and hippocampus but far less effective in the amygdala (Schwab et al., 2000). cGKIα smooth muscle rescue mice used in this study are characterized by a complete lack of the cGKI protein in neuronal tissue, including the amygdala. Intriguingly, this difference between the two mouse models was paralleled by a difference in their behavioral performance: a defect in associative fear learning was observed exclusively in the cGKIα smooth muscle rescue. More precisely, this defect was selective for auditory-cued fear memory, a discriminatory correlate of amygdaloid function (Phillips and LeDoux, 1992), whereas contextual fear memory was normal. These data in concert with the expression pattern of cGKI in the amygdala and the impairment of synaptic plasticity in the LA of cGK-deficient mice reinforce the view that the LA serves an exclusive function for auditory-cued fear, a view based on previous findings that the LA represents the locus receiving auditory projections from the thalamus and cortex and exhibiting LTP-like changes of synaptic transmission during auditory-cued fear conditioning (for review, see LeDoux, 2007; Sigurdsson et al., 2007). Conversely, intact contextual fear memory in cGKIα smooth muscle rescue mice reflects the fact that it primarily depends on the hippocampal formation and the basolateral nucleus of the amygdala. Therefore, it is not surprising that an impairment of synaptic plasticity in the LA of these mutants had a negligible functional impact on contextual fear memory.
cGKI may alter associative fear learning by modulating elementary behavioral aspects. To address this issue, we examined cGKIα smooth muscle rescue mice in the open field, the elevated plus maze, and the light/dark box. None of these tests yielded a significant difference between cGKIα smooth muscle rescue mice and their controls, arguing against a critical role of cGKI in regulating locomotive activity and innate fear response. Noteworthy, a slight tendency of cGKIα smooth muscle rescue mice to enter the open arm of the elevated plus maze less frequently than controls would, oppositely to our observation, even imply increased freezing during various phases of fear conditioning. According to a cellular model, auditory-cued fear memory results from potentiation of auditory inputs to the LA attributable to simultaneous strong activity of neighboring somatosensory inputs. The latter is triggered by the aversive unconditioned stimulus, in our experimental setup a painful foot shock. Hence, reduced freezing may also occur if cGKIα smooth muscle rescue mice had a defect in nociception. However, a universal function of cGKI in acute pain processing is unlikely. First, cGKI null mutants show normal acute thermal nociception (Tegeder et al., 2004). Second, cGKIα smooth muscle rescue exhibited intact short-term auditory-cued contextual fear memory. Third, the minimal current intensity evoking vocalizing and jumping was the same for cGKIα smooth muscle rescue and control mice. Fourth, there was no genotype difference in freezing to the conditioned tone immediately after its onset, but the difference became apparent solely with prolonged tone presentation. Impaired nociception would be expected to result in a different phenotype. The described time course of freezing in addition to the finding that control and mutant mice showed the same amount of freezing before tone presentation also argue against a major contribution of non-associative processes. Collectively, our data reinforce the view that decreased freezing of cGKIα smooth muscle rescue mice reflects a selective deficit of amygdala-associated auditory-cued fear memory that was not attributable to impaired processing of the tone and/or the shock in the mutants.
What are the implications of our findings with regard to various processes contributing to the freezing response, such as acquisition, consolidation, retrieval, reconsolidation, and extinction of conditioned fear? To target one of these processes selectively, a temporal breakdown of cGKI activity would be desirable. Although this condition is not met by our mouse model, we suggest that the phenotype reported was not attributable to altered reconsolidation or extinction. Fear memory was examined in separate matched cohorts only during a single trial, either 2 h, 1 d, or 7 d after conditioning to minimize contribution of these processes. Interestingly, statistical analysis of auditory-cued freezing depending on the time point revealed a significant deficit of cGKIα smooth muscle rescue mice in auditory-cued fear memory only with prolonged delay (starting from 1 d) between conditioning and testing trials. In contrast, freezing was not significantly reduced 2 h after conditioning. Such time dependency is suggestive of changes in fear memory consolidation, a protein synthesis-dependent process transforming short-term memory into more stable long-term memory (Nader et al., 2000; Schafe and LeDoux, 2000). Indeed, the phenotype of associative fear memory in cGKIα smooth muscle rescue mice developed within a time window similar to that required for consolidation of fear memories in the rat amygdala (Sacchetti et al., 1999). Assuming that the cGKI acts as a downstream effector of NO signaling in the amygdala, our results fit well with previous findings that NOS inhibitors and NO scavengers impair long-term fear memory without affecting short-term and contextual fear memory (Maren, 1998; Schafe et al., 2005). Intriguingly, cGK can link NO signals to protein synthesis in neurons via the transcription factor cAMP response element-binding protein (CREB) (Lu et al., 1999; Lu and Hawkins, 2002; Nagai-Kusuhara et al., 2007), which has been widely implicated in long-term memory (Kaang et al., 1993; Lamprecht et al., 1997; Schafe et al., 2001; Izquierdo et al., 2002; Josselyn et al., 2004; Izquierdo et al., 2006; LeDoux, 2007). It has further been proposed for the LA that relative CREB activity during conditioning determines the number of neurons that become modified to encode a memory (Han et al., 2007). Future studies are needed to elucidate the role of cGKI in these processes.
Collectively, our findings suggest that cGKIβ represents an important downstream element of the NO/cGMP signaling pathway involved in regulation of associative long-term fear memory and synaptic plasticity in the LA.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant KL-1172 (T.K.).
References
- Apergis-Schoute AM, Debiec J, Doyère V, LeDoux JE, Schafe GE. Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: a role for presynaptic plasticity in the fear system. J Neurosci. 2005;25:5730–5739. doi: 10.1523/JNEUROSCI.0096-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci. 2001;21:143–149. doi: 10.1523/JNEUROSCI.21-01-00143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WL, Liang KC, Teng CM, Kuo SC, Lee FY, Fu WM. Enhancement of long-term potentiation by a potent nitric oxide-guanylyl cyclase activator, 3-(5-hydroxymethyl-2-furyl)-1-benzyl-indazole. Mol Pharmacol. 2003;63:1322–1328. doi: 10.1124/mol.63.6.1322. [DOI] [PubMed] [Google Scholar]
- Chien WL, Liang KC, Teng CM, Kuo SC, Lee FY, Fu WM. Enhancement of learning behaviour by a potent nitric oxide-guanylate cyclase activator YC-1. Eur J Neurosci. 2005;21:1679–1688. doi: 10.1111/j.1460-9568.2005.03993.x. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Bladen C, Vincent SR. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995;64:2814–2817. doi: 10.1046/j.1471-4159.1995.64062814.x. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Williams J, Reiner PB, Pelech S, Vincent SR. Localization of the cGMP-dependent protein kinases in relation to nitric oxide synthase in the brain. J Chem Neuroanat. 1999;17:45–55. doi: 10.1016/s0891-0618(99)00023-x. [DOI] [PubMed] [Google Scholar]
- Feil R, Hofmann F, Kleppisch T. Function of cGMP-dependent protein kinases in the nervous system. Rev Neurosci. 2005a;16:23–41. doi: 10.1515/revneuro.2005.16.1.23. [DOI] [PubMed] [Google Scholar]
- Feil S, Zimmermann P, Knorn A, Brummer S, Schlossmann J, Hofmann F, Feil R. Distribution of cGMP-dependent protein kinase type I and its isoforms in the mouse brain and retina. Neuroscience. 2005b;135:863–868. doi: 10.1016/j.neuroscience.2005.06.051. [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Bevilaqua LR, Rossato JI, Bonini JS, Medina JH, Cammarota M. Different molecular cascades in different sites of the brain control memory consolidation. Trends Neurosci. 2006;29:496–505. doi: 10.1016/j.tins.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Izquierdo LA, Barros DM, Vianna MR, Coitinho A, deDavid e Silva T, Choi H, Moletta B, Medina JH, Izquierdo I. Molecular pharmacological dissection of short- and long-term memory. Cell Mol Neurobiol. 2002;22:269–287. doi: 10.1023/a:1020715800956. [DOI] [PubMed] [Google Scholar]
- Jarchau T, Häusler C, Markert T, Pöhler D, Vanderkerckhove J, De Jonge HR, Lohmann SM, Walter U. Cloning, expression, and in situ localization of rat intestinal cGMP-dependent protein kinase II. Proc Natl Acad Sci U S A. 1994;91:9426–9430. doi: 10.1073/pnas.91.20.9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol Learn Mem. 2004;82:159–163. doi: 10.1016/j.nlm.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Kaang BK, Kandel ER, Grant SG. Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 1993;10:427–435. doi: 10.1016/0896-6273(93)90331-k. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Wolfsgruber W, Feil S, Allmann R, Wotjak CT, Goebbels S, Nave KA, Hofmann F, Feil R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J Neurosci. 2003;23:6005–6012. doi: 10.1523/JNEUROSCI.23-14-06005.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krömer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L, Panhuysen M, Pütz B, Deussing JM, Holsboer F, Landgraf R, Turck CW. Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci. 2005;25:4375–4384. doi: 10.1523/JNEUROSCI.0115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Hazvi S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long- but not short-term conditioned taste aversion memory. J Neurosci. 1997;17:8443–8450. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J. The amygdala. Curr Biol. 2007;17:R868–R874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lu YF, Hawkins RD. Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J Neurophysiol. 2002;88:1270–1278. doi: 10.1152/jn.2002.88.3.1270. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Effects of 7-nitroindazole, a neuronal nitric oxide synthase (nNOS) inhibitor, on locomotor activity and contextual fear conditioning in rats. Brain Res. 1998;804:155–158. doi: 10.1016/s0006-8993(98)00668-4. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nagai-Kusuhara A, Nakamura M, Mukuno H, Kanamori A, Negi A, Seigel GM. cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp Eye Res. 2007;84:152–162. doi: 10.1016/j.exer.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Ota KT, Pierre VJ, Ploski JE, Queen K, Schafe GE. The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn Mem. 2008;15:792–805. doi: 10.1101/lm.1114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer A, Aszódi A, Seidler U, Ruth P, Hofmann F, Fässler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 1996;274:2082–2086. doi: 10.1126/science.274.5295.2082. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszódi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fässler R, Hofmann F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Lorenzini CA, Baldi E, Tassoni G, Bucherelli C. Auditory thalamus, dorsal hippocampus, basolateral amygdala, and perirhinal cortex role in the consolidation of conditioned freezing to context and to acoustic conditioned stimulus in the rat. J Neurosci. 1999;19:9570–9578. doi: 10.1523/JNEUROSCI.19-21-09570.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20(RC96):1–5. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Bauer EP, Rosis S, Farb CR, Rodrigues SM, LeDoux JE. Memory consolidation of Pavlovian fear conditioning requires nitric oxide signaling in the lateral amygdala. Eur J Neurosci. 2005;22:201–211. doi: 10.1111/j.1460-9568.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Del Turco D, Schmidtko A, Sausbier M, Feil R, Hofmann F, Deller T, Ruth P, Geisslinger G. Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc Natl Acad Sci U S A. 2004;101:3253–3257. doi: 10.1073/pnas.0304076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler MD. Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J Biol Chem. 1993;268:13586–13591. [PubMed] [Google Scholar]
- Weber S, Bernhard D, Lukowski R, Weinmeister P, Wörner R, Wegener JW, Valtcheva N, Feil S, Schlossmann J, Hofmann F, Feil R. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ Res. 2007;101:1096–1103. doi: 10.1161/CIRCRESAHA.107.154351. [DOI] [PubMed] [Google Scholar]
- Wegener JW, Nawrath H, Wolfsgruber W, Kühbandner S, Werner C, Hofmann F, Feil R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ Res. 2002;90:18–20. doi: 10.1161/hh0102.103222. [DOI] [PubMed] [Google Scholar]
- Werner C, Raivich G, Cowen M, Strekalova T, Sillaber I, Buters JT, Spanagel R, Hofmann F. Importance of NO/cGMP signalling via cGMP-dependent protein kinase II for controlling emotionality and neurobehavioural effects of alcohol. Eur J Neurosci. 2004;20:3498–3506. doi: 10.1111/j.1460-9568.2004.03793.x. [DOI] [PubMed] [Google Scholar]
- Wernet W, Flockerzi V, Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]