

Abstract
Charcot-Marie-Tooth (CMT) type 2 neuropathies are a group of autosomal-dominant axonal disorders genetically and clinically heterogeneous. In particular, CMT type 2B (CMT2B) neuropathies are characterized by severe sensory loss, often complicated by infections, arthropathy, and amputations. Recently, four missense mutations in the small GTPase Rab7 associated with the Charcot-Marie Tooth type 2B phenotype have been identified. These mutations target highly conserved amino acid residues. However, nothing is known about whether and how these mutations affect Rab7 function. We investigated the biochemical and functional properties of three of the mutant proteins. Interestingly, all three proteins exhibited higher nucleotide exchange rates and hydrolyzed GTP slower than the wild-type protein. In addition, whereas 23% of overexpressed wild-type Rab7 was GTP bound in HeLa cells, the large majority of the mutant proteins (82–89%) were in the GTP-bound form, consistent with the data on GTP hydrolysis and exchange rates. The CMT2B-associated Rab7 proteins were also able to bind the Rab7 effector RILP (Rab-interacting lysosomal protein) and to rescue Rab7 function after silencing. Altogether, these data demonstrate that all tested CMT2B-associated Rab7 mutations are mechanistically similar, suggesting that activated forms of the Rab7 are responsible for CMT2B disease.
Keywords: Charcot-Marie-Tooth, neurodegeneration, axon degeneration, Rab7, endocytosis, Rab proteins
Introduction
The inherited neuropathies of the peripheral nervous system called Charcot-Marie-Tooth (CMT) are clinically and genetically very heterogeneous (Auer-Grumbach et al., 2006; Niemann et al., 2006; Schroder, 2006; Zuchner and Vance, 2006). More than 30 genes have been linked to CMT, and many of them are associated with mitochondria and membrane traffic. Historically, CMT diseases type 2 (CMT2) are defined as axonal and show chronic axonal degeneration with loss of nerve fibers. Among these neuropathies, the CMT2B is clinically characterized by marked distal muscle weakness, high frequency of foot ulcers, infections, and amputations of the toes because of recurrent infections.
Recently, four missense mutations in the small GTPase Rab7 have been identified in autosomal dominant ulcero-mutilating neuropathy families (Verhoeven et al., 2003; Houlden et al., 2004; Meggouh et al., 2006). These missense mutations are associated with the CMT2B phenotype and target highly conserved amino acid residues in the GTP-binding and hydrolysis domains of Rab7. Up to now, nothing is known about whether and how these mutations affect specifically sensory and/or motor neurons and lead to an axonal pathology in CMT2B. This issue is particularly challenging considering that Rab7 is a ubiquitous protein.
Rab GTPases localize to the cytosolic face of specific intracellular organelles, regulating distinct steps of membrane traffic (Grosshans et al., 2006; Cai et al., 2007). In addition, recent data have discovered direct links between Rab proteins and signal transduction (Miaczynska et al., 2004; Bucci and Chiariello, 2006; Ceresa, 2006).
Rab7, in particular, controls various aspects of late endocytic traffic in mammalian cells. Indeed, it regulates transport from early to late endosomes, biogenesis of lysosomes and phagolysosomes, and maturation of autophagosomes (Press et al., 1998; Bucci et al., 2000; Harrison et al., 2003; Jager et al., 2004). In addition, it has been demonstrated that Rab7 has a fundamental role in growth factor-regulated cell nutrition and apoptosis (Snider, 2003). Internalization and retrograde axonal transport of neurotrophin receptors are essential for controlling neuronal differentiation, plasticity, and survival. Many neurodegenerative diseases, such as Alzheimer's disease and Niemann-Pick disease type C, show alterations of vesicular transport. Therefore, it is tempting to speculate that altered neurotrophin trafficking is one of the factors that could heavily influence neurodegeneration in these diseases (Bronfman et al., 2007). Interestingly, Rab7 has been recently involved in the control of retrograde axonal transport of neurotrophin receptors (Saxena et al., 2005; Deinhardt et al., 2006). In addition, Rab-interacting lysosomal protein (RILP), a Rab7 effector, is required for formation of multivesicular bodies involved in receptor degradation (Progida et al., 2007). These data support the hypothesis that CMT2B disease could be caused by alteration of neurotrophin receptor trafficking as a consequence of a mutated Rab7 gene. It is therefore of interest to understand how CMT2B-associated Rab7 mutations alter the functionality of this GTPase. Understanding how dysfunction of Rab7 causes sensory and/or motor neuropathy in patients with CMT2B will be instrumental in gaining insights into the pathogenesis of this group of disorders, and will open the way to identify therapeutic approaches.
Materials and Methods
Cells and reagents.
Restriction and modification enzymes were from New England Biolabs (Ipswich, MA), chemicals were from Sigma-Aldrich (St. Louis, MO), and radioactive compounds were from Perkin-Elmer (Waltham, MA). Tissue culture reagents were from Sigma-Aldrich. HeLa cells were grown in DMEM supplemented with 10% FBS, 2 mm glutamine, 100 U/ml penicillin, and 10 μg/ml streptomycin, in a 5% CO2 incubator at 37°C.
Mutagenesis and plasmid construction.
Most constructs used in this study have been described previously (Vitelli et al., 1997; Bucci et al., 2000; Cantalupo et al., 2001; Colucci et al., 2005). Rab7 Charcot-Marie-Tooth mutants were constructed by PCR-mediated mutagenesis (Landt et al., 1990). Oligonucleotides used to generate the Rab7 L129F, Rab7 N161T, and Rab7 V162M mutations were, respectively, 5′-GTTGGGAAACAAGATTGACTTCGAAAACAGACAAGTGG-3′, 5′-GTGCCAAGGAGGCCATCACTGTGGAGCAGGCGTTCCAG-3′, and GCCAAGGAGGCCATCAATATGGAGCAGGCGTTCCAGAC-3′. The mutagenesis was performed on wild-type Rab7 (Rab7 wt) cDNA previously cloned in pEGFPC1 vector. pEGFPfor (5′-GATCACTCTCGGCATGGA-3′) and pEGFPrev (5′-CATTTTATGTTTCAGGTTCAGGG-3′) were used as outer primers. Subsequently, the mutated Rab7 cDNAs were cloned in pEGFP-C1, pET16b, pCDNA3-Myc, pCDNA3–2xHA, or pBTM116 vectors in frame, respectively, with green fluorescent protein (GFP), poly-His, Myc, hemagglutinin (HA) tags, or LexA-binding domain.
Transfection and RNA interference.
Transfection was performed using Metafectene Pro from Biontex (Karlsruhe, Germany), as indicated by manufacturer. After 20 h of transfection, cells were processed for immunofluorescence or biochemical assays. For RNA interference, small interfering RNAs (siRNAs) were purchased from MWG-Biotech (Ebersberg, Germany). We used the following oligonucleotides: siRNA Rab7-1, sense sequence 5′-GGAUGACCUCUAGGAAGAATT and antisense sequence 5′-UUCUUCCUAGAGGUCAUCCTT-3′; Rab7-2, sense sequence 5′-GAACACACGUAGGCCUUCATT and antisense sequence 5′-UGAAGGCCUACGUGUGUUCTT-3′. As a negative control we used a scrambled sequence: sense scrambled control 5′-ACUUCGAGCGUGCAUGGCUTT-3′ and antisense scrambled control 5′-AGCCAUGCACGCUCGAAGUTT-3′. Both Rab7 siRNAs were efficient in silencing Rab7, as mRNA levels were reduced by ∼80% and the Rab7 endogenous protein was not anymore detectable by Western blot analysis. For all subsequent experiments, we used siRNA Rab7-1. HeLa cells were plated 1 d before transfection in tissue culture dishes (6 cm diameter). Cells were transfected with siRNAs using Oligofectamine from Invitrogen (Milan, Italy) for 72 h, replated, and left 48 h before performing further experiments.
Antibodies.
Anti-RILP antibodies have been previously described (Cantalupo et al., 2001). Mouse monoclonal 9E10 anti-Myc and rabbit polyclonal anti-HA antibodies were from Abcam (Cambridge, UK), rabbit polyclonal anti-Rab7 and mouse monoclonal anti-tubulin were from Sigma-Aldrich, whereas sheep polyclonal antibody against epidermal growth factor receptor (EGFR) was from Fitzgerald (Concord, MA). Secondary antibodies conjugated with fluorochromes or HRP were from Abcam or Fitzgerald. All primary antibodies were used at a 1:1000 dilution.
Western blotting.
Separated proteins were transferred onto polyvinylidene difluoride membrane from Millipore (Milan, Italy). The filter was blocked in 5% milk in PBS for 30 min at room temperature and incubated with the appropriate antibody and then with a secondary antibody conjugated with HRP (diluted 1:5000). Bands were visualized using enhanced chemiluminescence system from GE (Milan, Italy).
Confocal immunofluorescence microscopy.
Cells grown on 11 mm round glass coverslips were permeabilized, fixed, and incubated with the antibodies as described previously (Bucci et al., 1992). Cells were viewed with Zeiss (Thornwood, NY) LSM 510 confocal microscope.
GTP overlay and GTPase assay.
Cells were lysed in standard SDS sample buffer and extracts were electrophoresed on 12% SDS-polyacrylamide gels. For GTP overlay, after electrophoresis, the gel was treated, transferred to nitrocellulose filter, and incubated with [α-32P] GTP as described (Bucci et al., 1992). The GTPase assay was performed as previously described (Stenmark et al., 1994). Briefly, aliquots of Ni2+ agarose were incubated with His-tagged Rab7 wt or mutant purified proteins for 20 min at 25°C in buffer A (20 mm Tris-Cl, pH 7.8, 100 mm NaCl, 5 mm MgCl2, 1 mm Na-phosphate, and 10 mm 2-mercaptoethanol). Bound nucleotide was eluted with 1 m guanidine-HCl. [α-32P] GTP was bound to the immobilized protein for 10 min at 0°C in buffer A, and then the protein was incubated at 37°C for various time periods (0–8 h). After incubation, samples were washed rapidly at 4°C, and buffer B (0.2% SDS, 2 mm EDTA, 10 mm GDP, and 10 mm GTP, pH 7.5) was added. Samples were heated at 70°C for 2 min and then spotted on polyethyleneimine (PEI) cellulose plates. GTP hydrolysis was quantified as GDP signal relative to GDP+GTP signal.
Nucleotide dissociation assay.
His-tagged proteins were expressed and purified from Escherichia coli as previously described (Chiariello et al., 1999). Nucleotide exchange assay was performed essentially as described previously (Shapiro et al., 1993). Proteins were bound to NiNTA resin for 20 min at 25°C in buffer A (20 mm Tris-Cl, pH 7.8, 100 mm NaCl, 5 mm MgCl2, 1 mm Na-phosphate, pH 7.4, and 10 mm 2-mercaptoethanol), washed, and then incubated for 6 h at 37°C with [3H]GTP or [3H]GDP. After binding, 100-fold molar excess of unlabeled GTP or GDP was added, and samples were incubated for various times (from 0 to 60 min). After incubation, samples were washed extensively in buffer A. The radioactivity associated with the proteins was determined by scintillation counting. Nucleotide dissociation constants (Koff) were determined as described (Shapiro et al., 1993).
Determination of GTP/GDP ratio.
The analysis was performed as described (Stenmark et al., 1994). Briefly, HeLa cells in tissue culture dishes (3 cm diameter) were transfected with HA-tagged Rab7 wt and mutant constructs. Transfection was performed in phosphate-free DMEM containing 0.5 mCi/ml [32P]orthophosphate and 5% dialyzed serum. Sixteen hours after transfection, cells were washed three times with PBS and lysed. Nuclei and debris were removed by centrifugation, and the supernatant was subjected to immunoprecipitation using EZview Red anti-HA affinity gel from Sigma-Aldrich according to the manufacturer's instructions. The bound nucleotides were eluted in sample buffer, and samples were spotted onto PEI cellulose thin-layer chromatography (TLC) plates and developed with 0.6 m Na-phosphate, pH 3.4. After drying, plates were subjected to autoradiography. Quantification was made using a PhosphorImager, taking into account that the specific activity of [32P]GDP is 2/3 of [32P]GTP.
Two-hybrid assay.
pBTM116-Rab7 wt and mutant constructs were used together with the pGADGH-RILPC33 to test the interaction using the Saccharomyces cerevisiae L40 reporter strain (Schiestl and Giest, 1989; Hill et al., 1991; Bartel et al., 1993; Bucci et al., 1999). Transformants were plated onto synthetic medium lacking His, Leu, and Trp; after 4 d of growth at 30°C, we performed a liquid β-galactosidase assay using o-nitrophenyl-β-d-galactoside as a substrate to quantify the interactions (Guarente, 1983; Bartel et al., 1993).
EGF and EGFR degradation assay.
HeLa cells were incubated overnight in starvation medium (0.5% BSA, 20 mm HEPES, pH 7.3, in DMEM). For EGF degradation assay, cells were subsequently incubated for 1 h at 4°C with 0.8 mg/ml rhodamine-labeled EGF (Invitrogen) in starvation medium and then washed several times with starvation medium. After incubation at 37°C in complete DMEM medium for different time points (from 15 min up to 3 h), cells were fixed, mounted on slides, and processed for quantitative confocal microscopy. Transfected cells were selected, and the amount of red signal (rhodamine-labeled EGF) in each entire cell was quantified using Zeiss LSM510 software. For EGFR degradation assay, HeLa cells were treated with 10 μg/ml cycloheximide for 1 h, preventing synthesis of new EGFR during the stimulation by EGF. Immediately after, cells were continuously stimulated with 50 ng/ml EGF for 15, 60, 120, or 180 min. The levels of undegraded EGFR were determined by Western blotting. The intensity of EGF or EGFR at each time point was calculated relative to the intensity measured at 15 min (which was set to 100%).
Results
The CMT2B-associated Rab7 mutant proteins bind GTP
To start investigating the molecular mechanism responsible for the development of the disease, we decided to analyze the biochemical properties of the Rab7 mutant proteins associated with CMT2B. To this purpose, we constructed the following mutants: Rab7 L129F, Rab7 N161T, and Rab7 V162M. We started analyzing the ability of the mutant proteins to bind GTP performing a classical GTP overlay. We expressed Myc-tagged constructs in HeLa cells. Proteins were loaded onto an SDS-polyacrylamide gel, renatured during transfer onto nitrocellulose, and allowed to bind radioactive GTP. As a control, we loaded also Rab7 wt, the dominant-negative mutant Rab7 T22N (impaired in nucleotide exchange and with a reduced affinity for GTP) and the constitutively active mutant Rab7 Q67L (impaired in its intrinsic GTP hydrolysis but not in GTP binding). The Myc-tagged Rab7 wt and mutant proteins were expressed at comparable levels as judged by Western blot analysis made with a monoclonal anti-Myc antibody (Fig. 1B). As expected, both Rab7 wt and Rab7 Q67L were binding GTP efficiently, whereas Rab7 T22N was not (Fig. 1A). The Rab7 L129F, Rab7 N161T, and Rab7 V162M mutants were all binding GTP (Fig. 1A).
Figure 1.

GTP overlay and Western blot analysis on HeLa cells expressing Myc-tagged Rab7 wt and mutant proteins. Cells were transfected with the indicated Myc-tagged Rab7 wt and mutant proteins and lysed, and the extracts were analyzed by SDS-PAGE. A, GTP overlay was performed as indicated in Materials and Methods. B, Western blot analysis with the same lysates was performed using monoclonal anti-Myc antibody as detailed in Materials and Methods.
The CMT2B-associated Rab7 mutant proteins have impaired GTPase activity
We then decided to analyze the GTPase activity of the mutants. Rab7 wt, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M were tagged N-terminally with His residues and expressed in E. coli. Proteins were purified and immobilized onto Ni2+ beads, loaded with [α-32P] GTP, and subsequently incubated at 37°C for various time intervals to allow GTP hydrolysis to occur. The estimated hydrolysis rate constant for His-Rab7 wt at 37°C was 0.0023 min−1, in agreement with a previous finding made on Rab7 wt (Shapiro et al., 1993). The results of this experiment are shown in Figure 2A. As expected, the constitutively active mutant Rab7 Q67L hydrolyzed GTP much slower than Rab7 wt. Interestingly, all three mutants showed slower GTP hydrolysis, similar to the Rab7 Q67L mutant.
Figure 2.
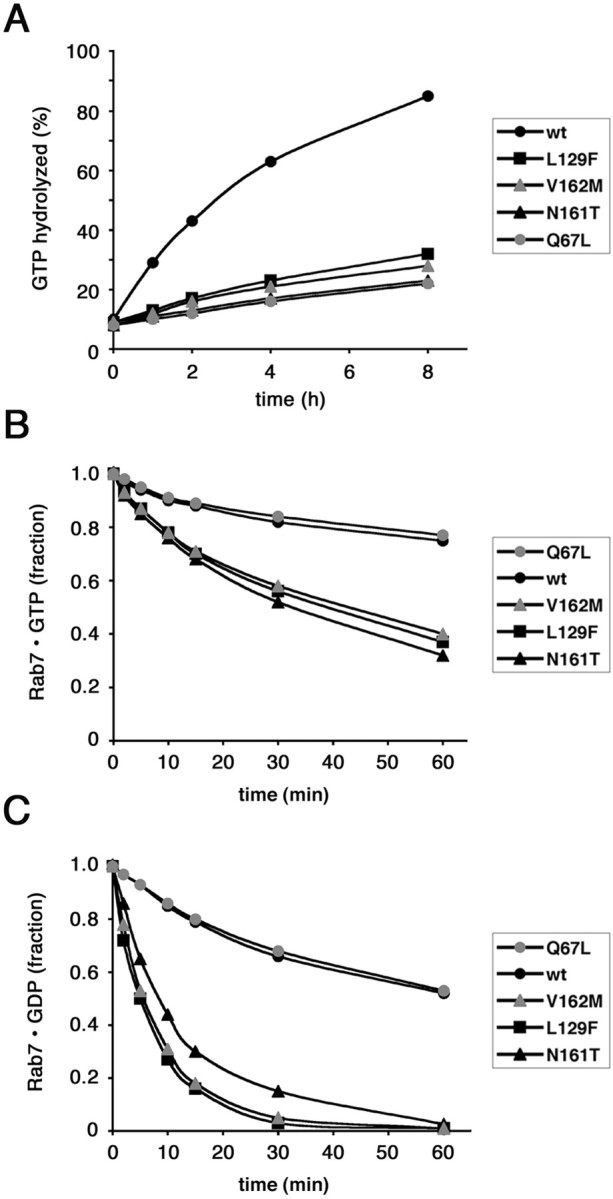
Biochemical characterization of CMT2B-associated Rab7 mutant proteins. A, Measurement of the intrinsic GTPase activity of His-tagged Rab7 wt, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M, as indicated. Proteins were loaded with labeled GTP, and then 100-fold molar excess of GTP or GDP was added. GTP hydrolysis was monitored during 8 h. Differences between the values of all Rab7 mutated proteins and those of Rab7 wt are statistically significant (p value <0.01) at 1, 2, 4, and 8 h. B, C, Dissociation of guanine nucleotides from Rab7 wt, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M. Purified proteins were incubated with [3H]GTP (B) or [3H]GDP (C). In the experiment shown, prebinding was allowed to proceed to 30% saturation. One hundred-fold molar excess of cold competitor (GDP or GTP) was then added, and dissociation of nucleotide was monitored for 1 h. In B, differences between the values of all Rab7 CMT2B mutated proteins and those of Rab7 wt are statistically significant (p value <0.01) at 15, 30, and 60 min. In C, differences between the values of all Rab7 CMT2B mutated proteins and those of Rab7 wt are statistically significant (p value <0.01) at 5, 10, 15, 30, and 60 min.
Nucleotide dissociation rates are higher in the CMT2B-associated Rab7 mutant proteins
We then investigated the nucleotide dissociation activity of the Rab7 mutants compared with Rab7 wt. Dissociation rate constants (Koff) were determined by prebinding radiolabeled nucleotide ([3H] GTP or [3H] GDP) to His-tagged proteins, adding 100-fold molar excess of unlabeled competitor nucleotide and then assaying for loss of radioactivity bound to proteins over time (Fig. 2B,C). As a competitor, we used GDP or GTP with similar results, in agreement with previous studies (Shapiro et al., 1993). Rate constants were calculated from data fit to first-order exponential functions; Koff values of His-Rab7 wt at 37°C were ∼0.0045 for GDP (mol GDP released per mol protein min−1) and 0.0025 for GTP (mol GTP released per mol protein min−1), very close to previously published results (Shapiro et al., 1993). As expected, Rab7 Q67L exhibited Koff values similar to those of Rab7 wt, confirming that the mutation affects only GTP hydrolysis and not the affinity for guanine nucleotides (Fig. 2B,C). In contrast, all three CMT2B-associated Rab7 mutant proteins exhibited Koff values significantly higher than Rab7 wt (Fig. 2B,C). In particular, Koff values for GDP (0.0179, 0.0150, and 0.0175, respectively, for Rab7 L129F, Rab7 N161T, and V162M) were up to fourfold higher than those measured with Rab7 wt. Koff values for GTP were less affected (0.0064, 0.0068, and 0.0062, respectively, for Rab7 L129F, Rab7 N161T, and V162M), slightly more than twofold higher than those of Rab7 wt. These results demonstrate that all CMT2B-associated Rab7 mutant proteins have reduced affinity for guanine nucleotides, in particular for GDP.
The CMT2B-associated Rab7 mutant proteins are prevalently in the GTP-bound form in HeLa cells
Cellular factors heavily modify nucleotide exchange and GTP hydrolysis of Rab proteins, and therefore we decided to determine the ratios between the GDP- and the GTP-bound forms of the wild-type and mutant proteins when expressed in HeLa cells. For this purpose, we transfected HeLa with HA-tagged Rab7 wt, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M. During transfection, cells were incubated with 32PO43− to label the endogenous pool of GDP and GTP. Cells were lysed, and the expressed proteins were immunoprecipitated with a polyclonal anti-HA antibody. [32P]-labeled nucleotides were then analyzed by TLC as described (Stenmark et al., 1994) (Fig. 3).
Figure 3.

GTP:GDP ratio of Rab 7 wt and mutant proteins in transfected HeLa cells. Cells were transfected with the indicated HA-tagged constructs and lysed, and immunoprecipitation was performed using an anti-HA antibody. The amounts of nucleotides bound to the immunoprecipitated proteins were determined by TLC, as described in Materials and Methods. The positions of GTP and GDP are indicated.
Immunoprecipitated HA-tagged Rab7 was predominantly in the GDP-bound form, in agreement with literature data on other Rab GTPases; indeed, only 23% was GTP bound (Fig. 3). In contrast, as expected, the constitutively active mutant Rab7 Q67L, defective in GTP hydrolysis, was predominantly in the GTP-bound form (70%) (Fig. 3). Interestingly, all three Rab7 mutant proteins associated with CMT2B were >80% in the GTP-bound form (82% for Rab7 L129F, 89% for Rab7 N161T, and 87% for Rab7 V162M) (Fig. 3). This finding was coherent with the biochemical properties of these proteins in vitro (Fig. 2).
The mutant proteins interact with RILP and colocalize with RILP on membranes
Next, we wanted to investigate whether the CMT2B-associated Rab7 mutants were able to interact with RILP, one of the Rab7 effectors. This is particularly relevant because, using our polyclonal antibodies against RILP, Houlden et al. (2004) observed a significant reduction of RILP expression in the sural nerve biopsy specimen from a patient with a CMT2B form of ulcero-mutilating neuropathy compared with control.
Using the two-hybrid assay, we tested the ability of the mutants to interact with RILP, and in particular with the C-terminal half of the protein, which contains the Rab7-binding domain (Cantalupo et al., 2001). As control, we used Rab7 wt, Rab7 T22N, and Rab7 Q67L (Fig. 4). Rab7 wt and Rab7 Q67L showed a strong interaction with RILP that interacts preferentially with the GTP-bound form of Rab7, whereas, as expected, the interaction of the dominant-negative mutant Rab7 T22N was extremely weak (Fig. 4). All three CMT2B-associated Rab7 mutants were interacting strongly with RILP (Fig. 4).
Figure 4.

Interaction of Rab7 wt and mutant proteins with RILP. L40 yeast cells were cotransformed with pGADGH-RILPC33 and the indicated pLexA-Rab7 wt or mutant constructs. The β-galactosidase activity of double transformants was measured using o-nitrophenyl-β-d-galactoside as substrate as detailed in Materials and Methods. Activities are measured as arbitrary relative units and represent mean ± SEM values of six independent transformants. Differences between the values of Rab7 CMT2B mutated proteins and that of Rab7 wt are statistically significant at p values <0.05 (*) or <0.01 (**).
Next, we tested whether RILP expression was decreased in cells transfected with the CMT2B mutants compared with Rab7 wt, similarly to what has been observed in sural nerve biopsy of a CMT2B patient (Houlden et al., 2004). We transfected HeLa cells for 20 h with HA-tagged Rab7 wt, Rab7 L129F, Rab7 N161T, and Rab7 V162M and checked RILP expression by Western blot analysis (Fig. 5). Equal loadings were verified using anti-tubulin antibody (Fig. 5). The Rab7 wt and mutant proteins were expressed at similar levels as demonstrated by Western blot analysis made with an anti-HA antibody (Fig. 5). In these conditions, we could not reveal any significant decrease in RILP expression (Fig. 5).
Figure 5.

Transient expression of CMT2B-associated Rab7 mutant proteins does not alter RILP expression. HeLa cells were nontransfected (NT) or transfected with HA-tagged Rab7 wt, Rab7 L129F, Rab7 N161T, or Rab7 V162M for 20 h and then lysed. Lysates were analyzed by Western blot using anti-RILP antibody. Anti-tubulin was used to verify equal loadings, and anti-HA antibody was used to check expression of the different Rab7 constructs.
We then decided to analyze whether these CMT2B-associated Rab7 proteins colocalized with the Rab7 effector RILP in HeLa cells. Therefore, we transfected GFP-tagged Rab7 wt, Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M together with RILP, and we analyzed their intracellular localization. Overexpression of RILP causes clustering of late endosomal/lysosomal structures around the MTOC (microtubule-organizing center) (Cantalupo et al., 2001; Johansson et al., 2007). Rab7 wt and the constitutively active mutant Rab7 Q67L colocalized with RILP on these clustered structures, whereas the Rab7 T22N dominant-negative mutant showed a more disperse localization (Fig. 6 A–L) (Cantalupo et al., 2001). When coexpressed with RILP, the CMT2B-associated Rab7 mutant proteins were present on these clustered endosomal structures, similarly to Rab7 wt and Rab7 Q67L (Fig. 6 M–X).
Figure 6.
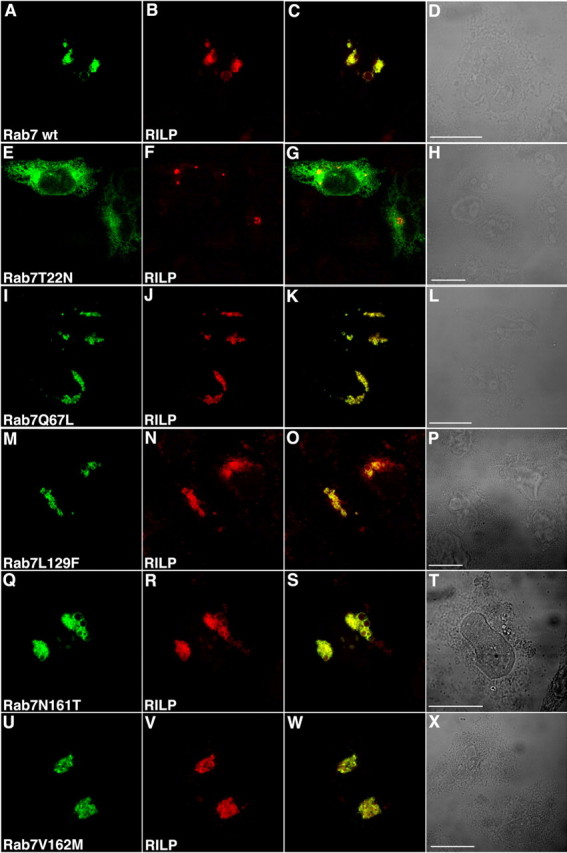
Colocalization of CMT2B-associated Rab7 wt and mutant proteins with RILP. HeLa cells were cotransfected with RILP and GFP-tagged Rab7 wt and mutants as indicated. A, E, I, M, Q, U, GFP signal. B, F, J, N, R, V, TRITC signal of secondary anti-rabbit antibody used after a primary anti-RILP antibody. C, G, K, O, S, W, Overlays of the two colors. D, H, L, P, T, X, Phase-contrast images. Scale bars, 10 μm.
Expression of the mutant proteins is able to rescue Rab7 function after silencing
After having established the biochemical properties of the CMT2B-associated Rab7 mutant proteins in terms of GTP/GDP binding and GTP hydrolysis, intracellular localization, and interaction with RILP, we decided to analyze their functional properties in HeLa cells.
It has been established that Rab7, together with RILP, controls transport to degradative compartments and, in particular, degradation of EGF/EGF receptor complex (Vitelli et al., 1997; Cantalupo et al., 2001; Ceresa, 2006; Wang and Hong, 2006). Thus, we decided to measure EGF degradation after expression of Rab7 wt and mutant proteins. To this purpose, we transfected cells to express GFP-tagged Rab7, Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M, and then we starved the cells and incubated them with rhodamine-labeled EGF. After washing, we incubated the cells for various time intervals (from 15 min to 3 h) to follow EGF degradation. Transfected cells were selected, and the red signal (rhodamine-labeled EGF) present in each selected cell was quantified. Expression of the dominant-negative mutant Rab7 T22N inhibited dramatically EGF degradation, whereas no effect was observed after transfection of Rab7 wt or Rab7 Q67L (Fig. 7), in accord with previously published results (Vitelli et al., 1997; Cantalupo et al., 2001). Indeed, at 3 h time point, EGF signal was barely visible in control cells and in cells transfected with Rab7 wt and Rab7 Q67L, in contrast to cells transfected with Rab7 T22N, where most of the label was still present (>70%) (Fig. 7). Interestingly, also the expression of the CMT2B-associated Rab7 mutant proteins did not alter EGF degradation significantly (Fig. 7).
Figure 7.
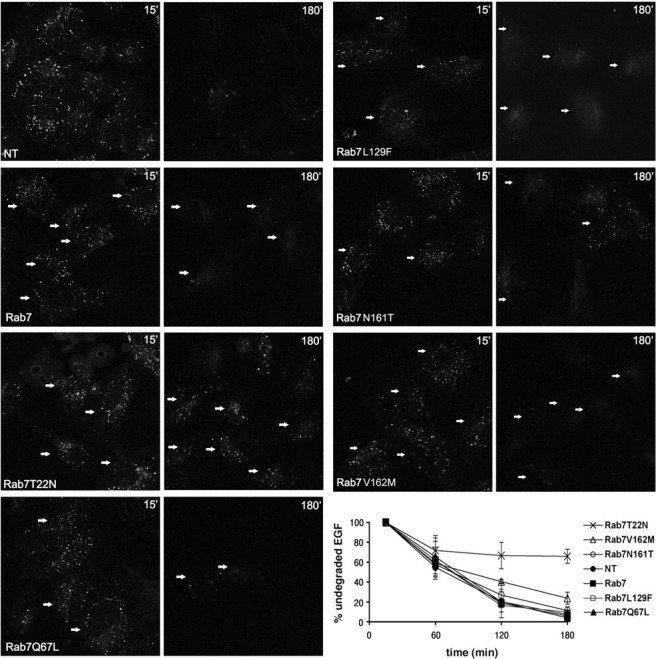
Effect of CMT2B-associated Rab7 mutations on EGF degradation. HeLa cells were transfected with GFP-tagged Rab7 wt, Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 N161T, Rab7 V162M, or nontransfected (NT) as indicated. Then cells were incubated 1 h at 4°C with rhodamine-labeled EGF, washed, and incubated at 37°C for 15 min or 3 h. Finally, they were processed for confocal fluorescent microscopy. Transfected cells were selected, and the total red signal in the entire cell (representing undegraded EGF) was quantified. Staining of EGF at 15 and 180 min is shown. Arrows indicate transfected cells. Intensities of EGF were quantified and plotted as percentage of the respective intensities after 15 min incubation at 37°C (bottom right). Values are calculated on 30 cells for each construct and represent means ± SEM. Differences between the values of Rab7 CMT2B mutated proteins and that of Rab7 T22N are statistically significant (p value <0.01) at 180 min. Differences between the values of Rab7 V162M and those of Rab7 wt or the other Rab7 CMT2B mutated proteins are not statistically significant.
We then decided to look at degradation of EGFR using Western blot analysis (Fig. 8). HA-tagged Rab7 wt and mutant proteins were transfected in HeLa cells. Cells were starved and incubated with EGF for various times as described in Materials and Methods. The amount of EGFR was revealed using a polyclonal anti-EGFR antibody, and anti-tubulin antibody was used to verify equal loadings (Fig. 8A). In agreement with the data obtained on EGF degradation, a strong inhibition of EGFR degradation was detected when the Rab7 T22N mutant was expressed (Fig. 8). In contrast, expression of Rab7 wt, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M did not significantly alter EGFR degradation (Fig. 8).
Figure 8.

Effect of CMT2B-associated Rab7 mutations on EGFR degradation. HeLa cells were transfected for 20 h with HA-tagged Rab7 wt, Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 N161T, Rab7 V162M, or nontransfected (NT) as indicated. Cells were then treated with cycloheximide for 1 h and subsequently with EGF for 15 or 180 min. A, Lysates were subjected to Western blot analysis using an anti-EGFR antibody. Western blot with anti-tubulin antibody on the same membrane was used to verify equal loadings. B, The intensities of EGFR staining were quantified and plotted as a percentage of the respective intensities after 15 min of EGF stimulation. Values at 15 min were set to 100%. Values are means of four independent experiments ± SEM (error bars). Differences between the values of cells expressing Rab7 T22N and the other samples are statistically significant (p value <0.01); differences between the values of cells expressing Rab7 wt and cells expressing CMT2B associated Rab7 mutants are not statistically significant.
To establish whether the CMT2B-associated Rab7 mutant proteins were able to rescue Rab7 function after silencing, we used RNA interference to shut down the expression of endogenous Rab7. By this methodology, Rab7 mRNA expression was decreased by ∼80% (Fig. 9A), and we were not able to detect the Rab7 protein by Western blot (Fig. 9B). Degradation of EGF was inhibited by ∼80% after silencing (Fig. 9C), similarly to what happened when the dominant-negative mutant Rab7 T22N was expressed (Fig. 8B). Cells silenced for Rab7 were then transfected with HA-tagged Rab7 wt, Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 N161T, and Rab7 V162M. As expected, both Rab7 wt and Rab7 Q67L were able to rescue Rab7 function, although partially, whereas Rab7 T22N mutant was not. Interestingly, all three CMT2B-associated Rab7 mutant proteins rescued Rab7 function, although to a different extent. These data demonstrate that these mutant proteins, prevalently bound to GTP, are functionally active.
Figure 9.

Expression of CMT2B-associated Rab7 mutant proteins restores Rab7 function. A, HeLa cells were treated with control RNA (scr) and two different siRNAs against Rab7 (Rab7-1 or Rab7-2). Rab7 mRNA levels were determined by real-time PCR as indicated in Materials and Methods. Decreases in Rab7 mRNA levels in siRNA-treated samples are statistically significant at p value <0.01 (**). B, HeLa cells were treated with control siRNA (scr) or siRNA against Rab7 (Rab7-1), lysed, and subjected to Western blot analysis using anti-Rab7 antibody. Anti-tubulin antibody was used to verify equal loadings. C, HeLa cells were treated with control RNA (scr) or with Rab7-1 siRNA. Cells silenced for Rab7 were then transfected with the different Rab7 wt or mutant constructs as indicated. All cells were incubated for 1 h at 4°C with rhodamine-labeled EGF, washed, and incubated at 37°C for 15 min or 3 h. Transfected cells were selected, and the red signal (representing undegraded EGF) was quantified by confocal microscopy. The intensities of EGF labeling were quantified and plotted as percentage of the respective intensities after 15 min of incubation at 37°C. Values at 15 min were set to 100%. Values were calculated on 30 cells for each construct and represent means ± SEM. Differences between the values of cells expressing Rab7 CMT2B mutated proteins and that of siRNA-treated control cells are statistically significant at p value <0.01 (**). Differences between the values of cells transfected with the different CMT2B mutated proteins are not statistically significant. D, The amount of expression of Rab7 wt and mutant proteins was checked by Western blot analysis using anti-Rab7 antibody. Anti-tubulin antibody was used to verify equal loadings.
Discussion
Recently, four Rab7 mutations have been associated with CMT2B neuropathies (Verhoeven et al., 2003; Houlden et al., 2004; Meggouh et al., 2006). This association is particularly puzzling, because Rab7 is a ubiquitous protein, important for transport to degradative compartment in endocytosis. We have started to characterize the properties of three of these mutant proteins. Interestingly all three mutant proteins show the same features, having altered guanine nucleotide dissociation and GTP hydrolysis, which result in a strong accumulation of the GTP-bound form in the cells.
The biochemical properties of the three CMT2B-associated Rab7 mutant proteins are consistent with structure–function relationship studies and crystallographic data on Rab proteins (Segev, 2001; Pfeffer, 2005; Grosshans et al., 2006). All three mutations map to putative structural domains that are thought to participate in the formation of the nucleotide-binding site. In all GTP-binding proteins, this site is characterized by four conserved sequence motifs (Σ1–Σ4) (Kjeldgaard et al., 1996). The first motif, GxxxxGK(S/T) (Σ1), is common to many proteins that bind purine nucleoside triphosphates and is often referred to as the phosphate-binding loop. The second motif, DxxG (Σ2), is located close to Σ1 and is thought to be involved in the conformational change taking place between the GDP and GTP forms. The third, NKxD (Σ3), and the fourth, (C/S)A[x]x (Σ4), motifs are situated, respectively, within the β5/α4 (also known as λ8) and the b6/a5 (λ10) loops, which form a hydrophobic pocket on the surface of the core domain accommodating the guanine base (Fig. 10A).
Figure 10.

Amino acid sequence and secondary structure of wild-type and CMT2B-associated Rab7 proteins. A, Amino acid sequence of the human wild-type Rab7 protein. Asterisks mark the amino acid substitutions in the mutated proteins Rab7 T22N, Rab7 Q67L, Rab7 L129F, Rab7 K157N, N161T, and V162M. Amino acids in the four sequence motifs (Σ1–Σ4) conserved in all GTP-binding proteins are underlined. Secondary structure above the amino acid sequence has been predicted on the basis of the available structural data for Rab proteins (Merithew et al., 2001). α1–α5, α helices; β1–β6, β sheets; λ1–λ10, loops. B, Secondary structure prediction in the C-terminal region of wild-type and CMT2B-associated Rab7 proteins. The software Secondary Structure Prediction at the Pôle Bioinformatique Lyonnais (http://pbil.univ-lyon1.fr/) has been used with the methods DSC, MLRC, and PHD (with default parameters) that generated the secondary consensus shown below each amino acid sequence. h, α helix; e, extended strand; c, random coil; ?, ambiguous state.
The Rab7 mutation L129F affects a Leu residue that maps within the λ8 loop, immediately downstream of the NKxD (Σ3) motif (Fig. 10A). In Ras-p21, the Asp119 (Asp128 in Rab7) in the motif NKxD forms hydrogen bonds with N1 and N2 of the guanine base (Kjeldgaard et al., 1996; Paduch et al., 2001). It is reasonable that substitution of bulky Phe for Leu at position 129 may disturb the correct positioning of the adjacent Asp128 with respect to the guanine moiety of GDP or GTP, resulting in an increased Koff for both nucleotides and in a decreased GTPase activity (Fig. 2). N161T and V162M affects two evolutionarily conserved amino acids in all exocytic and endocytic Rab GTPases (Merithew et al., 2001). These residues (Asn161 and Val162) map within the λ10 loop immediately upstream of the α5 helix (Fig. 10A). Interestingly, the fourth CMT2B-associated Rab7 mutation, K157N (Meggouh et al., 2006), is located in the same region and affects the third amino acid of the (C/S)A[x]x motif, immediately downstream of the Ala156, which is thought to bind the exocyclic O6 of the guanine moiety of both GDP and GTP (Kjeldgaard et al., 1996; Paduch et al., 2001). An in silico analysis of secondary structure led to the prediction that the conformational changes induced by these three mutations may propagate to the upstream β6 sheet (Fig. 10B). This effect is dramatic, with K157N and N161T causing regional loss of secondary structure. The more severe GTPase defect of N161T with respect to V162M (Fig. 2A) may be associated with this change.
One of the major findings of this study is that all known CMT2B-associated Rab7 mutations are mechanistically similar. The more reduced affinity for GDP than for GTP exhibited by the mutated proteins may be attributable to the fact that the GTP-bound conformation is stabilized by additional hydrogen-bonding interactions with the γ phosphate of GTP, mediated by Ser residues in the phosphate loop and switch region I, as well as an extensive hydrophobic interface between the switch I and II regions, which are also involved in effector binding. Because of impaired GTPase activity and reduced affinity for guanylic nucleotides, in particular for GDP, CMT2B-associated Rab7 proteins were found within the cell almost completely associated with GTP, whereas in our assay conditions, the wild-type protein was detected predominantly in the inactive GDP-bound form (Fig. 3). The ratios between the GTP- and GDP-bound forms were even higher than those observed with Rab7 Q67L (Fig. 3), a constitutively active protein harboring a mutation mapping immediately downstream of the DxxG (Σ2) motif involved in GTP hydrolysis (Fig. 10A) but exhibiting a Koff for GTP and GDP close to that of the wild-type protein (Fig. 2B,C). One might thus be led to suppose that all CMT2B-associated Rab7 mutations produce constitutively activated effects.
Transient expression of the CMT2B-associated mutations did not produce significant functional change along the Rab7-controlled endocytic pathway in HeLa cells. Indeed, expression of the mutant proteins neither altered significantly EGF or EGFR degradation (Figs. 7, 8) nor impaired RILP effect on clustering of late endosomal/lysosomal compartment (Fig. 6). To explain such a lack of functional effect in the cellular system, we have to consider that, whereas Rab dominant-negative mutants display strong phenotypes, activated Rab mutant proteins show only minor functional alterations along trafficking routes. This behavior can be explained considering that Rab proteins exert their function by interacting with multiple effectors. One or more of these factors might be rate limiting in the assays, therefore not allowing us to see effects of the expression of the activated mutant proteins. Clearly, future functional assays on these mutations will have to be done on different cell types (as for instance PC12 cells or, even better, DRG neurons) following trafficking of relevant molecules (for instance, neurotrophins), but the presence of limiting factors will probably not allow easy identification of a phenotype.
Altogether, these results raise an interesting question about the causative role of the Rab7 mutations in the CMT2B disease. Clearly, long-term expression in perennial sensory neurons could determine the CMT2B phenotype. Indeed, continued subtle dysfunction, not easily detected using transient expression, might negatively affect, in the long-term, specific neuronal cells. We have to consider that sensory neurons have particularly long axons, and endocytosis occurs only at the tip of the axon. Therefore material (including signaling molecules) has to make a long trip to the soma. A subtle dysfunction in traffic might be detrimental for a sensory neuron without affecting other cell types.
Interestingly, Houlden et al. (2004) demonstrated in a sural nerve biopsy from a CMT2B patient that RILP expression was reduced. Possibly to balance a stronger interaction between RILP and the Rab7 mutant proteins (because of the fact that they are more often in the GTP bound form) and to try to restore a normal situation, expression of RILP is downregulated in these cells. This finding suggests an additional (or alternative) causative mechanism of the activated Rab7 mutations. Because many Rab effector proteins bind more than one Rab, an explanation for the onset of this pathology will be the titration of some shared interactor. RILP, for instance, has been demonstrated to bind Rab7 and Rab34 (Wang and Hong, 2002). These CMT2B-associated Rab7 mutant proteins, being mostly in the GTP-bound form and with a reduced GTPase activity, bind RILP more strongly. This could limit the availability of RILP to Rab34, impairing the step of transport regulated by Rab34.
An alternative hypothesis to explain why dysfunction in a ubiquitously expressed protein affects only the peripheral nervous system and, in particular, sensory and/or motor neurons is the existence of specific effector(s) in these kind of cells. The altered biochemical properties of the CMT2B-associated Rab7 mutant proteins could affect positively or negatively the interaction with such a specific effector determining the altered phenotype.
Footnotes
The financial support of Telethon-Italy (Grant GGP05160 to C.B.) is gratefully acknowledged.
The authors declare no competing financial interests.
References
- Auer-Grumbach M, Mauko B, Auer-Grumbach P, Pieber TR. Molecular genetics of hereditary sensory neuropathies. Neuromolecular Med. 2006;8:147–158. doi: 10.1385/nmm:8:1-2:147. [DOI] [PubMed] [Google Scholar]
- Bartel P, Chien CT, Sternglanz R, Fields S. Elimination of false positives that arise in using the two-hybrid system. Biotechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- Bronfman FC, Escudero CA, Weis J, Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev Neurobiol. 2007;67:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- Bucci C, Chiariello M. Signal transduction gRABs attention. Cell Signal. 2006;18:1–8. doi: 10.1016/j.cellsig.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Bucci C, Chiariello M, Lattero D, Maiorano M, Bruni CB. Interaction cloning and characterization of the cDNA encoding the human prenylated rab acceptor (PRA1) Biochem Biophys Res Commun. 1999;258:657–662. doi: 10.1006/bbrc.1999.0651. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresa BP. Regulation of EGFR endocytic trafficking by rab proteins. Histol Histopathol. 2006;21:987–993. doi: 10.14670/HH-21.987. [DOI] [PubMed] [Google Scholar]
- Chiariello M, Bruni CB, Bucci C. The small GTPases Rab5a, Rab5b and Rab5c are differentially phosphorylated in vitro. FEBS Lett. 1999;453:20–24. doi: 10.1016/s0014-5793(99)00686-9. [DOI] [PubMed] [Google Scholar]
- Colucci AMR, Campana MC, Bellopede M, Bucci C. The Rab-interacting lysosomal protein, a Rab7 and Rab34 effector, is capable of self-interaction. Biochem Biophys Res Commun. 2005;334:128–133. doi: 10.1016/j.bbrc.2005.06.067. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo GP. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Yeast promoters and LacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983;101:181–189. doi: 10.1016/0076-6879(83)01013-7. [DOI] [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23:6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J, Donald KA, Griffiths DE. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 1991;19:5791. doi: 10.1093/nar/19.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, King RH, Muddle JR, Warner TT, Reilly MM, Orrell RW, Ginsberg L. A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann Neurol. 2004;56:586–590. doi: 10.1002/ana.20281. [DOI] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, Neefjes J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol. 2007;176:459–471. doi: 10.1083/jcb.200606077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldgaard M, Nyborg J, Clark BF. The GTP binding motif: variations on a theme. FASEB J. 1996;10:1347–1368. [PubMed] [Google Scholar]
- Landt O, Grunert HP, Hahn U. A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene. 1990;96:125–128. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- Meggouh F, Bienfait HM, Weterman MA, de Visser M, Baas F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology. 2006;67:1476–1478. doi: 10.1212/01.wnl.0000240068.21499.f5. [DOI] [PubMed] [Google Scholar]
- Merithew E, Hatherly S, Dumas JJ, Lawe DC, Heller-Harrison R, Lambright DG. Structural plasticity of an invariant hydrophobic triad in the switch regions of Rab GTPases is a determinant of effector recognition. J Biol Chem. 2001;276:13982–13988. doi: 10.1074/jbc.M009771200. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:217–242. doi: 10.1385/nmm:8:1-2:217. [DOI] [PubMed] [Google Scholar]
- Paduch M, Jelen F, Otlewski J. Structure of small G proteins and their regulators. Acta Biochim Pol. 2001;48:829–850. [PubMed] [Google Scholar]
- Pfeffer SR. Structural clues to Rab GTPase functional diversity. J Biol Chem. 2005;280:15485–15488. doi: 10.1074/jbc.R500003200. [DOI] [PubMed] [Google Scholar]
- Press B, Feng Y, Hoflack B, Wandinger-Ness A. Mutant Rab7 causes the accumulation of cathepsin D and cation-independent mannose 6-phosphate receptor in an early endocytic compartment. J Cell Biol. 1998;140:1075–1089. doi: 10.1083/jcb.140.5.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Progida C, Malerød L, Stuffers S, Brech A, Bucci C, Stenmark H. RILP is required for the proper morphology and function of late endosomes. J Cell Sci. 2007;120:3729–3737. doi: 10.1242/jcs.017301. [DOI] [PubMed] [Google Scholar]
- Saxena S, Bucci C, Weis J, Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J Neurosci. 2005;25:10930–10940. doi: 10.1523/JNEUROSCI.2029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Giest RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Schroder JM. Neuropathology of Charcot-Marie-Tooth and related disorders. Neuromolecular Med. 2006;8:23–42. doi: 10.1385/nmm:8:1-2:23. [DOI] [PubMed] [Google Scholar]
- Segev N. Ypt and Rab GTPases: insight into functions through novel interactions. Curr Opin Cell Biol. 2001;13:500–511. doi: 10.1016/s0955-0674(00)00242-8. [DOI] [PubMed] [Google Scholar]
- Shapiro AD, Riederer MA, Pfeffer SR. Biochemical analysis of rab9, a ras-like GTPase involved in protein transport from late endosomes to the trans Golgi network. J Biol Chem. 1993;268:6925–6931. [PubMed] [Google Scholar]
- Snider MD. A role for rab7 GTPase in growth factor-regulated cell nutrition and apoptosis. Mol Cell. 2003;12:796–797. doi: 10.1016/s1097-2765(03)00401-5. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, Van Gerwen V, Wagner K, Hartung HP, Timmerman V. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli R, Santillo M, Lattero D, Chiariello M, Bifulco M, Bruni CB, Bucci C. Role of the small GTPase Rab7 in the late endocytic pathway. J Biol Chem. 1997;272:4391–4397. doi: 10.1074/jbc.272.7.4391. [DOI] [PubMed] [Google Scholar]
- Wang T, Hong W. Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol Biol Cell. 2002;13:4317–4332. doi: 10.1091/mbc.E02-05-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Hong W. RILP interacts with VPS22 and VPS36 of ESCRT-II and regulates their membrane recruitment. Biochem Biophys Res Commun. 2006;350:413–423. doi: 10.1016/j.bbrc.2006.09.064. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Vance JM. Mechanisms of disease: a molecular genetic update on hereditary axonal neuropathies. Nat Clin Pract Neurol. 2006;2:45–53. doi: 10.1038/ncpneuro0071. [DOI] [PubMed] [Google Scholar]