

Abstract
Neuronal nicotinic acetylcholine receptors (nAChRs), ligand-gated ion channels implicated in a variety of cognitive, motor, and sensory behaviours, are targeted to compartments rich in mitochondria, particularly postsynaptic domains and presynaptic terminals, exposing these receptors to reactive oxygen species (ROS) generated by oxidative phosphorylation. In addition, these receptors can become exposed to ROS during the progression of certain neurodegenerative diseases. Because ROS are known to modify several membrane proteins, including some types of ion channels, it raises the question of whether elevations in cytosolic ROS alter the function of nAChRs. To address this, we elevated ROS in cultured sympathetic neurons, directly by perfusing neurons intracellularly with ROS, indirectly by blocking the mitochondrial electron transport chain, or noninvasively by transient NGF removal; we then simultaneously measured changes in cytosolic ROS levels and whole-cell ACh-evoked currents. In addition, we elevated cytosolic ROS in postganglionic neurons in intact ganglia and measured changes in nerve-evoked EPSPs. Our experiments indicate that mild elevations in cytosolic ROS, including that produced by transient interruption of NGF signaling, induce a use-dependent, long-lasting rundown of ACh-evoked currents on cultured sympathetic neurons and a long-lasting depression of fast nerve-evoked EPSPs. We show that these effects of cytosolic ROS are specific to nAChRs on neurons and do not cause rundown of ACh-evoked currents on muscle. Our results demonstrate that elevations in cytosolic ROS inactivate neuronal nAChRs in a use-dependent manner and suggest that mild oxidative stress impairs mechanisms mediated by cholinergic nicotinic signaling at neuronal–neuronal synapses.
Keywords: nerve growth factor, sympathetic neurons, desensitization, autonomic nervous system, oxidative stress, antimycin-A, Fenton reaction, mitochondrial ROS
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs), members of a large family of neurotransmitter-gated ion channels, are expressed widely throughout the nervous system and are implicated in several physiological mechanisms, including sensory processing, pain perception, and reenforcing behavior to nicotine (Picciotto, 2003; Champtiaux and Changeux, 2004; Laviolette and van der Kooy, 2004; Dani and Bertrand, 2007). That nAChR signaling is crucial for these behaviors has been confirmed by recent studies on animals carrying gain-of-function or loss-of-function mutations in neuronal nAChR subunit genes (Cordero-Erausquin et al., 2000; Picciotto et al., 2001; Wang et al., 2002; Tapper et al., 2004; Maskos et al., 2005). How neuronal nAChRs alter the function of neural circuits depends primarily on where the receptors are located: on some neurons, such as those in peripheral sympathetic ganglia, nAChRs are targeted to the soma-dendritic domain where they mediate fast excitatory synaptic transmission (De Biasi, 2002: Skok, 2002); on other neurons, particularly those in the CNS, nAChRs are targeted to preterminal axons or presynaptic terminals where they modify GABA or glutamate transmitter release (Gray et al., 1996; Role and Berg, 1996; Broide and Leslie, 1999; MacDermott et al., 1999; Sher et al., 2004; Gotti et al., 2006). Because of the difficulties in recording directly from nAChRs on nerve terminals, however, we know little about the precise manner by which nAChR-mediated depolarizations modulate presynaptic transmitter release. However, considerable progress has been made in understanding these receptors at the molecular level, and the structural determinants involved in ligand-binding, ion permeation through the receptor pore, and receptor desensitization are becoming clearer (Corringer et al., 2000; Karlin, 2002; Unwin, 2005; Purohit et al., 2007). Moreover, a number of studies indicate that second messenger pathways modify the desensitization properties of the receptor, partly by phosphorylating receptor subunits (Swope et al., 2006; Quick and Lester, 2002; Giniatullin et al., 2005; Guo and Lester, 2007); however, the intracellular regulatory pathways that produce these receptor-modifying actions are not fully understood.
Because most nAChRs on cholinoceptive neurons are targeted to compartments rich in mitochondria, such as presynaptic terminals and postsynaptic compartments, potentially, these receptors are exposed to reactive oxygen species (ROS) generated by oxidative phosphorylation, particularly during periods of strong electrical activity. Our interest is whether increases in the oxidative strength of the cytosol, as occurs during the overproduction of ROS, modify receptor function. Elevations in cytosolic ROS levels are associated with several diseases (Coyle and Puttfarcken, 1993; Smith et al., 1996; Mattson, 2004; Lin and Beal, 2006; Mancuso et al., 2006; Savitt et al., 2006) and are known to alter the function of several membrane proteins including certain ion channels (Park et al., 1995; Ichinari et al., 1996; Annunziato et al., 2002; Dröge, 2002; Liu and Gutterman, 2002; Tang et al., 2004; Gamper et al., 2006); yet, we have no information whether elevations in cytosolic ROS alter the function of neuronal nAChRs. Given the connection between ROS and disease, if elevations in cytosolic ROS alter the function of neuronal nAChRs, the resulting effects on synaptic transmission could have serious consequences for ROS-related diseases, including associated dysautonomias, particularly if fast nicotinic synapses in the autonomic nervous system are impaired. Consequently, we set out to determine whether changes in the oxidative status of the cytosol produce changes in nAChR function and cholinergic nicotinic synaptic transmission.
Our study started with an unexpected finding: while recording ACh-evoked currents on cultured sympathetic neurons, we observed that nAChRs on neurons in short-term cultures entered into a long-lasting, nonconducting state that caused a specific rundown of the ACh-evoked currents, whereas on neurons in long-term cultures the ACh-evoked currents were stable. This finding suggested that the functional state of nAChR on these neurons depends on some intracellular regulatory process. Here, we demonstrate that elevated mitochondrial ROS, achieved either by blocking mitochondrial electron transport chain complex III or by transiently withdrawing growth factors, induce nAChRs to adopt a long-lasting, inactivated state in a use-dependent manner and cause long-term depression of synaptic transmission in intact sympathetic ganglia. Our findings link the functional state of nAChRs on neurons to cytosolic ROS levels; equally relevant, our results imply that, during diseases involving mitochondrial dysfunction and/or oxidative stress, cholinergic nicotinic signaling mechanisms and autonomic regulation of homeostasis are impaired.
Materials and Methods
Primary cultures.
Superior cervical ganglion (SCG) neurons were cultured from neonatal [postnatal day 1 (P1) to P3] mice (C57 black or CD1 strains; Charles River, St. Constant, Quebec, Canada) and rats (Sprague Dawley; Charles River) as previously described (McFarlane and Cooper, 1992). Briefly, ganglia were removed under sterile conditions and enzymatically dissociated at 37°C in HBSS containing trypsin (180–200 U/ml; Worthington, Freehold, NJ) and buffered with HEPES (adjusted to pH 7.4 with NaOH). The resulting cell suspension was washed twice in serum-containing medium to inactivate the trypsin and plated on laminin-coated coverslips attached to modified 35 mm tissue culture dishes. The neurons were grown in media consisting of L-15 supplemented with vitamins, cofactors, penicillin–streptomycin, 5% rat serum, and NGF (40 ng/ml). Cultures were maintained at 37°C in a humidified atmosphere of 95% air–5% CO2 and fed every 3 d with growth media. To eliminate non-neuronal cells, cultures were treated with cytosine arabinoside (10 μm; Sigma, St. Louis, MO) from day 2 to 4. For NGF withdrawal experiments, cultures were washed twice with growth media without NGF and maintained in growth media without NGF for 6–72 h.
To culture skeletal myotubes, we removed pectoral muscles from neonatal mice under sterile conditions, cut them into 1–2 mm pieces, and enzymatically dissociated them at 37°C in HBSS containing trypsin (180–200 U/ml; Worthington), buffered with HEPES (adjusted to pH 7.4 with NaOH). The resulting cell suspension was washed twice in serum-containing medium and plated on laminin-coated coverslips. The myoblasts were grown in media consisting of L-15 supplemented with vitamins, cofactors, penicillin–streptomycin, 10% horse serum, and maintained at 37°C in a humidified atmosphere of 95% air–5% CO2 and fed every 3 d with growth media. After 3–5 d, the myoblasts fused to form myotubes; we selected the smallest myotubes for electrophysiological experiments.
Whole-cell recording.
ACh-evoked currents were measured with whole-cell patch-clamp techniques. Membrane currents were recorded with a VE-2 amplifier (Alembic Instruments, Montreal, Quebec, Canada) at room temperature, sampled at 500 Hz, and stored on a Pentium II-based personal computer and filtered off-line. Recording electrodes had resistances of 2–5 MΩ, and series resistance was compensated from 70 to >98%. Patchkit software (Alembic) was used for stimulation and data acquisition and currents were filtered and analyzed off-line with Origin (OriginLab, Northampton, MA) or Igor (WaveMetrics, Lake Oswego, OR) software. Recording electrodes were filled with the following solution (in mm): 65 KF, 55 KAc, 5 NaCl, 0.2 CaCl2, 1 MgCl2, 10 EGTA, 2 MgATP, and 10 HEPES, and pH was adjusted to 7.2 with KOH. Cultured neurons were perfused continuously at 1 ml/min with control perfusion solution consisting of the following (in mm): 140 NaCl, 5.4 KCl, 0.33 NaH2PO4, 0.44 KH2PO4, 2.8 CaCl2, 0.18 MgCl2, 10 HEPES, 5.6 glucose, 2 glutamine, and 5 μg/ml phenol red; pH was adjusted to 7.4 with NaOH. ACh (100 μm) was dissolved in the perfusion solution and applied by pressure ejection from pipettes with tip diameters of 2–5 μm positioned 20–30 μm from the cell body (Mandelzys et al., 1995). In some experiments, atropine (1 μm; Sigma) was added to the perfusion solution. Voltage-gated Na+ currents were measured in control perfusion solution; we acquired the data at 50 kHz and filtered off-line. When measuring voltage-gated K+ currents, we added 1 μm TTX (Alomone Labs, Jerusalem, Israel) and 200 μm CdCl2 (Sigma) to the perfusion solution. For K+ currents, we acquired the data at 5 kHz and filtered off-line. All activators and inhibitors of second messenger pathways were applied to individual neurons through the patch pipette.
To increase cytosolic ROS directly in individual neurons, we used two methods: (1) we dissolved 1 μm antimycin-A (Sigma) in 100% EtOH [at 1:1000 (v/v) dilution] and added it to neurons through the recording electrode; or (2) we generated ROS from a Fenton reaction and introduced them into neurons through the recording electrode. To generate ROS from the Fenton reaction, we added 10 mm H2O2 to a solution containing 44 mm-ascorbate and 1 mm FeCl3·6H2O at a 1:20 (v/v) dilution; this solution was added to the recording electrode solution (see above) at a 1:10 (v/v) dilution. We remade solutions for the Fenton reaction every 30 min from powdered ingredients at room temperature. To control for the EtOH in the antimycin-A experiments, we recorded from neurons with electrodes containing EtOH at 1:1000 (v/v) dilution but without the antimycin-A, and to control for the low ionic strength of the Fenton-containing electrode solution, we diluted the solution in the recording electrode with H2O at 1:10 (v/v); neither the added EtOH (n = 6) nor the lower ionic strengthen of the Fenton-containing solution (n = 6) had any detectable effects on the ACh-evoked currents.
As a measure of rundown of the ACh-evoked currents we plotted the ratio of the peak current (I) in response to the 30th application (I30) in a series to the peak current in response to the first application (I1) in the series. To quantify receptor desensitization, we fit the falling phase of the ACh-evoked currents during the ACh application to a single or double exponential function using Igor software (WaveMetrics).
Whole-cell recordings from neuronal α4β2 receptors and muscle α1β1δε receptors in Xenopus oocytes.
Xenopus oocytes were defolliculated and prepared as described by Bertrand et al. (1991). We injected 1–3 ng of cDNAs coding for neuronal nAChR subunits α4 and β2, or muscle α1, β1, δ, and ε subunits into the nucleus of oocytes. Oocytes were incubated at 19°C for 2–5 d before recording.
To measure the macroscopic ACh-evoked currents on oocytes, we used two-electrode voltage-clamp techniques (Bertrand et al., 1991). These experiments were performed at room temperature (22–24°C) using a standard voltage-clamp amplifier (Alembic Instruments). During the recordings, oocytes were perfused with control perfusion solution or agonist solutions at 10–20 ml/min; the switch from one solution to another was performed manually. Currents were sampled at 100–350 Hz on-line with a Pentium-based PC computer (with a 64 kilobyte cache and A/D card; Omega, Stamford, CT) and filtered off-line. The program Patchkit (Alembic) was used for stimulation and data acquisition. Recording electrodes had tip diameters of 10–15 μm and were filled with 1 m KAc. To test the effects of mitochondrial ROS, antimycin-A (100 μm) was added to the electrode-filling solution. Control experiments were performed by adding 100% EtOH [at 1:1000 (v/v) dilution] to the electrode solution (n = 6). The external perfusion solution contained 96 mm NaCl, 2 mm KCl, 1 mm NaH2PO4, 1 mm BaCl2, 10 mm HEPES, and 1 μm atropine; pH was adjusted with NaOH to 7.4–7.5.
Intracellular ROS measurement.
To monitor changes in cytosolic ROS, we used the redox-sensitive dye CM-H2DCFDA (Molecular Probes, Burlington, Ontario, Canada), an acetoxymethyl (AM) ester. Cultures were incubated for 40 min at 37°C with medium containing CM-H2DCFDA (10 μm) and subsequently washed five times with control perfusion solution (see above). The cultures were then placed on the stage of an inverted microscope (Axiovert 200 M; Zeiss, Oberkochen, Germany) and viewed through a 40× (1.3 numerical aperture) or 63× (1.4 numerical aperture) Plan Neofluor oil-immersion objective (Zeiss) at room temperature and continuously perfused throughout the experiment as described for whole-cell recordings. To obtain fluorescent images, we excited the cultures with 450–480 nm wavelength for 200 ms from a 150 W xenon arc lamp (LAMBDA DG-4; Sutter Instruments, Novato, CA) and collected 510–550 nm wavelength emissions (filter set 31001; Chroma Technology, Brattleboro, VT) with a cooled CCD camera (CoolSnap HQ; PhotoMetrics, Tucson, AZ) controlled by Metafluor or MetaMorph softwares (Universal Imaging, West Chester, PA). For neurons treated with antimycin-A or Fenton reaction, we quantified changes in CM-H2DCFDA fluorescence over time by acquiring images every 25 s; for each image, we defined regions of interest (neuronal cell body, excluding the nucleus) on a differential interference contrast image and transferred these regions to the fluorescent images of the same field. The control fluorescence intensity (minus the background), F, was determined from neurons before establishing whole-cell patch configurations and from neighboring untreated neurons. For each neuron, we subtracted its mean fluorescent intensity (minus background) from F to obtain the change in fluorescent intensity (ΔF) and expressed it as ΔF/F. To monitor changes in cytosolic ROS during NGF withdrawal experiments, we defined regions of interest as above, acquired Z stack planes at 1 μm intervals to find the plane with the maximum mean pixel intensity for each neuron, and then calculated ΔF/F; in these experiments, F was the mean fluorescent intensity (after subtracting background) for neurons in control cultures with NGF. A total of >450 neurons were measured, >50 neurons in each condition.
Antioxidant treatment.
To load neurons with ascorbate, we incubated neurons with ascorbate oxidase and l-ascorbate to circumvent in vitro artifacts associated with the poor transport and prooxidant effects of ascorbate (Buettner and Jurkiewicz, 1996; Clement et al., 2001). To do this, neurons were incubated for 30 min at 37°C in freshly prepared ascorbate (0.55 mm) and 0.5 U/ml ascorbate oxidase (Frank et al., 2006). Ascorbate oxidase converts l-ascorbate to dehydroascorbate (DHA), which is transported through the facilitative glucose transporter and quickly converted to ascorbate. In other experiments, the antioxidant α-lipoic acid (100 μm) was dissolved in intracellular solution, treated with 1000 U/ml catalase for 1 h at 37°C (Vincent et al., 2005), and used as the recording solution during whole-cell recording.
Extracellular and intracellular recordings from intact SCGs.
SCGs were dissected rapidly from neonatal rat and mice pups (P3–P7), pinned down with minutia pins in a recording chamber (1.5 ml volume) mounted on a fixed stage, and viewed with a dissecting microscope (SMZ-10; Nikon, Tokyo, Japan); the tissue was perfused continuously at 3–4 ml/min with oxygenated Ringer's solution at 36–37°C. To stimulate the preganglionic nerve, the cervical sympathetic trunk was connected to a stimulator (4710 ORTEC dual channel; EG&G, Gaithersburg, MD) with a suction electrode and stimulated with brief (0.1–0.3 ms) voltage pulses. For extracellular compound action potentials, the postganglionic trunk was connected to an AC differential amplifier (DP-301; Warner Instruments, Hamden, CT) with a suction electrode; the postganglionic compound action potentials were amplified, filtered at 100 Hz (low-pass cutoff) and 1 kHz (high-pass cutoff), digitized at 10 kHz, and stored on a Pentium II-based computer. To record intracellularly from individual SCG neurons, we used 50–70 MΩ glass microelectrodes (G150F-4; Warner Instruments) made with a DMZ universal puller (Zeitz Instruments, Munich, Germany) as described previously (Rassadi et al., 2005). Stable intracellular recordings were achieved with a high inertial precision microdrive (Inchworm 8200; EXFO, Vanier, Quebec, Canada) attached to a micromanipulator (SM11; Narshige, Tokyo, Japan) that drove the electrode through the ganglion. The recording electrode was filled with 1 m KAc and connected by a thin silver chlorided wire to the head stage of an Axoclamp 2A amplifier (Molecular Devices, Sunnyvale, CA) used in current-clamp mode; depolarizing or hyperpolarizing constant current pulses were applied through the recording electrode. Membrane potentials were sampled, displayed, and stored on a Pentium II-based personal computer. Stimulation and acquisition was done with Patchkit software (Alembic), and the data were analyzed off-line with Igor. To elevate mitochondrial ROS in individual neurons, we added antimycin-A (100 μm) to the recording electrode; as control, we recorded from neurons with the identical solution without antimycin-A. To test the effects of transient interruption in NGF signaling on ganglionic transmission, we injected neonatal mice intraperitoneally with anti-NGF antibodies (Sigma) (2.5 μl of 3 mg/ml stock per gram of body weight) or control IgG (Cederlane Lab, Burlington, Ontario, Canada) (2.5 μl of 2.5 mg/ml stock per gram of body weight). Usually, the evoked EPSPs were suprathreshold, and therefore to measure the changes in EPSP amplitudes accurately, in some experiments, we added curare (10 μm) to the perfusion solution. Only neurons with membrane potentials greater than −40 mV were included in this study.
Statistics.
To determine the statistical significance for differences in (1) the I30/I1 ratios and (2) the mean fluorescent intensities, we used a nonparametric test (Mann–Whitney). To determine the statistical significance for differences in cell numbers among different conditions in the NGF withdrawal experiments, we used ANOVA, and for EPSP amplitudes, we used Student's t test.
All procedures for animal handling were performed according to the guidelines of the Canadian Council on Animal Care.
Results
Our overall interests are in cytoplasmic factors that influence the functional properties of nAChRs on neurons. The present study began by observing that the ACh-evoked currents on sympathetic neurons in short-term cultures (1–3 d) run down irreversibly, as if the AChRs were trapped in a long-lasting inactivated state. This unexpected rundown of ACh-evoked currents has not been well described and suggested to us that some unappreciated regulatory mechanism controls the function of nAChRs on neurons. Therefore, first we characterized this rundown, and then we focused our investigation on its underlying cause.
Rundown of ACh-evoked currents on sympathetic neurons in short-term cultures
Repeated application of ACh (100 μm) to rodent sympathetic neurons in short-term cultures (1–3 d) resulted in a steady decline of the ACh-evoked currents; when ACh was applied for 1 s at 15 s intervals, the peak ACh-evoked inward current declined to <10% of the initial response in ∼2–3 min (Fig. 1A). In contrast, when repeatedly applying ACh to neurons in long-term cultures (>10 d), we observed no significant decline in the ACh-evoked currents (Fig. 1A). α-Bungarotoxin had no detectable effect on these currents (n = 6) (data not shown), indicating that the currents in Figure 1A are likely mediated by α3β4-containing receptors. Equally noteworthy, once the ACh-evoked current on neurons in short-term cultures had run down, it never recovered, even after washing with ACh-free solution for 30 min (Fig. 1A).
Figure 1.
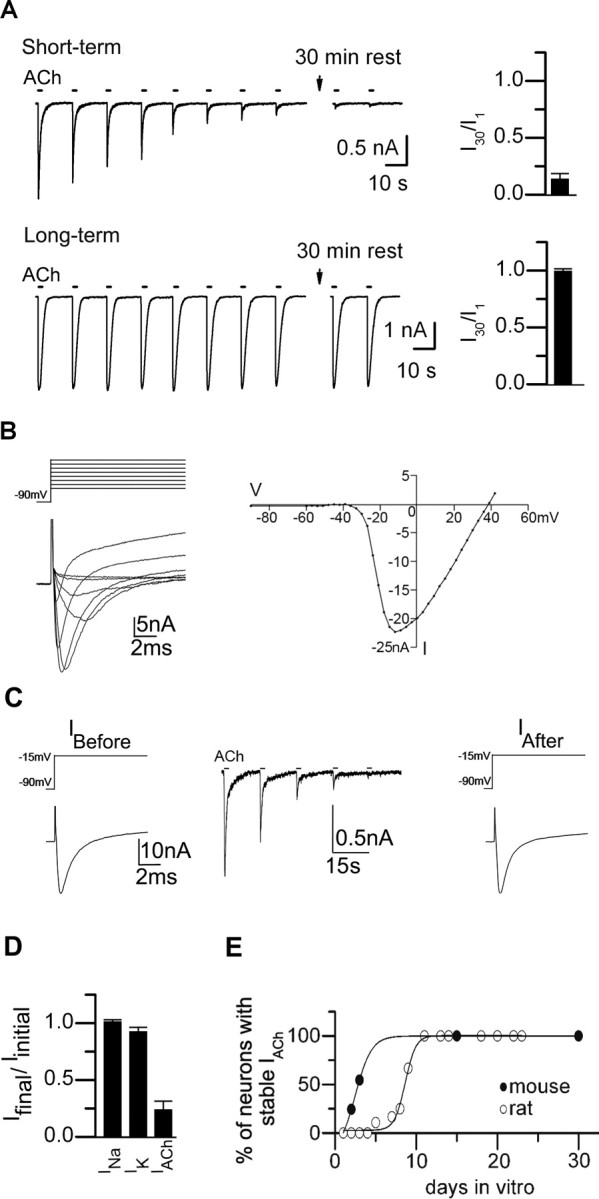
Rundown of ACh-evoked currents on sympathetic neurons in short-term cultures. A, ACh-evoked currents from neonatal mouse SCG neurons in culture for 2 d (short-term) or in culture for 10 d (long-term) in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals. The bar graph on the right shows the mean ratio of the 30th ACh-evoked current to the first in the series (I30/I1) for neurons in short-term culture (n = 7) and neurons in long-term culture (n = 6). Each bar is the mean I30/I1 ± SE. The ACh-evoked currents on neurons in short-term culture run down rapidly, and never recovered, even after washing with ACh-free solution for 30 min; whereas those on neurons in long-term culture are stable. B, Left, Superimposed voltage-gated inward Na+ currents (INa) and outward K+ currents (IK) measured from a neuron in short-term culture in response to a series of voltage steps from −90 mV holding potential delivered at 3 mV increments to +40 mV. Only every fourth trace starting at −60 mV is shown for clarity. The currents have not been corrected for leakage. Right, The corresponding peak INa–voltage relationship. C, The peak INa recorded from a neuron in short-term culture in response to voltage steps from −90 to −15 mV before (left) and after (right) rundown of ACh-evoked currents (middle). The ACh-evoked currents run down but the sodium currents do not. D, Bar graph shows the ratio of the peak INa and IK on neurons in short-term culture before and after rundown of the ACh-evoked currents (mean ± SE; n = 7). There was no significant difference in the peak INa or IK before and after rundown of the ACh-evoked currents. E, The proportion of neurons with stable ACh-evoked currents on rat (○) and mouse (●) SCG neurons in sister cultures as they developed in culture over time. ACh-evoked currents in response to a series of ACh applications were measured on at least 10 neurons selected at random in a given culture; each point represents the proportion of neurons in a given culture with stable ACh-evoked currents.
This striking and unexpected long-lasting rundown of ACh-evoked currents from neurons in short-term cultures was not caused by a generalized change in the neurons. We observed no significant difference in the voltage-gated Na+ currents before and after rundown of the ACh-evoked currents (Fig. 1 B–D), or in the voltage-gated K+ currents (Fig. 1D). Importantly, these results indicate that rundown of the ACh-evoked currents was not attributable to a generalized, nonspecific change in the neurons during whole-cell recording.
Over a few days in culture, the proportion of neurons with stable ACh-evoked currents increased. To quantify this conversion, we sampled neurons in sister cultures for stable ACh-evoked currents at different times after plating. To judge whether the ACh-evoked currents were stable, we delivered a series of ACh applications at 15 s intervals: If the response to the 30th application (I30) was at least 80% of the initial response (I1), we classified the neuron as having stable ACh-evoked currents. For the first 24 h after plating, the proportion of neurons with stable ACh-evoked currents was very low (<5%); thereafter, the proportion increased sigmoidally, and by 10 d after plating, 100% of the neurons had stable ACh-evoked currents (Fig. 1E). Moreover, the transition to neurons with stable ACh-evoked currents occurred significantly faster on sympathetic neurons from mice (within 2–3 d) compared with those from rats (7–10 d) (Fig. 1E). These results suggest that, as neonatal rodent sympathetic neurons develop in culture, some maturation process stabilizes their ACh-evoked currents.
The rundown of ACh-evoked currents from neurons in short-term cultures depends on receptor activation. When ACh was applied at 15 s intervals, the ACh-evoked current decreased by 50% in 1.3 ± 0.66 min (n = 6) (Fig. 2A); whereas the ACh-evoked current took approximately fourfold longer to decrease by 50% when ACh was applied consecutively at 60 s intervals (4.7 ± 0.81 min; n = 6) (Fig. 2A). However, when the peak ACh-evoked currents were plotted against the absolute number of ACh applications for both the 15 s series and 60 s series, the decline of the ACh-evoked currents were comparable; the plot of the IACh versus agonist applications decayed as a single exponential function, indicating that it requires approximately six consecutive ACh applications to inactivate the current by two-thirds (Fig. 2B). These results demonstrate a use dependence to the rundown: the more the receptors are activated, the greater the rundown of the ACh-evoked currents.
Figure 2.

Use-dependent rundown of ACh-evoked currents on sympathetic neurons in short-term cultures. A, ACh-evoked currents were measured in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals (♦) (n = 9) and at 60 s intervals (■) (n = 8) for neurons in culture for 2 d (short-term) and at 15 s intervals (▼) (n = 6) for neurons in culture for 10 d (long-term). Ix/I1 is the ratio of the xth response to the first response in a series. Each point is the mean; only every second ratio of the time series is shown for clarity. The solid lines are exponential fits to the data and the gray shaded area is the SE. The Ix/I1 ratio decreases by 50% in 78 s when ACh is delivered at 15 s intervals and 282 s when delivered at 60 s intervals. B, The average Ix/I1 ratio versus application number from the data in A and D: 1 s ACh (100 μm) at 15 s intervals (♦) (n = 9), and at 60 s intervals (■) (n = 8), and 10 s ACh (100 μm) at 60 s intervals (●) (mean ± SE). The solid line is an exponential fit to the data and the gray shaded area is the SE. The Ix/I1 ratio decreases by 1/e in approximately six applications independent of the interval between ACh applications or ACh duration. C, ACh-evoked currents on neurons in short-term culture desensitize more rapidly than those in long-term cultures. Examples of ACh-evoked currents from neonatal mouse SCG neurons in short-term or long-term culture in response to ACh (100 μm) applied for 10 s. The inset shows the current at a faster time base in response to a 1 s application. For the neuron in short-term culture, the current evoked by a 10 s ACh application was fit with a double exponential function of τf = 1.15 s and τs = 3.6 s; for the neuron in long-term culture, the current was fit with a single exponential function of τ = 3.4 s (solid lines). D, ACh-evoked currents from neonatal mouse SCG neurons in short-term culture in response to a series of 10 s ACh (100 μm) applications delivered at 60 s intervals. The ACh-evoked current in response to the first application (I1) desensitized by over 95% and recovered by >70% in 60 s (second response; I2) but ran down by the 10th application (I10). E, Average responses to a series of 10 s ACh (100 μm) applications delivered at 60 s intervals for neurons in short-term (●) (n = 6) and neurons in long-term culture (▶◀) (n = 6). Each point is the mean Ix/I1 ratio. The solid lines are exponential fits to the data, and the gray shaded area is the SE. There is no significant difference in the time course for the decrease in ratio when ACh is applied at for 10 s at 60 s intervals compared with ACh applied for 1 s at 60 s intervals (A). ST refers to short-term cultures, and LT refers to long-term cultures.
This rundown of ACh-evoked currents does not result from an inability to recover from desensitization. The ACh-evoked currents on neurons in short-term cultures desensitized approximately three times faster than those on neurons in long-term cultures (Fig. 2C); with 10 s applications to neurons in short-term cultures, 85% of the evoked current decayed as a single exponential function with a time constant (τd) of 1.36 ± 0.29 s (n = 8), and the remaining 15% decayed with a τd of 3.95 ± 0.32 s (n = 8); whereas for neurons in sister cultures for >2 weeks (long-term cultures), the ACh-evoked currents decayed with a single exponential time course (τd = 4.13 ± 0.68 s; n = 10) (Fig. 2C). Most nAChRs on neurons in short-term cultures recover rapidly from desensitization; a second ACh application given 60 s after a long (10 s) desensitizing pulse evoked ACh currents that were ∼70% of those evoked by the first (Fig. 2D). With successive ACh applications, however, ACh-evoked currents ran down with a time course similar to that induced by successive 1 s ACh applications (compare Fig. 2E), indicating that rundown does not depend on the extent of desensitization. However, successive long, desensitizing ACh applications to neurons in long-term cultures did not induce any significant rundown of the ACh-evoked currents (Fig. 2E). These results indicate that the long-lasting rundown of ACh-evoked currents depends on a mechanism other than receptor desensitization.
Increasing the antioxidant content of the cytosol prevents rundown of ACh-evoked currents on neurons in short-term cultures
Rundown of ACh-evoked currents on neurons in short-term cultures was not affected by activating protein kinase A (PKA) or PKC, inhibiting PKA, PKC, tyrosine phosphorylation, or preventing elevations in intracellular Ca2+ (Fig. 3 A–C). In addition, none of these treatments induced rundown of ACh-evoked currents on neurons in long-term cultures (Fig. 3 D–F). Together, these results suggest that the rundown of ACh-evoked currents involves a process that does not depend directly on PKA, PKC, tyrosine kinase, or Ca2+ second messenger pathways.
Figure 3.
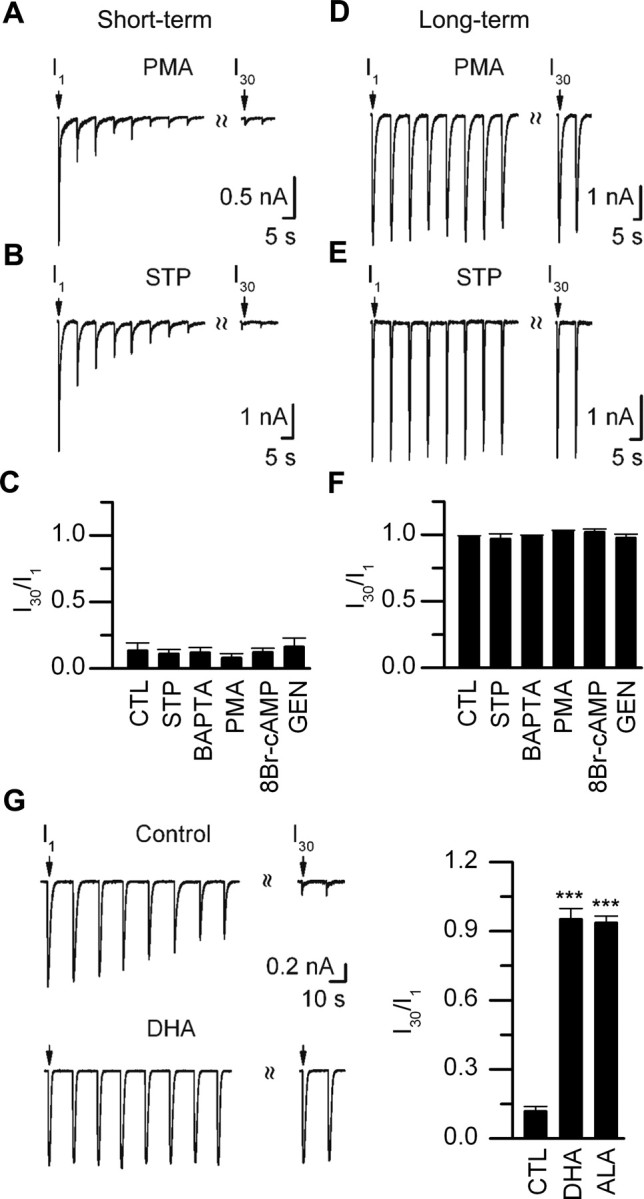
Rundown of ACh-evoked currents is prevented by antioxidants. A, B, ACh-evoked currents from neonatal mouse SCG neurons in culture for 2 d (short-term) in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals when PMA (5 μm) (A) or staurosporine (STP; 100 nm) (B) was included in the recording electrode. C, I30/I1 ratio for neurons in short-term culture for control (CTL) (n = 6), staurosporine (STP) (100 nm; n = 6), BAPTA (10 mm; n = 7), PMA (5 μm; n = 6), genistein (10 μm; n = 6), or 8-bromo-cAMP (250 μm; n = 7). Each bar is the mean I30/I1 ± SE. D, E, ACh-evoked currents from neonatal mouse SCG neurons in culture for 10 d (long-term) in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals when PMA (5 μm) (D) or staurosporine (STP; 100 nm) (E) was included in the recording electrode. F, The I30/I1 ratio for neurons in long-term culture for control (CTL) (n = 6), staurosporine (STP) (100 nm; n = 7), BAPTA (10 mm; n = 6), PMA (5 μm; n = 7), genistein (10 μm; n = 6), or 8-bromo-cAMP (250 μm; n = 7). Each bar is the mean I30/I1 ± SE; for control, BAPTA, and PMA experiments, the SEs are too small to resolve. G, ACh-evoked currents from two neonatal rat SCG neurons in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals, one in control culture for 4 d and the other in a sister culture for 4 d but treated with antioxidants (DHA) (see Materials and Methods) 24 h before. The graph on the right shows the mean I30/I1 ratio ± SE for control (CTL) (n = 11) and neurons in sister cultures treated with the antioxidants, DHA (n = 8), or α-lipoic acid (ALA) plus catalase (n = 6). ***p < 0.001.
Next, we asked whether shifts in the intracellular thio/disulfide redox state toward more oxidative conditions could underlie the rundown of the ACh-evoked currents from neurons in short-term cultures (Park et al., 1995; Annunziato et al., 2002; Tang et al., 2004; Gamper et al., 2006). To test this, we cultured neurons in media containing ascorbate and ascorbate oxidase to increase the reducing power of the cytosol (Frank et al., 2006). Unlike ACh-evoked currents on neurons in control sister cultures, all (eight of eight) neurons in short-term cultures incubated with ascorbate and ascorbate oxidase had stable ACh-evoked currents (i.e., I30/I1 was >0.80) (Fig. 3G). In addition, we found that adding the antioxidants α-lipoic acid and catalase to the recording electrode (see Materials and Methods) also prevented rundown of ACh-evoked currents on neurons in short-term cultures (Fig. 3G). These results suggest strongly that oxidative conditions of the cytosol induce long-lasting, use-dependent rundown of ACh-evoked currents on neurons.
Elevations in cytosolic ROS induce long-lasting, use-dependent inactivation of neuronal nAChRs
To test whether elevations in cytosolic ROS induce rundown of the ACh-evoked currents on neurons in long-term cultures, we generated ROS from a Fenton reaction (see Materials and Methods) and added them to neurons through the patch electrode. To monitor changes in cytosolic ROS over time, we loaded neurons with the fluorescent ROS indicator dye, CM-H2DCFDA. In all neurons recorded with ROS-containing electrodes, we observed an increase in ROS-induced fluorescence; the fluorescence started to increase within minutes after establishing whole-cell recording and reached a plateau by 10–15 min (Fig. 4A,B). On average, ΔF/F increased to 0.55 ± 0.05 (n = 7) (Fig. 4C), whereas for neighboring neurons recorded with control intracellular solution, ΔF/F was 0.12 ± 0.03 (n = 6) (Fig. 4C).
Figure 4.

Cytosolic ROS induces rundown of ACh-evoked currents. A, SCG neuron in culture for 14 d was loaded with CM-H2DCFDA and fluorescent images were obtained every 25 s and expressed as ΔF/F; the increase in fluorescence was described by a Hill function (solid line). At the arrow, ROS generated from a Fenton reaction was introduced into the neuron through the recording electrode, and a series of 1 s ACh (100 μm) applications were delivered to the neuron at 15 s intervals to measure the ACh-evoked currents. The inset shows six selected ACh-evoked currents from the series, each obtained at different times indicated by the numbers and lines next to the curve showing the increase in ROS-induced fluorescence. B, Example of the change in ROS-induced fluorescence in neurons in culture for 16 d when loaded acutely through the recording electrode with either ROS generated from the Fenton reaction, antimycin-A (1 μm) or control (CTL). The increase in fluorescence was described by a Hill function (solid line). C, The average increase in ROS-induced fluorescence for neurons in culture for 15–18 d in control (n = 6), or loaded with ROS generated from a Fenton reaction (n = 7), or antimycin-A (1 μm; n = 7). The bars show the mean ΔF/F plateau ± SE recorded from neurons with control solution or when generating ROS from a Fenton reaction or antimycin-A. ***p < 0.001.
To test the effects of cytosolic ROS on the ACh-evoked currents, we recorded from neurons in long-term cultures with electrodes containing ROS generated from a Fenton reaction and simultaneously imaged changes in fluorescence and monitored ACh-evoked currents. Delivering a series of ACh applications, we found that, as ROS increased, the responses to ACh decreased (Fig. 4A), whereas for neighboring neurons recorded with electrode containing control solution, the ACh-evoked currents were stable. These results indicate that elevating ROS in the cytosol induces rundown of ACh-evoked currents.
In the experiment above, however, we applied ACh repeatedly to neurons as cytosolic ROS was increasing, making it difficult to know whether this ROS-induced rundown of ACh-evoked currents required receptor activation. To address this, we recorded from neurons with ROS-containing electrodes, waited 15 min for cytosolic ROS to reach its plateau, and then applied ACh repeatedly. The first response in the series was not significantly different from evoked currents on the neighboring control neurons, indicating that elevations in ROS by themselves do not have a detrimental affect on the receptors. However, when we delivered a series of ACh pulses to neurons with elevated ROS, the evoked current ran down rapidly (Fig. 5A); the ACh-evoked current amplitude in response to the 30th application was <15% of the response to the first (Fig. 5A,B). This ROS-induced rundown of ACh-evoked current did not recover for at least an additional 20–30 min. In contrast, the ACh-evoked currents were stable on neighboring neurons recorded with electrodes containing control solution (Fig. 5A,B). These results indicate that elevations in cytosolic ROS must be paired with receptor activation to induce a long-lasting rundown of the ACh-evoked currents.
Figure 5.
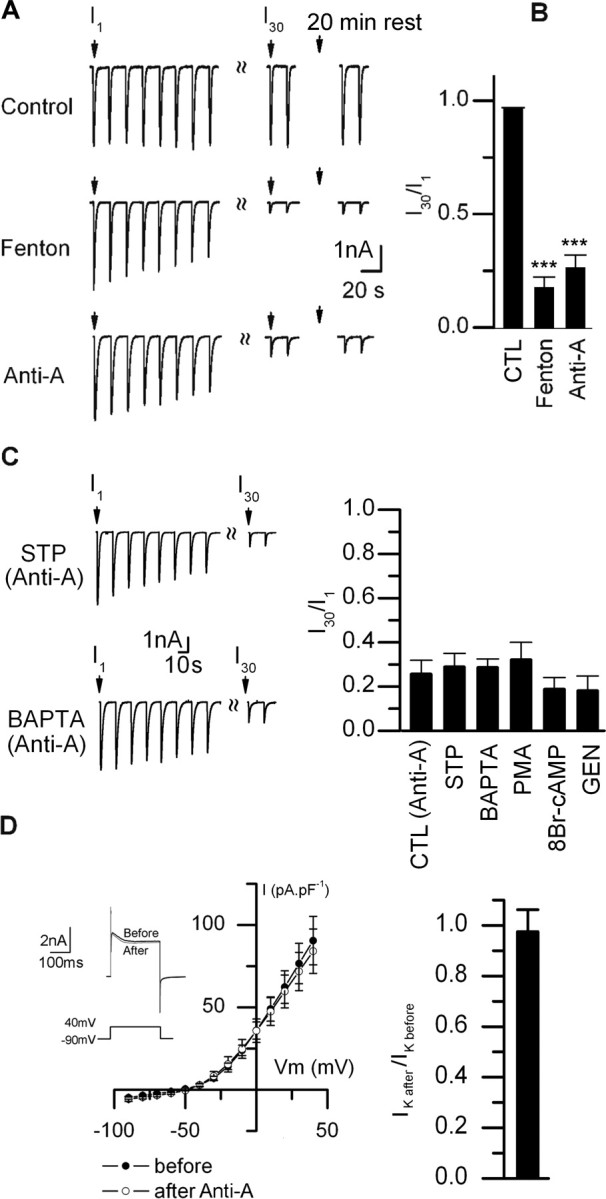
Mitochondrial ROS induces rundown of ACh-evoked currents. A, ACh-evoked currents in response to repetitive ACh (100 μm) applications at 15 s intervals from the three neurons in Figure 4B, control (top), Fenton reaction (middle), or antimycin-A (lower). B, I30/I1 ratio (mean ± SE) for neurons loaded with control (CTL) (n = 12), Fenton reaction (n = 6), or antimycin-A (1 μm; n = 12). The SE for CTL are too small to resolve. C, Left, ACh-evoked currents from two neurons in culture for 16 d recorded with electrodes containing antimycin-A (1 μm) and either staurosporine (STP) (100 nm) or BAPTA (10 mm). Right, I30/I1 ratio for neurons in long-term cultures recorded with electrodes containing antimycin-A (1 μm) plus control [CTL (anti-A)] (n = 12), staurosporine (STP) (100 nm; n = 6), BAPTA (10 mm; n = 7), PMA (5 μm; n = 5), genistein (10 μm; n = 6), or 8-bromo-cAMP (250 μm; n = 6). None of these treatments prevented rundown by antimycin-A. D, Left, The current–voltage relationship for voltage-gated K+ currents (IK) before and after inducing rundown of ACh-evoked currents. The inset shows outward K+ currents from a neuron perfused intracellularly with antimycin-A; the outward K+ currents were evoked by voltage steps from −90 to +40 mV within 2 min after establishing whole-cell recording (before) and 20 min later (after). The currents were not corrected for leakage. On the right is the ratio of IK at +40 mV before and after inducing rundown of ACh-evoked currents with antimycin-A (mean ± SE; n = 6). There is no significant difference in the amplitude or voltage dependence of IK or the holding current before and after inducing rundown of ACh-evoked currents. ***p < 0.001.
Mitochondrial ROS induces long-lasting, use-dependent rundown of neuronal nAChRs
Physiologically, an important source of ROS comes from the mitochondrial electron transport chain generated as by-products of oxidative phosphorylation (Nicholls and Budd, 2000). Therefore, we asked whether mitochondrial ROS inactivate neuronal nAChRs. To address this, we inhibited complex III of the electron transport chain by adding antimycin-A (1 μm) through the recording electrode to neurons in long-term cultures, and simultaneously monitored changes in CM-H2DCFDA fluorescence and membrane currents over time. After blocking complex III with antimycin-A, ROS increased with a delay of 3–5 min after establishing whole-cell recording and reached a plateau by 15–20 min (Fig. 4B); on average, ΔF/F increased to 0.58 ± 0.12 (n = 12), comparable with that in neurons perfused intracellularly with ROS from the Fenton reaction (Fig. 4C).
To determine whether elevations in mitochondrial ROS induced rundown of the ACh-evoked currents, we gave a series of ACh pulses to neurons after ΔF/F had reached a plateau (15–20 min). The ACh-evoked current in response to the first ACh pulse was not significantly different from those on control neurons; however, we observed rundown of the ACh-evoked currents with subsequent applications; the ACh-evoked current amplitude in response to the 30th application was ∼25% of the response to the first (Fig. 5A,B). These results indicate that mild elevations in ROS from mitochondria are sufficient to induce an irreversible, use-dependent rundown of ACh-evoked currents. Moreover, the rundown of ACh-evoked currents induced by antimycin-A does not depend directly on PKA, PKC, tyrosine kinase, or Ca2+ second messenger pathways (Fig. 5C).
To rule out the possibility that elevating cytosolic ROS produced nonspecific changes in membrane currents, we measured voltage-gated K+ currents before (within 2 min of whole-cell recording) and after elevations in ROS and rundown of ACh-evoked currents (>20 min). We observed no change in either the voltage-gated K+ currents, or in the holding current before and after increase in cytosolic ROS (Fig. 5D). These results indicate that elevations in ROS do not cause a generalized rundown of other membrane currents.
Mitochondrial ROS induce long-lasting, use-dependent rundown of α4β2 nAChRs
To determine whether other nAChR subtypes expressed on neurons are inactivated by elevations in mitochondrial ROS, we examined α4β2 receptors expressed in Xenopus oocytes. When recording from oocytes expressing α4β2 receptors, brief (∼2–3 s) applications of ACh (0.5 μm) evoked large 3–4 μA inward currents (Fig. 6A). These currents were stable for at least 30 min when recording from oocytes with electrodes containing control solution and ACh (0.5 μm; 2–3 s) was applied at 1 min intervals (Fig. 6A). In contrast, when recording from oocytes with electrodes containing antimycin-A (100 μm), the ACh-evoked currents ran down in a use-dependent manner after a delay of 10–15 min (Fig. 6A). These results suggest that elevations in mitochondrial ROS lead to the inactivation of α4β2 receptors, similar to endogenous nAChRs on sympathetic neurons.
Figure 6.
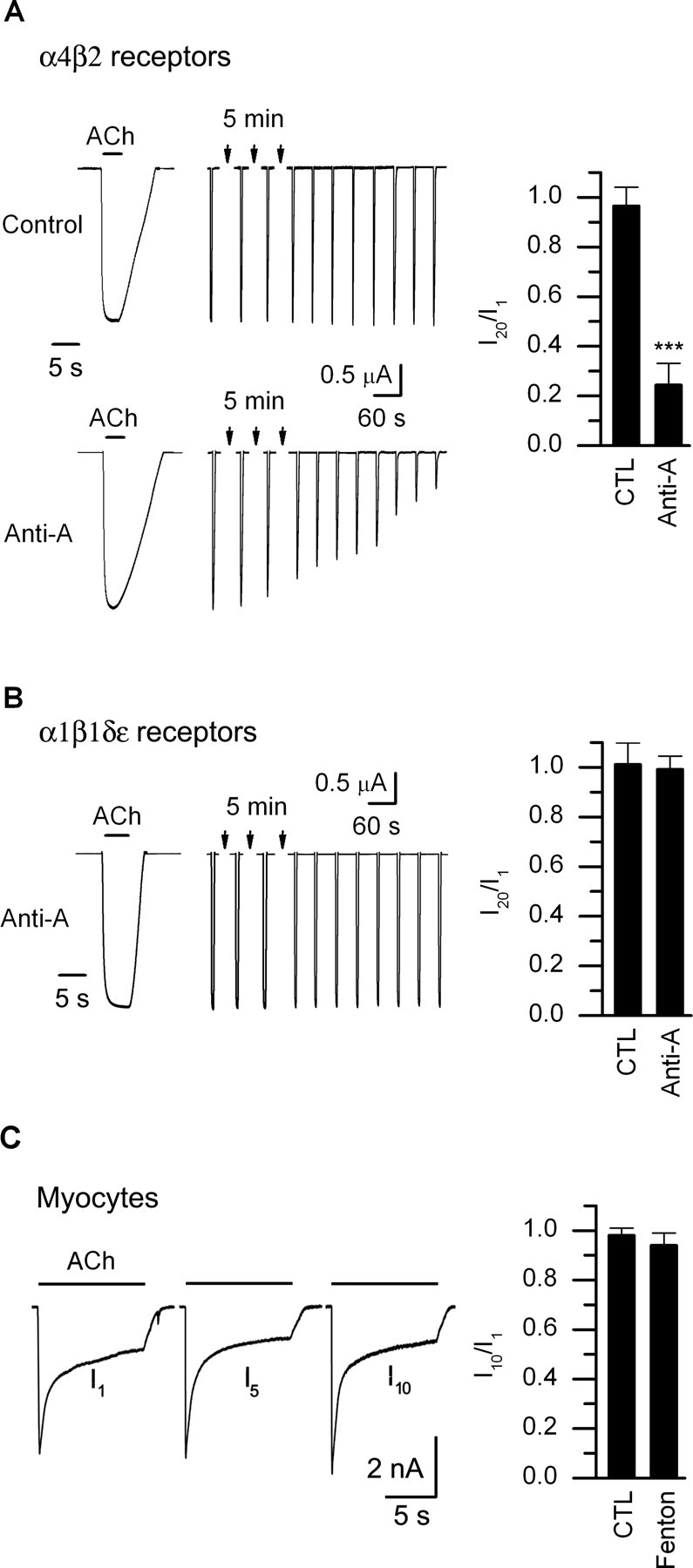
ROS induced rundown of ACh-evoked currents is specific for nAChRs on neurons and does not inactivate muscle nAChRs. A, α4β2 receptors. On the left are ACh-evoked currents from α4β2-expressing oocytes recorded with an electrode containing control (top) or antimycin-A (lower). ACh (0.5 μm) was applied repeatedly every 1 min. The left-most traces are the ACh-evoked currents on an expanded timescale. On the right is the I20/I1 ratio for α4β2-expressing oocytes recorded with electrodes containing control solutions (n = 7) or electrodes containing antimycin A (n = 8). Currents were obtained after 20–30 min to allow time for antimycin-A to act. B, α1β1δε receptors. Left, ACh-evoked currents (10 μm every min) from α1β1δε-expressing oocytes recorded with an electrode containing antimycin-A. Right, the I20/I1 ratio for α1β1δε-expressing oocytes recorded with electrodes containing antimycin-A (n = 8). Currents were obtained after 20–30 min to allow time for antimycin-A. α1β1δε receptors do not rundown in oocytes containing antimycin-A. C, Myotubes. ACh-evoked currents from a mouse myotube in culture for 5 d and recorded with an electrode containing ROS generated from the Fenton reaction. ACh (10 μm) was applied for 10 s every 5 min. The figure shows the first, fifth, and 10th records of the series. On the right is the I10/I1 ratio for myotubes recorded with electrodes containing control solutions (n = 5) or electrodes containing ROS from the Fenton reaction (n = 6). ***p < 0.001.
Muscle nAChRs are not inactivated by cytosolic ROS
To determine whether nAChRs on muscle were similarly affected by elevations in cytosolic ROS, we examined muscle α1β1δε receptors expressed in Xenopus oocytes. In contrast to ACh-evoked currents from neuronal nAChRs when recording with electrodes containing antimycin-A, the ACh-evoked currents from oocytes expressing α1β1δε receptors were stable for at least 30 min (Fig. 6B).
In addition, we recorded endogenous ACh-evoked currents from cultured mice myotubes. First, we loaded myotubes with CM-H2DCFDA, and then introduced ROS into individual myotubes through the recording electrode. The resting ROS-induced fluorescence was considerably higher in myotubes compared with neurons; nonetheless, we observed no significant rundown of the ACh-evoked currents in response to a series of 10 s ACh applications every 5 min, even after applying ACh repeatedly for up to 50 min (Fig. 6C). These results indicate that ROS does not inactivate muscle nAChRs.
Transient elevation of mitochondrial ROS by disruption in NGF signaling
In the above experiments, we elevated cytosolic ROS by introducing ROS directly into neurons or by inhibiting electron transport chain complex III with antimycin-A. Next, we asked whether we could induce a long-lasting inactivation of nAChRs on SCG neurons by elevating mitochondrial ROS noninvasively by transiently disrupting NGF signaling (Kirkland and Franklin, 2001; Kirkland et al., 2002).
To address this, first we withdrew NGF from neonatal sympathetic neurons in long-term cultures for various times and measured elevations in cytosolic ROS with CM-H2DCFDA fluorescence. ROS-induced fluorescence changed little during the first 6 h without NGF; by 18 h without NGF, however, ΔF/F increased to 0.81 ± 0.03 (n = 152) (Fig. 7B), comparable with that achieved by antimycin-A or ROS from a Fenton reaction (Fig. 4C). This increase in cytosolic ROS at 18 h occurred without detectable change in neuronal morphology (when viewed with phase optics), or affect on neuronal survival (Fig. 7A,C). Moreover, when NGF was added back to these cultures, the neurons continued to grow for at least 2 weeks. In contrast, at 48 h without NGF, ΔF/F increased to 3.12 ± 0.16 (n = 162) (Fig. 7B), and after 72 h without NGF these cultures had significantly fewer neurons and many of those that remained were in poor health when examined 1 week after adding back NGF (Fig. 7C). These results indicate that a 18 h transient disruption in NGF signaling causes relatively mild elevations in cytosolic ROS, elevations comparable with those achieved with antimycin-A, but importantly, not sufficient to affect neuronal growth or survival.
Figure 7.
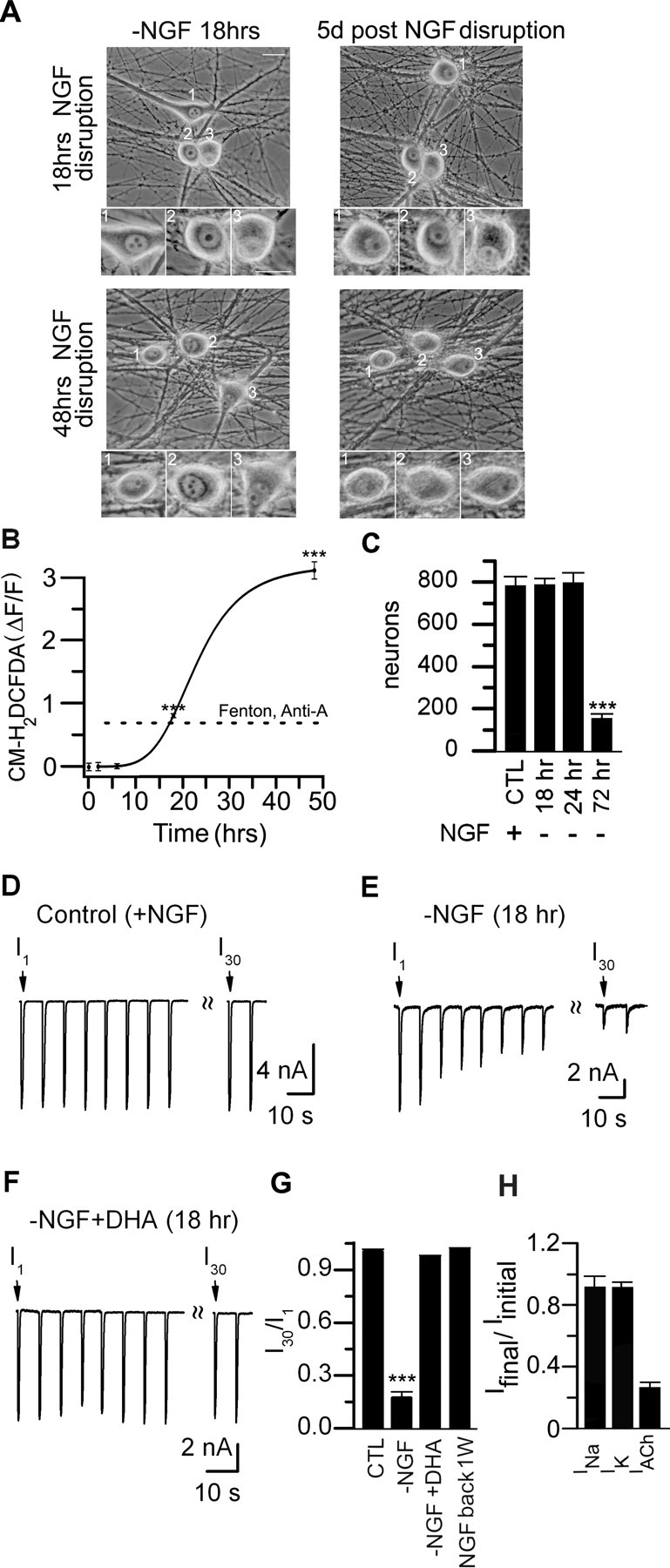
Transient interruption in nerve growth factor signaling increases cytosolic ROS and causes rundown of ACh evoked currents. A, Phase micrographs of sympathetic neurons in culture for 2 weeks. NGF was removed for 18 h (top) or 48 h (bottom). The micrographs were taken at 18 h after removing NGF (left) and the same field retaken 5 d (right) after replacing NGF. The insets are magnified images of the cell bodies in each field. After 18 h without NGF followed by 5 d with NGF, the neurons have phase-bright nuclei, clearly visible nucleoli; however, after 48 h without NGF, the neurons are in poorer health. B, ΔF/F measurements from sympathetic neurons before and after being deprived of NGF for 2, 6, 18, and 48 h (>50 neurons were measured at each time point). Neurons were loaded with CM-H2DCFDA for 1 h and then imaged. The fluorescence remained near basal levels for the first 6 h; after 18 h of NGF removal, the increase in ΔF/F was comparable with that induced by ROS from the Fenton reaction or antimycin-A (dotted line). The solid line is a Hill function. C, Number of neurons per culture before (CTL) (n = 80) and 5 d after a transient removal of NGF for 18 h (n = 150), 24 h (n = 53), or 48–72 h (n = 109). A transient interruption in NGF signaling for 18 h had no significant effect on neuronal survival. D–F, ACh-evoked currents in response to a series of 1 s ACh (100 μm) applications delivered at 15 s intervals from a control (+NGF) neonatal mouse SCG neuron (D) in culture for 10 d, one in a sister culture deprived of NGF for 18 h (E), and one in a sister culture deprived of NGF for 18 h but pretreated with antioxidants (DHA) for 24 h at 6 h before NGF removal (F). G, I30/I1 ratio for neurons from control (CTL) (n = 7), 18 h after NGF removal (−NGF) (n = 14), 18 h after NGF removal with antioxidants (−NGF + DHA) (n = 6), and 1 week after a transient 18 h NGF withdrawal (n = 6). The SEs are too small to resolve. H, Bar graph showing the ratio of the peak INa and IK (mean ± SE; n = 7) before and after rundown of the ACh-evoked currents on neurons 18 h after removal of NGF. There was no significant difference in the peak INa or IK before and after rundown of the ACh-evoked currents. ***p < 0.001.
To address whether this mild increase in ROS after 18 h of NGF withdrawal induced a use-dependent rundown of ACh-evoked currents, we simultaneously measured ROS-induced fluorescence and ACh-evoked currents. The first ACh-evoked current on neurons without NGF for 18 h was not significantly different from that on neurons in sister cultures maintained continuously in NGF; however, when we delivered a series of consecutive ACh applications, the ACh-evoked currents ran down rapidly: the response to the 30th application (I30) was <20% of the first (I1) (Fig. 7E,G). However, for neurons in sister cultures maintained continuously in NGF, the ACh-evoked currents were stable (Fig. 7D,G). In addition, when we measured ACh-evoked currents on neurons 5 d after a transient 18 h disruption, we observed that the ROS-induced fluorescence had returned to control levels, and that the ACh-evoked currents were stable (Fig. 7G). These results indicate that a disruption in growth factor signaling can induce a long-lasting, use-dependent inactivation of ACh-evoked currents on neonatal sympathetic neurons.
Moreover, we observed no significant difference in the voltage-gated inward Na+ or outward K+ currents before and after rundown of the ACh-evoked currents (Fig. 7H). These results indicate that an 18 h disruption in NGF signaling does not cause a generalized, nonspecific change in membrane currents.
To determine that transient disruptions in NGF signaling cause rundown of the ACh-evoked currents through a mechanism involving ROS, we withdrew NGF for 18 h and simultaneously increased the antioxidant activity of the cytosol by adding ascorbate and ascorbate oxidase to the cultures. Treating these NGF-deprived neurons with ascorbate and ascorbate oxidase prevented any significant change in ROS-induced fluorescence; equally relevant, ACh-evoked currents on these 18 h NGF-deprived neurons were stable (Fig. 7F,G). These results indicate that the transient removal of NGF causes a use-dependent rundown of the ACh-evoked currents by elevating cytosolic ROS.
Elevation of mitochondrial ROS leads to long-term depression of fast nicotinic synaptic transmission in intact ganglia
Our results with cultured sympathetic neurons indicate that elevations in mitochondrial ROS induce a long-lasting, use-dependent rundown of ACh-evoked currents. If extended to neurons in vivo, these findings predict that acute elevation in cytosolic ROS in sympathetic neurons in intact ganglia would cause a use-dependent, long-lasting inactivation of the postsynaptic nicotinic receptors and depress fast nicotinic synaptic transmission. To test this prediction, we used intracellular recordings to measure nerve-evoked fast nicotinic EPSPs on sympathetic neurons in intact ganglia from neonatal mice after inhibiting complex III in the recorded neuron with antimycin-A.
In control recordings, a maximal stimulus to the preganglionic nerve evoked large (∼15 mV) suprathreshold EPSPs on sympathetic neurons. The evoked action potential obscures the peak of the EPSPs; therefore, for accurate measurements of changes in the peak amplitude of the nerve-evoked EPSPs, we partially blocked the postsynaptic nAChRs with curare (10 μm) to make the EPSPs subthreshold. In control neurons, when stimulating the preganglionic nerve at 1 Hz for 60 s we observed no significant change in the amplitude of the nerve-evoked EPSPs (Fig. 8A). To test whether acute elevations in cytosolic ROS in sympathetic neurons affect synaptic transmission, we recorded from neurons in intact ganglia with electrodes containing antimycin-A and waited 20 min for it to diffuse into the neuron before stimulating the preganglionic nerve. In all 20 neurons recorded with electrodes containing antimycin-A, the initial EPSPs were not significantly different from control neurons; however, when stimulating the preganglionic nerve at 1 Hz for 60 s, we observed a steady depression of nerve-evoked EPSPs which leveled off at ∼50% of the initial amplitude (Fig. 8A,B). There was no change in resting potential or input resistance during this 60 s stimulation; moreover, this depression lasted for as long as we could record from the neuron (usually at least ∼50 min). In contrast, we observed no depression of the nerve-evoked EPSPs on neurons recorded without antimycin-A in the electrode solution (all other components were identical) (Fig. 8A,B). These results indicate that acute elevations in mitochondrial ROS in postsynaptic sympathetic neurons can produce a long-lasting depression of the nerve-evoked EPSPs in sympathetic ganglia.
Figure 8.

Elevated ROS in sympathetic neurons in intact ganglia depress fast synaptic transmission. A, The first and last five consecutive EPSPs produced by stimulating the preganglionic nerve at 1 Hz for 60 s and recording intracellularly from neurons with electrodes containing control solution (top) or antimycin-A (100 μm) (bottom); the expanded trace at the end of each series represents the average EPSP. The EPSPs were made subthreshold by adding curare (10 μm). Note: The timescale for the average EPSPs is 10 ms and 50 ms for the five consecutive EPSPs. B, The average peak EPSP for the last five EPSPs recorded with electrodes containing control (n = 20) or antimycin-A (n = 20). C, EPSPs from sympathetic neurons in ganglia from P28 mice injected with control IgG or anti-NGF antibodies (20 ng/ml; 2.5 μl/g) for 2 d. D, The average peak EPSP for sympathetic neurons from control (n = 20) and anti-NGF-treated (n = 20) mice. ***p < 0.001.
Transient disruption in NGF signaling leads to long-term depression of fast nicotinic synaptic transmission in intact ganglia
We have shown above that transient withdrawal of NGF signaling elevates cytosolic ROS and causes a use-dependent rundown of the ACh-evoked currents on cultured sympathetic neurons. Therefore, we asked whether a transient disruption in NGF signaling in vivo would depress fast EPSPs in sympathetic ganglia. To address this, we injected neonatal mice with either anti-NGF antibodies or control IgG antibodies and measured the nerve-evoked fast EPSPs on sympathetic neurons in intact ganglia 48 h later. In ganglia from mice injected with control IgG antibodies, stimulating the preganglionic nerve evoked suprathreshold EPSPs that were not significantly different in amplitude from those on neurons in ganglia from control mice (Fig. 8C,D). However, in ganglia from mice injected with anti-NGF antibodies 48 h earlier, the nerve-evoked EPSPs were all subthreshold and their amplitudes were ∼50% smaller than in control ganglia (Fig. 8C,D). We observed no significant difference in resting potential or input resistance among neurons in ganglia from mice injected with anti-NGF compared with those in control ganglia. These results suggest that transient disruption of NGF signaling depresses cholinergic–nicotinic synaptic transmission in sympathetic ganglia.
Discussion
In this study, we uncovered a new mechanism that inactivates nAChRs on neurons. We show that relatively mild shifts in the intracellular thiol/disulfide redox state toward more oxidative conditions induce a rundown of ACh-evoked currents. Once the ACh-evoked currents run down, they do not recover for at least 1 h; it is as if the receptors become trapped in a long-lasting inactivated state. This ROS-induced rundown of ACh-evoked currents is specific to neuronal nAChRs, because elevating ROS had no detectable effect on muscle nAChRs.
In previous work (De Koninck and Cooper, 1995), we measured gene expression of sympathetic neurons developing in culture. We found that these neurons express five nicotinic receptor transcripts: mRNA for α3 and β4 are the most abundant and increase over the first week in culture; mRNA for β2 and α5 are severalfold lower and remain constant, whereas mRNA for α7 is initially high but falls rapidly within 1–2 d in culture and remains low. Although there are no dramatic changes in nAChR subunit gene expression in rodent sympathetic neurons in culture, we cannot conclude definitively that the subunit composition of the receptors stays constant. Nonetheless, whatever the exact subunit composition, nAChRs on neurons both in short-term cultures and in long-term cultures are inactivated by cytosolic ROS in a use-dependent manner.
Cytosolic ROS, by themselves, do not trap resting receptors in an inactivated state: elevated ROS must be paired with receptor activation to induce rundown of ACh-evoked currents. We are uncertain of the underlying mechanisms. Our preliminary evidence suggests that cysteine residues in the M1–M2 linker are oxidized by ROS and lock the receptor in an inactive configuration (Campanucci and Cooper, 2007); we speculate that these residues are buried or hidden when the receptor is in the resting state and only become exposed to ROS during activation as the receptor undergoes the conformational change to the open state. Importantly, this relationship between increases in cytosolic ROS and nAChR inactivation extends to fast nicotinic transmission in sympathetic ganglia; increasing ROS in sympathetic neurons depresses nerve-evoked EPSPs in intact ganglia.
Fast nicotinic EPSPs in sympathetic ganglia link the activity of various CNS nuclei to sympathetic nerve activity and are crucial for maintaining peripheral homeostasis. Therefore, our findings that elevated ROS in sympathetic neurons depress ganglionic transmission have relevance to sympathetic insufficiencies experienced during diseases associated with mitochondrial dysfunction and oxidative stress, such as Parkinson's disease and diabetes. For example, ∼20–30% of patients with diabetes have cardiovascular autonomic neuropathy (Vinik et al., 2003; Savitt et al., 2006), and ∼20–50% of patients with Parkinson's disease experience dysautonomia, especially orthostatic hypotension, which increases the incidence of accidental trauma (Goldstein, 2003). The cause of dysautonomia in patients with Parkinson's disease or diabetes is not known, but if mitochondrial dysfunction and/or oxidative stress associated with these diseases occurs in sympathetic neurons, our results suggest that the elevated cytosolic ROS would disrupt ganglionic synaptic transmission, thereby producing dysautonomic symptoms.
Sympathetic neurons, in particular, are vulnerable to oxidative damage because tyrosine hydroxylase and monoamine oxidase produce H2O2 as normal by-products of their activities (Coyle and Puttfarcken, 1993), and because catecholamines auto-oxidize to produce H2O2 which decomposes to the highly reactive hydroxyl radical, a process accelerated by Fe2+ (Halliwell, 2006). Our results predict that if ROS are elevated in sympathetic neurons of patients with diseases related to mitochondrial dysfunction and/or oxidative stress, ganglionic transmission will be depressed, and these patients will experience sympathetic insufficiencies, such as poorly regulated blood pressure, cardiac arrhythmias, and perturbations in other homeostatic control processes.
The major symptoms of Parkinson's disease result from a selective loss of dopaminergic neurons in the substantia nigra pars compacta (Beal, 2003; Dauer and Przedborski, 2003; Abou-Sleiman et al., 2006). Many of these substantia nigra neurons express α4β2-containing and α6β2-containing nAChRs (Zoli et al., 2002; Champtiaux et al., 2003), and our results indicate that elevated ROS trap α4β2 nAChRs in a long-lasting inactivated state, similar to nAChRs on sympathetic neurons. We speculate that, during the early progression of Parkinson's disease, oxidative stress in dopaminergic nigral neurons induces long-lasting inactivation of these α4β2-containing and α6β2-containing nAChRs, and contributes, in part, to the movement disorders and cognitive deficits experienced by Parkinson's patients.
In the CNS, most nAChRs are located presynaptically where they enhance transmitter release, an important process that underlies many forms of synaptic plasticity (MacDermott et al., 1999; Dani and Bertrand, 2007). These presynaptic compartments are rich in mitochondria, and therefore presynaptic nAChRs could be subjected to elevated levels of cytosolic ROS, particularly generated by the mitochondrial electron transport chain during periods of high activity (Nicholls and Budd, 2000). Because of the difficulties in recording directly from nAChRs on nerve terminals, we know little about the exact manner that nAChR-mediated depolarization and Ca2+ influx, in concert with normal impulse activity, modulate presynaptic transmitter release, or whether fluctuations in cytosolic ROS occur. But if cytosolic ROS levels do increase, they would inactivate these presynaptic receptors and reduce the effects of ACh at these synapses.
We show that transient interruption of NGF signaling induces rundown of ACh-evoked currents on sympathetic neurons by increasing cytosolic ROS without affecting neuronal survival. It is unclear how much circulating NGF fluctuates in vivo, or whether and by how much circulating NGF decreases during disease. Our results suggest that a transient drop in circulating NGF during disease, or therapeutic treatment with anti-NGF antibodies, would depress synaptic transmission in sympathetic ganglia and lead to sympathetic insufficiencies and exacerbate the disease condition. This unexplored area deserves additional investigation.
In young cultured neurons, ROS out balances the antioxidant activities of ROS scavengers, shifting the thio/disulfide redox state of the cytosol toward oxidative conditions and causes the ACh-evoked currents to run down. It is known that oxidative conditions trigger redox signaling mechanisms, including increased signaling through tyrosine kinase pathways by suppressing tyrosine phosphatase activity (Dröge, 2002). Tyrosine kinase pathways are crucial for the growth and survival of neurons (Huang and Reichardt, 2003). Therefore, we speculate that the mild shifts in the intracellular thiol/disulfide redox state toward more oxidative conditions in young neurons may help them to survive in culture, particularly during the early stages when recovering from the trauma of axotomy and enzymatic dissociation. However, ACh-evoked depolarizations, and the related increase in calcium influx, together with elevated ROS could trigger excitotoxicity and cell death. Therefore, the ROS-induced rundown of ACh-evoked currents could act as a protective mechanism to prevent overexcitation, excess calcium influx, and cell death.
Our results have uncovered a cellular mechanism that regulates the function of neuronal nAChRs; however, they also raise a number of questions. For example: How do ROS inactivate neuronal nAChRs? Do ROS act on the receptors directly, or do they act through intermediators? How do nAChRs recover from ROS-induced rundown? Are the receptors removed from the membrane and destroyed, or are receptors restored to their resting state by intracellular reducing agents. Answers to these questions must await additional work.
Footnotes
This work was supported by Canadian Institutes of Health Research (E.C.) and McGill Faculty of Medicine awards (V.A.C., A.K.). We thank Brigitte Pié for technical assistance and Linda Cooper for comments on this manuscript.
References
- Abou-Sleiman PM, Muqit MMK, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- Annunziato L, Pannaccione A, Cataldi M, Secondo A, Castaldo P, Di Renzo G, Taglialatela M. Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol Aging. 2002;23:819–834. doi: 10.1016/s0197-4580(02)00069-6. [DOI] [PubMed] [Google Scholar]
- Beal MF. Bioenergetic approaches for neuroprotection in Parkinson's disease. Ann Neurol. 2003;53(Suppl 3):S39–S47. doi: 10.1002/ana.10479. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Cooper E, Valera S, Rungger D, Ballivet M. Electrophysiology of neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes following nuclear injection of genes or cDNAs. Methods Neurosci. 1991;4:174–193. [Google Scholar]
- Broide RS, Leslie FM. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol. 1999;20:1–16. doi: 10.1007/BF02741361. [DOI] [PubMed] [Google Scholar]
- Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- Campanucci VA, Cooper E. Inactivation of neuronal nicotinic ACh receptors by oxidation of intracellular cysteine residues. Soc Neurosci Abstr. 2007;33:677–8. [Google Scholar]
- Champtiaux N, Changeux J-P. Knockout and knockin mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res. 2004;145:235–251. doi: 10.1016/s0079-6123(03)45016-4. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David D, Przybylski C, Léna C, Le Novère N, del Mar Arroyo-Jimenez M, Clementi F, Moretti M, Rossi FM, Le Novère N, McIntosh JM, Gardier AM. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knockout mice. J Neurosci. 2003;23:7820–7829. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement MV, Ramalingam J, Long LH, Halliwell B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid Redox Signal. 2001;3:157–163. doi: 10.1089/152308601750100687. [DOI] [PubMed] [Google Scholar]
- Cordero-Erausquin M, Marubio LM, Klink R, Changeux JP. Nicotinic receptor function: new perspectives from knockout mice. Trends Pharmacol Sci. 2000;21:211–217. doi: 10.1016/s0165-6147(00)01489-9. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Le Novère N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- De Biasi M. Nicotinic mechanisms in the autonomic control of organ systems. J Neurobiol. 2002;53:568–579. doi: 10.1002/neu.10145. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Cooper E. Differential regulation of neuronal nicotinic ACh receptor subunit genes in cultured neonatal rat sympathetic neurons: specific induction of alpha 7 by membrane depolarization through a Ca2+/calmodulin-dependent kinase pathway. J Neurosci. 1995;15:7966–7978. doi: 10.1523/JNEUROSCI.15-12-07966.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Frank J, Flaccus A, Schwarz C, Lambert C, Biesalski HK. Ascorbic acid suppresses cell death in rat DS-sarcoma cancer cells induced by 5-aminolevulinic acid-based photodynamic therapy. Free Radic Biol Med. 2006;40:827–836. doi: 10.1016/j.freeradbiomed.2005.10.034. [DOI] [PubMed] [Google Scholar]
- Gamper N, Oleg Zaika O, Yang Li Y, Martin P, Hernandez CC, Perez MC, Wang AYC, Jaffe DB, Shapiro MS. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25:4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Dysautonomia in Parkinson's disease: neurocardiological abnormalities. Lancet Neurol. 2003;2:669–676. doi: 10.1016/s1474-4422(03)00555-6. [DOI] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Guo X, Lester RA. Regulation of nicotinic acetylcholine receptor desensitization by Ca2+ J Neurophysiol. 2007;97:93–101. doi: 10.1152/jn.01047.2005. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ichinari K, Kakei M, Matsuoka T, Nakashima H, Tanaka H. Direct activation of the ATP-sensitive potassium channel by oxygen free radicals in guinea-pig ventricular cells: its potentiation by MgADP. J Mol Cell Cardiol. 1996;28:1867–1877. doi: 10.1006/jmcc.1996.0179. [DOI] [PubMed] [Google Scholar]
- Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Franklin JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci. 2001;21:1949–1963. doi: 10.1523/JNEUROSCI.21-06-01949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002;22:6480–6490. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gutterman DD. The coronary circulation in diabetes: influence of reactive oxygen species on K+ channel-mediated vasodilation. Vascul Pharmacol. 2002;38:43–49. doi: 10.1016/s1537-1891(02)00125-8. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Coppede F, Migliore L, Siciliano G, Murri L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J Alzheimers Dis. 2006;10:59–73. doi: 10.3233/jad-2006-10110. [DOI] [PubMed] [Google Scholar]
- Mandelzys A, De Koninck P, Cooper E. Agonist and toxin sensitivities of ACh-evoked currents on neurons expressing multiple nicotinic ACh receptor subunits. J Neurophysiol. 1995;74:1212–1221. doi: 10.1152/jn.1995.74.3.1212. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane S, Cooper E. Postnatal development of voltage-gated K currents on rat sympathetic neurons. J Neurophysiol. 1992;67:1291–1300. doi: 10.1152/jn.1992.67.5.1291. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Park MK, Lee SH, Lee SJ, Ho WK, Earm YE. Different modulation of Ca-activated K channels by the intracellular redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflügers Arch. 1995;430:308–314. doi: 10.1007/BF00373904. [DOI] [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of behavior: beyond the inverted U. Trends Pharmacol Sci. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL. Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacol Ther. 2001;92:89–108. doi: 10.1016/s0163-7258(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Purohit P, Mitra A, Auerbach A. A stepwise mechanism for acetylcholine receptor channel gating. Nature. 2007;446:930–933. doi: 10.1038/nature05721. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Rassadi S, Krishnaswamy A, Pie B, McConnell R, Jacob MH, Cooper E. A null mutation for the alpha3 nicotinic acetylcholine (ACh) receptor gene abolishes fast synaptic activity in sympathetic ganglia and reveals that ACh output from developing preganglionic terminals is regulated in an activity-dependent retrograde manner. J Neurosci. 2005;25:8555–8566. doi: 10.1523/JNEUROSCI.1983-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher E, Chen Y, Sharples TJ, Broad LM, Benedetti G, Zwart R, McPhie GI, Pearson KH, Baldwinson T, De Filippi G. Physiological roles of neuronal nicotinic receptor subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr Top Med Chem. 2004;4:283–297. doi: 10.2174/1568026043451393. [DOI] [PubMed] [Google Scholar]
- Skok VI. Nicotinic acetylcholine receptors in autonomic ganglia. Auton Neurosci. 2002;97:1–11. doi: 10.1016/s1566-0702(01)00386-1. [DOI] [PubMed] [Google Scholar]
- Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer's. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- Swope SL, Qu Z, Huganir RL. Phosphorylation of the nicotinic acetylcholine receptor by protein tyrosine kinases. Ann NY Acad Sci. 1995;757:197–214. doi: 10.1111/j.1749-6632.1995.tb17476.x. [DOI] [PubMed] [Google Scholar]
- Tang XD, Garcia ML, Heinemann SH, Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–178. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Vincent AM, McLean LL, Backus C, Feldman EL. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005;19:638–640. doi: 10.1096/fj.04-2513fje. [DOI] [PubMed] [Google Scholar]
- Vinik AL, Freeman R, Erbas T. Diabetic autonomic neuropathy. Semin Neurol. 2003;23:365–372. doi: 10.1055/s-2004-817720. [DOI] [PubMed] [Google Scholar]
- Wang N, Orr-Urtreger A, Korczyn AD. The role of neuronal nicotinic acetylcholine receptor subunits in autonomic ganglia: lessons from knockout mice. Prog Neurobiol. 2002;68:341–360. doi: 10.1016/s0301-0082(02)00106-5. [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh MJ, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]