

Abstract
Several studies report microglial accumulation and activation in the CA1 area in response to transient forebrain ischemia (TFI). Here we examine the possibility that free radicals and chemokines mediate the transient activation of microglia. Free radicals are produced primarily in CA1 pyramidal neurons within 2 h of TFI. Administration of trolox, a vitamin E analog, led to the inhibition of free radical production and recruitment of microglia in the CA1 area. In addition, intrahippocampal injection of Fe2+ triggered free radical production in CA1 neurons, followed by the recruitment and activation of microglial cells into this area. TFI-induced expression of macrophage inflammatory protein-1α (MIP-1α) was increased in CA1 neurons before microglial recruitment, and blocked by trolox. Moreover, the MIP-1α level was upregulated in cultured hippocampal neurons exposed to Fe2+, suggesting an essential role of free radicals in TFI-induced expression of MIP-1α. Intracerebroventricular injection of vMIP-2 (viral macrophage inflammatory protein-2), a broad-spectrum peptide antagonist of chemokine receptors, attenuated microglial recruitment and delayed CA1 neuronal degeneration after TFI. Our data suggest that free radicals produced in CA1 neurons contribute to the recruitment and activation of microglia and neurodegeneration through MIP-1α expression.
Keywords: CA1, microglia, free radicals, MIP-1α, vMIP-2, transient forebrain ischemia
Introduction
Accumulating evidence shows that microglia, the major immunocompetent cells of the CNS, contribute to delayed neuronal death in the hippocampal CA1 area after transient forebrain ischemia (TFI). Recruitment and activation of microglial cells gradually increase within the CA1 area over 24 h after TFI, before the degeneration of neurons (Morioka et al., 1991). Microglia subsequently produce inflammatory mediators, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and nitric oxide (NO), which contribute to delayed neuronal death (Sairanen et al., 1997; Uno et al., 1997; Yamashita et al., 2000). Consistent with these findings, immunosuppressants, such as minocycline and FK506, prevent NO production and neuronal death in the CA1 sector after forebrain ischemia (Yrjanheikki et al., 1998; Sasaki et al., 2004).
Activated microglial cells with an amoeboid macrophage phenotype are observed rapidly after nervous system injuries and appear to migrate toward the lesion site (Kreutzberg, 1996). Chemokines that regulate cells in the peripheral immune system may mediate microglial cell activation and migration after forebrain ischemia. For example, hypoxic ischemia stimulates the expression of monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α), and macrophage inflammatory protein-1β (MIP-1β) to attract leukocytes and microglia into the lesion site in rat brain (Ivacko et al., 1997; Gourmala et al., 1999; Cowell et al., 2002; Cartier et al., 2005). MCP-1 overexpression in the brain leads to increased recruitment of inflammatory cells and brain lesions after focal cerebral ischemia (Chen et al., 2003). Expression of MIP-1α receptor (CCR5) is elevated in microglia during hypoxia-ischemia in neonatal rat brain (Cowell et al., 2006). Moreover, endangered neurons release chemokines to recruit and activate microglia after brain lesions (Rappert et al., 2004; de Jong et al., 2005).
Free radicals play an essential role in neuronal death after hypoxic-ischemic injury (Chan, 2001). Copper/zinc-superoxide dismutase (SOD1) suppresses MIP-1α and MCP-1 expression after transient focal cerebral ischemia (Nishi et al., 2005). Mitochondrial free radicals, produced in CA1 neurons as early as 2 h, are required for delayed neuronal death after forebrain ischemia (Park et al., 2005). This raises the possibility that free radicals mediate the recruitment and activation of microglial cells via regulation of chemokine expression after TFI, which possibly contributes to the delayed death of CA1 pyramidal neurons. This hypothesis was examined using a model of TFI in adult rat and primary hippocampal neuronal cell cultures.
Materials and Methods
Materials
Neuronal-specific nuclear protein (NeuN) antibody was purchased from Millipore (Billerica, MA). MIP-1α antibody, viral macrophage inflammatory protein-2 (vMIP-2), and MC148 were from R & D Systems (Minneapolis, MN). Mouse anti-rat CD11b (OX-42) was obtained from Serotec (Oxford, UK). MitoTracker Red CM-H2XROs (MT Red CM-H2XROs) was purchased from Invitrogen (Carlsbad, CA). Microtubule-associated protein-2 (MAP-2) antibody, cresyl violet, Hoechst 33258, and trolox were acquired from Sigma (St. Louis, MO), whereas fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG, FITC-conjugated anti-mouse IgG, aminomethylcoumarin acetate (AMCA)-conjugated streptavidin, and biotinylated anti-mouse IgG were from Vector Laboratories (Burlingame, CA).
Transient forebrain ischemia
TFI was induced by four-vessel occlusion in male Sprague Dawley rats weighing 280–310 g, according to the method of Pulsinelli and Brierley (1979). In brief, rats were anesthetized with chloral hydrate (400 mg/kg, i.p.), and bilateral vertebral arteries were exposed and electrocauterized, followed by isolation of both common carotid arteries (CCAs) and placing polyethylene tube (PE-10) loosely around the CCAs for later CCA occlusion. After 24 h, the CCAs were occluded bilaterally using aneurysm clips for 10 min. Arteries were visually inspected to ensure adequate reflow. The body temperature was maintained at 37.5 ± 0.5°C with heating pads until animals recovered from surgery. Thereafter, animals were housed with ad libitum access to food and water. Sham-operated animals were treated identically, except that vertebral arteries and common carotid arteries were not occluded. All animal procedures were performed in accordance with institutional guidelines.
Stereotaxic surgery and chemical injection
Male Sprague Dawley rats (280–310 g) were anesthetized with chloral hydrate (400 mg/kg, i.p.), and positioned in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA). FeCl2 (4 nmol in 2 μl of PBS) was administrated by intrahippocampal injection into stratum radiatum using the following stereotaxic coordinates: 4.0 mm posterior to the bregma, 2.5 mm lateral to the midline, and 3.0 mm below pial surface, and for controls, PBS was injected. vMIP-2 (1 μg in 5 μl of PBS) was administrated by intracerebroventricular injection using the following coordinates: 0.8 mm posterior to the bregma, 1.4 mm lateral to the midline, and 3.9 mm below pial surface, according to the atlas of Paxinos and Watson (1998), at the onset of TFI reperfusion. All injections were made using a 10 μl Hamilton syringe attached to a syringe pump (KDScientific, New Hope, PA) at the rate of 0.5 μl/min. After injections, the syringe remained in the injection site for 5 min before being withdrawn. Trolox was dissolved in saline and administrated by single intraperitoneal injection (50 mg/kg) at the onset of TFI reperfusion.
Detection of free radicals
In vivo.
Our group found that MT Red CM-H2XROs, a fluorescence dye for mitochondrial free radicals, was retained after fixation, compatible with immunocytochemistry or nuclear staining. Therefore, experiments were performed as described previously (Kim et al., 2002). MT Red CM-H2XROs (0.4 nmol) dissolved in equal volume of dimethylsulfoxide and saline was injected before administration of FeCl2 at the following stereotaxic coordinates: 0.8 mm posterior to the bregma, 1.4 mm lateral to the midline, and 3.9 mm below pial surface for the ventricular injections. To monitor free radicals in TFI, MT Red CM-H2XROs was injected after electrocauterization of bilateral vertebral arteries. For each injection, a volume of 8 μl was delivered via a 10 μl Hamilton syringe at the rate of 1.0 μl/min. The fluorescence signal of MT Red CM-H2XROs was analyzed using the IX71 confocal laser-scanning microscope kept constant for all captured images (Olympus, Tokyo, Japan).
In vitro.
Hippocampal neuronal cell cultures grown on coverslips were loaded with 25 nm MT Red CM-H2XROs in HEPES-buffered control salt solution (HCSS) containing 120 mm NaCl, 5 mm KCl, 1.6 mm MgCl2, 2.3 mm CaCl2, 15 mm glucose, 20 mm HEPES, and 10 mm NaOH. Cultures were incubated for 30 min at 37°C and washed three times with HCSS buffer. The fluorescence signal of MT Red CM-H2XROs was analyzed using the IX71 confocal laser-scanning microscope (Olympus).
Tissue preparation and immunohistochemistry
Animals were anesthetized with chloral hydrate and transcardially perfused with PBS and then with 4% paraformaldehyde dissolved in 0.1 m phosphate buffer (PB). Brains were removed, postfixed overnight at 4°C in 4% paraformaldehyde, and immersed in 30% sucrose solution at 4°C until they sank. Frozen perfusion coronal sections of brain were cut into thickness of 30 μm using Cryocut Microtome (Leica Microsystems, Wetzlar, Germany). Sections were then stored in stock solution [0.1 m phosphate buffer (pH 7.4), 30% (v/v) glycerol, 30% ethylene glycol] at 4°C until use. Free-floating serial sections were washed three times for 5 min in PBS at room temperature (RT) and pretreated for 5 min at RT in PBS containing 1% H2O2. Sections were washed with PBS and incubated with PBS containing 0.25% Triton X-100 for 5 min. Next, sections were preincubated in 10% normal goat serum (NGS) for 1 h and incubated overnight at 4°C with one of the following primary antibodies: mouse anti-NeuN (1:200) for neurons; mouse anti-complement receptor type 3 (OX-42; 1:200) for microglia; and rabbit anti-MIP-1α (1:500). After incubation, sections were rinsed three times for 5 min in PBS and reacted with secondary antibodies: FITC-conjugated anti-mouse IgG (1:200) and FITC-conjugated anti-rabbit IgG (1:200) for 2 h at RT. For hippocampal pyramidal neuron (dendrite) staining, sections were reacted with mouse anti-MAP-2 (1:500) overnight at 4°C. The sections were washed three times with PBS, incubated with biotinylated anti-mouse IgG (1:400) for 3 h, and then reacted with AMCA-conjugated streptavidin (1:300) for 2 h. This was followed by rinsing in PBS and mounting on gelatin-coated slides. Finally, sections were viewed using an IX71 confocal laser-scanning microscope (Olympus).
Hippocampal neuronal cell culture and free radical injury
Embryonic day 16–17 fetal rat hippocampus was dissected and incubated in Dulbecco's PBS (JBI, Taegu, Korea) containing 0.1% trypsin and 40 μg/ml DNase I (Sigma) for 15 min at 37°C. Hippocampal tissues were dissociated into single cells by gentle pipetting through a fire-polished Pasteur pipette. The plating medium consisted of Neurobasal medium (Invitrogen) and insulin-transferrin-sodium selenite medium supplement (1 mg/ml bovine insulin, 0.55 mg/ml transferrin, 0.5 μg/ml sodium selenite, 100 U/ml penicillin, and 100 μg/ml streptomycin) (Sigma). Cells were plated on coverslips precoated with 100 μg/ml poly-d-lysine and 4 μg/ml laminin (1 × 105 cells per coverslip). And maintained at 37°C in 5% CO2/95% air atmosphere. Free radical injury was induced in hippocampal neuronal cell cultures (days in vitro 6–7) by exposure to 30 μm Fe2+, and cultures were loaded with MT Red CM-H2XROs for free radical detection. After the removal of medium, cells were washed with PBS and fixed with 4% paraformaldehyde in 0.1 m PB for 10 min. Next, cells were incubated in 0.25% Triton X-100 in PBS for 10 min and washed three times with PBS. Cells were preincubated with 10% NGS in PBS for 1 h at RT and incubated overnight at 4°C with rabbit anti-MIP-1α antibody (1:500). After rinsing three times for 5 min in PBS, cells were reacted with FITC-conjugated anti-rabbit IgG (1:200) for 2 h at RT. Cells were also incubated with 0.1 m Hoechst 33258 for 5 min for staining nuclei. Cells were then washed three times with PBS, and coverslips were mounted using Gel Mount (Biomeda, Foster City, CA) and viewed using an IX71 confocal laser-scanning microscope (Olympus).
Western blot analysis
Brain tissues from the CA1 region were homogenized in lysis buffer containing 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1% Triton X-100, 30 mm sodium pyrophosphate, 50 mm NaF, 5 μm ZnCl2, 2 mm PMSF, 100 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mm DTT. Lysates were centrifuged at 13,000 × g for 10 min at 4°C. Supernatant fractions were collected, subjected to electrophoresis on a 12% SDS-polyacrylamide gel, and transferred to a nitrocellulose membrane. The blots were washed with Tris-buffered saline solution containing 2.5 mm EDTA (TNE) and then blocked with 5% nonfat dry milk in TNE for 30 min at RT. The blots were then incubated overnight with gentle shaking at 4°C with a primary rabbit polyclonal antibody against MIP-1α (1:500). Next, the blots were incubated with a biotinylated anti-rabbit secondary antibody (1:400) for 2 h, followed by an ABC elite system (Vector Laboratories). After washing, blots were developed with enhanced chemiluminescence detection reagents (GE Healthcare, Buckinghamshire, UK). The intensities of bands on immunoblots were analyzed by Image gauge 3.12 (Fujifilm, Tokyo, Japan).
Counting of hippocampal CA1 microglia and neurons
Microglial cells and neurons within the CA1 layer were counted by using five coronal sections from each animal, beginning 4.0 mm posterior to bregma and spaced 150 μm apart. The number of OX-42-positive microglial cells within the CA1 layer was counted within a 250 × 250 μm square box, using an IX71 confocal laser-scanning microscope (Olympus) at a magnification of 400×, and microscopic field was applied at the center of the CA1 region in the hippocampus. To quantify viable CA1 neurons, tissue was stained with 0.5% cresyl violet and then counted under a bright-field microscope (Olympus) at a magnification of 200×, and microscopic field was centered over the CA1 cell layers. The mean number of CA1 microglia and neurons for both hemispheres was calculated for each group. We routinely performed counting of microglial cells and neurons in a double-blind manner.
Statistical analysis
All statistical comparisons were assessed by ANOVA using Instat 3.05 (GraphPad Software, San Diego, CA), followed by Student–Newman–Keuls analyses. Data were expressed as means ± SEM. A value of *p < 0.05 was considered statistically significant.
Results
Free radicals are required for microglial cell activation in the CA1 region after TFI
Microglial cells with ramified morphology retract their processes, and those with an amoeboid macrophage phenotype were predominantly identified in the hippocampal CA1 area at 3 d after TFI (Fig. 1A). The number of microglial cells was significantly increased (approximately up to twofold of the sham-operated control within 1d), and further elevated over the next 3 d, after ischemic injury (Fig. 1B). Because the OX-42 antibody detects CR3 complement receptors on leukocytes as well as microglia, some of OX-42-immunoreactive cells in the CA1 area may represent leukocytes derived from blood. The intensity of MT Red CM-H2XROs, a fluorescence dye for mitochondrial free radicals, was increased selectively in CA1 pyramidal neurons within 2 h after forebrain ischemia (FI) (Fig. 1C). Administration of trolox, a scavenger of peroxyl and alkoxyl radicals, led to the blockage of free radical production and reduction in the number of microglial cells in the CA1 area down to the sham-operated control level (p < 0.05). Activated microglia with amoeboid morphology were rarely detectable in the ischemic CA1 area of animals treated with trolox. However, some microglial cells revealed a less ramified form with short processes in trolox-treated animals compared with control (Fig. 1D,E). The results imply that free radicals generated in CA1 neurons are required for the recruitment and activation of microglial cells after TFI.
Figure 1.

Free radicals are required for microglial activation in the CA1 region after TFI. A, Fluorescent photomicrographs of the hippocampal section immunolabeled with the OX-42 antibody 3 d after sham operation (top) or TFI (bottom). Resting and activated microglia in the CA1 area are presented in the inlet. Scale bar, 500 μm. B, Quantitative analysis of OX-42-immunopositive (ip) microglial cells in the square (dotted lines) underlying the CA1 area at the indicated time points after forebrain ischemia. Data are presented as means ± SEM (n = 4 for each condition). *Significantly different from the sham control at p < 0.05 with ANOVA and Student–Newman–Keuls analyses. C, D, Fluorescent photomicrographs of the hippocampal CA1 area labeled with MT Red CM-H2XROs (red) and Hoechst 33258 (blue) at 2 h (C), immunolabeled with OX-42 antibody at 3 d (D) after sham operation (left), forebrain ischemia alone (middle), or in the presence of trolox (right; 50 mg/kg, i.p.) immediately after reperfusion. Scale bars, 200 μm. E, Quantitative analysis of OX-42-ip microglial cells in the square (dotted lines) underlying the CA1 area at 3 d after the sham operation (Ctrl) or forebrain ischemia alone or in the presence of trolox immediately after reperfusion. Data are presented as means ± SEM (n = 4 animals for each condition). *Significantly different from the relevant control at p < 0.05, using ANOVA and Student–Newman–Keuls analyses.
Injection of FeCl2 into the CA1 region induces neuronal free radical production and microglial cell activation
Additional experiments were performed to determine whether free radicals are sufficient to recruit and activate microglial cells. Intrahippocampal injection of 4 nmol of FeCl2 (Fe2+), a hydroxyl radical-producing agent through the Fenton reaction, into the CA1 area led to the generation of mitochondrial free radicals in the CA1 neurons within 2 h (Fig. 2A). Neurites and neuronal cell bodies in CA1 remained intact at 1d after Fe2+ injection (Fig. 2 B–D). Activated microglia displaying the amoeboid phenotype were not observed within 2 h, but were evident in the CA1 pyramidal cell layer and stratum radiatum at 1 d. The number of activated microglia was further increased in the CA1 pyramidal cell layer undergoing degenerative changes 3 d later (Fig. 2E). This implies that free radicals produced in the CA1 neurons recruit and activate microglial cells.
Figure 2.
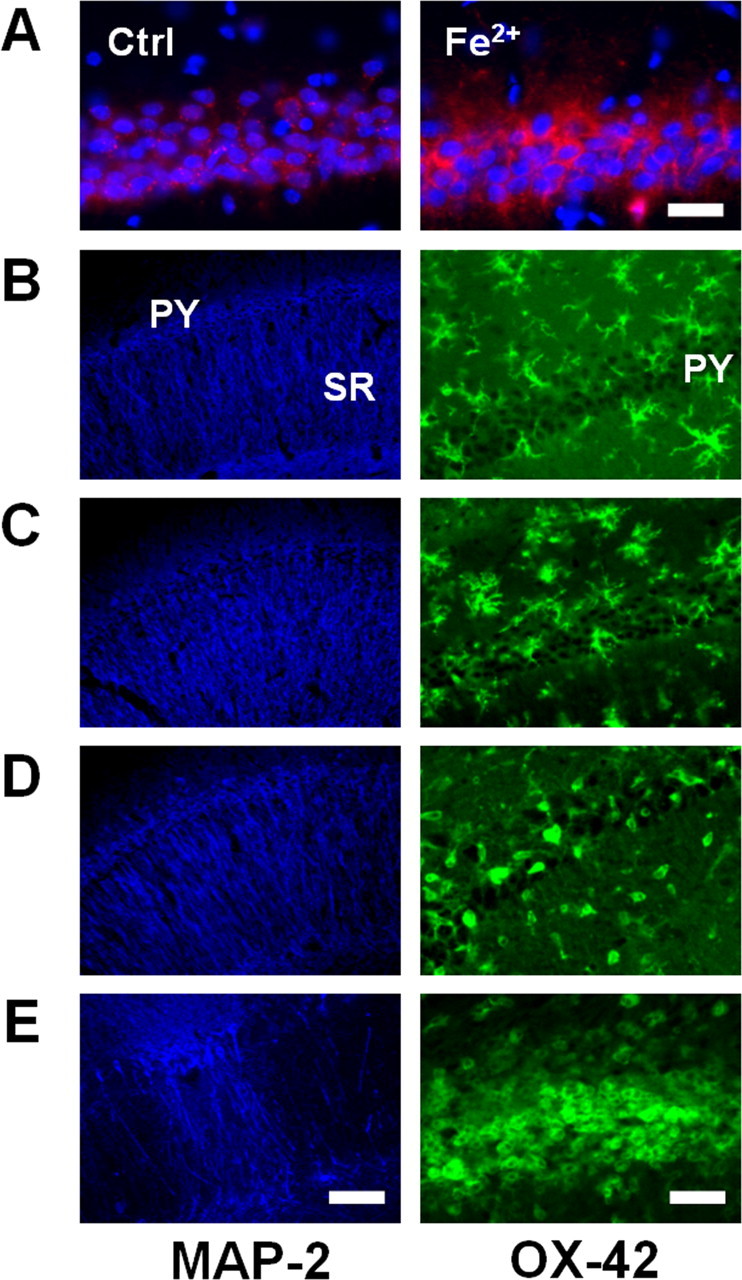
Intrahippocampal injections of Fe2+ induce early neuronal free radical production and delayed microglial activation in the CA1 region. A, Fluorescent photomicrographs of the hippocampal CA1 area labeled with MT Red CM-H2XROs (red) and Hoechst 33258 (blue) at 2 h after injection of vehicle (left) or 4 nmol of Fe2+ (right) into the stratum radiatum. Scale bar, 100 μm. B–E, Fluorescent photomicrographs of the hippocampal CA1 area immunolabeled with a MAP-2 (left) or OX-42 antibody (right) before (B) or 2 h (C), 1 d (D), or 3 d (E) after Fe2+ injections. PY, Pyramidal cell layer; SR, stratum radiatum. Scale bars: left, 250 μm; right, 100 μm.
MIP-1α expression is increased in CA1 pyramidal neurons before microglial recruitment
Next, we examined the possible roles of chemoattractants, such as MIP-1α, MIP-1β and MCP-1, in the recruitment and activation of microglia. MIP-1α expression in the CA1 area increased within 1 d after TFI. The MIP-1α level was further increased up to threefold after 2 d (p < 0.05) (Fig. 3A). In sham-operated animals, MIP-1α was localized in the cell bodies and dendrites of CA1 neurons. The MIP-1α expression was decreased in the dendrites but markedly increased in the CA1 pyramidal cell body layer at 2 d after FI, which was accompanied by recruitment of OX-42-positive microglia in the cell body layer (Fig. 3B). At this time, no increase in MIP-1β or MCP-1 was observed in the CA1 area (data not shown). These findings suggest that MIP-1α may mediate microglial recruitment to the CA1 region.
Figure 3.
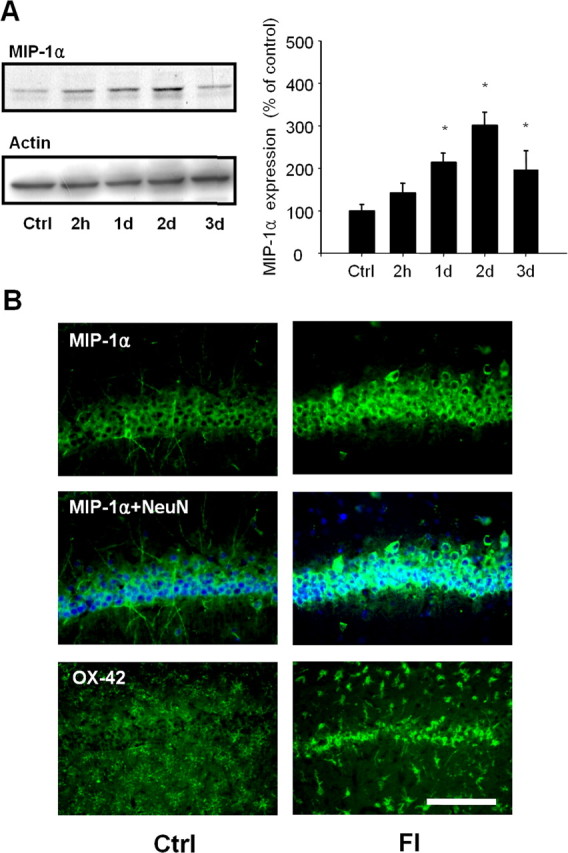
Upregulation of MIP-1α expression in CA1 pyramidal neurons is accompanied by microglial recruitment after TFI. A, Western blot analysis of MIP-1α and actin in the CA1 hippocampal layer after sham operation (Ctrl) or at the indicated time points after forebrain ischemia. MIP-1α levels were analyzed and presented as means ± SEM (n = 4 animals for each condition). *Significantly different from the relevant control at p < 0.05 using ANOVA and Student–Newman–Keuls analyses. B, Fluorescent photomicrographs of CA1 sections immunolabeled with MIP-1α alone (top; green), NeuN (middle; blue), or OX-42 (bottom) antibody after sham operation (Ctrl) or 2 d after forebrain ischemia (FI). Scale bar, 200 μm.
Free radicals induce MIP-1α expression in hippocampal neurons in vitro and in vivo
Next, we examined whether free radicals mediate MIP-1α expression. TFI-induced expression of MIP-1α in the hippocampus was prevented by administration of trolox that blocks free radical production in CA1 pyramidal neurons (Fig. 4A), indicating that free radicals are required for the upregulation of MIP-1α. Cultured hippocampal neurons displayed marked and transient production of free radicals within 2 h after administration of a subtoxic dose of Fe2+ (30 μm) (Fig. 4B). Low MIP-1α expression was observed in cultured hippocampal neurons, which significantly increased at 8 h after exposure to Fe2+ (Fig. 4B). The temporal patterns of Fe2+-induced free radical production and MIP-1α expression imply that free radicals directly induce MIP-1α in the hippocampal neurons in vivo and in vitro.
Figure 4.

Free radicals mediate MIP-1α expression in CA1 neurons after TFI and in cultured hippocampal neurons. A, Levels of MIP-1α in the CA1 area were analyzed by Western blot analysis after sham operation (Ctrl), 2 d after forebrain ischemia alone (FI), or in the presence of trolox immediately after reperfusion. Levels of MIP-1α were analyzed and presented as means ± SEM (n = 4 animals for each condition). *Significantly different from the relevant control at p < 0.05 using ANOVA and Student–Newman–Keuls analyses. B, Fluorescent photomicrographs of cultured hippocampal neurons labeled with Hoechst 33258 (blue), MT Red CM-H2XROs (red), and MIP-1α (green), after exposure to sham control (Ctrl) or 30 μm Fe2+ for the indicated time points. Scale bar, 100 μm.
The MIP-1α receptor antagonist, vMIP-2, prevents recruitment of microglia and neuronal loss in the CA1 area
Additional experiments were performed to establish a putative role of MIP-1α in microglial recruitment and delayed neuronal death in the CA1 area after TFI, using vMIP-2, a broad-spectrum peptide antagonist of chemokine receptors. The number of OX-42-positive microglial cells increased in the CA1 hippocampal cell layer (approximately up to threefold) at 3 d after TFI. This increase was prevented by administration of vMIP-2 in the lateral ventricle (p < 0.05) (Fig. 5A,B). Administration of MC148, a CCR8 antagonist showing sequence homology with vMIP-2 (Lüttichau et al., 2001), did not prevent the microglial accumulation and neuronal loss in the CA1 layer. Delayed neuronal loss in the CA1 was attenuated by administration of vMIP-2 or trolox at a dose that blocked free radical production and MIP-1α expression in CA1 hippocampal neurons at 3 d after TFI (Fig. 6 A–C). Survival of the CA1 neurons remained increased 7 d after TFI in rats treated with vMIP-2 (Fig. 6B). Delayed administration of vMIP-2 at 12 h after reperfusion significantly attenuated the number of microglial cells and delayed neuronal death in the CA1 field after TFI (Figs. 5B, 6B).
Figure 5.
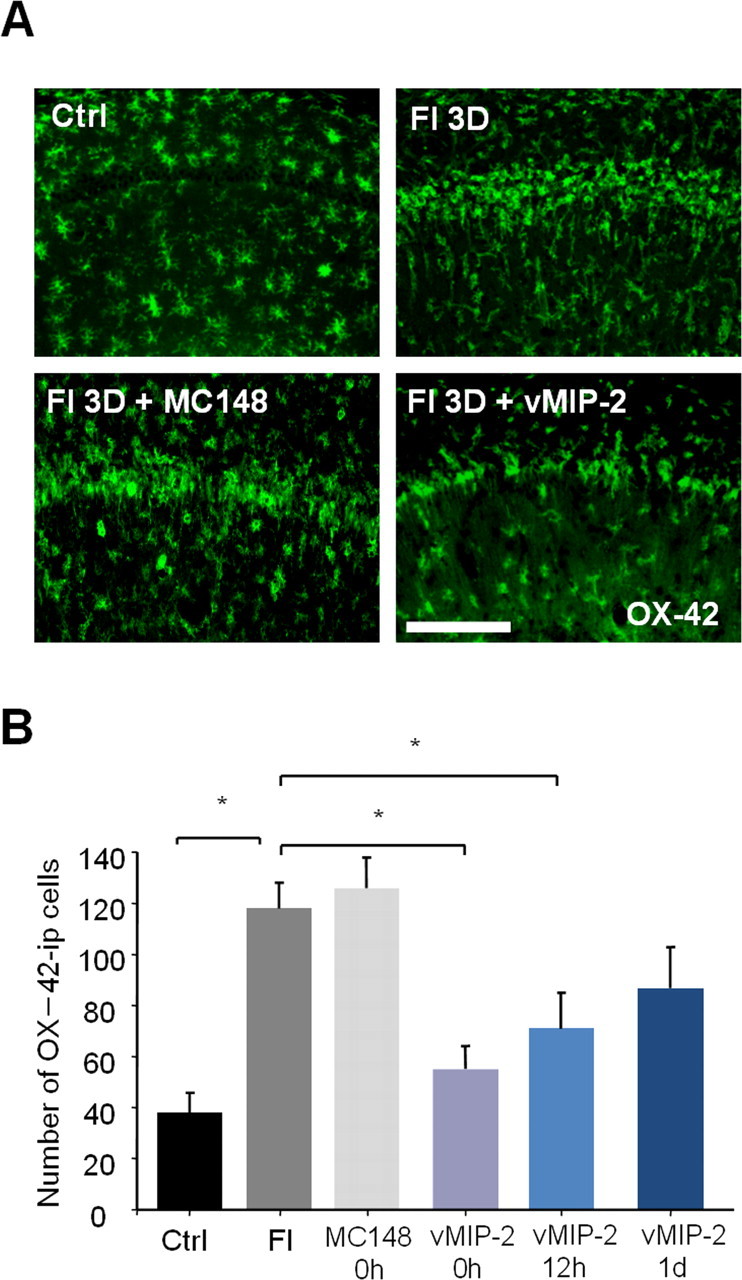
vMIP-2, a broad-spectrum chemokine receptor antagonist, prevents microglial recruitment in the CA1 region after TFI. A, Fluorescent photomicrographs of CA1 sections labeled with an OX-42 antibody after sham operation (Ctrl), 3 d after forebrain ischemia alone (FI), or with an intracerebroventricular injection of 1 μg of vMIP-2 or MC148. Scale bar, 200 μm. B, The number of OX-42-immunopositive (ip) microglial cells at 3 d after FI with an intracerebroventricular injection of 1 μg of vMIP-2 at 0, 12, and 24 h after reperfusion was analyzed and presented as means ± SEM (n = 4 animals for each condition). *Significantly different from the relevant control at p < 0.05 using ANOVA and Student–Newman–Keuls analyses.
Figure 6.
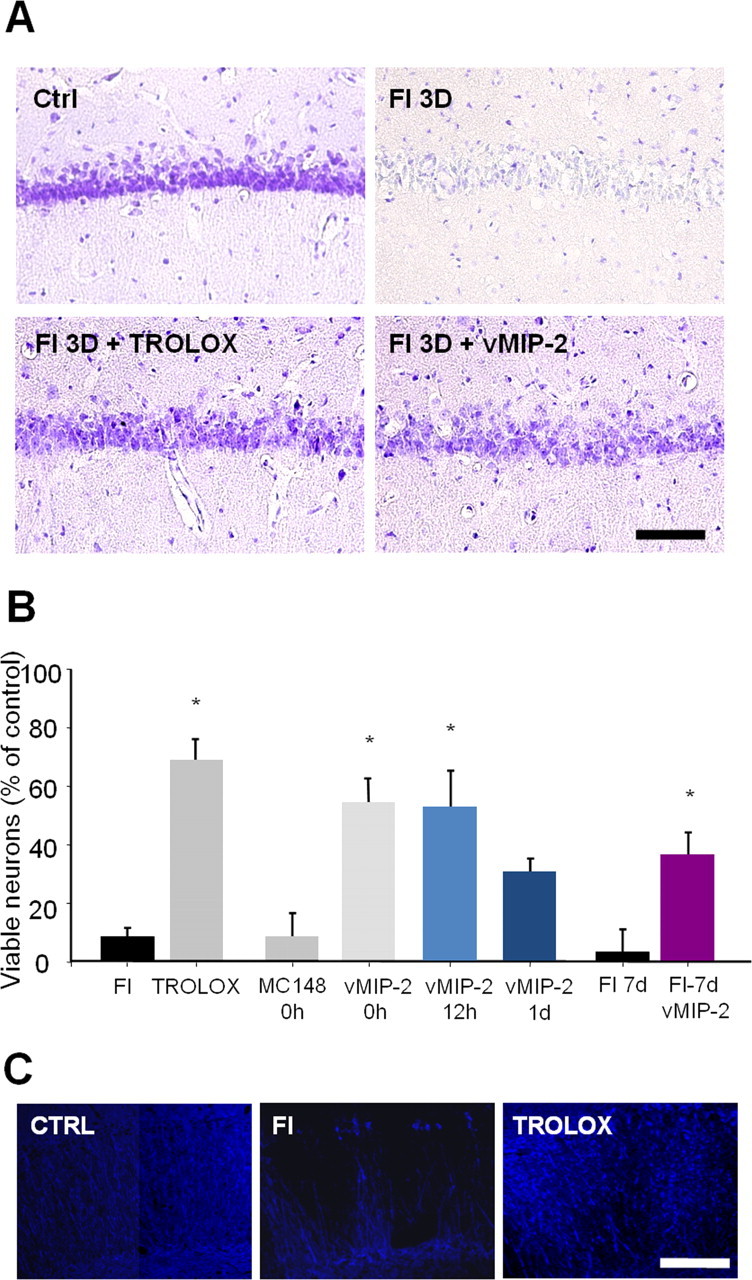
vMIP-2, a broad-spectrum chemokine receptor antagonist, prevents neuronal cell death in the CA1 region after TFI. A, Bright-field photomicrographs of the CA1 section stained with cresyl violet after sham operation (Ctrl), at 3 d after forebrain ischemia alone (FI), or in the presence of trolox or vMIP-2. Scale bar, 200 μm. B, Viable neurons at 3 d after FI with an intracerebroventricular injection of 1 μg of vMIP-2 at 0, 12, and 24 h after reperfusion in the CA1 region and at 7 d after FI with vMIP-2 treatment were analyzed and presented as means ± SEM (n = 4 animals for each condition). *Significantly different from the relevant control at p < 0.05 using ANOVA and Student–Newman–Keuls analyses. C, Fluorescent photomicrographs of CA1 sections labeled with a MAP-2 antibody after sham operation (Ctrl), 3 d after forebrain ischemia alone (FI), or in the presence of trolox (50 mg/kg, i.p.) immediately after reperfusion. Scale bar, 250 μm.
Discussion
Free radicals generated in CA1 hippocampal neurons are essential for the recruitment and activation of microglia after TFI or intrahippocampal injections of Fe2+. Free radical production is accompanied by elevated expression of MIP-1α in the CA1 hippocampal neurons. This upregulation of MIP-1α contributes to microglial recruitment and delayed neuronal death in the CA1 area.
Although activation and recruitment of microglia in the CA1 area after TFI are well documented (Morioka et al., 1991, 1992), the underlying mechanisms are largely unknown. We observed a biphasic pattern of free radical production in CA1 hippocampal neurons after TFI (Park et al., 2005). In particular, rapid TFI-induced free radical production in CA1 neurons was evident within 2 h before microglial activation, suggesting a putative role of free radicals in the recruitment and activation of microglial cells in the CA1 field. In the present study, TFI-induced recruitment and activation of microglia were prevented by trolox, a vitamin E analog that blocks early free radical production in CA1 neurons. Furthermore, intrahippocampal injections of Fe2+ induced free radical generation in CA1 neurons within 2 h, accompanied by recruitment and activation of microglial cells into the area 24 h later. The results imply that free radicals produced in CA1 neurons are essential for recruiting and activating microglia after TFI. However, some microglia in trolox-treated ischemic rats revealed less ramification and shorter processes than those of control rats. This implies that morphology and activity of microglia are influenced to some extent by other factors in addition to free radicals after TFI.
Endangered neurons can synthesize and release chemokines, such as MCP-1, IP-10 (interferon-inducible protein), and fractalkine, which act as signaling molecules for recruitment of microglia (Flugel et al., 2001; Rappert et al., 2004; de Jong et al., 2005). Expression of MCP-1 or SLC (secondary lymphoid-tissue chemokine) is increased in neurons after focal cerebral ischemia (Biber et al., 2001; Che et al., 2001). Overexpression of antioxidant enzymes, such as GPx1 (glutathione peroxidase) or SOD1, prevents upregulation of MIP-1α and MCP-1 under ischemic conditions (Ishibashi et al., 2002; Nishi et al., 2005). This raises the possibility that TFI-induced free radical production in CA1 neurons facilitates microglial recruitment by modulating the expression of chemokines. Our finding that TFI induces MIP-1α expression in CA1 neurons in a manner sensitive to trolox suggests an essential role of free radicals in chemokine expression. Free radicals are sufficient for MIP-1α upregulation, as confirmed from data obtained with cultured hippocampal neurons exposed to Fe2+.
MIP-1α expression in CA1 neurons was accompanied by the recruitment of OX-42-positive microglial cells to this area after TFI. Immunocytochemical study showed marked increase of MIP-1α expression primarily in the CA1 cell body layer with decreased expression in the dendrites at 1–2 d after TFI. The latter appears to be associated with rapidly evolving ischemic damage occurring in the dendrites of the CA1 neurons (Kitagawa et al., 1989). The MIP-1α expression in the neuronal cell body may contribute to microglial recruitment in the hippocampal CA1 cell layer after TFI, as evident from the reduced number of microglia in this region after intracerebroventricular injection of vMIP-2. The morphological, histochemical, and temporal patterns of activated microglia appearing in the CA1 after TFI suggest that the recruited microglial cells are derived from brain parenchyma adjacent to the hippocampal region (Morioka et al., 1991, 1992; Ábrahám and Lázár, 2000). However, we cannot exclude the possibility that microglia in the CA1 region represent blood-derived leukocyte infiltration and accumulation into ischemic lesions, because OX-42 does not differentiate between microglia, monocytes, and macrophages. MIP-1α expressed in CA1 neurons after TFI may be released, in turn acting as a chemoattractant for microglial cells with CCR5, a chemokine receptor for the protein.
Direct evidence for the deleterious role of chemokines in cerebral ischemia was obtained from studies showing that mice with genetic deletion of MCP-1 or fractalkine display a reduction in infarct size after focal cerebral ischemia (Hughes et al., 2002; Soriano et al., 2002). In addition, intracerebroventricular injection of vMIP-2, a broad-spectrum chemokine receptor antagonist, attenuated brain infarction after permanent occlusion of the middle cerebral artery, whereas MIP-1α injection exacerbated the infarction (Takami et al., 2001). In our experiments, microglial recruitment was inhibited by an intracerebroventricular injection of vMIP-2, which prevented neuronal cell loss in the CA1 region after TFI. Although the mechanisms of chemokine-mediated neuronal death are not well defined at present, previous studies on transient forebrain ischemia suggest that early production of inflammatory mediators in activated microglial cells, including IL-1β, TNF-α, and NO, may contribute to ischemic neuronal death (Sairanen et al., 1997; Uno et al., 1997; Yamashita et al., 2000). As a matter of fact, ischemic brain damage is attenuated by administration of anti-inflammatory drugs (Yrjanheikki et al., 1998; Arvin et al., 2002; Wang et al., 2003), but potentiated by maneuvers enhancing IL-1β, TNF-α, and NO (Barone et al., 1997; Dirnagl et al., 1999; Touzani et al., 1999; Zhao et al., 2003). We found free radical production in the CA1 neurons 2 d as well as 2 h after TFI (Park et al., 2005). Delayed administration of trolox 24 h after TFI markedly reduced the delayed neuronal death in the CA1 field. Thus, it is conceivable to hypothesize that TFI-induced microglial cell activation in the CA1 field may contribute to the delayed free radical production and neuronal death.
In conclusion, our study provides preliminary evidence that free radicals that are rapidly produced in CA1 neurons induce microglial activation after TFI. Moreover, free radical-mediated MIP-1α expression in CA1 pyramidal neurons contributes to microglial recruitment and neuronal cell death in the CA1 region. The finding that MIP-1α is expressed by neurons and activates microglia supports the novel concept that the protein acts as a molecular signal from neurons, contributing to neuronal–microglial communication in the early phase of forebrain ischemia.
Footnotes
This work was supported by the Korea Science and Engineering Foundation through the Brain Disease Research Center at Ajou University (Y.B.L., B.J.G.) and the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea Grant 03-PJ1-PG10-21300-0006 (Y.B.L.).
References
- Ábrahám H, Lázár G. Early microglial reaction following mild forebrain ischemia induced by common carotid artery occlusion in rats. Brain Res. 2000;862:63–73. doi: 10.1016/s0006-8993(00)02072-2. [DOI] [PubMed] [Google Scholar]
- Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52:54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW. Ischemia-induced neuronal expression of the microglia attracting chemokine secondary lymphoid-tissue chemokine (SLC) Glia. 2001;34:121–133. doi: 10.1002/glia.1047. [DOI] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Che X, Ye W, Panga L, Wu DC, Yang GY. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001;902:171–177. doi: 10.1016/s0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, Vogel SN. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003;23:748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Parent JM, Silverstein FS. Microglial expression of chemokine receptor CCR5 during rat forebrain development and after perinatal hypoxia-ischemia. J Neuroimmunol. 2006;173:155–165. doi: 10.1016/j.jneuroim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Flugel A, Hager G, Horvat A, Spitzer C, Singer GM, Graeber MB, Kreutzberg GW, Schwaiger FW. Neuronal MCP-1 expression in response to remote nerve injury. J Cereb Blood Flow Metab. 2001;21:69–76. doi: 10.1097/00004647-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Gourmala NG, Limonta S, Bochelen D, Sauter A, Boddeke HW. Localization of macrophage inflammatory protein: macrophage inflammatory protein-1 expression in rat brain after peripheral administration of lipopolysaccharide and focal cerebral ischemia. Neuroscience. 1999;88:1255–1266. doi: 10.1016/s0306-4522(98)00295-4. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002;22:308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Ishibashi N, Prokopenko O, Reuhl KR, Mirochnitchenko O. Inflammatory response and glutathione peroxidase in a model of stroke. J Immunol. 2002;168:1926–1933. doi: 10.4049/jimmunol.168.4.1926. [DOI] [PubMed] [Google Scholar]
- Ivacko J, Szaflarski J, Malinak C, Flory C, Warren JS, Silverstein FS. Hypoxic-ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J Cereb Blood Flow Metab. 1997;17:759–770. doi: 10.1097/00004647-199707000-00006. [DOI] [PubMed] [Google Scholar]
- Kim DY, Won SJ, Gwag BJ. Analysis of mitochondrial free radical generation in animal models of neuronal disease. Free Radic Biol Med. 2002;33:715–723. doi: 10.1016/s0891-5849(02)00968-1. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Niinobe M, Mikoshiba K, Hata R, Ueda H, Handa N, Fukunaga R, Isaka Y, Kimura K. Microtubule-associated protein 2 as a sensitive marker for cerebral ischemic damage-immunohistochemical investigation of dendritic damage. Neuroscience. 1989;31:401–411. doi: 10.1016/0306-4522(89)90383-7. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lüttichau HR, Gerstoft J, Schwartz TW. MC148 encoded by human molluscum contagiosum poxvirus is an antagonist for human but not murine CCR8. J Leukoc Biol. 2001;70:277–282. [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1991;11:966–973. doi: 10.1038/jcbfm.1991.162. [DOI] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathol (Berl) 1992;83:149–157. doi: 10.1007/BF00308474. [DOI] [PubMed] [Google Scholar]
- Nishi T, Maier CM, Hayashi T, Saito A, Chan PH. Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:1312–1324. doi: 10.1038/sj.jcbfm.9600124. [DOI] [PubMed] [Google Scholar]
- Park U, Lee Y, Kim K, Yoon S, Gwag B. Iron-mediated ROS production causes degeneration of the hippocampal CA1 neurons following the transient global ischemia: delayed neuroprotection with NEU2000. Soc Neurosci. 2005 Abstr 31:97.9. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sairanen TR, Lindsberg PJ, Brenner M, Siren AL. Global forebrain ischemia results in differential cellular expression of interleukin-1beta (IL-1beta) and its receptor at mRNA and protein level. J Cereb Blood Flow Metab. 1997;17:1107–1120. doi: 10.1097/00004647-199710000-00013. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Hamada J, Shibata M, Gotoh J, Araki N, Fukuuchi Y. FK506 abrogates delayed neuronal death via suppression of nitric oxide production in rats. Brain Res. 2004;1009:34–39. doi: 10.1016/j.brainres.2004.01.088. [DOI] [PubMed] [Google Scholar]
- Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- Takami S, Minami M, Nagata I, Namura S, Satoh M. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1430–1435. doi: 10.1097/00004647-200112000-00007. [DOI] [PubMed] [Google Scholar]
- Touzani O, Boutin H, Chuquet J, Rothwell N. Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J Neuroimmunol. 1999;100:203–215. doi: 10.1016/s0165-5728(99)00202-7. [DOI] [PubMed] [Google Scholar]
- Uno H, Matsuyama T, Akita H, Nishimura H, Sugita M. Induction of tumor necrosis factor-alpha in the mouse hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:491–499. doi: 10.1097/00004647-199705000-00002. [DOI] [PubMed] [Google Scholar]
- Wang CX, Yang T, Shuaib A. Effects of minocycline alone and in combination with mild hypothermia in embolic stroke. Brain Res. 2003;963:327–329. doi: 10.1016/s0006-8993(02)04045-3. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Kataoka Y, Sakurai-Yamashita Y, Shigematsu K, Himeno A, Niwa M, Taniyama K. Involvement of glial endothelin/nitric oxide in delayed neuronal death of rat hippocampus after transient forebrain ischemia. Cell Mol Neurobiol. 2000;20:541–551. doi: 10.1023/A:1007007710703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ross ME, Iadecola C. L-Arginine increases ischemic injury in wild-type mice but not in iNOS-deficient mice. Brain Res. 2003;966:308–311. doi: 10.1016/s0006-8993(02)04223-3. [DOI] [PubMed] [Google Scholar]