

Abstract
Behaviorally relevant stimuli prompt midbrain dopamine (DA) neurons to switch from tonic to burst firing patterns. Similar shifts to burst activity are thought to contribute to the addictive effects of opiates and nicotine. The nucleus accumbens DA overflow produced by these drugs is a key element in their pathological effects. Using electrochemical techniques in brain slices, we explored the effects of opioids on single-spike and burst stimuli-evoked DA overflow in the dorsal and ventral striatum. In specific subregions of the nucleus accumbens, μ-opioids inhibit DA overflow elicited with single-spike stimuli while leaving that produced by burst stimuli unaffected. This is similar to published effects of nicotinic receptor blockade or desensitization, and is mediated by opioid receptor-induced inhibition of cholinergic interneurons. Whereas δ-opioids have similar effects, κ-opioids inhibit evoked DA overflow throughout the striatum in a manner that is not overcome with high-frequency stimuli. These observations reveal remarkable mechanistic overlap between the effects of nicotine and opiates within the dopamine reward pathway.
Keywords: opiate, addiction, electrochemistry, voltammetry, acetylcholine, cholinergic
Introduction
Midbrain dopamine (DA) neurons fire action potentials at low frequency (∼4 Hz) during quiescent periods and transition to burst firing patterns in response to rewarding or reward-predicting stimuli (Grace and Bunney, 1984; Hyland et al., 2002; Dahan et al., 2007). These tonic and phasic activity patterns make distinct contributions to both extracellular DA levels and behavior (Schultz, 2007). Specifically, because DA transporters (DATs) can be overwhelmed and saturated by high DA release rates, burst firing patterns make more significant contributions to extrasynaptic DA concentrations than single-spike activity (Gonon, 1988; Chergui et al., 1994). Behaviorally, tonic firing patterns are associated with motivational state and phasic firing patterns are associated with discrete learning events (Cagniard et al., 2006; Schultz, 2007).
Although the role of tonic and phasic firing patterns to the pathology of drug addiction is unclear, the addictive nature of many drugs of abuse has been linked to the enhancement of extracellular nucleus accumbens (NAc) DA levels (Di Chiara and Imperato, 1988). Opiates and nicotine enhance extracellular DA primarily by inducing burst firing of ventral tegmental area (VTA) neurons (Grenhoff et al., 1986; Kiyatkin and Rebec, 2001; Erhardt et al., 2002), but there is also evidence for direct actions of these drugs in the NAc (Nisell et al., 1994; Hirose et al., 2005). It was demonstrated previously throughout the dorsal and ventral striatum that nicotine differentially alters the DA overflow produced by tonic and phasic activity patterns (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). By desensitizing nicotinic acetylcholine receptors (nAChRs), nicotine inhibits the DA overflow elicited with low-frequency firing patterns, although the impact of burst firing activity is either augmented or unchanged.
Within the striatum, opioid receptors are expressed both presynaptically and postsynaptically on glutamatergic, GABAergic, dopaminergic, and cholinergic afferents (Gerfen, 1992; Svingos et al., 1999, 2001a,b; Williams et al., 2001). Although the overall influence of striatal opioid receptors is complex, we have addressed their effects on DA overflow elicited by single-spike and burst stimuli. Our findings show that in subregions of the striatum, particularly within the NAc, μ- and δ-opioids produce effects similar to nAChR disruption, inhibiting DA overflow produced with single-spike activity. This inhibition can be overcome with burst stimulation and, at least for μ-opioids, is mediated via cholinergic interneuron inhibition. κ-Opioids profoundly inhibit single-spike-evoked DA overflow throughout the striatum, but this inhibition is more absolute and cannot be overcome by high-frequency stimuli.
Materials and Methods
Slice preparation.
Brain slices from adult [postnatal day 60 (P60)–P90] Sprague Dawley rats were used for all electrochemical experiments. For investigating the electrophysiology of striatal cholinergic interneurons, young rats (P20–P25) were used to optimize cell identification and viability of these cells. The rats were anesthetized with isoflurane (Abbott Laboratories, North Chicago, IL) and aortically perfused (Martin et al., 2006) with 25 ml of cold, sucrose-artificial CSF (ACSF) containing the following (in mm): 200 sucrose, 25 NaHCO3, 20 glucose, 10 ascorbic acid, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, and 1 NaH2PO4, pH 7.4, saturated with 95% O2 and 5% CO2 (Fagen et al., 2007). They were then rapidly decapitated and their brains were removed into the same solution. The brains were sliced into 300-μm-thick coronal slices that included the shell region of the NAc on a vibratome (VT100S; Leica, Nussloch, Germany). Slices were incubated for at least 1 h in bath circulated at 20 ml/min with normal, 32°C ACSF containing (in mm) 125 NaCl, 25 NaHCO3, 20 glucose, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, 1 NaH2PO4, and 1 ascorbic acid, pH 7.4, saturated with 95% O2 and 5% CO2 (Fagen et al., 2007). During recording, slices were superfused (2 ml/min) with this same ACSF at ∼34°C, but without the ascorbic acid, because it oxidizes within the voltage range tested and interferes with the dopamine signal. Slices were visualized using an upright microscope (Axioskop; Carl Zeiss, Thornwood, NY).
Electrochemistry.
Carbon fiber microelectrodes were fabricated by threading a 7 μm diameter carbon fiber (Fortafil Fibers, Knoxville, TN) through borosilicate glass capillary tubing (G150-4, Warner Instruments, Hamden, CT) that was then pulled on a Flaming/Brown micropipette puller (Model P-97; Sutter Instruments, Novato, CA) as described previously (Zhang and Sulzer, 2004). The carbon fiber was subsequently cut to restrict the length of the exposed fiber in the slice to ∼50 μm. Recording electrodes were placed in the NAc shell ∼150 μm from a bipolar stimulating electrode with a 250 μm tip separation (MS 303/3; Plastics One, Roanoke, VA). Dopamine release was evoked by single-pulse stimulations (400 μA, 1–6 V, 500 μs) delivered every 2 min with an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Currents were recorded using either a Mulitclamp 700A amplifier (Molecular Devices, Sunnyvale, CA) or an NPI VA-10X amplifier (ALA Scientific, Westbury, NY), both with a DigiData 1322A interface and pClamp 9.2 software (Molecular Devices). For fast-scan cyclic voltammetry (FSCV), the electrode voltage was ramped from the baseline voltage of −400 mV to + 1000 mV and then back again at 200–400 V/s versus Ag/AgCl at 100 ms intervals. Current was filtered at 10 kHz and digitized at 50 kHz. Background-subtracted FSCV was used to visualize oxidation and reduction currents, allowing for identification of the oxidized substance and for calibration with 5 μm dopamine at the end of each experiment. For amperometry, a constant voltage of +400 mV was applied using the same equipment and software. Amperometric traces were filtered at 1 kHz, digitized at 2.5 kHz and then digitally filtered at 100 Hz. All drugs were obtained from Tocris (Ellisville, MO) except for nicotine, which was obtained from Sigma (St. Louis, MO). None of the drugs used were electroactive at the relevant concentrations across the voltage range used.
Electrophysiology.
Recordings were obtained with pipettes made from borosilicate glass. On cell recording pipettes were filled with the ACSF described above and whole-cell, ruptured patch recording pipettes were filled with an internal solution containing the following (in mm): 154 K-gluconate, 1 KCL, 1 EGTA, 10 HEPES, 10 glucose, 5 ATP, and 0.1 GTP, and brought to a pH of 7.4 with KOH (Mansvelder and McGehee, 2000). Currents were recorded using a Mulitclamp 700A amplifier with a DigiData 1322A interface and pClamp 9.2 software (Molecular Devices). Cholinergic interneurons were identified by their large size, baseline firing rate and, in those cells where a rupture patch was obtained, the presence of an Ih current (Zhou et al., 2002). Action potential frequency was monitored in current-clamp mode in either on-cell or whole-cell recordings. Action potential frequency was monitored off-line by threshold detection of each spike using MiniAnalysis software (Synaptosoft, Decatur, GA). In the voltage-clamp studies, cholinergic interneurons were held at −60 mV. The current–voltage relationship plot was obtained by subtracting the currents elicited from a voltage ramp that went from −120 to −60 mV over 5 s before and after a bath application of 1 μm endomorphin-1 (EM-1).
Statistics and curve-fitting.
Paired t tests were used to compare one-, two-, four-, or six-pulse evoked DA levels in the presence and absence of drugs. ANOVA was used to compare the paired pulse ratios in the presence and absence of drugs. Statistical significance was determined by p < 0.05. All compiled results are presented as the mean ± SEM. Curve-fitting of concentration effect relationship for EM-1 effects on single-spike-evoked DA overflow was performed by SigmaPlot (SPSS, Chicago, IL). Curve-fitting of DA oxidation current decay was performed using MiniAnlysis software (Synaptosoft).
Results
μ-Opioids and δ-opioids inhibit low-frequency evoked DA overflow
Axon-terminal DA overflow was measured using fast-scan cyclic voltammetry in 300-μm-thick coronal slices from adult rats (P60–P90). A 7-μm-diameter carbon fiber microelectrode and a bipolar stimulating electrode were placed ∼150 μm apart in several areas throughout the dorsal and ventral striatum. Immediately after placing the carbon fiber recording electrode in the slice, large DA oxidation currents were observed, which declined after 1–10 stimulations to highly reproducible baseline currents evoked with single 0.4 mA pulses every 2 min. These DA oxidation currents are TTX-sensitive, Ca2+-dependent, and, as demonstrated in mice, are substantially altered by genetic manipulations to the DA transporter (Cagniard et al., 2006).
Throughout the striatum, we tested the effects of μ-opioid receptor activation on evoked DA overflow. In ∼47% of recordings from the NAc shell, a saturating concentration of EM-1 (1 μm, Harrison et al., 1998) inhibited peak dopamine oxidation currents elicited by single stimulus pulses to 41 ± 3% of control (t = 17.6, p < 0.001, n = 24) (Fig. 1a). This inhibition was largely reversed by subsequent application of a μ-opioid receptor specific antagonist, naloxonazine (1 μm), which had no effect on evoked DA overflow in recordings where no effect of EM-1 was observed. In addition, application of naloxonazine alone had no effect on evoked DA overflow (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). We then tested EM-1 effects on DA overflow evoked with 25 Hz burst stimuli, which was chosen to mimic the burst frequency exhibited by rat DA neurons in vivo (Grace and Bunney, 1984; Hyland et al., 2002). Burst stimulation with four or six pulses elicited DA oxidation currents that were similar before and after EM-1 exposure (Fig. 1b). Higher-frequency stimuli (100 Hz) did not significantly augment DA overflow beyond that produced with 25 Hz (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material) and 100 Hz bursts also induced similar magnitude oxidation currents in control and with μ-opioid agonists (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material). Experiments using other μ-opioid agonists, including EM 2, produced comparable effects on DA overflow to that obtained with EM-1 (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material).
Figure 1.

μ- and δ-opioid receptor agonists inhibit single-spike-evoked DA overflow. a, Single-spike-evoked DA responses constructed using fast-scan cyclic voltammetry before and after successive 10 min bath applications of the μ-opioid EM-1 (1 μm) and the μ-opioid receptor antagonist naloxonazine (1 μm). The insets are subtracted voltammograms obtained at the peak of evoked DA responses in each condition. b, Summary of the effect of 1 μm EM-1 on evoked DA responses in the dorsal medial NAc shell (n = 24). Each data point reflects a single stimulus pulse, unless noted by 2P, 4P, or 6P, indicating two, four, or six stimulus pulses, respectively, delivered at 25 Hz (in this and subsequent figures). Average DA concentration elicited by single spikes during control conditions was normalized to 1 for each recording. EM-1 significantly inhibited single-spike-evoked DA overflow (t = 17.6, p < 0.001, n = 24) and naloxonazine induced significant recovery (t = −12.1, p < 0.001, n = 24). Baseline single-spike dopamine levels during control averaged 0.59 ± 0.10 μm. c, The difference between DA release from burst and single stimuli were determined by subtracting the peak DA overflow responses to single spikes from that measured after two, four, or six stimuli at 25 Hz. Statistical comparison by repeated-measures ANOVA revealed significant effects of pulse number (F(2,69) = 25.6, p < 0.0001), EM-1 (F(1,69) = 31.1, p < 0.0001), and an interaction (F(2,69) = 4.4, p = 0.0157). d, Summary of the effect of 1 μm deltorphin II, a δ-opioid receptor agonist, followed by 1 μm BNTX, an δ1-opioid receptor antagonist, on evoked DA responses in the NAc shell (n = 6). The 25 Hz burst stimuli of two, four, or six stimulus pulses are indicated. Deltorphin II induced significant inhibition of single-spike-evoked DA overflow (t = 8.9, p < 0.001, n = 6) and BNTX induced significant recovery (t = −2.5, p < 0.05, n = 6).
When DA overflow induced by low-frequency tonic activity is suppressed in the presence of μ-opioid agonists, the effects of phasic activity are comparatively larger. To represent this contrast, we plotted the difference between peak responses to tonic and phasic stimulation for two, four, and six pulses (Fig. 1c). This measure reflects the idea that the impact of burst firing patterns on DA overflow must be measured against that produced by single, low-frequency stimuli.
In additional experiments, we examined δ-opioid receptor effects on evoked DA overflow throughout the striatum. In ∼55% of recordings collected from the NAc shell, a saturating concentration of deltorphin II (1 μm) that is selective for δ-opioid receptors (Watson and Lanthorn, 1993; Chen et al., 2000) produced effects similar to that obtained with μ-opioids (Fig. 1d). In these recordings, the DA overflow elicited from single pulse stimuli was inhibited to 45 ± 7% of control (t = 8.9, p < 0.001, n = 6), but four- and six-pulse stimuli overcame this inhibition and produced similar oxidation currents. The inhibition was partially reversed by application of the δ1-opioid receptor anatagonist 7-benzylidenenaltrexone hydrochloride (BNTX) (1 μm) (Fig. 1d), which had no effects on its own (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material). The lack of effect of the opioid antagonists on DA overflow induced by single or burst stimulation suggests that endogenous opioid release is low in our slice preparation. This is consistent with previous observations that medium spiny neurons (MSNs) do not fire spontaneously in slice preparations.
The μ- and δ-opioid receptor effects on DA overflow are similar to those seen when nAChRs are blocked or desensitized in the NAc (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). Consistent with these observations, application of low concentrations of nicotine (1 μm) created comparable effects to those obtained with μ- or δ-opioid agonists (Fig. 2a). As in previous studies, the nicotine effect was caused by desensitization of nAChRs, as similar effects were seen with nicotinic antagonists (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 2.
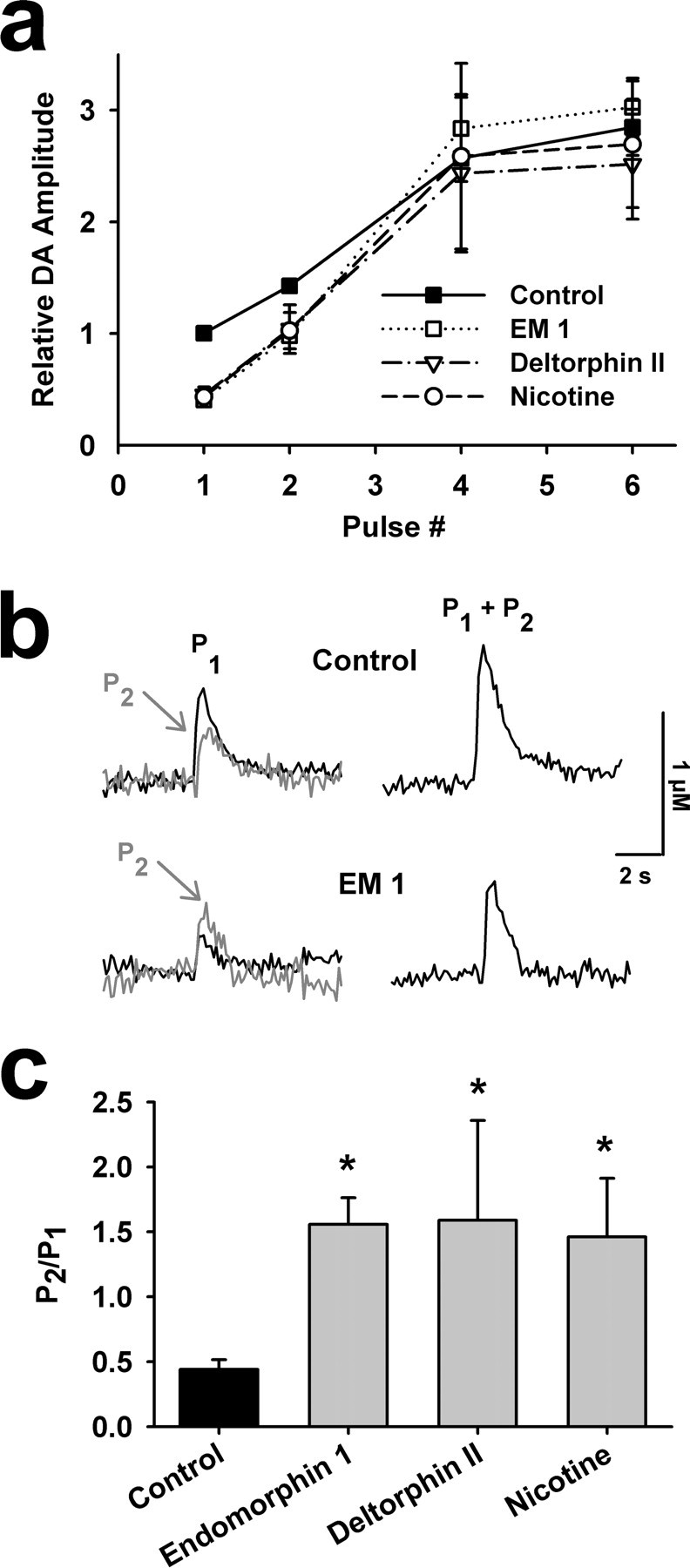
μ-Opioid receptor effects on single and burst stimulation induced DA overflow, as well as short-term presynaptic facilitation of DA overflow. a, Effect of the number of 25 Hz stimulus pulses on peak DA overflow under control conditions and after treatment with EM-1, deltorphin II, or nicotine (n = 24, 24, 6, 8, respectively). b, DA responses to single and 25 Hz paired pulses demonstrate EM-1-induced transformation of paired-pulse depression to paired-pulse facilitation. P2 is calculated by subtracting P1 from P1 + P2. c, Paired-pulse ratios in control conditions and in the presence of 1 μm EM-1, deltorphin II, or nicotine (n = 24, 6, 8, respectively). A one-way ANOVA followed by Fisher's PLSD post hoc test found the control condition was significantly different from each drug condition (F(3,57) = 6.7, *p < 0.02).
nAChRs on DA neuron axon terminals promote transmitter release by activating Ca2+ conductances (Zhang and Sulzer, 2004). Consequently, blocking or desensitizing these receptors will diminish Ca2+ influx and lower the probability of transmitter release. The impact of this effect can be observed as an increase in the paired-pulse ratio of evoked dopamine overflow (Rice and Cragg, 2004; Zhang and Sulzer, 2004). We similarly examined paired-pulse ratios in the presence and absence of the μ-opioid agonist EM-1. EM-1 converted the paired-pulse depression observed in control conditions to facilitation (Fig. 2b,c), which is consistent with a decrease in release probability (Dobrunz and Stevens, 1997). We found that the change in paired-pulse ratio induced by μ- and δ-opioid agonists in the NAc shell were comparable with the effects of nicotine (Fig. 2c). These data suggest similar or overlapping mechanisms of action between these drugs.
In contrast to effects of nicotine on DA overflow, which was observed throughout the dorsal and ventral striatum, responses to μ- and δ-opioid agonists were much less prevalent. Our first hypothesis was that the sporadic nature of the μ-opioid effects might correspond to the patchy expression of μ-opioid receptors seen throughout the striatum, which has been used to delineate the patch-matrix divisions (Gerfen, 1992). However, EM-1 had no effect in 12 recordings in the dorsal striatum (Fig. 3a,b). Likewise, only one recording of 19 in the NAc core showed significant effects with μ-opioid agonists (Fig. 3a,b). Within the NAc shell, however, we identified a region that consistently exhibited μ-opioid effects on DA overflow. Within the dorsal half of the NAc shell and between 2.0 and 3.0 mm rostral to bregma, drug-induced effects were found in ∼89% of recordings (Fig. 3a,d). This area roughly corresponds to an area recently identified as a being important for the effects of μ-opioid agonists on the hedonic value of food (Pecina and Berridge, 2005). Although we found considerable overlap between the region with high μ-opioid response prevalence and this “hot spot” described by Pecina and Berridge (2005), it is important to note that our discrete point measurement do not provide the same spatial resolution as the c-fos staining used in that study.
Figure 3.
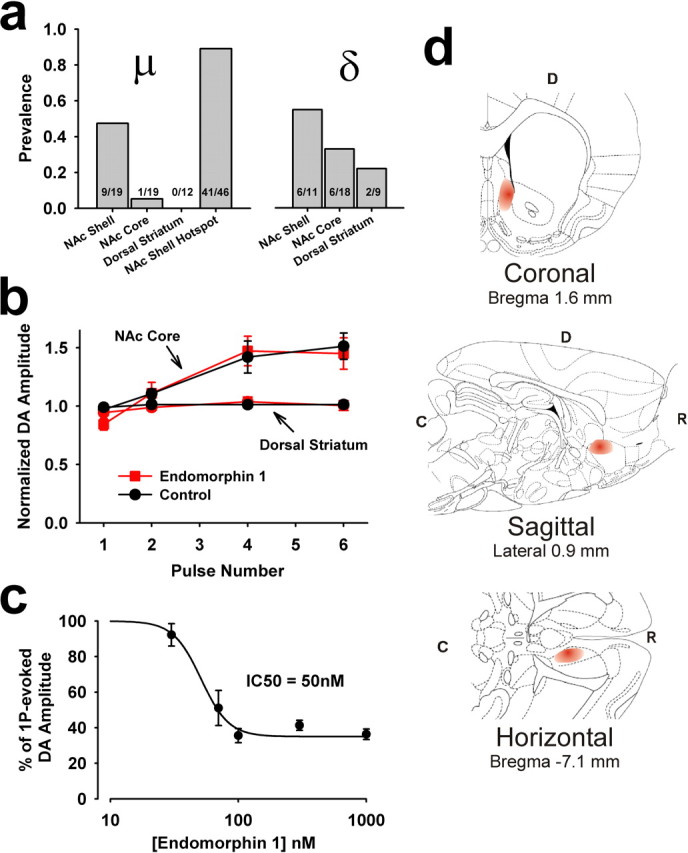
μ- and δ-opioid responses are not uniform throughout the striatum. a, The prevalence of μ- and δ-opioid effects on DA responses in the NAc shell, NAc core, and the dorsal striatum. The highest prevalence of the μ-opioid effect was seen in a subregion of the NAc shell, identified here as the “hotspot,” which is located in the dorsal half of the NAc shell between 2 and 3 mm rostral to bregma. b, In the NAc core and dorsal striatum, EM-1 is ineffective in modulating evoked DA overflow (n = 19 and 12, respectively). Note that in these areas, burst stimuli only slightly augment the DA overflow over that produced with single pulses. Baseline single-spike dopamine levels during control averaged 1.57 ± 0.28 μm in the NAc Core, and 2.50 ± 0.24 μm in the dorsal striatum. c, Concentration–effect relationship for the effects of EM-1 on single-spike-evoked DA overflow from recordings in the NAc shell hotspot region (n = 4–15). d, Schematic of the NAc hot spot, where EM-1 most consistently affected DA overflow. The red portion of each diagram denotes the hot spot region. D, Dorsal; R, rostral; C, caudal. Adapted from Paxinos and Watson (1998).
We took advantage of the high prevalence of response in this area to investigate the mechanism of action of μ-opioids on DA overflow. The effect appears to be μ-opioid receptor-specific because the inhibition mostly recovers after application of a specific μ-opioid antagonist. In addition, the IC50 is ∼50 nm (Fig. 3c), which is similar to the potency of EM-1 on μ-opioid receptor effects in SH-SY5Y human neuroblastoma cells (Harrison et al., 1998). The drugs actions are likely to be indirect, however, because μ-opioid receptors are not expressed on DA axon terminals (Trovero et al., 1990).
The robust similarities between the effects on evoked DA overflow elicited by μ-opioid agonists and nicotine suggested a common mechanism of action. Consistent with this idea, nicotine produced negligible effects on DA release when applied subsequent to a saturating dose of EM-1 (Fig. 4a). Thus, we hypothesized an indirect action where μ-opioids altered evoked DA overflow by reducing the local, ambient ACh concentrations (Trovero et al., 1990; Svingos et al., 2001a). To test this, we measured the effects of μ-opioid agonists on firing rate of cholinergic interneurons located within the region of the NAc shell where DA overflow was most responsive to these drugs. Cholinergic interneurons were identified by their large soma diameter and baseline firing activity. In extracellular recordings, EM-1 robustly inhibited the firing rate of these cells, with several recordings exhibiting a complete cessation of action potentials in the presence of the drug (Fig. 4b). The firing rate recovered after application of the μ-opioid specific antagonist naloxonazine. Whole-cell patch recordings from these neurons showed similar effects (Fig. 4c), although a gradual decrease in the firing rate under control conditions was observed in most of the whole-cell experiments. Voltage-clamp studies from cholinergic interneurons revealed an EM-1-induced outward current at a holding potential of −60 mV (Fig. 4d). Investigation of the current–voltage relationship indicates an inwardly rectifying current with a reversal potential near K+ Nernst potential (Fig. 4e), suggesting activation of a GIRK (G-protein-gated potassium) conductance. These inhibitory effects likely contribute to μ-opioid receptor mediated inhibition of ACh release. It is important to note that opioid receptors on cholinergic axon terminals may also contribute to reduced ACh release, although this was not investigated.
Figure 4.

μ-Opioids modulate DA responses by inhibiting cholinergic interneuron activity. a, A saturating concentration of EM-1 (1 μm) occludes further modulation of DA overflow by nicotine. Peak DA responses elicited by single pulses or, where labeled, 25 Hz burst stimuli showing that nicotine (1 μm) is not effective in modulating evoked DA overflow when applied subsequent to EM-1. b, On-cell electrophysiological recordings revealed that EM-1 (1 μm) suppresses the firing rate of cholinergic interneurons in the NAc shell hotspot region (n = 6). Frequency data from each cell was normalized to baseline for averaging (average baseline firing rate, 3.9 ± 1.7 Hz). c, An example whole-cell recording from a cholinergic interneuron in the NAc shell hotspot region before and after a 2 min bath application of 1 μm EM-1. d, Whole-cell voltage-clamp recordings from cholinergic interneurons in the NAc shell hotpot region reveal that EM-1 produces an outward current. Top, Example trace from a single cell. Bottom, Summary data (n = 5). e, Voltage ramps from −120 to −60 mV were applied during current response to EM-1 in cholinergic interneurons from the NAc shell hotspot. Currents were averaged and the difference current was obtained by subtracting control from EM-1 to produce the current–voltage relationship for each cell. The largest inward current was normalized to −1 and the traces were averaged across all cells. The reversal potential (−104 ± 10 mV) was similar to calculated Nernst potential for potassium (−107 mV; n = 5).
κ-Opioid receptor activation inhibits DA release presynaptically
As shown above, μ- and δ-opioids as well as nicotine have similar effects on dopamine overflow in the NAc, and all three classes of drug enhance NAc shell DA levels and are readily self-administered in rats (Goldberg and Henningfield, 1988; Pentney and Gratton, 1991; Devine and Wise, 1994; Yoshida et al., 1999; You et al., 1999; Ferrari et al., 2002; Hirose et al., 2005; Okutsu et al., 2006). In light of these observations, we were interested in assessing the effects of κ-opioid agonists on evoked DA overflow (Schmidt et al., 2002; Woolley et al., 2007). Both rats and humans find κ-opioids aversive and in microdialysis studies they inhibit DA levels in the NAc shell in rats (Pfeiffer et al., 1986; Heijna et al., 1992; Ronken et al., 1993; You et al., 1999; Thompson et al., 2000; Walsh et al., 2001).
We tested the effects of κ-opioids on evoked DA overflow throughout the dorsal and ventral striatum. Bath application of a saturating concentration of BRL52537 (1 μm) (Dortch-Carnes and Potter, 2002), a specific κ-opioid agonist, significantly inhibited DA overflow evoked with single stimulus pulses in every recording (Fig. 5a). Application of the specific κ-opioid receptor antagonist nor-binaltorphimine dihydrochloride (nBI; 1 μm) mostly restored the DA signal, but alone had no direct effect on DA overflow (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material). The inhibition produced by κ-opioids, to 28 ± 5% of control, was comparable in magnitude to that produced with μ- or δ-opioids, but in contrast it could not be overcome with high-frequency burst stimuli (Fig. 5b). In fact, the impact of high-frequency bursts relative to single pulse stimuli on DA overflow was similar between control and κ-opioid conditions (Fig. 5c). The averaged responses to burst stimuli were smaller because of the pooling of data from shell and core, which had similar κ-opioid effects. The paired-pulse ratio of transmitter release also did not differ in the presence of the drug (0.11 ± 0.06 vs 0.12 ± 0.16 for control and drug treatment, respectively; t = −0.06, p = 0.95, n = 10)
Figure 5.
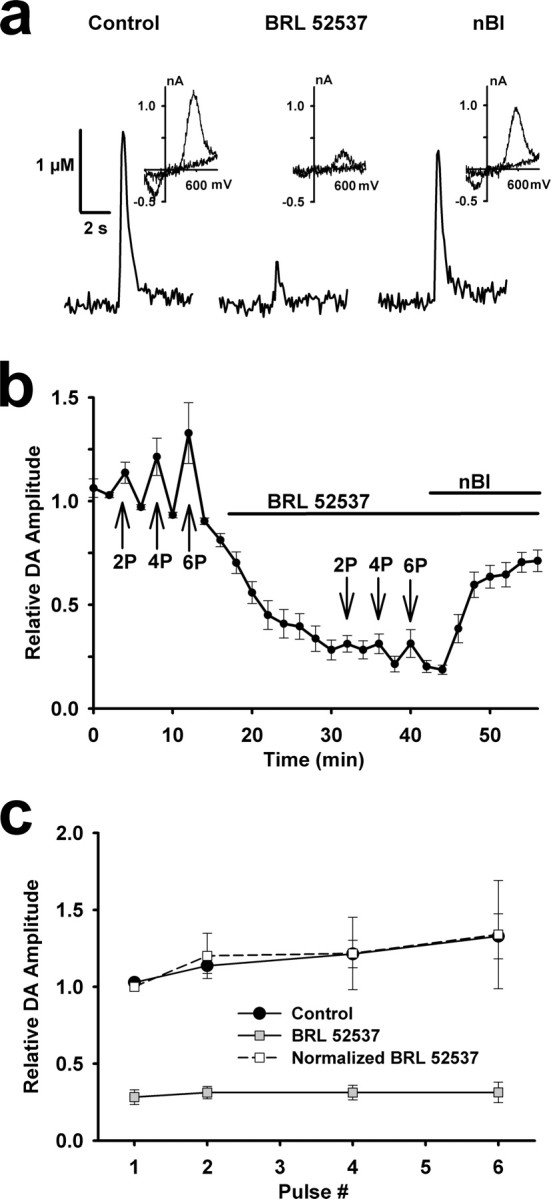
κ-Opioid receptor activation uniformly inhibits evoked DA overflow. a, Single-spike-evoked DA responses constructed using fast-scan cyclic voltammetry before and after successive 10 min bath applications of a κ-opioid receptor agonist, BRL52537 (1 μm), and antagonist nBI (1 μm). The insets are subtracted voltammograms obtained at the peak of evoked DA responses in each condition. b, Summary of the effect of 1 μm BRL52537 on evoked DA responses in recordings throughout the NAc (n = 10). Each data point reflects a single stimulus pulse, unless noted by 2P, 4P, or 6P, indicating two, four, or six stimulus pulses, respectively, delivered at 25 Hz. BRL52537 inhibited single-spike-evoked DA overflow (t = 14.2, p < 0.001, n = 10) and nBI induced recovery (t = −8.2, p < 0.001, n = 10). Baseline single-spike dopamine levels during control averaged 1.17 ± 0.17 μm. c, Effect of number of 25 Hz stimulus pulses on peak DA responses under control and BRL52537-treated conditions. To compare the relative effects of multiple stimuli on DA overflow, single-spike-evoked DA levels in the presence of BRL52537 were normalized to 1.
κ-Opioid receptors are located on DA axon terminals (Svingos et al., 2001b), so their influence on DA overflow is likely to be direct and completely independent of nAChR function. To confirm this, we bath applied BRL52537 subsequent to a saturating concentration of nicotine. To accurately assay any effects of BRL52537 on the already suppressed DA levels in the presence of nicotine, we used amperometry, which has a greater DA sensitivity relative to voltammetry. BRL52537 inhibited DA overflow to a similar degree after nicotine pretreatment, indicating that nAChR activity does not contribute to κ-opioid modulation of DA release (Fig. 6a).
Figure 6.
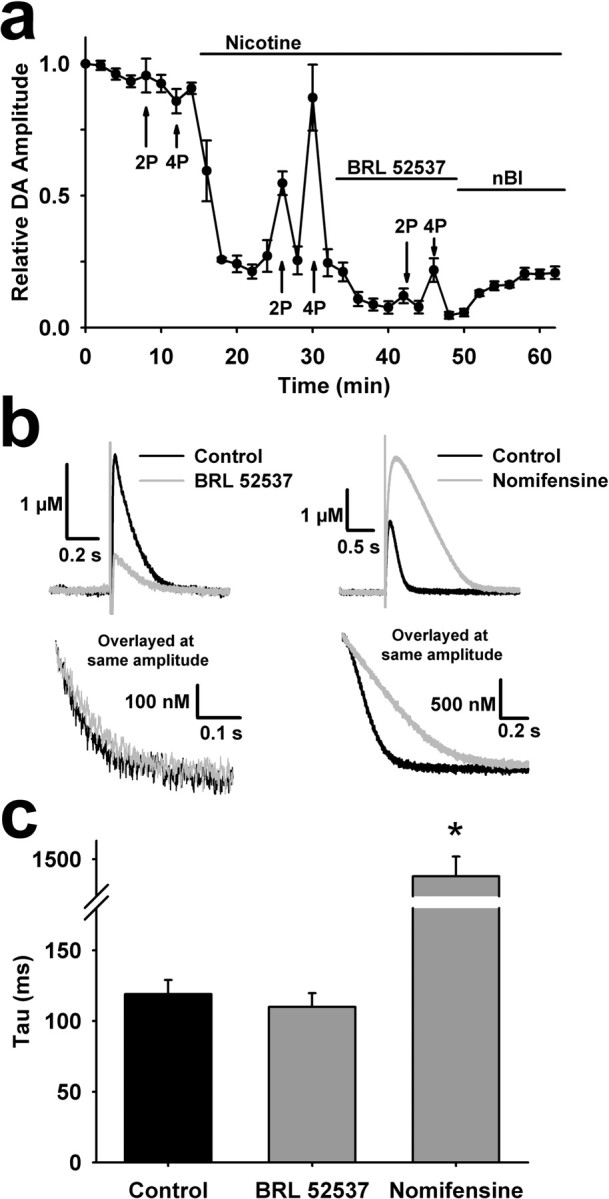
Effects of κ-opioids on DA overflow are independent of nAChR function and DA reuptake. a, A saturating concentration of nicotine (1 μm) does not occlude further modulation of DA overflow by κ-opioid receptor activation. Peak DA responses elicited by single pulses or, where labeled, 100 Hz burst stimuli from amperometric recordings show that BRL52537 (1 μm) modulates evoked DA overflow despite a preceding application of nicotine (n = 5). Amperometric recordings have faster kinetics and the stimulus artifact leads to underestimation of the peak magnitude during burst stimuli. b, Representative amperometric traces obtained with single pulse stimuli in the presence and absence of BRL52537 (1 μm) and the DAT inhibitor nomifensine (1 μm) (top). To assess drug-induced changes in decay rates, the portion of the currents with similar magnitudes were overlayed (bottom). c, Summary of the effects of BRL52537 (n = 8) and nomifensine (n = 3) on DA clearance rates, measured by fitting the current decay with a single exponential (τ). *p < 0.001.
We then tested whether the κ-opioid effects were mediated by alterations to DAT function, as κ-opioid receptor activation in the NAc shell can augment DAT activity (Chefer et al., 2005, 2006). Although this effect develops after an hour of drug exposure, we wanted to test its possible contribution to the changes in evoked DA release measured in our experiments. To assess possible differences in uptake we examined DA clearance rates, as reflected in the τ value obtained by fitting the decay of the amperometric oxidation current with a single exponential. The decay time constants of these currents, obtained before and after BRL52537 application (1 μm) were almost identical (119 ± 10 ms vs 110 ± 10 ms, t = 0.65, p = 0.52, n = 8). These findings contrasted sharply with the effects of 1 μm nomifensine, which dramatically prolonged the amperometric current decay (1409 ± 106 ms, n = 3, t = −21.37, p < 0.001) (Fig. 6b,c). Overlaying the amperometric traces obtained in the presence and absence of both BRL52537 and nomifensine clearly demonstrates no effect on dopamine clearance rates by brief application of this κ-opioid agonist (Fig. 6b,c).
Discussion
In response to behaviorally relevant stimuli, midbrain DA neurons switch from slow, single-spike firing patterns to pronounced brief bursts of activity (Dahan et al., 2007; Schultz, 2007). These two modes of activity differentially contribute to extrasynaptic DA concentrations and have also been associated with distinct behaviors (Gonon, 1988; Chergui et al., 1994; Cagniard et al., 2006; Schultz, 2007). Our results demonstrate that in certain areas of the striatum, μ- and δ-opioids selectively inhibit the DA overflow produced by single-spike activity. For μ- and very likely the δ-opioids, this effect is indirect, mediated by an inhibition of cholinergic interneuron activity (Fig. 7). The data support the idea that suppression of cholinergic interneurons inhibits ACh release, which decreases nAChR activation. The endpoint is diminished current flux through the nAChRs on DA terminals, and is similar to that obtained by blocking or desensitizing nAChRs directly (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). This loss of excitatory drive and Ca2+ influx in the DA axon terminal lowers DA release probability, which is reflected in an increased paired-pulse ratio of DA release. Thus, burst stimuli can overcome the suppression of single-spike-evoked DA overflow. These effects contrast those produced by κ-opioid receptor activation, which inhibits the DA overflow produced from single-spike and burst activity equally. The paired-pulse ratio is not affected by κ-opioid receptor agonists, so burst stimuli do not overcome the inhibition (Fig. 7). Although the mechanisms underlying this modulation remain unclear, the lack of recovery of release with high-frequency stimulation suggests a modification of the molecular interactions of vesicular trafficking or fusion (Ford et al., 2007).
Figure 7.
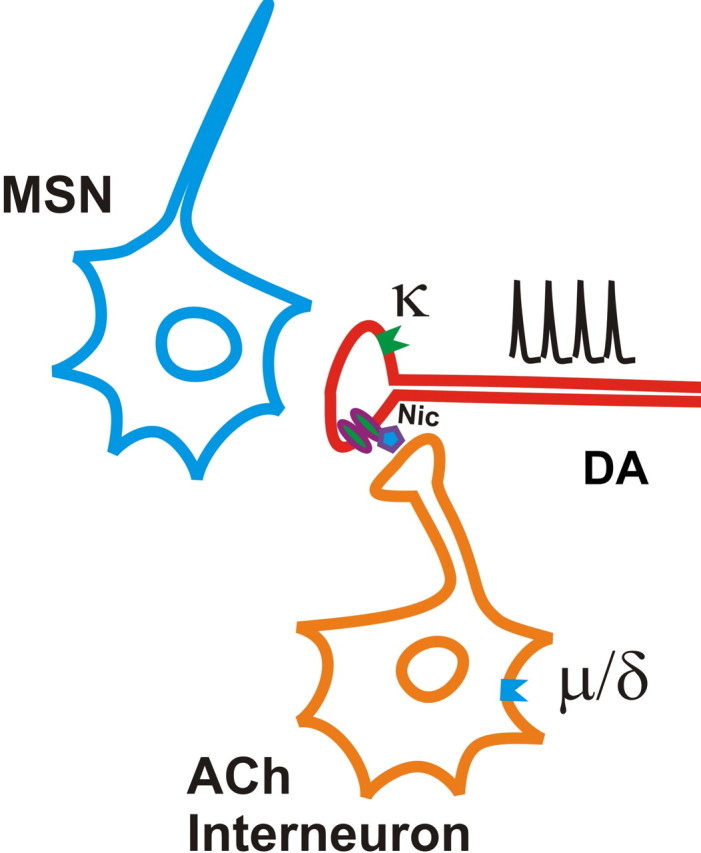
Schematic representation of opioid receptor modulation of DA release in the NAc shell. Activation of μ-opioid receptors on cholinergic interneurons suppresses ACh release and, in turn, decreases nAChR activity on DA terminals. κ-Opioid receptors are located directly on DA terminals where they suppress DA release directly, but their downstream effectors are unknown.
In the NAc shell, as well as other regions of the striatum, μ-opioid receptors are not expressed on DA axon terminals (Trovero et al., 1990). Cholinergic interneurons within the NAc shell do express μ-opioid receptors, and microdialysis experiments in vivo have demonstrated that their activation lowers extracellular ACh concentrations (Rada et al., 1996; Svingos et al., 2001a). Previous research highlighted the potential role of cholinergic interneurons in the physiological effects of opiate drugs, as lesion of cholinergic neurons with cholinotoxins eliminated the changes in DA levels produced by NAc infusion of opiates (Dourmap et al., 1997). Our results extend these studies by directly demonstrating the critical role of nAChRs in μ-opioid receptor modulation of DA release.
It is noteworthy that the suppression of single-spike-evoked DA overflow by μ- and δ-opioids is not associated with a significant change in the DA overflow evoked by burst stimuli. On the surface, this may seem to have minimal functional impact for drugs that induce burst firing. As suggested previously, however, this has a profound effect on the magnitude of the burst-evoked dopamine levels relative to baseline (Zhang and Sulzer, 2004). Modifying the ratio of a critical signal relative to background noise increases salience and potential impact. Similar modifications in signal-to-noise ratio have been observed in other parts of the reward circuitry and may be a common mechanism for enhancing perception of important environmental stimuli (Couey et al., 2007).
The mechanistic overlap between μ-opioids and nicotine modulation of DA overflow is intriguing when considered in the context of the other similarities of the drugs. Both μ-opioids and nicotine are considered addictive drugs that are readily self-administered by rats and humans (Thompson and Pickens, 1970; Goldberg and Henningfield, 1988). They also both have analgesic properties when applied directly to the NAc (Schmidt et al., 2001; Berrendero et al., 2005). Importantly, changes in NAc shell DA levels have been associated with each of these phenomena (Di Chiara and Imperato, 1988; Schmidt et al., 2001; Berrendero et al., 2005). In fact, whether taken systemically or microinjected focally into either the VTA or NAc, these drugs raise extracellular NAc DA levels as measured by microdialysis (Devine et al., 1993; Hemby et al., 1995; Ferrari et al., 2002; Hirose et al., 2005). VTA microinjections are effective because nicotine and μ-opioids modulate the afferent inputs to VTA DA neurons (nicotine also activates somatic receptors on these cells), causing them to transition from tonic to burst firing patterns (Grenhoff et al., 1986; Pidoplichko et al., 1997; Mansvelder and McGehee, 2000; Kiyatkin and Rebec, 2001; Williams et al., 2001; Erhardt et al., 2002; Mansvelder et al., 2002; Fagen et al., 2003).
It is less clear how opiates or nicotine administration directly into the NAc lead to enhanced extracellular DA levels, as measured by microdialysis (Ferrari et al., 2002; Hirose et al., 2005). Many have argued that microdialysis measures extrasynaptic DA tone, which reflects the overall population activity or burst firing of DA neurons (Floresco et al., 2003; Schultz, 2007). One possibility is that NAc administration of these drugs enhances DA neuron firing and/or burst activity through effects on the circuits that connect this nucleus to the VTA. The circuitry of the system supports this possibility, as GABAergic accumbal MSNs project to the VTA where they likely inhibit DA neuron activity (Kalivas et al., 1993). A major effect of opiates in the NAc is an inhibition of the excitatory drive to MSNs (Martin et al., 1997; Hoffman and Lupica, 2001; Brundege and Williams, 2002), which could lead to persistent disinhibition of VTA DA neurons.
This idea does not explain why cholinotoxin administration into the NAc eliminates increased DA levels produced by local NAc infusion of opiates (Dourmap et al., 1997). Thus, we propose an alternate hypothesis that a reduction in DA release produced from single-spike or tonic firing alters network activity to increase DA neuron burst firing. This theory presumes that between bursts of DA neuron activity the steady-state DA tone is negligible (Roitman et al., 2006). There is evidence that concerted activation of D1 and D2 receptors by DA enhances glutamate-induced excitation of MSNs (Hu and White, 1997). Consequently, suppression of tonic DA release between bursts could disrupt the coordinated activation of D1 and D2 receptors, leading to a decrease in activity of the MSNs that project to the VTA. This could disinhibit DA neurons and increase burst firing to enhance DA levels in the NAc.
The modulation of DA overflow by nicotine was found ubiquitously throughout the dorsal and ventral striatum. In contrast, μ- and -δ-opioids had more sporadic effects, with the μ-opioid receptors showing profound spatial segregation within the NAc shell. Because nAChR disruptions affect DA overflow everywhere in the striatum, our hypothesis is that the opioids limited effectiveness reflects variations in the opioid receptor expression or signaling cascades in striatal cholinergic interneurons. In the dorsal half of the NAc shell, between 2.0 and 3.0 mm rostral to bregma, μ-opioids were particularly effective in modulating DA overflow. This area generally corresponds to a region of the NAc shell identified previously as the DAMGO ([D-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin) hotspot in rats, because it is associated with the μ-opioid receptor enhancement of the hedonic impact of food (Pecina and Berridge, 2005).
There is considerable evidence that DA does not mediate hedonic reactions. Rats with dopamine depletion or injected with DA receptor antagonists still elicit hedonic reactions (Treit and Berridge, 1990; Berridge and Robinson, 1998; Barbano and Cador, 2006). Also, hyperdopaminergic mice show no difference in hedonic reactions to sucrose compared with wild-type animals (Pecina et al., 2003). One concern when using the techniques used in these studies is that they affect both tonic and phasic DA signaling. These alterations scale DA release up or down, but may not change the ratio of tonic versus burst-evoked DA release. One possibility is that hedonic reactions are modulated by changes in the signal-to-noise ratio of burst to single-spike-evoked DA release, but this remains speculative. We favor the hypothesis that these effects on DA signaling contribute to compulsive behaviors associated with drug addiction.
The actions of κ-opioids differ from and in many instances actively oppose those of μ- and δ-opioids, particularly in the NAc shell (Schmidt et al., 2002; Woolley et al., 2007). Both rats and humans find systemic administration of κ-opioids aversive and, in the rat, there is a corresponding decrease in extracellular DA concentrations in the NAc (Pfeiffer et al., 1986; Donzanti et al., 1992; Thompson et al., 2000; Walsh et al., 2001). Our data show that κ-opioid receptor activation in striatal slices inhibits evoked DA overflow in a manner that cannot be overcome by high-frequency burst stimuli, which is consistent with in vivo effects of κ-opioids on extracellular DA concentrations. The receptors that mediate this effect are located directly on DA terminals (Svingos et al., 2001b), but the downstream targets are unknown. We found independence between nAChR activity and the effects of κ-opioid receptor activation, as well as no acute effect of κ-opioids on DA reuptake. Furthermore, κ-opioid receptor activation did not alter the paired-pulse ratio of DA release, suggesting that the effects are not mediated by changes in either calcium influx or axon terminal membrane potential. These findings are complementary to recent investigations into κ-opioid inhibition of somatodendritic DA release in the VTA. These studies revealed that κ-opioid-induced membrane hyperpolarization did not account for the concomitant suppression of DA release (Ford et al., 2007).
Endogenous opioids are expressed throughout the striatum, and their role in striatal physiology remains speculative. MSNs express several endogenous opioid peptides including enkephalin, dynorphin, and endomorphins 1 and 2 (Gerfen et al., 1991; Svingos et al., 1996; Schreff et al., 1998; Martin-Schild et al., 1999; Pierce and Wessendorf, 2000). In fact, MSNs are often grouped into two populations based on their differential expression of both DA receptors and opioid peptides (Gerfen, 1992). D1 receptor-containing MSNs contribute to the anatomically defined direct pathway and express the endogenous opioid dynorphin, a selective agonist for κ receptors. D2 receptor-containing MSNs contribute to the indirect pathway and express the endogenous opioid enkephalin, an agonist that preferentially activates μ- and δ-opioid receptors. It is tempting to speculate, based on our findings, that by locally releasing different opioid peptides from axon collaterals D1 and D2 receptor-containing MSNs may differentially modulate DA release.
Footnotes
This work is supported by the National Institutes of Health Grants T32GM07839 and F31DA023340 (J.P.B.), and DA015918 and DA019695 (D.S.M.). We thank Hui Zhang and David Sulzer for their generous instruction regarding voltammetry and amperometry techniques. We thank Aaron Fox for comments on previous versions of this manuscript and Cristianne Frazier for experimental assistance.
References
- Barbano MF, Cador M. Differential regulation of the consummatory, motivational and anticipatory aspects of feeding behavior by dopaminergic and opioidergic drugs. Neuropsychopharmacology. 2006;31:1371–1381. doi: 10.1038/sj.npp.1300908. [DOI] [PubMed] [Google Scholar]
- Berrendero F, Mendizabal V, Robledo P, Galeote L, Bilkei-Gorzo A, Zimmer A, Maldonado R. Nicotine-induced antinociception, rewarding effects, and physical dependence are decreased in mice lacking the preproenkephalin gene. J Neurosci. 2005;25:1103–1112. doi: 10.1523/JNEUROSCI.3008-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Brundege JM, Williams JT. Differential modulation of nucleus accumbens synapses. J Neurophysiol. 2002;88:142–151. doi: 10.1152/jn.00766.2001. [DOI] [PubMed] [Google Scholar]
- Cagniard B, Beeler JA, Britt JP, McGehee DS, Marinelli M, Zhuang X. Dopamine scales performance in the absence of new learning. Neuron. 2006;51:541–547. doi: 10.1016/j.neuron.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, Shippenberg TS. Endogenous κ-opioid receptor systems regulate mesoaccumbal dopamine dynamics and vulnerability to cocaine. J Neurosci. 2005;25:5029–5037. doi: 10.1523/JNEUROSCI.0854-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Zapata A, Shippenberg TS, Bungay PM. Quantitative no-net-flux microdialysis permits detection of increases and decreases in dopamine uptake in mouse nucleus accumbens. J Neurosci Methods. 2006;155:187–193. doi: 10.1016/j.jneumeth.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Chen X, Zidichouski JA, Harris KH, Jhamandas JH. Synaptic actions of neuropeptide FF in the rat parabrachial nucleus: interactions with opioid receptors. J Neurophysiology. 2000;84:744–751. doi: 10.1152/jn.2000.84.2.744. [DOI] [PubMed] [Google Scholar]
- Chergui K, Suaud-Chagny MF, Gonon F. Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience. 1994;62:641–645. doi: 10.1016/0306-4522(94)90465-0. [DOI] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Dahan L, Astier B, Vautrelle N, Urbain N, Kocsis B, Chouvet G. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology. 2007;32:1232–1241. doi: 10.1038/sj.npp.1301251. [DOI] [PubMed] [Google Scholar]
- Devine DP, Wise RA. Self-administration of morphine, DAMGO, and DPDPE into the ventral tegmental area of rats. J Neurosci. 1994;14:1978–1984. doi: 10.1523/JNEUROSCI.14-04-01978.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine DP, Leone P, Pocock D, Wise RA. Differential involvement of ventral tegmental mu, delta and kappa opioid receptors in modulation of basal mesolimbic dopamine release: in vivo microdialysis studies. J Pharmacol Exp Ther. 1993;266:1236–1246. [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Donzanti BA, Althaus JS, Payson MM, Von Voigtlander PF. Kappa agonist-induced reduction in dopamine release: site of action and tolerance. Res Commun Chem Pathol Pharmacol. 1992;78:193–210. [PubMed] [Google Scholar]
- Dortch-Carnes J, Potter DE. Inhibition of cAMP accumulation by kappa-receptor activation in isolated iris-ciliary bodies: role of phosphodiesterase and protein kinase C. J Pharmacol Exp Ther. 2002;301:599–604. doi: 10.1124/jpet.301.2.599. [DOI] [PubMed] [Google Scholar]
- Dourmap N, Clero E, Costentin J. Involvement of cholinergic neurons in the release of dopamine elicited by stimulation of mu-opioid receptors in striatum. Brain Res. 1997;749:295–300. doi: 10.1016/S0006-8993(96)01319-4. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Schwieler L, Engberg G. Excitatory and inhibitory responses of dopamine neurons in the ventral tegmental area to nicotine. Synapse. 2002;43:227–237. doi: 10.1002/syn.10044. [DOI] [PubMed] [Google Scholar]
- Fagen ZM, Mansvelder HD, Keath JR, McGehee DS. Short- and long-term modulation of synaptic inputs to brain reward areas by nicotine. Ann NY Acad Sci. 2003;1003:185–195. doi: 10.1196/annals.1300.011. [DOI] [PubMed] [Google Scholar]
- Fagen ZM, Mitchum R, Vezina P, McGehee DS. Enhanced nicotinic receptor function and drug abuse vulnerability. J Neurosci. 2007;27:8771–8778. doi: 10.1523/JNEUROSCI.2017-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Le Novere N, Picciotto MR, Changeux JP, Zoli M. Acute and long-term changes in the mesolimbic dopamine pathway after systemic or local single nicotine injections. Eur J Neurosci. 2002;15:1810–1818. doi: 10.1046/j.1460-9568.2001.02009.x. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, McGinty JF, Young WS., III Dopamine differentially regulates dynorphin, substance P, and enkephalin expression in striatal neurons: in situ hybridization histochemical analysis. J Neurosci. 1991;11:1016–1031. doi: 10.1523/JNEUROSCI.11-04-01016.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg SR, Henningfield JE. Reinforcing effects of nicotine in humans and experimental animals responding under intermittent schedules of i.v. drug injection. Pharmacol Biochem Behav. 1988;30:227–234. doi: 10.1016/0091-3057(88)90450-9. [DOI] [PubMed] [Google Scholar]
- Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiologica Scandinavica. 1986;128:351–358. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- Harrison LM, Kastin AJ, Zadina JE. Differential effects of endomorphin-1, endomorphin-2, and Tyr-W-MIF-1 on activation of G-proteins in SH-SY5Y human neuroblastoma membranes. Peptides. 1998;19:749–753. doi: 10.1016/s0196-9781(98)00022-9. [DOI] [PubMed] [Google Scholar]
- Heijna MH, Hogenboom F, Mulder AH, Schoffelmeer AN. Opioid receptor-mediated inhibition of 3H-dopamine and 14C-acetylcholine release from rat nucleus accumbens slices. A study on the possible involvement of K+ channels and adenylate cyclase. Naunyn Schmiedebergs Arch Pharmacol. 1992;345:627–632. doi: 10.1007/BF00164575. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Martin TJ, Co C, Dworkin SI, Smith JE. The effects of intravenous heroin administration on extracellular nucleus accumbens dopamine concentrations as determined by in vivo microdialysis. J Pharmacol Exp Ther. 1995;273:591–598. [PubMed] [Google Scholar]
- Hirose N, Murakawa K, Takada K, Oi Y, Suzuki T, Nagase H, Cools AR, Koshikawa N. Interactions among mu- and delta-opioid receptors, especially putative delta1- and delta2-opioid receptors, promote dopamine release in the nucleus accumbens. Neuroscience. 2005;135:213–225. doi: 10.1016/j.neuroscience.2005.03.065. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol. 2001;85:72–83. doi: 10.1152/jn.2001.85.1.72. [DOI] [PubMed] [Google Scholar]
- Hu XT, White FJ. Dopamine enhances glutamate-induced excitation of rat striatal neurons by cooperative activation of D1 and D2 class receptors. Neurosci Lett. 1997;224:61–65. doi: 10.1016/s0304-3940(97)13443-7. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–1060. doi: 10.1016/0306-4522(93)90048-k. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Impulse activity of ventral tegmental area neurons during heroin self-administration in rats. Neuroscience. 2001;102:565–580. doi: 10.1016/s0306-4522(00)00492-9. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Martin G, Nie Z, Siggins GR. μ-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci. 1997;17:11–22. doi: 10.1523/JNEUROSCI.17-01-00011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Martin-Schild S, Gerall AA, Kastin AJ, Zadina JE. Differential distribution of endomorphin 1- and endomorphin 2-like immunoreactivities in the CNS of the rodent. J Comp Neurol. 1999;405:450–471. [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH. Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacol Toxicol. 1994;75:348–352. doi: 10.1111/j.1600-0773.1994.tb00373.x. [DOI] [PubMed] [Google Scholar]
- Okutsu H, Watanabe S, Takahashi I, Aono Y, Saigusa T, Koshikawa N, Cools AR. Endomorphin-2 and endomorphin-1 promote the extracellular amount of accumbal dopamine via nonopioid and mu-opioid receptors, respectively. Neuropsychopharmacology. 2006;31:375–383. doi: 10.1038/sj.npp.1300804. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pecina S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do μ-opioids cause increased hedonic impact of sweetness? J Neurosci. 2005;25:11777–11786. doi: 10.1523/JNEUROSCI.2329-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecina S, Cagniard B, Berridge KC, Aldridge JW, Zhuang X. Hyperdopaminergic mutant mice have higher “wanting” but not “liking” for sweet rewards. J Neurosci. 2003;23:9395–9402. doi: 10.1523/JNEUROSCI.23-28-09395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentney RJ, Gratton A. Effects of local delta and mu opioid receptor activation on basal and stimulated dopamine release in striatum and nucleus accumbens of rat: an in vivo electrochemical study. Neuroscience. 1991;45:95–102. doi: 10.1016/0306-4522(91)90106-x. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Pierce TL, Wessendorf MW. Immunocytochemical mapping of endomorphin-2-immunoreactivity in rat brain. J Chem Neuroanat. 2000;18:181–207. doi: 10.1016/s0891-0618(00)00042-9. [DOI] [PubMed] [Google Scholar]
- Rada PV, Mark GP, Taylor KM, Hoebel BG. Morphine and naloxone, i.p. or locally, affect extracellular acetylcholine in the accumbens and prefrontal cortex. Pharmacol Biochem Behav. 1996;53:809–816. doi: 10.1016/0091-3057(95)02078-0. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Roitman MF, Day JJ, Seipel A, Carelli RM, Wightman RM. A steady-state concentration of dopamine is comprised of time-averaged, phasic dopamine release events. In: Di Chiara G, Carboni E, Valentini V, Acquas E, Bassareo V, Cadoni C, editors. Proceedings of the 11th International Conference on In Vivo Methods; Cagliari, Italy: University of Cagliari; 2006. pp. 95–97. [Google Scholar]
- Ronken E, Van Muiswinkel FL, Mulder AH, Schoffelmeer AN. Opioid receptor-mediated inhibition of evoked catecholamine release from cultured neurons of rat ventral mesencephalon and locus coeruleus. Eur J Pharmacol. 1993;230:349–355. doi: 10.1016/0014-2999(93)90572-y. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Gear RW, Levine JD. Nicotine withdrawal hyperalgesia and opioid-mediated analgesia depend on nicotine receptors in nucleus accumbens. Neuroscience. 2001;106:129–136. doi: 10.1016/s0306-4522(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Levine JD, Gear RW. mu/delta Cooperativity and opposing kappa-opioid effects in nucleus accumbens-mediated antinociception in the rat. Eur J Neurosci. 2002;15:861–868. doi: 10.1046/j.1460-9568.2002.01915.x. [DOI] [PubMed] [Google Scholar]
- Schreff M, Schulz S, Wiborny D, Hollt V. Immunofluorescent identification of endomorphin-2-containing nerve fibers and terminals in the rat brain and spinal cord. NeuroReport. 1998;9:1031–1034. doi: 10.1097/00001756-199804200-00014. [DOI] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Moriwaki A, Wang JB, Uhl GR, Pickel VM. Ultrastructural immunocytochemical localization of μ-opioid receptors in rat nucleus accumbens: extrasynaptic plasmalemmal distribution and association with Leu5-enkephalin. J Neurosci. 1996;16:4162–4173. doi: 10.1523/JNEUROSCI.16-13-04162.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Clarke CL, Pickel VM. Localization of the delta-opioid receptor and dopamine transporter in the nucleus accumbens shell: implications for opiate and psychostimulant cross-sensitization. Synapse. 1999;34:1–10. doi: 10.1002/(SICI)1098-2396(199910)34:1<1::AID-SYN1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Colago EE, Pickel VM. Vesicular acetylcholine transporter in the rat nucleus accumbens shell: subcellular distribution and association with mu-opioid receptors. Synapse. 2001a;40:184–192. doi: 10.1002/syn.1041. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Chavkin C, Colago EE, Pickel VM. Major coexpression of kappa-opioid receptors and the dopamine transporter in nucleus accumbens axonal profiles. Synapse. 2001b;42:185–192. doi: 10.1002/syn.10005. [DOI] [PubMed] [Google Scholar]
- Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. κ-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T, Pickens R. Stimulant self-administration by animals: some comparisons with opiate self-administration. Fed Proc. 1970;29:6–12. [PubMed] [Google Scholar]
- Treit D, Berridge KC. A comparison of benzodiazepine, serotonin, and dopamine agents in the taste-reactivity paradigm. Pharmacol Biochem Behav. 1990;37:451–456. doi: 10.1016/0091-3057(90)90011-6. [DOI] [PubMed] [Google Scholar]
- Trovero F, Herve D, Desban M, Glowinski J, Tassin JP. Striatal opiate mu-receptors are not located on dopamine nerve endings in the rat. Neuroscience. 1990;39:313–321. doi: 10.1016/0306-4522(90)90270-e. [DOI] [PubMed] [Google Scholar]
- Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001;157:151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- Watson GB, Lanthorn TH. Electrophysiological actions of delta opioids in CA1 of the rat hippocampal slice are mediated by one delta receptor subtype. Brain Res. 1993;601:129–135. doi: 10.1016/0006-8993(93)91703-u. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Woolley JD, Lee BS, Kim B, Fields HL. Opposing effects of intra-nucleus accumbens mu and kappa opioid agonists on sensory specific satiety. Neuroscience. 2007;146:1445–1452. doi: 10.1016/j.neuroscience.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Koide S, Hirose N, Takada K, Tomiyama K, Koshikawa N, Cools AR. Fentanyl increases dopamine release in rat nucleus accumbens: involvement of mesolimbic mu- and delta-2-opioid receptors. Neuroscience. 1999;92:1357–1365. doi: 10.1016/s0306-4522(99)00046-9. [DOI] [PubMed] [Google Scholar]
- You ZB, Herrera-Marschitz M, Terenius L. Modulation of neurotransmitter release in the basal ganglia of the rat brain by dynorphin peptides. J Pharmacol Exp Ther. 1999;290:1307–1315. [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]