

Abstract
Extracellular proteolysis plays an essential role in synaptic remodeling that is indispensable for cognitive function. The extracellular serine protease neurotrypsin was implicated in cognitive function, because humans lacking a functional form of neurotrypsin suffer from severe mental retardation. By immunoelectron microscopy, neurotrypsin has been localized to presynaptic terminals, suggesting a local proteolytic function after its synaptic release. Here, we studied axonal trafficking and synaptic exocytosis of neurotrypsin by live imaging of hippocampal neurons expressing neurotrypsin fused with enhanced green fluorescent protein or its pH-sensitive variant, superecliptic pHluorin. In differentiated neurons, we identified neurotrypsin in mobile transport vesicles along axons and in both an intracellular and an extracellular pool at synapses. Short depolarization triggered rapid synaptic exocytosis of neurotrypsin. Once externalized, neurotrypsin lingered at its synaptic release site for several minutes before it disappeared. Cell depolarization also enhanced synaptic capture of intracellular neurotrypsin transport vesicles, and elevated synaptic activity increased both number and motility of mobile axonal neurotrypsin vesicles. We further observed trading of neurotrypsin vesicles between adjacent synapses. These activities may support the replenishment of neurotrypsin after activity-induced synaptic exocytosis. Together, the activity-dependent recruitment of neurotrypsin to synapses and its exocytosis and transient persistence at its synaptic release site argue for a spatially and temporally restricted proteolytic action at the synapse. Thereby, neurotrypsin may play a role in activity-dependent remodeling of the synaptic circuitry that is key to adaptive synaptic changes in the context of cognitive functions, such as learning and memory.
Keywords: extracellular proteolysis, mental retardation, cognitive function, synaptic plasticity, activity-dependent secretion, synaptic capture
Introduction
Cognitive functions, including learning and memory, are based on adaptive changes in signal transmission and processing in the CNS. These are achieved by (1) structural and functional modifications of established synapses and (2) structural reorganizations of synaptic circuits involving the formation of new synapses and the elimination of existing ones (Chklovskii et al., 2004). Both mechanisms have been reported to involve proteolysis by extracellular proteases (Shiosaka, 2004). The neuronal serine protease neurotrypsin was recognized to play an indispensable role in cognitive processes in humans, because a four-nucleotide deletion in the coding region of neurotrypsin that resulted in the expression of a truncated protein lacking the catalytic domain caused a severe form of nonsyndromic mental retardation (Molinari et al., 2002). The affected children exhibited severe deficits in cognitive development, with first symptoms becoming evident at ∼18 months of age. The pathophysiological phenotype and the age of disease onset in neurotrypsin-deficient human subjects indicate that neurotrypsin does not play a critical role in morphogenic processes during early neural development or in the formation of synapses. Rather, neurotrypsin may be crucial for the reorganization of synapses and neuronal circuits, which are required to establish and maintain higher cognitive functions during later developmental stages and in the adult brain.
To reach temporally and spatially restricted substrate cleavage, the activity of extracellular proteases is subject to complex regulations. These include synthesis as inactive zymogens that require regulated activation (Khan and James, 1998), control of the active enzyme by specific inhibitors (Bode and Huber, 1992), and termination of the proteolytic activity by removal or degradation of the enzyme (Binder et al., 2007). Compartmentalization of proteases in intracellular storage vesicles and regulated delivery to the site of action is another way to achieve temporal and spatial control and to prevent inappropriate actions. In mature neurons, neurotrypsin was localized to the presynaptic compartment, in particular to the area lining the synaptic cleft and in a subpopulation of vesicles in the vicinity of the active zone of neurotransmitter release (Molinari et al., 2002). Therefore, regulated exocytosis from presynaptic nerve endings was a conceivable mechanism to control neurotrypsin-dependent substrate cleavage. To explore trafficking and exocytosis of neurotrypsin in neurons, we expressed neurotrypsin tagged with enhanced green fluorescent protein (EGFP) or its pH-sensitive variant, superecliptic pHluorin, which allows distinguishing the externalized form of neurotrypsin from the one contained intracellularly in secretory vesicles. We examined transport, synaptic targeting, and exocytosis of neurotrypsin in live neurons. We observed that recruitment as well as exocytosis of neurotrypsin at synapses was regulated by neuronal activity and that externalized neurotrypsin lingered at the synapse for several minutes before it disappeared because of diffusion or degradation. We also identified activity-induced enhancement of the mobility of neurotrypsin-containing vesicles and their redistribution between the internal pools of adjacent synapses as mechanisms for rapid refilling of synaptic neurotrypsin stores after exocytosis. These findings suggest a role of neurotrypsin in activity-dependent synaptic plasticity via a local extracellular proteolytic activity at the synapse.
Materials and Methods
Antibodies.
Rabbit serum sz177 was raised against a peptide corresponding to the proline-rich basic domain of neurotrypsin. The following commercially available primary antibodies were used: mouse anti-postsynaptic density 95 (PSD95) (clone K28/43; Upstate/Millipore, Billerica, MA), rabbit anti-vesicular glutamate transporter (vGlut) and rabbit anti-synapsin (both from Synaptic Systems, Goettingen, Germany), and mouse anti-microtubule-associated protein 2 (MAP2) (clone HM-2; Sigma, St. Louis, MO). Secondary fluorescence-coupled antibodies were purchased from Jackson ImmunoResearch (Newmarket, UK).
Expression constructs and cDNAs.
To obtain expression vectors for neurotrypsin and neurotrypsin–EGFP, the coding sequences of mouse neurotrypsin and mouse neurotrypsin fused at its 3′ end in frame with the sequence of EGFP were inserted into the pcDNA3.1(−)A vector. The cDNA of ecliptic synapto-pHluorin (Miesenbock et al., 1998) was generously provided by Dr. J. E. Rothman (Cellular Biochemistry and Biophysics Program, Memorial Sloan-Kettering Cancer Center, New York, NY). The sequence was modified by PCR as published previously to obtain the superecliptic variant of pHluorin (Sankaranarayanan et al., 2000). Superecliptic pHluorin was fused to the 3′ end of the coding sequence of mouse neurotrypsin.
Neuronal cultures.
Primary dissociated hippocampal cultures were prepared from embryonic day 18 NMRI mice as described previously (Banker, 1980). Briefly, cell suspensions obtained after trypsin treatment and trituration were plated onto round glass coverslips (diameter of 18 mm; Menzel-Glaeser, Braunschweig, Germany) coated with poly-l-lysine (Sigma). After 1–2 h at 37°C, the coverslips were transferred into 12-well plates containing a 70–80% confluent monolayer of astrocytes in DMEM supplemented with B27, 5 mm glutamine, 1 mm sodium pyruvate, and 0.5 mg/ml Albumax. Cultures were maintained at 37°C in a humidified incubator with an atmosphere of 95% air and 5% CO2. All chemicals used for neuronal cultures were obtained from Invitrogen (Basel, Switzerland), unless indicated otherwise.
Transfection of cultured neurons.
Neurons were transfected at 4 d in vitro (DIV) using Effectene transfection reagent (Qiagen, Hombrechtikon, Switzerland) according to the protocol of the manufacturer.
Preparation of adenoviruses and infection of neurons.
An adenoviral expression system was used (Mohanty et al., 2005). Briefly, the expression cassettes of untagged neurotrypsin, neurotrypsin–EGFP, and neurotrypsin–pHluorin were inserted into a transfer vector. Cre-mediated recombination into the adenoviral vector was performed in Escherichia coli strain BM25.8. The infectious viral particles were produced in a helper cell line (human embryonic kidney HEK293). For large-scale production, over-infected cells were collected and resuspended in 10 mm Tris-HCl, pH 8. The viral particles were released by three cycles of freezing and thawing and 5 min sonification on ice. For transfection of neurons, the viruses were purified by centrifugations over a CsCl step gradient and two continuous CsCl gradients. The CsCl step gradient contained (from bottom to top) 2 ml of 1.4 g/ml CsCl in TD (in mm: 137 NaCl, 6 KCl, 0.7 Na2HPO4, and 25 Tris-HCl, pH 7.5), 2 ml of 1.25 g/ml CsCl in TD, and 4 ml of cell lysate containing the virus. The banded virus from the step gradient was mixed with 1.35 g/ml CsCl in TD and centrifuged in a Beckman SW41 rotor (Beckman Coulter, Nyon, Switzerland) at 30,000 rpm for 16 h. The virus was collected and purified on the continuous gradient once more. After purification, the virus was dialyzed against TD. Virus titers were determined by plaque assays on 911 cells or by cytopathic effect assays on HEK293 cells (Payment and Trudel, 1993). For infection of primary neurons, 1000 plaque-forming units per neuron were used.
Immunocytochemistry.
Cells were fixed after 20 DIV in 4% paraformaldehyde, 4% sucrose in PBS, pH 7.4, for 10 min at room temperature. Blocking was done for 1 h in PBS containing 10% fetal calf serum, 0.1% glycine, and 0.3% Triton X-100. The primary and secondary antibodies were diluted in PBS containing 3% fetal calf serum. Images were taken with a Leica SP2 confocal microscope with LCS software (Leica, Wetzlar, Germany).
Live imaging microscopy.
For live imaging, hippocampal neurons were infected with adenoviral vectors containing expression cassettes for the respective EGFP fusion proteins three days before the experiment. Live imaging was performed at 37°C using an inverted microscope (Eclipse TE300; Nikon, Badhoevedorp, The Netherlands) equipped with a Plexiglas box to maintain a stable atmosphere (Life Imaging Services, Reinach, Switzerland). Images were collected with a SensiCAM QE camera (Cooke Corporation, Romulus, MI) controlled by MetaMorph Imaging software (Molecular Devices, Sunnyvale, CA), using a 100× objective (numerical aperture 1.4), a standard FITC filter set, and a Lambda DG-4 light source (Sutter Instruments, Novato, CA). Images were captured every 2 s for the evaluation of vesicle movement and every 3–5 s for synaptic recruitment experiments for a period of 5–20 min. Experiments were done under constant perfusion with the following (in mm): 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, and 25 HEPES, pH 7.4). Ammonium chloride solution to alkalize vesicles was prepared by replacing 50 mm NaCl with NH4Cl, with all other components remaining unchanged. The solution with pH 5.5 was prepared by replacing HEPES with 2-[N-morpholino]ethane sulfonic acid, with all other components remaining unchanged. For experiments with disinhibited neurons, 100 μm picrotoxin (Tocris Bioscience, Ellisville, MO) was added to the culture medium 12–24 h before imaging. For experiments with inhibited neurons, 50 μm APV and 10 μm CNQX (both from Tocris Bioscience) were added 12–24 h before imaging.
Data analysis.
Data were analyzed using NIH ImageJ or MetaMorph software. Statistical analyses were performed with Prism 4 software (GraphPad Software, San Diego, CA).
Results
Neurotrypsin is sorted into axons and accumulates at presynaptic boutons
We used dissociated hippocampal neurons to study intracellular trafficking and exocytosis of the serine protease neurotrypsin, because neurotrypsin is expressed in the murine hippocampus throughout life, with peak expression during the first two postnatal weeks (Gschwend et al., 1997; Wolfer et al., 2001). Neurotrypsin mRNA expression in cultured dissociated hippocampal neurons could only be detected by reverse transcription-PCR but not by in situ hybridization (data not shown). Our attempts to visualize endogenous neurotrypsin protein with specific antibodies failed. Therefore, we transfected dissociated hippocampal neurons with a plasmid encoding a neurotrypsin–EGFP fusion protein to study the localization of neurotrypsin. Cells were transfected after 4 DIV. At 20 DIV, cells were fixed, stained, and inspected with a confocal microscope. At this time in culture, the neurons had adopted their typical polarized morphology with one axon and many dendrites. Neurotrypsin–EGFP was found in a punctate pattern (Fig. 1A1,B1). Costaining for synapsin, a general presynaptic marker, and PSD95, a marker of excitatory postsynapses, indicated the synaptic localization of many neurotrypsin–EGFP puncta (Fig. 1A, arrowheads). Under the conditions used here, only a minor fraction of the synapses exhibited neurotrypsin. Neurotrypsin–EGFP puncta were also found at extrasynaptic locations (Fig. 1A, arrows). The largest neurotrypsin–EGFP puncta tended to be synaptic, whereas extrasynaptic neurotrypsin–EGFP puncta were often small and faint. Neurotrypsin–EGFP puncta were often also costained with vGlut, a selective marker of excitatory presynapses (Fig. 1B, arrowheads in high-magnification images). Again, only a minority of the vGlut-labeled synapses contained neurotrypsin, and neurotrypsin–EGFP puncta were also found at extrasynaptic locations (Fig. 1B, arrows in high-magnification images). Also, with vGlut as a synaptic marker, the largest neurotrypsin–EGFP puncta tended to be synaptic, whereas faint neurotrypsin–EGFP puncta in general tended to be extrasynaptic (Fig. 1B, arrows in high-magnification images). Absence of costaining of neurotrypsin–EGFP and MAP2, a marker of dendrites, indicated sorting of neurotrypsin to the axonal compartment (Fig. 1B, arrow in the low-magnification images). Together, these results indicated that neurotrypsin–EGFP was targeted to axons and presynaptic boutons. This is in accordance with a previous study that demonstrated a presynaptic location of endogenous neurotrypsin using immunoelectron microscopy on brain sections (Molinari et al., 2002). Because we detected neurotrypsin also in extrasynaptic puncta that were often of lower intensity and of smaller size than synaptic puncta, we speculated that these were transport vesicles carrying neurotrypsin along axons toward their synaptic destination.
Figure 1.

Confocal microscopy confirms localization of neurotrypsin to axons and presynaptic boutons. A, Costaining of neurotrypsin–EGFP-expressing hippocampal neurons (A1) with the PSD95 (A2) and the presynaptic protein synapsin (A3). The overlay is shown in A4. Pairwise colocalizations of neurotrypsin–EGFP puncta with either PSD95 or synapsin are indicated by arrowheads in the high-magnification images. Examples of extrasynaptic neurotrypsin–EGFP, most likely representing vesicular transport packages, are indicated by arrows in the high-magnification images of A1–A4. B, Neurites positive for neurotrypsin–EGFP are not costained with the dendritic protein MAP2 (B2), suggesting axonal localization of neurotrypsin (arrows in B1–B4). Many neurotrypsin–EGFP puncta are colocalized with vGlut, a presynaptic marker of excitatory synapses (arrowheads in high-magnification images). Faint extrasynaptic neurotrypsin–EGFP puncta are indicated by arrows in the high-magnification images. The regions of the high-magnification images are boxed in A1–A4 and B1–B4, respectively. Scale bars: B4, 15 μm; high-magnification images, 2 μm.
Synaptic neurotrypsin is found on the cell surface and in intracellular pools
To study the exocytosis of neurotrypsin, we produced an adenovirus carrying neurotrypsin fused to superecliptic pHluorin, a pH-sensitive variant of EGFP (Miesenbock et al., 1998; Sankaranarayanan et al., 2000). The fluorescence emission of superecliptic pHluorin is minimal at pH < 6.0 compared with pH > 7. Therefore, superecliptic pHluorin can only be detected when it resides in a compartment with neutral pH (e.g., cytoplasm or extracellular space), although it remains undetected when contained in secretory vesicles with acidic pH (Miesenbock et al., 1998). Thus, the fusion of neurotrypsin with superecliptic pHluorin allowed for discrimination between its intracellular and extracellular pools. In a first set of experiments, we infected cultured hippocampal neurons at 15 DIV and performed live imaging at 18 DIV. As expected, neurotrypsin was distributed in a punctate pattern along neurites. We labeled active synapses by K+-induced uptake of the styryl dye FM 4-64 [N-(3-triethylammoniumpropyl)-4-(6-(4-diethylamino)phenyl)hexatrienyl)pyridinium dibromide] (Fig. 2A). Most extracellular neurotrypsin–pHluorin puncta, as detected at pH 7.4, showed synaptic localization (Fig. 2B1,B2). Notably, the fluorescent signal of extracellular neurotrypsin was restricted to a small area of the dye-filled synapses (Fig. 2B2). Imaging medium containing NH4Cl was used to visualize intracellular pools of neurotrypsin. Ammonia, in equilibrium with ammonium ions, diffuses across cell membranes, enters membranal organelles, such as secretory vesicles, and neutralizes their normally acidic pH (Roos and Boron, 1981; Miesenbock et al., 1998). As shown in Figure 2, C1 and C2, most synapses exhibited additional fluorescence puncta after a brief exposure of the cells to 50 mm NH4Cl. These results indicated the presence of neurotrypsin in synaptic intracellular pools. Moreover, neurotrypsin fluorescence was now also found at nonsynaptic places that were devoid of fluorescence before NH4Cl addition. These neurotrypsin puncta probably represented transport packets (Fig. 2C1,C2, arrowheads).
Figure 2.

Synaptic neurotrypsin is found in intracellular and extracellular pools by live imaging of cells infected with an adenovirus expressing neurotrypsin–pHluorin. A, Labeling of active synapses with FM 4-64. B, In normal buffer (pH 7.4), only extracellular neurotrypsin–pHluorin is visible (B1). Most extracellular neurotrypsin colocalizes with synapses labeled with FM 4-64 (B2). C, Addition of NH4Cl unmasks intracellular pools of neurotrypsin (C1). Note the additional neurotrypsin–pHluorin dots within FM 4-64-positive areas (synapses) in the presence of NH4Cl (C2) compared with normal imaging buffer (B2). Boxed areas in B2 and C2 are shown at higher magnification. In addition, nonsynaptic dots representing transport vesicles appear in NH4Cl-containing buffer (arrowheads in C1 and C2). Scale bars: overviews, 5 μm; high-magnification images, 1 μm.
For a quantification of the cellular distribution of neurotrypsin and the degree of its synaptic localization, we determined the colocalization of neurotrypsin–pHluorin puncta with FM 4-64 puncta in normal buffer and in the presence of NH4Cl. In normal buffer, revealing only extracellular neurotrypsin–pHluorin, we found that 87.6 ± 3.7% of the neurotrypsin–pHluorin puncta were colocalized with FM 4-64-labeled active synapses. In contrast, in the presence of NH4Cl, revealing total (intracellular and extracellular) neurotrypsin–pHluorin, only 27.5 ± 2.6% of the neurotrypsin–pHluorin puncta were colocalized with synapses (mean ± SEM; n = 7 cells). This observation indicated that exocytosis of neurotrypsin occurred preferentially at synaptic sites. We also measured the fluorescence intensities of synaptic and extrasynaptic neurotrypsin–pHluorin puncta and determined the intensity ratio between synaptic puncta and extrasynaptic puncta. We found that the fluorescence intensity of synaptic puncta was 3.5 ± 0.14 times higher than that of extrasynaptic puncta (mean ± SEM; n = 60; p < 0.001). These results indicated that the intracellular synaptic pool of neurotrypsin was formed by accumulation of several transport vesicles.
Neuronal activity drives the translocation of mobile neurotrypsin transport packages to synapses
During our initial transfection experiments, we characterized neurotrypsin as a presynaptic component of a small fraction of synapses. To study mechanisms of synaptic targeting of neurotrypsin and a possible impact of neuronal activity on this process, we cotransfected hippocampal neurons at 4 DIV with plasmids encoding untagged neurotrypsin and the presynaptic marker vesicle-associated membrane protein (VAMP2)–EGFP. To alter the overall network activity, either the GABA receptor blocker picrotoxin (PTX) or the glutamate receptor blockers APV and CNQX were added to the culture medium at 17 DIV, i.e., 24 h before analysis. Application of PTX increases network activity attributable to the blockage of inhibitory input, whereas APV/CNQX decreases network activity by inhibiting glutamatergic excitatory input. At 18 DIV, the cells were fixed and stained with anti-neurotrypsin antibodies. Microscopic analysis showed a similar pattern of neurotrypsin immunoreactivity as found previously with the neurotrypsin–EGFP fusion construct. Neurotrypsin was located predominantly in axons of transfected cells, and both synaptic and extrasynaptic localizations were found (Fig. 3A1,B1). However, the fraction of neurotrypsin-containing synapses was dramatically increased in disinhibited (PTX-treated) versus silenced (APV/CNQX-treated) cultures. Quantification indicated that, in APV/CNQX-treated cultures, only 19.6 ± 4.4% of the VAMP2–EGFP-labeled synapses also contained neurotrypsin immunoreactivity. In marked contrast, neurotrypsin immunoreactivity was found in 74.2 ± 1.9% of the VAMP2–EGFP-labeled synapses of PTX-treated cultures (mean ± SEM; n = 17 cells; p < 0.001). These results indicated an activity-mediated translocation of neurotrypsin from extrasynaptic locations to synapses.
Figure 3.

Chronic disinhibition of hippocampal cultures with PTX enhances the synaptic localization of neurotrypsin. Hippocampal neurons were cotransfected with neurotrypsin and VAMP2–EGFP and stained with anti-neurotrypsin antibody. Cells were treated with APV/CNQX (A) or PTX (B) 24 h before fixation. In APV/CNQX-treated cells, only a small fraction of the VAMP2–EGFP puncta contained neurotrypsin (arrows in A3). Chronic neuronal activity led to a higher occupation of synapses by neurotrypsin, as indicated by the high degree of colocalization of neurotrypsin with VAMP2–EGFP in PTX-treated cells (arrows in B3). Scale bar, 5 μm.
To directly observe the translocation of intracellular neurotrypsin transport vesicles to synapses, we performed live imaging of hippocampal neurons infected with an adenovirus expressing neurotrypsin–EGFP. Cultures were depolarized with KCl (Fig. 4A2,A3). Active synapses were labeled by FM 4-64 uptake at the end of the experiment (Fig. 4A11). Already during KCl depolarization, we could observe increased trafficking of neurotrypsin-containing vesicles (Fig. 4A3, arrows). Within <5 min after depolarization, new synaptic pools were established (Fig. 4A4/A12, A5/A13, arrowheads). During the initial period, after first arrival at the synapse, the synapse association of neurotrypsin was often not stable, as demonstrated by the disappearance of neurotrypsin after a short synaptic visit (Fig. 4A6, arrowhead). After a resting time, which could last from several seconds up to 2 min, neurotrypsin-containing vesicles often moved to adjacent synaptic contacts (Fig. 4A6) (supplemental movie, available at www.jneurosci.org as supplemental material). Later, established synaptic pools remained stable during the observation period of 15 min (Fig. 4A6–A10, arrowheads). However, trading of small neurotrypsin transport packages between neighboring synapses could be observed (Fig. 4A6–A10, arrows) (supplemental movie, available at www.jneurosci.org as supplemental material). As shown in Figure 4B, the number of synapses containing neurotrypsin gradually increased during the first minutes after depolarization. It reached a plateau of twofold increase 5 min after depolarization (Fig. 4B, filled circles). Unstimulated cells exhibited a stable number of neurotrypsin-containing synapses (Fig. 4B, open circles). The amount of neurotrypsin at newly labeled synapses increased strongly during the time of observation (Fig. 4A, follow left arrow from A5 to A10). Together, we found that depolarization led to a recruitment of neurotrypsin to previously neurotrypsin-free synapses within the first 5 min after depolarization. Synapses once defined as neurotrypsin-containing continued to accumulate neurotrypsin throughout the time of observation. The synaptic pools were not entirely stationary but exchanged with the extrasynaptic mobile pool and other synaptic pools of neurotrypsin.
Figure 4.

K+-induced depolarization of hippocampal neurons results in translocation of neurotrypsin to synapses. A, Image sequence from a time-lapse experiment of hippocampal neurons expressing neurotrypsin–EGFP. Neurotrypsin–EGFP signals were recorded for a period of 1 min before and 12:20 min after a 90 s period of K+-induced depolarization (A1–A10). Synapses were stained with FM 4-64 at the end of the experiment (A11). The image of the FM 4-64-labeled synapses was used to generate overlays with selected images of neurotrypsin–pHluorin (A12–A14). Before depolarization, no synaptic neurotrypsin was observed in this particular segment (A1, A2). At the end of the depolarization period, we observed enhanced neurotrypsin trafficking (arrows in A3). Later, transport vesicles started to be captured at active synapses (arrowheads in A4/A12, A5/A13). Neurotrypsin was often redistributed in small transport vesicles after lingering for several minutes at a particular synapse (arrow in A6), remaining at the synapse in only small amounts (right arrowhead in A6). At other synapses, neurotrypsin was continuously accumulated (left arrowhead in A5–A10 and overlays A13, A14). Between neighboring synapses, an exchange of neurotrypsin transport packages could be observed (arrow in A6–A12). Scale bar, 1 μm. B, Synaptic neurotrypsin was quantified in 20 μm axonal segments before and after KCl depolarization. The number of synaptic puncta before depolarization was used for normalization. At 100 s after stimulation, significant increase of synaptic neurotrypsin was observed. During the following 300 s, the number of synaptic neurotrypsin increased gradually. At ∼400 s after KCl treatment, the occupation of the synapses with neurotrypsin was twofold compared with the control situation and did not increase further (***p < 0.001, two-way ANOVA, Bonferroni's post hoc test, n = 12 cells).
The mobility of neurotrypsin-containing vesicles is modulated by synaptic activity
During live imaging of hippocampal neurons expressing neurotrypsin–EGFP, we noticed many mobile fluorescent puncta that presumably represented neurotrypsin transport vesicles moving along axons. Depolarization of the neurons with KCl enhanced the mobility of vesicles (Fig. 5A, arrows). To further study to which extent the mobility of these structures was influenced by neural activity, we analyzed vesicle movement in neurons that were silenced with APV/CNQX or disinhibited with PTX. Under both conditions, the neurotrypsin–EGFP-containing structures moved with a broad range of velocities up to a maximal velocity of 6.5 μm/s. However, the PTX-treated cells exhibited a higher fraction of vesicles moving with a higher speed (Fig. 5B, open circles). The average velocities in silenced versus activated cultures were significantly different, with a velocity of 1.26 ± 0.06 μm/s in the presence of APV/CNQX and a velocity of 1.55 ± 0.05 μm/s in the presence of PTX (mean ± SEM; n = 340; p < 0.001). We did not observe a difference in vesicle directivity under the two conditions (data not shown). Surprisingly, we also registered that the number of vesicles counted in 20-μm-long axonal segments was significantly higher in disinhibited cells conditioned in PTX (13.89 ± 0.70) compared with silenced cells kept in APV/CNQX (4.08 ± 0.47) (mean ± SEM; n = 12 cells; p < 0.001). To estimate the time range of the mobilization of neurotrypsin-containing vesicles, we depolarized neurons by application of a short pulse of KCl-containing medium and counted the number of vesicles passing through an axonal segment of 20 μm length during a time window of 100 s (Fig. 5C). In cultures that were silenced with APV/CNQX, the vesicle number was increased by 50% between 200 and 300 s after stimulation and by 150% between 600 and 700 s after stimulation (Fig. 5C, filled circles). In a control experiment, in which no KCl was applied, the number of vesicles stayed unchanged (Fig. 5C, open triangles). Similarly, no change was observed in cells that were cultured in the presence of PTX (Fig. 5C, open circles). In summary, these results indicated that activity enhanced both the mobility and the number of neurotrypsin-containing transport vesicles. Absence of a K+-induced increase in vesicle number in cultures disinhibited with PTX before the addition of KCl suggested that vesicle mobilization in PTX-disinhibited neurons was maximal and could not be further increased.
Figure 5.

Vesicular transport of neurotrypsin–EGFP is enhanced after K+-induced depolarization and PTX-mediated disinhibition. A, Image series of an axonal segment before (0–8 s) and after (220–228 s) KCl application. Arrows indicate transport vesicles. Note the faster movement of the indicated vesicle after KCl. Also note the higher density of vesicles after stimulation. B, Frequency distribution of neurotrypsin–EGFP transport vesicle velocities from hippocampal neurons at DIV 18 treated with PTX or APV/CNQX for 24 h. Note the shift to higher velocities in PTX-treated cultures. C, Vesicles were counted in live neurons as they passed an axonal segment of 20 μm in 100 s time windows. Data were normalized to the number of vesicles counted during the first 100 s. If cells were not depolarized, the trafficking remained unchanged during the 800 s of observation (open circles). If cells were treated with PTX before imaging, depolarization had no effect on the trafficking (open triangles). However, if cells were kept in APV/CNQX, depolarization led to an increased trafficking within 200 s after KCl treatment (filled triangles). The increase in trafficking was up to 2.5-fold 600 s after onset of depolarization. **p < 0.01; ***p < 0.001.
Synapses of active neurons exhibit increased amounts of externalized neurotrypsin
To study whether the synaptic exocytosis of neurotrypsin was regulated by synaptic activity, we determined the ratio between intrasynaptic and extrasynaptic neurotrypsin in cultures after silencing with APV/CNQX or disinhibition with PTX. Cultured hippocampal neurons were infected with an adenovirus expressing neurotrypsin–pHluorin and treated with PTX or APV/CNQX for 24 h, and their synapses were labeled with FM 4-64. Extracellular neurotrypsin puncta were recorded in normal imaging buffer at pH 7.4, and the appearance of intracellular neurotrypsin puncta was monitored after the addition of NH4Cl (Fig. 6A,B). The fluorescence ratio in the absence (extracellular neurotrypsin) and in the presence (total neurotrypsin) of NH4Cl was determined. The ratio was 0.28 ± 0.02 when cells were cultivated for 24 h with APV/CNQX. When cultivated with PTX, the ratio was 0.57 ± 0.02 (mean ± SEM; n = 17 cells; p < 0.001). In addition, we observed that the absolute amount of total synaptic neurotrypsin was higher in cells treated with PTX (compare Fig. 6A,B). The increase in the ratio of extracellular versus total fluorescence at synapses after PTX disinhibition indicated an increased pool size of extracellular synaptic neurotrypsin in cultures with elevated network activity. These results suggested synaptic activity as a driving force for synaptic exocytosis of neurotrypsin.
Figure 6.

The synaptic extracellular pool of neurotrypsin is increased in PTX-disinhibited neurons. A, B, Axonal segment of neurons transfected with neurotrypsin–pHluorin. Synapses were labeled using FM 4-64 (A3, B3). In the cultures supplemented with APV/CNQX, there was only little synaptic extracellular neurotrypsin (arrow in A1). Addition of NH4Cl revealed intracellular pools of neurotrypsin. The intracellular pools located at a synapse are indicated by an arrow, an arrowhead, and an open arrow in A2 and A4. At numerous synaptic intracellular pools (visualized by NH4Cl), no extracellular neurotrypsin was observed (arrowhead and open arrow in A1). If cells were cultured in the presence of PTX, an overall increased fluorescence of both intracellular and extracellular pools was observed (B1–B4). At synapses, both extracellular and intracellular neurotrypsin were more prominent (arrows in B1, B2, B4). Scale bar, 2 μm.
Depolarization drives synaptic exocytosis of neurotrypsin and buildup of intracellular synaptic neurotrypsin stores
To further investigate the fate of synaptic neurotrypsin in response to activity, we monitored intracellular and extracellular neurotrypsin–pHluorin in individual synapses of hippocampal cultures before, during, and after a 90-s K+-induced depolarization period. In a typical experiment, we recorded images for 15 min before and after KCl stimulation. During this time, we changed the culture medium periodically to visualize intracellular neurotrypsin–pHluorin in the presence of NH4Cl and to transiently erase extracellular fluorescence by exposure to pH 5.5 (Fig. 7A). Active synapses were labeled with FM 4-64 at the end of the experiment.
Figure 7.
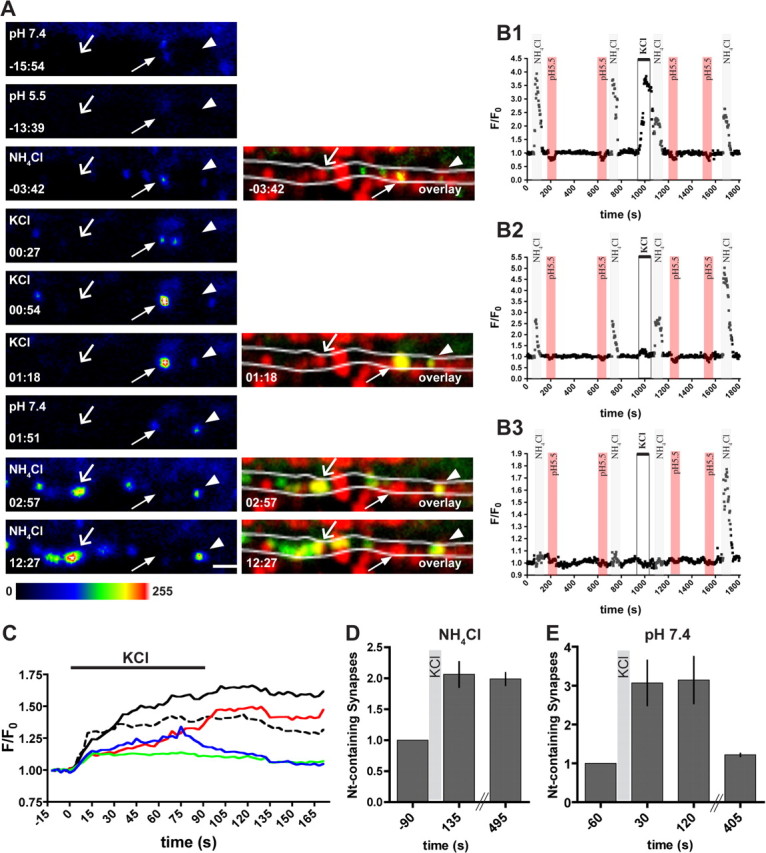
Depolarization leads to synaptic exocytosis of neurotrypsin and buildup of intracellular neurotrypsin stores at synapses. A, Live imaging of hippocampal neurons at DIV 18 infected with an adenovirus expressing neurotrypsin–pHluorin. Synapses were stained at the end of the experiment using FM 4-64. The image of the FM 4-64-stained synapses was used to generate overlays with selected images of neurotrypsin–pHluorin. At the beginning of the experiment, little synaptic extracellular neurotrypsin was found (arrow in −15:54). Short perfusion with pH 5.5 imaging buffer abolished all observed fluorescence (−13:39). Perfusion with NH4Cl-containing buffer revealed the presence of intracellular pools of neurotrypsin–pHluorin at synaptic (arrow at −03:42 and overlay at −03:42) and extrasynaptic sites. Starting from 0:00 min, cells were depolarized for 90 s with KCl. Shortly after depolarization, increased extracellular fluorescence could be observed at synapses in which intracellular neurotrypsin was present before depolarization (arrow in 00:27) and also at sites in which previously no neurotrypsin fluorescence was found (arrowhead at 01:18 and overlay at 01:18). At a number of synapses, externalized neurotrypsin could be observed for awhile after the end of depolarization (arrow and arrowhead in 01:51). In the minutes after KCl depolarization, new intracellular pools were formed at synapses that previously did not contain neurotrypsin (open arrow in 02:57, 12:27). Most intracellular pools were replenished after neurotrypsin–pHluorin exocytosis (arrowhead in 12:27), although a few were not (arrow in 02:57 and 12:27). Scale bar, 2 μm. B, Normalized fluorescence intensity of individual synaptic sites during a live-imaging experiment. B1 shows the course of fluorescence of a synaptic spot with an intracellular pool (gray bar at 100 s and 700 s) and little extracellular fluorescence (red bar) before depolarization. There was a strong increase in fluorescence when KCl was added (open bar, 1000 s). Note that the intracellular pool was diminished and not restored (gray bar, 1100 s and 1700 s). In B2, intracellular pools were present before depolarization, but only little was released during KCl depolarization (open bar at 1000 s in B2). Immediately after depolarization, the intracellular pool remained unaltered (gray bar, 1100 s). However, several minutes after KCl application, the intracellular fluorescence was more than doubled (gray column, 1700 s). Other synapses contained neither intracellular nor extracellular neurotrypsin before depolarization (B3). Also, there was no immediate increase of fluorescence when KCl was added (open bar at 1000 s in B3). Only several minutes after depolarization a large intrasynaptic pool was found (1700 s, gray bar). C, Time course of the change of fluorescence during and after depolarization at individual synapses containing neurotrypsin. The increase in fluorescence varied between individual synapses, ranging from 1.1-fold (blue line) to 1.65-fold (black line). Note that also the velocity of the signal change varied between individual synapses, in both the increasing and decreasing phase. D, The number of synapses containing intracellular or external neurotrypsin was increased 2.06 ± 0.20 times 135 s after onset of depolarization and stayed stable during the time of imaging. E, We observed a 3.07 ± 0.58-fold increase in the number of synapses with extracellular neurotrypsin 30 and 120 s after onset of depolarization. The number of synapses with extracellular neurotrypsin was back to basal level after 7 min (mean ± SEM, n = 6 cells).
The monitoring of external neurotrypsin fluorescence at individual synapses before, during, and after depolarization, together with intermittent visualization of the intracellular neurotrypsin pool revealed distinct patterns of response to depolarization. A first synaptic response pattern is represented by the synapse marked with an arrow in Figure 7A. At −15:54 min and pH 7.4, the presence of a synapse was indicated by a weak fluorescent spot (Fig. 7A, −15:54 min, arrow), which disappeared when the pH of the medium was changed to 5.5 (Fig. 7A, −13:39 min, arrow). After a change to NH4Cl-containing medium, intracellular neurotrypsin–pHluorin became visible and thus enhanced the fluorescent signal at this synapse (Fig. 7A, −3:42 min). Shortly after the onset of depolarization, externalized neurotrypsin–pHluorin was observed at this synapse (Fig. 7A, arrow, 00:27 min). The extracellular neurotrypsin–pHluorin signal then increased markedly during continued depolarization (Fig. 7A, 00:54 and 01:18 min). The extracellular synaptic neurotrypsin–pHluorin signal persisted for awhile after the end of the depolarization period (Fig. 7A, 01:51 min), but, 3 min after removal of KCl, neither extracellular nor intracellular neurotrypsin–pHluorin was found at this synapse (Fig. 7A, 02:57 min, arrow). This synapse did not refill its intracellular store during the entire observation period (Fig. 7A, 12:57 min). Thus, neurotrypsin–pHluorin was present at this synapse in an extracellular and intracellular pool. KCl-induced depolarization resulted in marked exocytosis of neurotrypsin–pHluorin. The depolarization-induced extracellular fluorescent signal at this synapse was considerably stronger than the combined signal for intracellular and extracellular neurotrypsin–pHluorin found before depolarization, indicating that neurotrypsin–pHluorin was delivered to and released from this synapse during the period of depolarization. After K+-induced depolarization, the intracellular pools of this synapse were not refilled. Thus, at this synapse, the intracellular neurotrypsin pool was externalized during depolarization but not replenished during the time of observation. In addition to its internal pool, this synapse also externalized neurotrypsin that was delivered during the period of depolarization.
Another synaptic response pattern with a pronounced depolarization-induced exocytosis of neurotrypsin–pHluorin without substantial replenishment during the following 15 min period is shown in the F/Fo tracing of Figure 7B1. Testing of this synapse with NH4Cl and pH 5.5 indicated a large intracellular store and a relatively small fraction of extracellular neurotrypsin–pHluorin at the beginning of the observation period. K+-induced depolarization resulted in a strong exocytosis, and subsequent NH4Cl testing indicated a marked reduction of the intracellular store. Additional NH4Cl testing showed that the intracellular store had not been refilled until the end of the observation period. In this synapse, a substantial fraction of the neurotrypsin–pHluorin that was externalized during depolarization was derived from intracellular stores, as indicated by the strong reduction of the intracellular stores found by NH4Cl testing shortly after depolarization. Because the reduction of the intracellular stores did not match the externalized neurotrypsin–pHluorin, we concluded that this synapse also showed synaptic delivery and instant exocytosis of neurotrypsin–pHluorin during the period of depolarization.
A second synaptic response pattern is represented by the synapse marked with the arrowhead in Figure 7A at 01:18 min and later time points. The external neurotrypsin–pHluorin signal at this synapse was first seen at 01:18 min. Thus, this synapse did not have internally stored neurotrypsin before KCl depolarization was induced. After KCl depolarization, external fluorescence appeared with some delay and was still observed 21 s after removal of KCl (Fig. 7A, 01:51 min). Subsequent tests of this synapse with NH4Cl revealed increasing amounts of intracellular neurotrypsin–pHluorin at 02:57 and 12:27 min. Thus, this synapse initially had no neurotrypsin, responded to depolarization by recruiting and immediately externalizing of a small amount of neurotrypsin, and by subsequent buildup of a large intracellular neurotrypsin pool.
A similar synaptic response pattern with a small stimulus-induced exocytosis and a large increase of the intracellular stores of neurotrypsin–pHluorin is represented by the F/Fo trace shown in Figure 7B2. This synapse exhibited a constant amount of internal neurotrypsin at two NH4Cl tests before KCl-induced depolarization. Depolarization resulted in the exocytosis of a relatively small fraction of the initial intracellular store with little reduction of the internal signal found in immediately after an NH4Cl test. A subsequent NH4Cl perfusion after 12 min revealed a markedly increased amount of intracellular neurotrypsin. Thus, this synapse had stored intracellular neurotrypsin but responded to depolarization by exocytosis of only a small fraction of it and subsequently increased its intracellular store during the following minutes.
A third synaptic response pattern is represented by the synapse marked with the open arrow in Figure 7A and the synapse shown in the F/Fo trace of Figure 7B3. These synapses were initially devoid of neurotrypsin–pHluorin and did not externalize during depolarization but responded to depolarization by formation of a new intracellular pool during the following 15 min observation period.
The time course of synaptic exocytosis and extracellular presence of neurotrypsin–pHluorin is shown in Figure 7C. Comparison of F/Fo traces of individual synapses indicated that the relative increase of extracellular neurotrypsin–pHluorin fluorescence at individual synapses was very variable, ranging from 20 to 65% in most synapses (Fig. 7C). However, we also observed several synapses exhibiting depolarization-induced increases up to 350% (Fig. 7B1). The onset of neurotrypsin exocytosis was observed in most cases during the first 20 s of the 90 s window of K+-induced depolarization, and peak levels of externalized neurotrypsin–pHluorin were reached before KCl was removed (Fig. 7C). However, at some synapses, neurotrypsin–pHluorin exocytosis started slowly, and full exocytosis was established after the end of the depolarization period (Fig. 7C, red and black lines). At the end of the depolarization, extracellular neurotrypsin–pHluorin signals persisted for variable times before disappearing. In some synapses, base levels were reached as early as 45 s after KCl removal, whereas in others the signal persisted for several minutes.
The monitoring of individual synapses further confirmed the depolarization-induced delivery of neurotrypsin–pHluorin to synaptic intracellular stores. Quantification indicated that the number of synapses containing intracellular neurotrypsin were doubled 45 s after the end of K+-induced depolarization and stayed stable during the time of observation (Fig. 7D).
The average increase in the number of synapses containing extracellular neurotrypsin was threefold during KCl depolarization and did not change within 120 s after onset of depolarization (Fig. 7E). However, 405 s after the onset of depolarization, the number of neurotrypsin-containing synapses decreased to the base level, confirming the notion of neurotrypsin removal from the synaptic space after a period of extracellular persistence at the synaptic release site (Fig. 7E). These measurements indicated an approximate lifetime of a few minutes for extracellular neurotrypsin at individual synapses.
Discussion
Suitability of the experimental system: methodological note
To study the intracellular transport and exocytosis of neurotrypsin, we chose dissociated hippocampal cultures as an experimental system (Kaech and Banker, 2006). First, dissociated hippocampal cultures are a well established, frequently used system for studies of protein and organelle translocation (Burack et al., 2000) and for analyses of synapse formation and protein targeting to synapses (Friedman et al., 2000; Shapira et al., 2003), as well as for studies of synaptic exocytosis and recycling (Darcy et al., 2006). Second, the hippocampus, together with the cerebral cortex and the amygdala, belongs to the brain regions with most pronounced expression of neurotrypsin mRNA (Gschwend et al., 1997; Wolfer et al., 2001). Unexpectedly, we failed to detect any neurotrypsin expression in cultures by staining with neurotrypsin-specific antibodies or by in situ hybridization. Possibly, the expression level of neurotrypsin decreased in response to the loss of synaptic targets caused by tissue dissociation. A recent report of neurotrypsin downregulation in motoneurons after interruption of their contact with target muscles by facial nerve axotomy supports this interpretation (Numajiri et al., 2006). To restore neurotrypsin expression and to introduce fluorescently tagged variants of neurotrypsin, we used highly purified recombinant adenoviruses. Using this expression system, we achieved moderate expression levels of neurotrypsin. Because of inevitable technical differences in the immunocytochemical methods used for the detection of neurotrypsin in cultured cells and intact tissue, we cannot claim that expression levels in our adenovirus-infected hippocampal neurons matched the expression levels of endogenous neurotrypsin in intact tissue. Yet, the suitability of our experimental system was supported by the observed axonal and synaptic targeting of fluorescent neurotrypsin, which is in accordance with the reported presynaptic localization of neurotrypsin in the intact brain (Molinari et al., 2002).
Neurotrypsin lingers at the synapse for several minutes after activity-dependent exocytosis from internal stores
Our experiments with neurotrypsin fused to pHluorin indicated that synaptic neurotrypsin is found both at the cell surface and in intracellular stores and that cell depolarization triggers the rapid synaptic exocytosis of neurotrypsin. K+-induced synaptic release of neurotrypsin was also observed at synapses that were devoid of neurotrypsin before depolarization, indicating that neurotrypsin exocytosis at synapses did not necessarily require a preexisting internal store. Apparently, newly recruited neurotrypsin was ready for immediate release.
Synaptic exocytosis of neurotrypsin–pHluorin did not result in immediate dispersal of its punctate fluorescence signal, as would be expected for a freely diffusible molecule. Rather, externalized neurotrypsin remained visible at the synaptic region for a period lasting up to several minutes before it disappeared. This indicates that externalized neurotrypsin may be transiently bound to extracellular synaptic components, before disappearing from the synapse by diffusion, degradation, or re-endocytosis. The exact molecular mechanism of transient retention of neurotrypsin at the synapse remains to be determined. Interactions with cell surface proteins or extracellular matrix components are possible. The noncatalytic domains found in neurotrypsin, i.e., the SRCR (scavenger receptor cysteine-rich) and kringle domains, have been characterized as mediators of protein–protein interactions in cell surface-associated complexes (Whitney et al., 1995; Gonzalez-Gronow et al., 2003). Likewise, the basic segment at the N terminus of neurotrypsin could exert a retaining function via interaction with glycosaminoglycans and other negatively charged surface components, as it was shown previously for other positively charged secreted molecules, such as the C-terminal basic segment of netrin (Kappler et al., 2000). The restricted diffusion achieved by cell surface and extracellular matrix association is crucial for a role as a chemotropic agent in axonal guidance for chemotactic molecules, such as netrin (Baker et al., 2006). Accordingly, the transient lingering at its synaptic site of exocytosis may argue for a spatially and temporally restricted proteolytic action of neurotrypsin in the extracellular space at or near the synapse.
Recently, we identified the proteoglycan agrin as the specific endogenous substrate of neurotrypsin (Reif et al., 2007). Neurotrypsin and agrin are both localized at CNS synapses (for neurotrypsin, Molinari et al., 2002; for agrin, Ksiazek et al., 2007). Lack of agrin has been reported recently to result in loss of CNS synapses, and it has been proposed that agrin plays a role in the formation and/or maintenance of excitatory synapses (Ksiazek et al., 2007). Binding of externalized neurotrypsin to the negatively charged glycan chains of synaptic agrin could enhance neurotrypsin-dependent cleavage of agrin and thus regulate its action at the CNS synapse.
The notion of a spatially and temporally restricted proteolytic action of neurotrypsin in the extracellular space at or near the synapse prompts speculations about mechanisms terminating extracellular neurotrypsin action. Dissociation from local macromolecular ligands and diffusion in the extracellular fluid phase, as well as lateral diffusion in the plane of the plasma membrane together with a membrane-bound interaction partner, would explain the disappearance of the local pHluorin signal after a period of extracellular lingering. Proteolytic degradation and reuptake by endocytosis, alone or in combination, might be additional conceivable mechanisms for terminating the extracellular action of neurotrypsin. At present, we do not have evidence favoring one of these processes.
The capture of neurotrypsin at synapses is regulated by neuronal activity
The activity-dependent synaptic exocytosis of neurotrypsin implies a need for replenishment. In our experiment with neurotrypsin–EGFP, long-time enhancement of network activity by application of PTX resulted in a significant rise of both number and velocity of neurotrypsin transport packages. The rapid mobilization of axonal neurotrypsin transport packages observed in axons within 3 min after K+-induced depolarization indicates a pool of preexisting stationary neurotrypsin vesicles. Whether the continuously increasing supply of vesicles observed after long-term elevation of network activity requires an additional contribution from enhanced transcription/translation remains to be determined.
An increased transport velocity after a K+-induced depolarization was reported recently for vesicles carrying the postsynaptic scaffold protein gephyrin (Maas et al., 2006). Interestingly, depolarization has the opposite effect on semaphorin3A-carrying vesicles. Axonal transport of these vesicles is arrested by depolarization (de Wit et al., 2006). Thus, activity-dependent enhancement of the motility of transport vesicle seems to be cargo specific. Although the underlying mechanisms are not fully understood, change in average speed of vesicular transport might be attributable to an activity-dependent regulation of molecular motors associated with vesicles (Morfini et al., 2004). Alternatively, vesicular motility might be influenced by microtubule-associated proteins (Sato-Harada et al., 1996).
Depolarization resulted in an immediate increase of the amount of intracellular neurotrypsin in both neurotrypsin-free and neurotrypsin-containing synapses. Our live imaging data revealed activity-induced synaptic recruitment of neurotrypsin-containing vesicles as a mechanism underlying this observation. However, a contribution to the formation of synaptic intracellular pools after depolarization might also result from reuptake of exocytosed neurotrypsin. Recently, transmembranal synaptic molecules such as VAMP were observed to spread throughout the plasma membrane of the presynaptic bouton and along the axon to be subsequently re-endocytosed (Fernandez-Alfonso et al., 2006; Wienisch and Klingauf, 2006). In analogy, surface-bound extracellular neurotrypsin might be distributed along the axonal membrane. Subsequent re-endocytosis might contribute to the regeneration of exocytosed synaptic pools as well as to the establishment of intracellular neurotrypsin pools at previously neurotrypsin-free synapses.
Activity-dependent recruitment to and from synapses has been reported for several other vesicular or cytoplasmic components of the presynapse (Shakiryanova et al., 2006) and the postsynapse (Neuhoff et al., 2005; Maas et al., 2006). However, a molecular mechanism for synaptic accumulation of transiting axonal transport packages has not been specified so far. It has been speculated that mobile transport packages transiting at a synaptic bouton may become retained by a constitutive affinity to a synapse-resident component or acquire this affinity via posttranslational modification received during synaptic transit. Because synapses represent preferred sites also for neurotrypsin, as indicated by the at least twofold higher neurotrypsin fluorescence intensity compared with mobile transport packages, a synaptic capture mechanism must also be assumed for neurotrypsin transport packages. The increase in vesicle number and motility in activated neurons, providing more frequent encounters between neurotrypsin transport packages and synaptic boutons, sufficiently explains our observation of enhanced recruitment of neurotrypsin to both neurotrypsin-free and neurotrypsin-containing synaptic sites by a constitutive synaptic capture mechanism. However, because activity-dependent synaptic exocytosis of neurotrypsin varied considerably between individual synapses, their need for replenishment of neurotrypsin may also be highly variable. Preferential delivery of neurotrypsin transport packages to activated synapses might be a conceivable way of satisfying this need. Recently, the activity-dependent enhancement of the capture of vesicles transiting through synaptic boutons has been characterized in detail for peptidergic vesicles destined for presynaptic boutons of motor neurons in Drosophila melanogaster (Shakiryanova et al., 2006). Likewise, activity might enhance the capture mechanism for neurotrypsin transport packages at synapses, contributing to their increased retention after encounter with an activated synaptic bouton. Our observations of an increased number of synapses containing neurotrypsin in cultures with elevated network activity are compatible with but do not prove a mechanism for activity-regulated enhancement of the synaptic capture mechanism. Alternatively, differential replenishment of synaptic neurotrypsin pools may be attributable to neurotrypsin transport between adjacent synapses. Exchange of vesicles between synapses has been reported previously for small synaptic vesicles in studies with hippocampal neurons (Krueger et al., 2003; Darcy et al., 2006) and has been suggested to serve as a substrate for presynaptic remodeling (Staras, 2007). Because of its leveling character, trading of neurotrypsin vesicles between adjacent synapses may add an adjusting element to the general delivery and capture mechanisms. Thus, enhanced translocation of vesicles between synapses may serve as an ancillary mechanism for establishing a balanced distribution of neurotrypsin to different synaptic sites after both activity-enhanced synaptic delivery and activity-induced synaptic exocytosis.
Conclusion
The severe mental retardation found in humans with a loss-of-function mutation in the neurotrypsin gene (Molinari et al., 2002) implies neurotrypsin as an essential regulator of adaptive synaptic plasticity that is crucial for establishment and maintenance of cognitive brain functions. Our results demonstrate that synaptic neurotrypsin is contained in intracellular stores and that both recruitment of neurotrypsin to synapses and synaptic exocytosis of neurotrypsin are enhanced by neuronal activity. After exocytosis, neurotrypsin transiently lingers at the synapse, before it disappears by diffusion or degradation. The selective activity-dependent exocytosis of neurotrypsin and its temporarily restricted presence at the synapse after exocytosis puts neurotrypsin in an ideal position as a regulator of structural and functional reorganizations of the synaptic circuitry that are key for adaptive synaptic plasticity in the context of cognitive functions, such as learning and memory.
Footnotes
A.F., A.S., and R.F. were supported by Swiss National Foundation, and R.F. was supported by Deutsche Forschungsgemeinschaft Gundelfinger 230-5/2. We thank Dr. Thomas Trüb for providing adenoviral vectors and helpful technical suggestions.
References
- Baker KA, Moore SW, Jarjour AA, Kennedy TE. When a diffusible axon guidance cue stops diffusing: roles for netrins in adhesion and morphogenesis. Curr Opin Neurobiol. 2006;16:529–534. doi: 10.1016/j.conb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Banker GA. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 1980;209:809–810. doi: 10.1126/science.7403847. [DOI] [PubMed] [Google Scholar]
- Binder BR, Mihaly J, Prager GW. uPAR-uPA-PAI-1 interactions and signaling: a vascular biologist's view. Thromb Haemost. 2007;97:336–342. [PubMed] [Google Scholar]
- Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- Burack MA, Silverman MA, Banker G. The role of selective transport in neuronal protein sorting. Neuron. 2000;26:465–472. doi: 10.1016/s0896-6273(00)81178-2. [DOI] [PubMed] [Google Scholar]
- Chklovskii DB, Mel BW, Svoboda K. Cortical rewiring and information storage. Nature. 2004;431:782–788. doi: 10.1038/nature03012. [DOI] [PubMed] [Google Scholar]
- Darcy KJ, Staras K, Collinson LM, Goda Y. Constitutive sharing of recycling synaptic vesicles between presynaptic boutons. Nat Neurosci. 2006;9:315–321. doi: 10.1038/nn1640. [DOI] [PubMed] [Google Scholar]
- de Wit J, Toonen RF, Verhaagen J, Verhage M. Vesicular trafficking of semaphorin 3A is activity-dependent and differs between axons and dendrites. Traffic. 2006;7:1060–1077. doi: 10.1111/j.1600-0854.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Kwan R, Ryan TA. Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron. 2006;51:179–186. doi: 10.1016/j.neuron.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gronow M, Kalfa T, Johnson CE, Gawdi G, Pizzo SV. The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J Biol Chem. 2003;278:27312–27318. doi: 10.1074/jbc.M303172200. [DOI] [PubMed] [Google Scholar]
- Gschwend TP, Krueger SR, Kozlov SV, Wolfer DP, Sonderegger P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol Cell Neurosci. 1997;9:207–219. doi: 10.1006/mcne.1997.0616. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kappler J, Franken S, Junghans U, Hoffmann R, Linke T, Muller HW, Koch KW. Glycosaminoglycan-binding properties and secondary structure of the C-terminus of netrin-1. Biochem Biophys Res Commun. 2000;271:287–291. doi: 10.1006/bbrc.2000.2583. [DOI] [PubMed] [Google Scholar]
- Khan AR, James MN. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM. The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron. 2003;40:945–957. doi: 10.1016/s0896-6273(03)00729-3. [DOI] [PubMed] [Google Scholar]
- Ksiazek I, Burkhardt C, Lin S, Seddik R, Maj M, Bezakova G, Jucker M, Arber S, Caroni P, Sanes JR, Bettler B, Ruegg MA. Synapse loss in cortex of agrin-deficient mice after genetic rescue of perinatal death. J Neurosci. 2007;27:7183–7195. doi: 10.1523/JNEUROSCI.1609-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas C, Tagnaouti N, Loebrich S, Behrend B, Lappe-Siefke C, Kneussel M. Neuronal cotransport of glycine receptor and the scaffold protein gephyrin. J Cell Biol. 2006;172:441–451. doi: 10.1083/jcb.200506066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mohanty S, Spinas GA, Maedler K, Zuellig RA, Lehmann R, Donath MY, Trub T, Niessen M. Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human beta-cells from hyperglycemia-induced apoptosis. Exp Cell Res. 2005;303:68–78. doi: 10.1016/j.yexcr.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J, Bacq D, Briault S, Vekemans M, Munnich A, Attie-Bitach T, Sonderegger P, Colleaux L. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff H, Sassoe-Pognetto M, Panzanelli P, Maas C, Witke W, Kneussel M. The actin-binding protein profilin I is localized at synaptic sites in an activity-regulated manner. Eur J Neurosci. 2005;21:15–25. doi: 10.1111/j.1460-9568.2004.03814.x. [DOI] [PubMed] [Google Scholar]
- Numajiri T, Mitsui S, Hisa Y, Ishida T, Nishino K, Yamaguchi N. The expression of a motoneuron-specific serine protease, motopsin (PRSS12), after facial nerve axotomy in mice. J Plast Reconstr Aesthet Surg. 2006;59:393–397. doi: 10.1016/j.bjps.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Payment P, Trudel M. Isolation and identification of viruses. In: Payment P, Trudel M, editors. Methods and techniques in virology. New York: Marcel Dekker; 1993. [Google Scholar]
- Reif R, Sales S, Hettwer S, Dreier B, Gisler C, Wolfel J, Luscher D, Zurlinden A, Stephan A, Ahmed S, Baici A, Ledermann B, Kunz B, Sonderegger P. Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J. 2007;21:3468–3478. doi: 10.1096/fj.07-8800com. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N. Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct. 1996;21:283–295. doi: 10.1247/csf.21.283. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Levitan ES. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shiosaka S. Serine proteases regulating synaptic plasticity. Anat Sci Int. 2004;79:137–144. doi: 10.1111/j.1447-073x.2004.00080.x. [DOI] [PubMed] [Google Scholar]
- Staras K. Share and share alike: trading of presynaptic elements between central synapses. Trends Neurosci. 2007;30:292–298. doi: 10.1016/j.tins.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Wienisch M, Klingauf J. Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat Neurosci. 2006;9:1019–1027. doi: 10.1038/nn1739. [DOI] [PubMed] [Google Scholar]
- Whitney GS, Starling GC, Bowen MA, Modrell B, Siadak AW, Aruffo A. The membrane-proximal scavenger receptor cysteine-rich domain of CD6 contains the activated leukocyte cell adhesion molecule binding site. J Biol Chem. 1995;270:18187–18190. doi: 10.1074/jbc.270.31.18187. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Lang R, Cinelli P, Madani R, Sonderegger P. Multiple roles of neurotrypsin in tissue morphogenesis and nervous system development suggested by the mRNA expression pattern. Mol Cell Neurosci. 2001;18:407–433. doi: 10.1006/mcne.2001.1029. [DOI] [PubMed] [Google Scholar]