

Abstract
Chronic treatments with selective serotonin reuptake inhibitors (SSRIs) have been shown to increase hippocampal neurogenesis. However, it is not known whether SSRIs impact the maturation and functional integration of newborn neurons. Here we examined the effects of subchronic and chronic fluoxetine on the structural and physiological properties of young granule cells. Our results show that doublecortin-positive immature neurons displayed increased dendritic arborization after chronic fluoxetine treatment. In addition, chronic but not subchronic fluoxetine elicited a decrease in the number of newborn neurons expressing immature markers and a corresponding increase in those expressing mature markers. These results suggest that chronic fluoxetine accelerates the maturation of immature neurons. We also investigated the effects of fluoxetine on a form of neurogenesis-dependent long-term potentiation (LTP) in the dentate gyrus. This form of LTP was enhanced by chronic fluoxetine, and ablation of neurogenesis with x-irradiation completely blocked the effects of chronic fluoxetine on LTP. Finally, we demonstrated that the behavioral effect of fluoxetine in the novelty-suppressed feeding test requires chronic administration and is blocked by x-irradiation. These results show that the effects of fluoxetine on LTP and behavior both require neurogenesis and follow a similar delayed time course. The effects of chronic fluoxetine on the maturation and functional properties of young neurons may therefore be necessary for its anxiolytic/antidepressant activity and contribute to its delayed onset of therapeutic efficacy.
Keywords: dentate gyrus, adult neurogenesis, dendrites, long-term potentiation, LTP, depression, selective serotonin reuptake inhibitors, SSRIs, hippocampus, antidepressants
Introduction
Selective serotonin reuptake inhibitors (SSRIs) have become the most commonly prescribed treatments for major depression (Millan, 2006). Nonetheless, the mechanisms underlying the action of antidepressants are still unclear: SSRIs require at least 2–4 weeks of administration before achieving therapeutic benefits (Wong and Licinio, 2001), despite the fact that serotonin levels rise rapidly after acute administration of SSRIs in both primates and rodents (Rutter et al., 1994; Kreiss and Lucki, 1995; Anderson et al., 2005). The paradox between a rapid increase in serotonin and the delayed onset of antidepressant action has led us to postulate that structural or functional changes that take place over time may be required for the therapeutic effects of SSRIs.
Administration of various antidepressants increases adult neurogenesis in the dentate gyrus (DG) of the hippocampus (Malberg et al., 2000; Santarelli et al., 2003). A chronic treatment is required to produce the increase in neurogenesis (Madsen et al., 2000; Malberg et al., 2000; Santarelli et al., 2003). Additionally, we and others have shown that some of the behavioral effects of SSRIs are dependent on hippocampal neurogenesis (Santarelli et al., 2003; Airan et al., 2007), indicating that neurogenesis plays a pivotal role in the mechanism of antidepressant action. Besides increasing the proliferation of neural progenitors, SSRIs also enhances survival of postmitotic granule cells (Malberg et al., 2000; Nakagawa et al., 2002). Studies have suggested that distinct mechanisms regulate proliferation and survival. For example, environmental enrichment enhances the survival of immature cells without affecting proliferation (Kempermann et al., 1997). In contrast, voluntary exercise increases proliferation and survival but does not alter the rate of maturation of newborn neurons (van Praag et al., 2005; Plumpe et al., 2006). Pilocarpine-induced seizures increase both proliferation and survival (Radley and Jacobs, 2003) and also improve dendritic outgrowth in newborn neurons (Overstreet-Wadiche et al., 2006). A recent study demonstrated that fluoxetine targets a class of amplifying neural progenitors by increasing the rate of symmetric divisions (Encinas et al., 2006). It is not clear, however, whether SSRIs also target immature neurons by influencing maturation and functional integration.
To assess the impact of fluoxetine on dendritic maturation, we examined the dendritic morphology of cells that express doublecortin (DCX). In the adult DG, DCX is exclusively expressed in immature neurons from 1 d to ∼4 weeks of age (Brown et al., 2003; Couillard-Despres et al., 2005) and thus has been widely used as an immature neuronal marker that reliably reflects the level of neurogenesis and its modulation (Couillard-Despres et al., 2005).
Recent studies have revealed that newborn neurons display enhanced long-term potentiation (LTP) (Wang et al., 2000; Schmidt-Hieber et al., 2004; Ge et al., 2007). In addition, ACSF–LTP, a form of DGLTP induced by a weak stimulation paradigm, has been shown to be completely blocked by manipulations that ablate hippocampal neurogenesis (Snyder et al., 2001; Saxe et al., 2006). Here we examined whether the SSRI-induced effects on newborn neurons will lead to enhanced synaptic plasticity in the hippocampus and, finally, produce improved behavioral outcome.
Materials and Methods
Animals and drugs.
SvEv129 age-matched adult male mice (12–25 weeks) were purchased from Taconic Farms (Germantown, NY). Mice were housed four to five per cage in a 12 h (6:00 A.M. to 6:00 P.M.) light/dark colony room at 22°C with available food and water ad libitum. All experiments were performed in compliance with the institutional regulations and guidelines for animal experimentation. Fluoxetine (18 mg · kg−1 · d−1; Anawa Biomedical Services and Products, Zurich, Switzerland) was given by gavage for behavior testing or in the drinking water for all other experiments. HPLC analysis of plasma levels of fluoxetine and its metabolite norfluoxetine were determined after chronic treatment (data not shown) (Suckow et al., 1992).
Immunohistochemistry and confocal imaging.
Mice were anesthetized with ketamine/xylazine (100 and 7 mg/kg, respectively) and transcardially perfused (cold saline, followed by 4% cold paraformaldehyde in PBS). All brains were postfixed overnight in 4% paraformaldehyde at 4°C, then cryoprotected in 30% sucrose, and stored at 4°C. Serial sections were cut through the entire hippocampus (Franklin and Paxinos, 1997) using a cryostat and stored in PBS. Immunohistochemistry was performed in the following steps: 2 h incubation in 1:1 formamide/2× SSC at 65°C, 5 min rinse in 2× SSC, 30 min incubation in 2N HCl at 37°C, and 10 min rinse in 0.1 m boric acid, pH 8.5, 2 h incubation in 0.1 m PBS with 0.3% Triton X-100, and 5% normal donkey serum. Sections were then incubated overnight at 4°C in primary antibodies for doublecortin (goat; 1:500; Santa Cruz Biotechnology, Santa Cruz, CA), bromodeoxyuridine (BrdU; rat; 1:100; Serotec, Oxford, UK), and neuronal-specific nuclear protein (NeuN) (mouse; 1:500; Chemicon, Temecula, CA). Biotinylated or fluorescent secondary antibodies were used. All secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). DCX staining for Sholl analysis was done as follows: sections were rinsed in PBS, treated with 1% H2O2 in 1:1 PBS and methanol for 15 min to quench endogenous peroxidase activity (and to enhance dendritic staining), incubated in 10% normal donkey serum and 0.3% Triton X-100 for 30 min, and then incubated overnight at 4°C in primary antibody for doublecortin. After secondary antibody incubation, sections were developed using Vector ABC kit and DAB kit. Bright-field images were taken with a Zeiss (Oberkochen, Germany) Axioplan-2 upright microscope. Stereological procedure was used to quantify labeled cells (Malberg et al., 2000). All cell counting for triple-stained sections were done using a Zeiss LSM 510 META confocal microscope.
Sholl analysis.
DCX-positive (DCX+) granule cells with tertiary, relatively untruncated dendritic branches or BrdU/DCX double-positive cells (one DCX+ cell was traced for each 35 μm hippocampal slice; n = 10–12 cells per brain for DAB-stained sections; n = 4–8 cells per brain for fluorescent staining, 5 mice per group) were traced using camera lucida at 40× magnification (Neurolucida; MicroBrightField, Williston, VT). Adult SvEvTac129 mice (16–20 weeks old) were used to obtain sparsely labeled DCX+ cells. DCX immunohistochemistry was done to maximize the labeling of dendrites (see above methods). Sholl analysis for dendritic complexity was performed using the accompanying software (NeuroExplorer; MicroBrightField), calculating dendritic complexity including dendritic length and number of intersections (branch points). All samples were number coded, and analysis was done blind to treatment. The dendritic complexity of DCX+ cells are likely to be underestimated because of the thickness of the slice (35 μm) used for DCX immunohistochemistry.
Irradiation procedure.
Mice were irradiated as described previously: three times in the course of 1 week (5 Gy per day), for a cumulative dose of 15 Gy (Santarelli et al., 2003). Mice were allowed 8–12 weeks to recover from irradiation, a time after which we no longer detected differences in inflammation markers between sham and x-ray animals (Meshi et al., 2006).
Electrophysiology.
Brains were collected from animals after deep anesthesia with halothane and decapitation, and transverse hippocampal slices (400 μm) were prepared using a vibratome. The slices were incubated in an interface chamber at 32°C and perfused with oxygenated artificial CSF (in mm: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose). Slices were allowed to equilibrate for 2 h before positioning the electrodes and beginning stimulation.
To record from the DG, the medial perforant path (MPP) was stimulated using a World Precision Instruments (Sarasota, FL) stimulation isolation unit and a bipolar tungsten electrode. Evoked potentials were recorded in the molecular layer above the upper blade of the DG using a glass capillary microelectrode filled with artificial CSF (tip resistance of 1–3 MΩ). Isolation of the MPP was confirmed by assessing paired-pulse depression (PPD) of the MPP/DG synaptic connection at 50 ms, which generated the highest level of depression (McNaughton, 1980). Input–output curves were obtained after 10 min of stable recordings. The stimulation intensity that produced one-third of the maximal response was used for the test pulses and tetanus. After 15 min of stable baseline response to test stimulation (once every 20 s), the ability to elicit LTP was assessed. LTP was induced with a weak stimulation paradigm consisting of four trains of 1 s each, 100 Hz within the train, repeated every 15 s (Saxe et al., 2006). Responses were recorded every 20 s for 60 min after LTP induction.
Novelty-suppressed feeding test.
The novelty-suppressed feeding (NSF) test is a behavior paradigm that is sensitive to chronic antidepressant treatments and acute treatments with anxiolytics (such as benzodiazepines) but not subchronic antidepressant treatments (Bodnoff et al., 1989). The test was performed as described previously (Santarelli et al., 2003): the testing apparatus consisted of a plastic box (50 × 50 × 20 cm). The floor was covered with ∼2 cm of wooden bedding. Twenty-four hours before behavioral testing, animals were deprived of all food in the home cage. At the time of testing, two food pellets were placed on a piece of round filter paper (12 cm diameter) positioned in the center of the box. The test began immediately after the animal was placed in a corner of the box. The latency to approach the pellet and begin feeding was recorded (maximum time, 5 min). Immediately afterward, the animal was transferred back to its home cage and the amount of food consumed in 5 min was measured. Each mouse was weighed before food deprivation and before testing to assess the percentage of body weight lost.
Statistical analysis.
Data were analyzed using StatView 5.0 software (SAS Institute, Cary, NC). For all experiments except the novelty-suppressed feeding test, two-way ANOVA was applied to the data. Significant interactions were resolved using post hoc ANOVAs with adjusted p values. Analyses specific to each experiment are described in Results. In the novelty-suppressed feeding test, we used the Kaplan–Meier survival analysis because of the lack of normal distribution of the data. Animals that did not eat during the 5 min testing period were censored. Mantel–Cox log-rank test was used to evaluate differences between experimental groups.
Results
Chronic fluoxetine increases cell proliferation and stimulates dendritic maturation of newborn cells
Mice were treated with vehicle, 5 d (subchronic) or 28 d (chronic) of fluoxetine. BrdU (150 mg/kg) was given 2 h before the animals were killed on the last day of treatment to label proliferating neural progenitors (Fig. 1 A). Proliferation and the number of immature neurons were assessed using BrdU and DCX immunohistochemistry, respectively (Fig. 1 D–G). Chronic, but not subchronic, fluoxetine treatment increased the number of BrdU+ cells in the granule cell layer (GCL) (F (2,12) = 4.11, p = 0.043) (Fig. 1 B). Fisher's post hoc analysis revealed significant differences between vehicle and chronic treatment groups (p = 0.015). Results are mean ± SEM of BrdU+ cells. In contrast, we did not detect a change in the total number of DCX+ cells after chronic or subchronic fluoxetine treatments (F (2,12) = 0.69, p = 0.52) (Fig. 1 C). Results are mean ± SEM of DCX+ cells.
Figure 1.
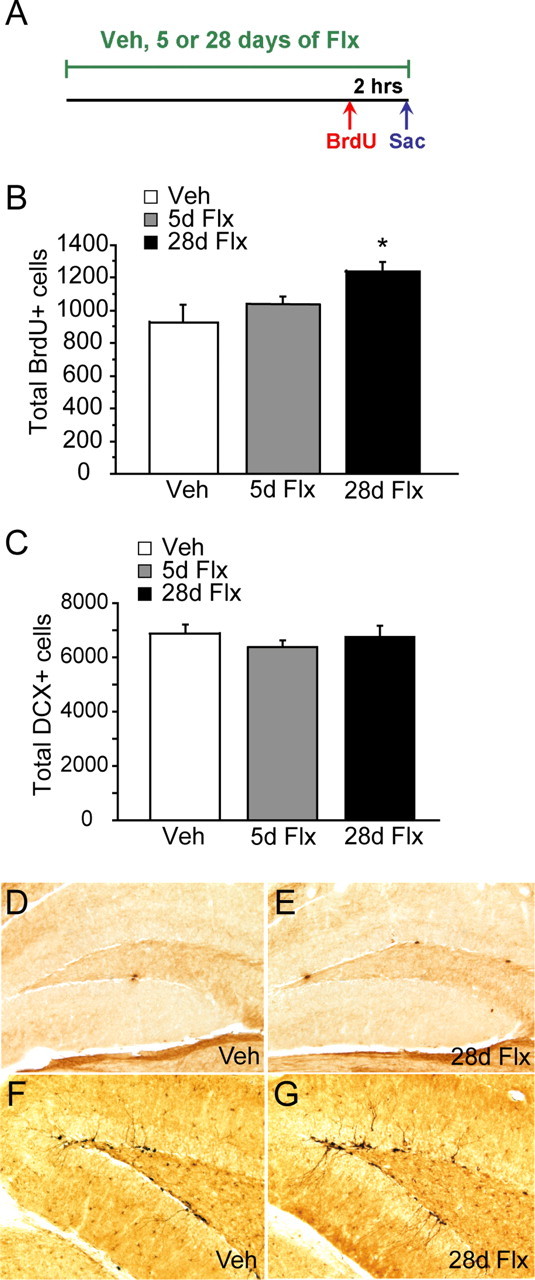
Chronic but not subchronic fluoxetine treatment increases cell proliferation but not the number of DCX+ immature granule cells in the dentate gyrus. A, Schematic diagram of BrdU administration protocol to examine cell proliferation (n = 5–6 per group). Mice were treated with vehicle (Veh), 5 d of fluoxetine (5d Flx), or 28 d of fluexeinte (28d Flx). BrdU (150 mg/kg) was given 2 h before they were killed (Sac). B, The number of BrdU+ cells increased significantly after chronic (28d Flx) but not subchronic (5d Flx) fluoxetine treatment compared with vehicle-treated animals (ANOVA, F (2,12) = 4.11, p = 0.043 for treatment). Fisher's post hoc analysis revealed significant differences between the chronic-treated group and both vehicle- and subchronic-treated groups (p < 0.05). The results are mean ± SEM of BrdU+ cells in the dentate gyrus. C, The total number of DCX+ cells did not change after subchronic and chronic fluoxetine treatment (ANOVA, F (2,12) = 0.69, p = 0.52 for treatment). The results are mean ± SEM of DCX+ cells. D–G, Images of BrdU (D, E) and DCX (F, G) immunohistochemistry after chronic fluoxetine treatment. Images were taken at 20× magnification. D, F, Vehicle-treated groups. E, G, Chronic fluoxetine-treated groups.
We next subcategorized the DCX+ cells according to their dendritic morphology: (1) DCX+ cells with no tertiary dendritic processes (Fig. 2 A), and (2) DCX+ cells with complex, tertiary dendrites (Fig. 2 B). We did not detect a change in the number of DCX+ cells with no tertiary dendrites after either chronic or subchronic fluoxetine treatment (F (2,12) = 0.98, p = 0.40) (Fig. 2 D). However, chronic but not subchronic fluoxetine significantly increased the number of DCX+ cells with tertiary dendrites (F (2,12) = 7.31, p = 0.008) (Fig. 2 C). Fisher's post hoc analysis revealed significant differences between the vehicle and chronic treated groups (p = 0.006), as well as between the subchronic and chronic treated groups (p = 0.007). The results are mean ± SEM of DCX+ cells with tertiary branches.
Figure 2.
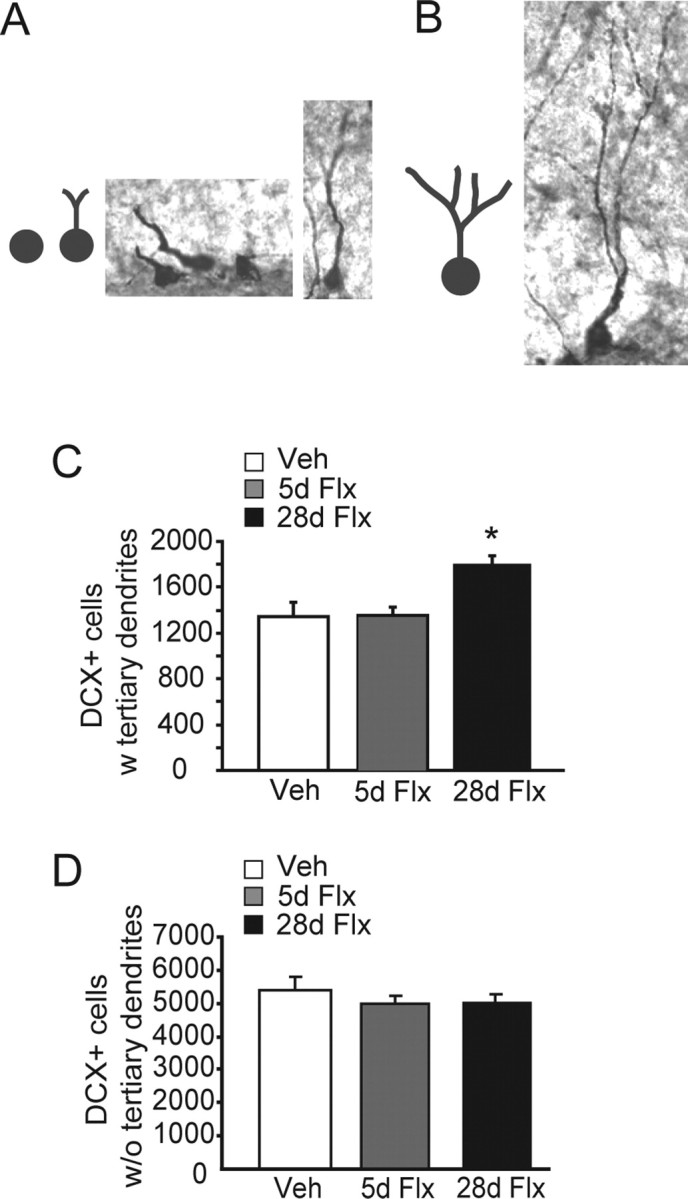
Chronic but not subchronic fluoxetine stimulates dendritic maturation of DCX+ cells. A, B, Categorization of DCX+ immature cells. We categorized DCX+ cells according to their dendritic morphology into DCX+ cells without tertiary dendrites (A) and DCX+ cells with tertiary dendrites (B) (n = 5–6 mice per group). C, Chronic [28 d of fluoxetine (28d Flx)] but not subchronic fluoxetine [5 d of fluoxetine (5d Flx)] increased the number of DCX+ cells with tertiary dendrites compared with vehicle (Veh)-treated animals (ANOVA, F (2,12) = 7.31, p = 0.008 for treatment). Fisher's post hoc analysis revealed significant differences between vehicle- and chronic-treated groups (p = 0.006), as well as subchronic- and chronic-treated groups (p = 0.007). The results are mean ± SEM of DCX+ cells with tertiary dendrites. D, Neither chronic nor subchronic fluoxetine changed the number of DCX+ cells without tertiary dendrites (ANOVA, F (2,12) = 0.98, p = 0.40 for treatment).
The dendrites of adult-born granule cells become progressively more complex during the 4 weeks after birth, a stage when the cells express DCX (Couillard-Despres et al., 2005). To further examine the effects of fluoxetine on the dendritic morphology of newborn cells, we performed Sholl analyses on DCX+ cells with tertiary dendrites (Fig. 3 A). Chronic but not subchronic fluoxetine-treated DCX+ cells displayed increased dendritic length (F (2,12) = 10.11, p = 0.003) (Fig. 3 B) and the number of intersections (F (2,12) = 9.13, p = 0.004) (Fig. 3 C). We also detected a significant treatment × radius interaction for both dendritic length (F (1,38) = 2.17, p < 0.001) and number of intersections (F (1,38) = 1.48, p = 0.043). Fisher's post hoc analysis revealed significant differences between all groups except for vehicle and subchronic group in both dendritic length and number of intersections (p < 0.05) (Fig. 3 B,C).
Figure 3.

Chronic but not subchronic fluoxetine enhances dendritic complexity of DCX+ cells. A, Representative image and traces from Sholl analyses of DCX+ cells with tertiary branches after vehicle (Veh), subchronic fluoxetine (5d Flx), and chronic fluoxetine (28d Flx) (n = 5 mice per group, 10–12 cells per mouse). B, Chronic but not subchronic fluoxetine increased dendritic length (ANOVA, F (2,12) = 10.11, p = 0.003 for treatment). We also detected a treatment × radius interaction (F (38,228) = 2.17, p < 0.001). Fisher's post hoc analysis revealed significant difference between vehicle- and chronic-treated groups (*p < 0.05). C, Chronic but not subchronic fluoxetine increased the number of intersections of DCX+ cells (ANOVA, F (2,12) = 9.13, p = 0.004 for treatment). We also detected a treatment × radius interaction (F (38,228) = 1.48, p < 0.001). Fisher's post hoc analysis revealed significant difference between vehicle- and chronic-treated groups (*p < 0.05). D–G, Representing images and traces from Sholl analysis of 3-week-old DCX+BrdU+ cells after 3 weeks of fluoxetine treatment. Hippocampal sections were double stained for DCX (D) and BrdU (E), and double-positive cells (F) were chosen to perform Sholl analysis on (n = 5 mice per group, 4–8 cells per mouse). H, Three weeks of fluoxetine (21d Flx) increased dendritic length compared with the vehicle group (Veh) (ANOVA, F (1,8) = 17.68, p = 0.003). We also detected a treatment × radius interaction (F (1,18) = 2.68, p = 0.0006). Fisher's post hoc analysis revealed significant difference between vehicle- and chronic-treated groups (*p < 0.05). I, Chronic fluoxetine also increased the number of intersections (ANOVA, F (1,8) = 21.68, p = 0.002). We also detected a treatment × radius interaction (F (1,18) = 2.34, p = 0.003). Fisher's post hoc analysis revealed significant difference between vehicle- and chronic-treated groups (*p < 0.05).
To compare dendritic morphology of DCX cells of a similar developmental stage, we injected animals with BrdU (75 mg/kg, four times over 8 h) on day 0, started fluoxetine treatment on day 1, and killed animals on day 21 (Fig. 4 A). Double fluorescent immunohistochemistry for BrdU and DCX were performed on hippocampal sections. We first identified BrdU cells that were also DCX+ and then performed Sholl analysis on the double-positive cells (Fig. 3 D–G). Three weeks of fluoxetine treatment, which is enough to achieve behavioral benefits in animal models of antidepressant action such as the novelty-suppressed feeding test (J.-W. Wang, unpublished data), enhanced both dendritic length and the number of intersections in 3-week-old granule cells (F (1,8) = 17.68, p = 0.003 for dendritic length; F (1,8) = 21.68, p = 0.002 for the number of intersections) (Fig. 3 H,I). We also detected a significant treatment × radius interaction for both dendritic length (F (1,18) = 2.68, p = 0.0006) and the number of intersections (F (1,18) = 2.34, p = 0.003). Fisher's post hoc analysis revealed significant differences between all groups except for vehicle and subchronic groups in both dendritic length and number of intersections (p < 0.05) (Fig. 3 H,I).
Figure 4.
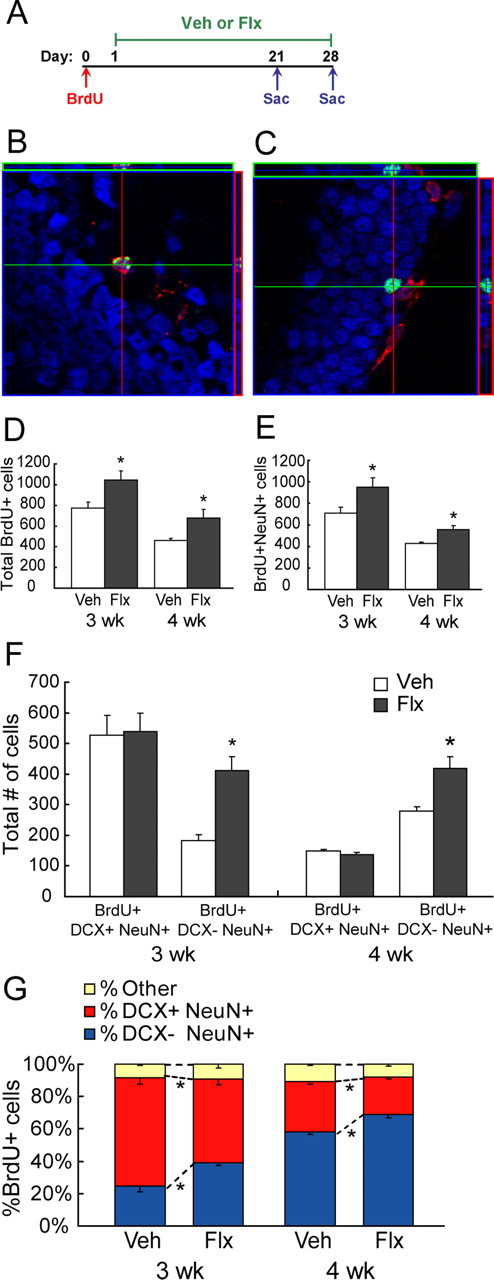
Chronic fluoxetine facilitates the maturation of newborn granule cells. A, Schematic diagram of BrdU administration protocol to examine survival of newborn cells (n = 5–6 per group). Mice were given four BrdU injections (75 mg/kg) over 8 h on day 0. Vehicle (Veh) or fluoxetine (Flx) treatment began on day 1, 24 h after the last BrdU injection. Mice were killed 3 or 4 weeks later (Sac). B, C, Confocal images of BrdU (green), DCX (red), and NeuN (blue) immunohistochemistry. D, Chronic fluoxetine increased the number of total BrdU+ cells 3 and 4 weeks later compared with vehicle-treated groups (ANOVA, F (1,16) = 12.63, *p = 0.003 for treatment; F (1,16) = 24.50, p < 0.0001 for time). E, Chronic fluoxetine increased the number of BrdU+NeuN+ cells 3 and 4 weeks later (ANOVA, F (1,16) = 8.89, *p = 0.01 for treatment; F (1,16) = 30.12, p < 0.0001 for time). F, Chronic fluoxetine increased the number of BrdU+DCX−NeuN+ cells (ANOVA, F (1,14) = 30.65, *p < 0.0001 for treatment; F (1,14) = 2.38, p = 0.14 for time) but not the number of BrdU+DCX+NeuN+ cells (ANOVA, F (1,14) = 1.47 × 10−4, p = 0.99 for treatment; F 1,14 = 62.52, p < 0.0001 for time). G, Chronic fluoxetine decreased the proportion of BrdU+NeuN+ cells that are DCX+ (percentage of BrdU cells) but increased the proportion that are DCX− (ANOVA, F (1,14) = 18.98, *p = 0.0007 for treatment; F (1,14) = 132.64, p < 0.0001 for time).
An alternative explanation to the increased dendritic complexity of DCX+ cells is that there is redistribution of DCX into dendritic processes after chronic fluoxetine. Although this is possible, other studies have demonstrated that the expression of DCX in immature granule cells is relatively stable (Couillard-Despres et al., 2005), and manipulations that either increase (voluntary exercise) or decrease (training in Morris water maze) neurogenesis do not always affect the dendritic structure of DCX cells (Couillard-Despres et al., 2005; Plumpe et al., 2006). Therefore, we favor our former explanation, which is that chronic fluoxetine stimulates dendritic maturation of newborn granule cells.
Chronic fluoxetine increases survival and facilitates maturation of newborn cells
We have demonstrated that, after chronic fluoxetine treatment, there is an increase in cell proliferation as shown by the number of BrdU+ cells, but we did not detect a difference in the number of immature granule cells using DCX immunohistochemistry. Two potential mechanisms may explain these seemingly paradoxical results: fluoxetine accelerates the maturation of immature cells, thereby shortening the DCX-expressing time window. In other words, newborn cells “mature/grow out of” the DCX-expressing stage faster, resulting in an unchanged number of DCX+ cells, or alternatively, cell death is increased in immature neurons after fluoxetine treatment, but the ones that do survive acquire more complex dendritic morphologies, thus resulting in an unchanged number of mature and immature neurons. To test these hypotheses, we designed a set of experiments to look at the effects of chronic fluoxetine on survival and maturation of newborn granule cells (Fig. 4).
As depicted in Fig. 4 A, BrdU (75 mg/kg) was given four times over 8 h on day 0 to achieve maximum labeling of proliferating progenitors over a restricted time window. Fluoxetine (18 mg · kg−1 · d−1) or vehicle administration began 24 h later and lasted for 3 or 4 weeks before the animals were killed. Hippocampal sections were triple stained for BrdU, DCX, and NeuN (Fig. 4 B,C). Image acquisition and cell counting were performed using a Zeiss LSM META 510 confocal microscopy. Consistent with previous literature, both 3 and 4 weeks of fluoxetine treatment significantly increased the total number of BrdU+ cells in the GCL (ANOVA, F (1,16) = 12.63, p = 0.003 for treatment) (Fig. 4 D). The total number of BrdU+ cells significantly decreased by ∼30% from 3 to 4 weeks after birth (ANOVA, F (1,16) = 24.50, p < 0.0001 for time), indicating that a significant number of newborn granule cells die within 4 weeks of birth. In addition, we found that the number of cells expressing both BrdU and the neuronal marker NeuN also increased after 3 and 4 weeks of fluoxetine (ANOVA, F (1,16) = 8.89, p = 0.01 for treatment; F (1,16) = 30.12, p < 0.0001 for time) (Fig. 4 E), indicating that the increase in BrdU+ cells is mostly contributed by an increase in neurons.
We then looked at the relative “maturity” of BrdU+NeuN+ cells according to whether or not they express DCX (Fig. 4 B,C). As expected, the number of immature BrdU+ granule cells (BrdU+NeuN+DCX+) decreased from 3 to 4 weeks after BrdU administration, indicating that the immature cells either die or progressively mature out of the DCX stage (Fig. 4 F). Fluoxetine did not have an effect on the number of BrdU+NeuN+DCX+ cells (ANOVA, F (1,14) = 1.47 × 10−4, p = 0.99 for treatment; F (1,14) = 62.52, p < 0.0001 for time). However, the pool of “mature” BrdU+NeuN+DCX− cells significantly increased after both 3 and 4 weeks of fluoxetine (ANOVA, F (1,14) = 30.65, p < 0.0001 for treatment; F (1,14) = 2.38, p = 0.14 for time) (Fig. 4 F). These results suggest that the increase in surviving BrdU+NeuN+ cells after chronic fluoxetine treatment mostly consists of DCX−, mature granule cells. This result is further validated by looking at the proportion of BrdU+NeuN+ cells that are either DCX+ or DCX− (Fig. 4 G). After 3 weeks of fluoxetine treatment, the proportion of BrdU+NeuN+ cells that express DCX significantly decreased from 67.04 ± 4.14% in the vehicle group to 51.64 ± 3.71% in the fluoxetine group, whereas the proportion of BrdU+NeuN+ cells that ceased to express DCX significantly increased from 24.72 ± 3.80 in the vehicle group to 39.24 ± 1.78 in the fluoxetine group (Fig. 4 G); similar effects are seen in the 4 week survival group (ANOVA, F (1,14) = 18.98, p = 0.0007 for treatment; F (1,14) = 132.64, p < 0.0001 for time) (Fig. 4 G). We did not detect an effect of fluoxetine on the proportion of BrdU+ cells that did not express the neuronal marker NeuN (ANOVA, F (1,14) = 0.235, p = 0.64 for treatment; F (1,14) = 0.183, p = 0.68 for time), indicating that chronic fluoxetine does not change the fate determination of early progenitors.
To determine the effect of subchronic fluoxetine treatment on maturation of immature neurons, we injected another group of mice with BrdU (150 mg/kg, one time) on day 0, started fluoxetine on day 1, and killed the animals on day 5 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Subchronic fluoxetine treatment did not change the survival of immature neurons as measured by BrdU (F (1,8) = 0.22, p = 0.65). In addition, 5 d of fluoxetine did not change the proportion of BrdU cells that are NeuN+ (F (1,8) = 0.047, p = 0.83) or the transition of BrdU+ immature cells from DCX+NeuN− stage (F (1,8) = 0.039, p = 0.85) to DCX+NeuN+ stage (F (1,8) = 0.28, p = 0.61). Therefore, our results demonstrate that chronic but not subchronic fluoxetine facilitates maturation of newborn granule cells.
Chronic and subchronic fluoxetine have differential effects on hippocampal synaptic plasticity
To determine whether or not the new neurons generated by chronic fluoxetine treatment functionally integrate into the local circuit and contribute to network plasticity, we performed field electrophysiological recordings on hippocampal slices from vehicle- or fluoxetine-treated animals. We used the previously developed focal x-irradiation protocol to completely and specifically ablate hippocampal neurogenesis (Santarelli et al., 2003). Animals were then treated with vehicle, 5 or 28 d of fluoxetine. We confirmed the successful ablation using BrdU and DCX immunohistochemistry and found that both the number of BrdU+ cells (ANOVA, F (1,15) = 353.85, p < 0.0001) as well as DCX+ cells (ANOVA, F (1,15) = 274.80, p < 0.0001) decreased dramatically after irradiation (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Consistent with our previous results, we detected an increase in the number of BrdU+ cells (ANOVA, F (1,15) = 8.17, p = 0.012) but not the number of DCX+ cells (ANOVA, F (1,15) = 0.556, p = 0.468) in sham animals after 28 d of fluoxetine (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Field EPSPs (fEPSPs) were evoked by stimulating the MPP and recording in the molecular layer of the upper blade of the DG. Paired-pulse depression (50 ms interstimulus interval) was assessed to confirm that recordings were done in the medial perforant path (McNaughton, 1980). Chronic fluoxetine suppressed paired-pulse depression at stimulation intensities that generated one-third of the maximal response (ANOVA, F (1,29) = 9.05, p = 0.005 for treatment; F (1,29) = 0.95, p = 0.34 for irradiation) (Fig. 5 B) and a constant stimulation intensity of 60 μA (ANOVA, F (1,27) = 7.06, p = 0.013 for treatment; F (1,23) = 0.28, p = 0.61 for irradiation) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We did not detect an effect of subchronic fluoxetine on paired-pulse depression at either stimulation intensities (ANOVA, F (1,23) = 0.17, p = 0.68 for treatment; F (1,23) = 0.31, p = 0.58 for irradiation at one-third of the maximum; F (1,23) = 0.014, p = 0.91 for treatment; F (1,23) = 1.49, p = 0.23 for irradiation at 60 μA) (Fig. 5 A) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Paired-pulse depression was not affected by x-irradiation (p > 0.05). The reduced PPD after chronic fluoxetine is likely attributable to changes in either the intrinsic properties of the release process (Mennerick and Zorumski, 1995) or a feedback of glutamate onto presynaptic terminals (Brown and Reymann, 1995). We then recorded input–output relationships. The input–output functions were fitted using a four-parameter logistic sigmoid function (DeLean et al., 1978). Both subchronic and chronic fluoxetine treatments significantly increased input–output functions in the MPP/DG (repeated-measures ANOVA, F (1,36) = 11.46, p = 0.0017 for treatment in the subchronic group; F (1,27) = 16.72, p = 0.0003 for treatment in the chronic group) (Fig. 5 C,D). The effects of fluoxetine on input–output relationships were not sensitive to x-irradiation (ANOVA, F (1,36) = 0.62, p = 0.44 for irradiation in the subchronic group; F (1,27) = 0.23, p = 0.63 for irradiation in the chronic group). Although chronic treatment was required to produce the effects on PPD, fluoxetine induced rapid changes on input–output functions after only 5 d of treatment. In addition, the fluoxetine-induced effects on PPD and input–output relationships were not sensitive to x-irradiation, suggesting that these effects were not dependent on the presence of newborn neurons.
Figure 5.
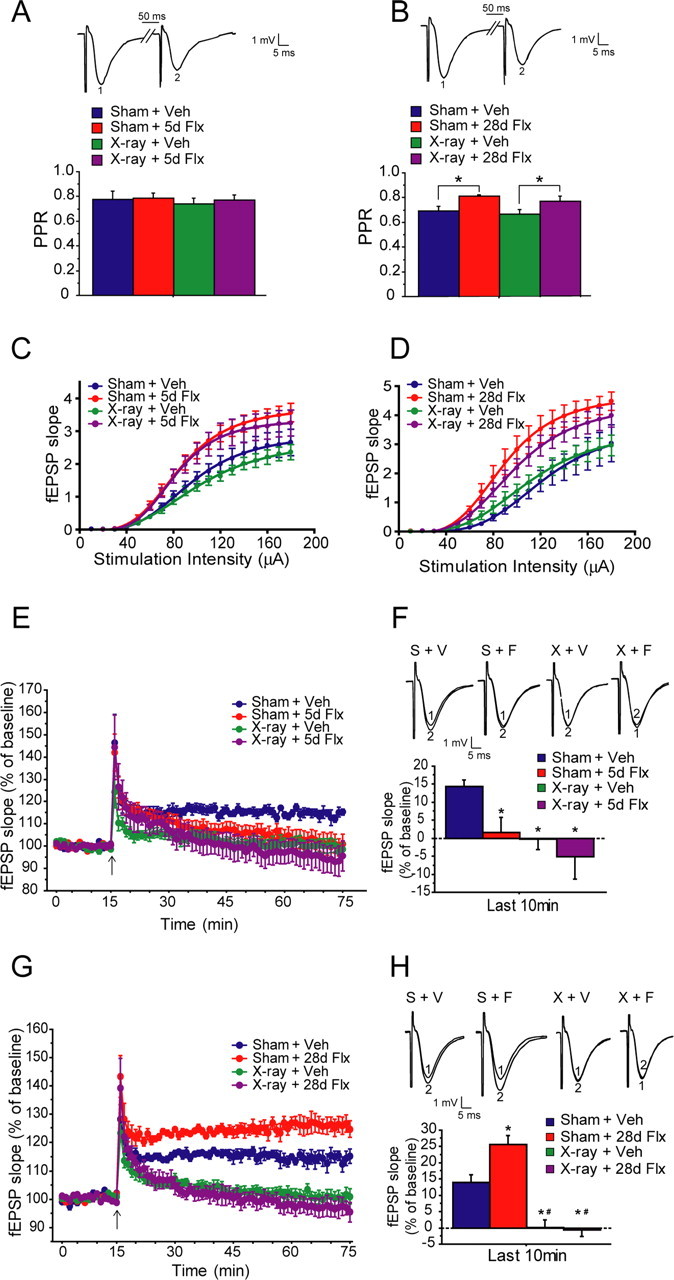
Effects of subchronic and chronic fluoxetine on hippocampal synaptic plasticity. A, B, Chronic fluoxetine (28d Flx; B) but not subchronic fluoxetine (5d Flx; A) reduces paired-pulse depression in both sham (Sham) and x-irradiated (x-ray) animals at stimulation intensity that elicited one-third of the maximal response compared with the vehicle group (Veh) (ANOVA, F (1,29) = 9.05, *p = 0.005 for chronic treatment; F (1,29) = 0.95, p = 0.34 for irradiation; F (1,23) = 0.17, p = 0.68 for subchronic treatment; F (1,23) = 0.31, p = 0.58 for irradiation). Inset, Representative traces of first response (1) and second response (2) (PPR, paired-pulse ratio, second response/first response). C, D, Both subchronic (C) and chronic (D) fluoxetine increased input–output relationships in both sham and x-irradiated animals (ANOVA, F (1,36) = 11.46, p = 0.0017 in subchronic group for treatment; F (1,36) = 0.62, p = 0.44 for irradiation; F (1,27) = 16.72, p = 0.0003 in chronic group for treatment; F (1,27) = 0.23, p = 0.63 for irradiation). Curves are fitted with a four-parameter logistic formula (McNaughton, 1980). E, Subchronic fluoxetine suppressed ACSF–LTP, and x-irradiation completely eliminates ACSF–LTP. F, ANOVA performed on the last 10 min of LTP recording revealed a significant main effect of irradiation (F (1,25) = 7.28, p = 0.012) as well as a main effect of subchronic fluoxetine (F (1,25) = 4.84, p = 0.037) (F). S, Sham; X, x-irradiation, V, vehicle; F, fluoxetine; Fisher's post hoc analysis were performed between individual groups (*p < 0.05). G, Chronic fluoxetine enhanced ACSF–LTP, and x-irradiation completely blocked LTP. Insets show averages of five consecutive fEPSPs at baseline (1) and in the last 10 min of LTP recordings (2). H, ANOVA performed on the last 10 min of LTP recording revealed a significant main effect of irradiation (F (1,27) = 63.01, p < 0.0001), a main effect of chronic fluoxetine (F (1,27) = 4.61, p = 0.041), as well as an irradiation × treatment interaction (F (1,27) = 6.21, p = 0.019). Fisher's post hoc analysis were performed between individual groups (*p < 0.05).
We and others have shown previously that a form of long-term potentiation elicited in the MPP/DG pathway using a weak stimulation paradigm in the absence of GABA blockers (ACSF–LTP) is sensitive to manipulations that block hippocampal neurogenesis (Snyder et al., 2001; Saxe et al., 2006). We hypothesized that, if the fluoxetine-induced new neurons functionally integrate into the local hippocampal circuit, we would see an enhancement of synaptic plasticity as assessed by ACSF–LTP. After subchronic fluoxetine treatment, we observed a suppression of ACSF–LTP in both sham and x-irradiated animals (Fig. 5 E,F). Two-way ANOVA performed on the average of the last 10 min of LTP recordings revealed a significant main effect of irradiation (F (1,25) = 7.28, p = 0.012), a main effect of subchronic fluoxetine (F (1,25) = 4.84, p = 0.037), but no irradiation × treatment interaction (F (1,25) = 0.99, p = 0.33). Fisher's post hoc analysis revealed significant differences between sham vehicle group and the other three groups (sham fluoxetine, x-ray vehicle, and x-ray fluoxetine, respectively) (p < 0.05). Therefore, we conclude that the suppression of LTP by subchronic fluoxetine does not depend on neurogenesis.
After chronic treatment with fluoxetine, however, we see the opposite effect. Chronic fluoxetine enhanced ACSF–LTP in sham animals. LTP was completely blocked in x-irradiated animals in both vehicle and chronic fluoxetine-treated groups (Fig. 5 G,H). Two-way ANOVA revealed a main effect of irradiation (F (1,27) = 63.01, p < 0.0001), a main effect of chronic fluoxetine (F (1,27) = 4.61, p = 0.041), as well as an irradiation × treatment interaction (F (1,27) = 6.21, p = 0.019). These results suggest that fluoxetine enhances ACSF–LTP in a time course that resembles the delayed onset of its antidepressant action. Because the fluoxetine-induced enhancing effect is not present in x-irradiated animals, it suggests that hippocampal neurogenesis is required to produce the increase in LTP. The inhibitory effects of subchronic fluoxetine on ACSF–LTP is likely the result of increased synaptic transmission that saturates the potential to further induce LTP (Stewart and Reid, 2000). However, after chronic fluoxetine treatment, increased neurogenesis and enhanced maturation of young cells may cause readjustments in the local circuitry, therefore counteracting the saturating effect and resulting in an increased ability to induce LTP, e.g., a net increase in ACSF–LTP.
Behavioral effects of fluoxetine require the presence of adult neurogenesis
Do the neurogenesis-dependent effects of fluoxetine on dendritic morphology, maturation, and LTP correlate with the behavioral effects of antidepressants? We irradiated another group of animals and looked at behavior after fluoxetine treatment. We used a chronic model of antidepressant/anxiolytic action, the NSF test (Santarelli et al., 2003), to examine the behavioral effects of fluoxetine on days 5 and 28 of the treatment. In the NSF paradigm, conflicting motivations are produced by presenting a food-deprived animal with a reward (food) within the context of a novel, aversive environment. The NSF test is among the few behavioral paradigms that can differentiate chronic versus subchronic responses to antidepressant treatments, using the latency to begin eating as an index of antidepressant/anxiety-like behavior.
After 5 d of fluoxetine, we did not detect an effect of treatment in either sham or x-irradiated animals (Fig. 6 A,B) (Kaplan–Meier survival analysis was used because of a lack of normal distribution of the data, Mantel–Cox log-rank test, p = 0.038 for treatment; p < 0.05 between sham fluoxetine and the other three groups; p > 0.05 between all other groups). Food consumption in the home cage was not different between groups (data not shown). These results indicate that chronic administration is required for the behavioral effects of fluoxetine and that neurogenesis is necessary to produce these effects. Our results confirmed therefore the conclusions from recent studies showing that the behavioral effects of fluoxetine in several models of antidepressant action are dependent on adult neurogenesis (Santarelli et al., 2003; Airan et al., 2007).
Figure 6.

Behavioral effects of fluoxetine depend on adult neurogenesis. Novelty-suppressed feeding test on day 5 (A, B) and day 28 (C, D) of vehicle (Veh) or fluoxetine treatment. A, Five days of fluoxetine (5d Flx) did not reduce latency to feed in sham (Sham) or x-irradiated (x-ray) animals (Cum. Survival, cumulative survival, percentage of animals that have not eaten) (Kaplan–Meier survival analysis, Mantel–Cox log-rank test, p > 0.05). B, Box plot of latency to feed after 5 d of vehicle or fluoxetine. C, Twenty-eight days of fluoxetine (28d Flx) reduced latency to feed in sham but not x-ray animals (Kaplan–Meier survival analysis, Mantel–Cox log-rank test, p = 0.038 for treatment; *p < 0.05 between sham fluoxetine and the other three groups; p > 0.05 for all other groups). D, Box plot of latency to feed after 28 d of fluoxetine treatment. The box plot displays 10, 25, 50, 75, and 90% percentiles.
Discussion
In the present study, we showed that chronic fluoxetine increased both proliferation of progenitors and survival of immature neurons in the adult DG of the hippocampus, which is consistent with several previous studies (Malberg et al., 2000; Santarelli et al., 2003; Encinas et al., 2006). We have demonstrated for the first time that chronic but not subchronic fluoxetine stimulates maturation of immature granule cells: first, a larger fraction of DCX+ cells possessed tertiary dendrites after chronic fluoxetine treatment; and second, these immature, DCX+ cells displayed more complex dendritic arborization after chronic fluoxetine. Overall, newborn neurons undergo an accelerated maturation after chronic fluoxetine treatment, as shown by the increased proportion of newborn cells that ceased to express the immature neuronal marker DCX (Fig. 7). The delayed effects of fluoxetine to stimulate maturation of young granule cells parallel the delayed onset of its behavioral effects. Interestingly, electroconvulsive therapy (ECT), one of the fastest and most effective antidepressant treatments (American Psychiatric Association, 1990), stimulates neurogenesis more rapidly than fluoxetine (Warner-Schmidt and Duman, 2007). In addition, the induction of seizures, a prerequisite for achieving therapeutic effects during ECT (American Psychiatric Association, 1990; Sackeim et al., 1996), stimulates dendritic development and maturation (Overstreet-Wadiche et al., 2006). Specifically, after seizure induction, newborn granule cells display increased dendritic outgrowth and start receiving glutamatergic synaptic input earlier than those from non-induced animals (Overstreet-Wadiche et al., 2006). These studies together with our results suggest that the processes that promote the maturation of newborn cells may be a target for future drug development.
Figure 7.
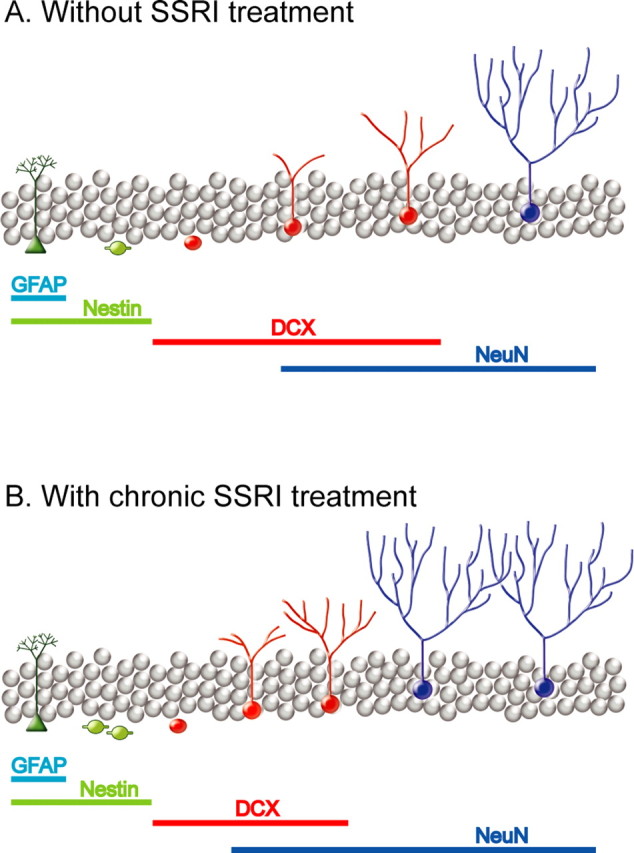
Chronic fluoxetine stimulates dendritic maturation and synaptic plasticity of newborn granule cells, a possible mechanism for antidepressant action. A and B, from left to right, shows anatomical and functional stages during neuronal differentiation and maturation, including quiescent, radial glia-like progenitors (green), rapidly amplifying neural progenitors (light green), immature granule cells (red), and mature granule cells. Bottom panels show immunohistochemical markers for each stage. We can conclude from this study and others that fluoxetine stimulate adult neurogenesis in a multifold manner. Chronic fluoxetine treatment: first, increases proliferation of neural progenitors; second, stimulates dendritic branching as well as facilitates maturation; third, enhances survival of immature granule cells; fourth, enables young neurons to functionally integrate into the local hippocampal circuit, resulting in an enhancement of long-term synaptic plasticity. Finally, these synergistic actions lead to an improved behavior outcome. (Malberg et al., 2000a; Encinas et al., 2006).
In addition, we also examined how fluoxetine modulates activity in the MPP/DG local circuitry. We demonstrated that fluoxetine dynamically regulates synaptic plasticity in the DG. We and others have shown that ACSF–LTP is dependent on the presence of adult neurogenesis (Snyder et al., 2001; Saxe et al., 2006). Here we have shown that chronic fluoxetine enhances ACSF–LTP in sham animals, whereas LTP is completely blocked in both vehicle- and fluoxetine-treated x-irradiated animals. We also observed effects of fluoxetine that were neurogenesis independent: chronic but not subchronic fluoxetine reduced PPD, whereas both subchronic and chronic fluoxetine led to increased input–output functions. The fluoxetine-induced effects on PPD and input–output functions were not blocked by x-irradiation, indicating that, although fluoxetine has effects on long-term plasticity that are neurogenesis dependent, there are also changes in short-term plasticity and synaptic transmission that may take place in both mature and immature granule cells. In the case of ACSF–LTP, the effect of an increase or a decrease in neurogenesis are readily detected probably because this phenomenon primarily recruits young neurons, which are more excitable and less inhibited by GABA (Wang et al., 2000; Snyder et al., 2001; Ge et al., 2006; Saxe et al., 2006). In contrast, in the case of PPD and input–output functions, both immature and mature neurons may be recruited, which would make the contribution of immature neurons negligible because they only represent a small fraction of the total granule cell population.
A recent study identified a critical period between 1 and 1.5 months after mitosis, a stage when the newborn neurons have ceased to express DCX, when newborn neurons exhibit enhanced LTP (Ge et al., 2007). Our results suggest that chronic fluoxetine facilitates the maturation of newborn granule cells. Specifically, only the number of BrdU+NeuN+DCX− neurons, but not the number of BrdU+NeuN+DCX+ neurons, increased after chronic fluoxetine. Therefore, along with results from the previous study by Ge et al., we can hypothesize that it is the more mature, BrdU+NeuN+DCX− neurons that make a major contribution to ACSF–LTP. It is also possible that it is not merely the cell number but also the increased dendritic complexity that plays a major role in the enhancement of ACSF–LTP. Another possibility is that chronic fluoxetine enables adult-born cells to enter the critical period earlier and expand the critical period during which these neurons are capable of exhibiting LTP. However, chronic fluoxetine may also push the newborn neurons out of the critical age quicker. Additional studies are needed to look at the effect of fluoxetine on LTP in individual newborn granule cells to distinguish between these possibilities.
To assess the behavioral consequences of the physiological actions of chronic fluoxetine, we investigated the effects of fluoxetine in the NSF test. Chronic fluoxetine decreased the latency to feed, a measure of anxiety/depression, and the effect disappeared after x-irradiation. Therefore, similar to ACSF–LTP, the effect of fluoxetine on the NSF test is dependent on both chronic treatment and the presence of young neurons. We conclude that the enhanced dendritic development and facilitated maturation of immature neurons, as well as enhanced ACSF–LTP, are good correlates of the behavioral effects of fluoxetine. This may be especially valuable because there has long been a paucity of chronic models of antidepressant action as well as cellular- and circuit-level readouts for chronic antidepressant action.
From these and other results, we can state that the effects of fluoxetine on adult neurogenesis are multifold (Fig. 7): first, chronic fluoxetine treatment increases proliferation of neural progenitors (Encinas et al., 2006); second, chronic fluoxetine stimulates dendritic branching and facilitates maturation of immature granule cells; third, chronic fluoxetine enhances the survival of young neurons; fourth, these young neurons functionally integrate into the local hippocampal circuit and produce an enhancement of long-term synaptic plasticity. Finally, these synergistic actions may lead to improved behavioral outcomes. However, additional studies are needed to dissect which of these effects are pivotal for the anxiolytic/antidepressant action of SSRIs.
It is likely that the fluoxetine-induced effects on dendritic morphology, maturation, and synaptic plasticity are mediated by neurotrophic factors. Growth factors, including brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF), as well as the activation of their corresponding receptors TrkB and Flk-1, are upregulated after chronic fluoxetine treatment (Castrén et al., 2007; Rantamäki et al., 2007; Warner-Schmidt and Duman, 2007). Activation of their downstream signaling pathways was shown to enhance maturation and dendritic development of granule cells (Fujioka et al., 2004; Chen et al., 2006; Warner-Schmidt and Duman, 2007). The growth-related effects we have described may contribute to the delayed onset of action of SSRIs. However, they do not exclude additional mechanisms such as the progressive desensitization of the 5-HT1A and 5-HT1B autoreceptors (Millan, 2006). In fact, it is possible that both phenomena contribute to the delayed onset of action of antidepressants: first, a desensitization of the autoreceptors; second, an activation of postsynaptic receptors (e.g., the hippocampal 5-HT1A receptors), followed by the release of growth factors (e.g., VEGF and BDNF), which in turn cause an increased proliferation of neural progenitors and facilitated maturation of young hippocampal neurons, and finally result in a normalization of the behavioral output compromised by depression. An elucidation of the relative contribution of these different mechanisms is likely to pave the way for the development of faster acting antidepressants.
Footnotes
This work was supported by National Alliance for Research on Schizophrenia and Depression (R.H.) and National Institute of Mental Health Grant R01 MH068542 (R.H.). We thank Dr. Jack Martin for the use of Camera Lucida, Drs. M. Drew and E. D. Leonardo for help with irradiating animals, and I. Mendez for behavioral testing. We thank Drs. H. Scharfman, A. DeCostanzo, A. Dranovsky, J. Gordon, and E. D. Leonardo for their insightful comments on this manuscript.
References
- Airan RD, Meltzer LA, Roy M, Gong Y, Chen H, Deisseroth K. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. The practice of ECT: recommendations for treatment, training and privileging. Convuls Ther. 1990;6:85–120. [PubMed] [Google Scholar]
- Anderson GM, Barr CS, Lindell S, Durham AC, Shifrovich I, Higley JD. Time course of the effects of the serotonin-selective reuptake inhibitor sertraline on central and peripheral serotonin neurochemistry in the rhesus monkey. Psychopharmacology (Berl) 2005;178:339–346. doi: 10.1007/s00213-004-2011-7. [DOI] [PubMed] [Google Scholar]
- Bodnoff SR, Suranyi-Cadotte B, Quirion R, Meaney MJ. A comparison of the effects of diazepam versus several typical and atypical anti-depressant drugs in an animal model of anxiety. Psychopharmacology (Berl) 1989;97:277–279. doi: 10.1007/BF00442264. [DOI] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci. 2003;17:2042–2046. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- Brown RE, Reymann KG. Metabotropic glutamate receptor agonists reduce paired-pulse depression in the dentate gyrus of the rat in vitro. Neurosci Lett. 1995;196:17–20. doi: 10.1016/0304-3940(95)11825-h. [DOI] [PubMed] [Google Scholar]
- Castrén E, Võikar V, Rantamäki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol. 2007;7:18–21. doi: 10.1016/j.coph.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci USA. 2006;103:8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Fujioka T, Fujioka A, Duman RS. Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J Neurosci. 2004;24:319–328. doi: 10.1523/JNEUROSCI.1065.03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007;54:559–566. doi: 10.1016/j.neuron.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kreiss DS, Lucki I. Effects of acute and repeated administration of antidepressant drugs on extracellular levels of 5-hydroxytryptamine measured in vivo. J Pharmacol Exp Ther. 1995;274:866–876. [PubMed] [Google Scholar]
- Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL. Evidence for two physiologically distinct perforant pathways to the fascia dentata. Brain Res. 1980;199:1–19. doi: 10.1016/0006-8993(80)90226-7. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshi D, Drew MR, Saxe M, Ansorge MS, David D, Santarelli L, Malapani C, Moore H, Hen R. Hippocampal neurogenesis is not required for behavioral effects of environmental enrichment. Nat Neurosci. 2006;9:729–731. doi: 10.1038/nn1696. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, Zhang YJ, Nestler EJ, Duman RS. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci. 2002;22:3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bromberg DA, Bensen AL, Westbrook GL. Seizures accelerate functional integration of adult-generated granule cells. J Neurosci. 2006;26:4095–4103. doi: 10.1523/JNEUROSCI.5508-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumpe T, Ehninger D, Steiner B, Klempin F, Jessberger S, Brandt M, Romer B, Rodriguez GR, Kronenberg G, Kempermann G. Variability of doublecortin-associated dendrite maturation in adult hippocampal neurogenesis is independent of the regulation of precursor cell proliferation. BMC Neurosci. 2006;7:77. doi: 10.1186/1471-2202-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Jacobs BL. Pilocarpine-induced status epilepticus increases cell proliferation in the dentate gyrus of adult rats via a 5-HT1A receptor-dependent mechanism. Brain Res. 2003;966:1–12. doi: 10.1016/s0006-8993(02)03989-6. [DOI] [PubMed] [Google Scholar]
- Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Männistö PT, Castrén E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- Rutter JJ, Gundlah C, Auerbach SB. Increase in extracellular serotonin produced by uptake inhibitors is enhanced after chronic treatment with fluoxetine. Neurosci Lett. 1994;171:183–186. doi: 10.1016/0304-3940(94)90635-1. [DOI] [PubMed] [Google Scholar]
- Sackeim HA, Luber B, Katzman GP, Moeller JR, Prudic J, Devanand DP, Nobler MS. The effects of electroconvulsive therapy on quantitative electroencephalograms. Relationship to clinical outcome. Arch Gen Psychiatry. 1996;53:814–824. doi: 10.1001/archpsyc.1996.01830090060009. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci USA. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Kee N, Wojtowicz JM. Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol. 2001;85:2423–2431. doi: 10.1152/jn.2001.85.6.2423. [DOI] [PubMed] [Google Scholar]
- Stewart CA, Reid IC. Repeated ECS and fluoxetine administration have equivalent effects on hippocampal synaptic plasticity. Psychopharmacology (Berl) 2000;148:217–223. doi: 10.1007/s002130050045. [DOI] [PubMed] [Google Scholar]
- Suckow RF, Zhang MF, Cooper TB. Sensitive and selective liquid-chromatographic assay of fluoxetine and norfluoxetine in plasma with fluorescence detection after precolumn derivatization. Clin Chem. 1992;38:1756–1761. [PubMed] [Google Scholar]
- van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Scott BW, Wojtowicz JM. Heterogenous properties of dentate granule neurons in the adult rat. J Neurobiol. 2000;42:248–257. [PubMed] [Google Scholar]
- Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA. 2007;104:4647–4652. doi: 10.1073/pnas.0610282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Licinio J. Research and treatment approaches to depression. Nat Rev Neurosci. 2001;2:343–351. doi: 10.1038/35072566. [DOI] [PubMed] [Google Scholar]