

Abstract
The cGMP/cGMP-dependent protein kinase I (cGKI) signaling pathway plays an important role in spinal nociceptive processing. However, downstream targets of cGKI in this context have not been identified to date. Using a yeast two-hybrid screen, we isolated cysteine-rich protein 2 (CRP2) as a novel cGKI interactor in the spinal cord. CRP2 is expressed in laminas I and II of the mouse spinal cord and is colocalized with cGKI, calcitonin gene-related peptide, and isolectin B4. Moreover, the majority of CRP2 mRNA-positive dorsal root ganglion (DRG) neurons express cGKI and peripherin. CRP2 is phosphorylated in a cGMP-dependent manner, and its expression increases in the spinal cord and in DRGs after noxious stimulation of a hindpaw. To elucidate the functional role of CRP2 in nociception, we analyzed mice with a targeted deletion of CRP2. CRP2-deficient (CRP2−/−) mice demonstrate normal behavioral responses to acute nociception and after axonal injury of the sciatic nerve, but increased nociceptive behavior in models of inflammatory hyperalgesia compared with wild-type mice. Intrathecal administration of cGMP analogs increases the nociceptive behavior in wild-type but not in CRP2−/− mice, indicating that the presence of CRP2 is important for cGMP-mediated nociception. These data suggest that CRP2 is a new downstream effector of cGKI-mediated spinal nociceptive processing and point to an inhibitory role of CRP2 in the generation of inflammatory pain.
Keywords: CRP2, cGKI, spinal cord, DRG, knock-out mice, pain behavior
Introduction
The perception of pain is initiated by activation of peripheral terminals of primary afferent nociceptive neurons. The cell bodies of these first-order neurons are localized in dorsal root and trigeminal ganglia, and their axons form synapses with second-order neurons in the dorsal horn of the spinal cord. The nociceptive sensory information is then processed and relayed to the brain, where the sensation of pain is ultimately experienced. Persistent activation of nociceptors, as in the case of peripheral tissue injury and inflammation, results in an increased pain sensitivity, which is mediated both by sensitization of nociceptors (peripheral sensitization) and by neuroplastic changes in the dorsal horn of the spinal cord and in the brain (central sensitization). This sensitization of the nociceptive system contributes to chronic pain, a major medical problem (Woolf, 1983; Woolf and Salter, 2000; Julius and Basbaum, 2001; Scholz and Woolf, 2002).
There is considerable evidence that cGMP signaling plays an important role in the modulation of nociceptive neuron sensitivity (Meller and Gebhart, 1993; Salter et al., 1996; Lin et al., 1997; Tao and Johns, 2002). A major target of cGMP in nociceptive processing is cGMP-dependent protein kinase I (cGKI, also known as PKG-I), of which the α-isoform is expressed in small- and medium-diameter dorsal root ganglion (DRG) neurons and in superficial laminas of the spinal cord (Qian et al., 1996; Tao et al., 2000). The expression of cGKI in the spinal cord is upregulated after noxious stimulation, and intrathecal administration of cGKI inhibitors provided profound antinociceptive effects in various animal models of pain (Tao and Johns, 2000; Tao et al., 2000; Schmidtko et al., 2003; Song et al., 2006). The essential contribution of cGKI to the mechanisms underlying pain hypersensitivity is further supported by strongly reduced inflammatory hyperalgesia in cGKI knock-out mice (Tegeder et al., 2004).
Several proteins have been identified to be phosphorylated by cGKI in vivo, most of them being expressed in smooth muscle cells and platelets. Neuronal cGKI substrates include G substrate and inositol-3-phosphate receptor type I in the cerebellum, and vasodilator-stimulated phosphoprotein in the hippocampus (Hofmann et al., 2006). In contrast to these tissues, to date no downstream targets of cGKI have been established in the spinal cord. To elucidate signaling proteins downstream of cGKI, we performed a yeast-two hybrid screen of spinal cord cDNA using cGKI as bait, thereby identifying cysteine-rich protein 2 (CRP2) (Okano et al., 1993) as cGKI interactor. Immunohistological stainings and in vivo phosphorylation experiments confirmed CRP2 as a target of cGMP/cGKI signaling in the spinal cord and in DRGs. To elucidate the functional role of CRP2 in nociception, we generated CRP2-deficient (CRP2−/−) mice by targeted deletion of the CRP2 gene and analyzed their nociceptive behavior in models of acute, inflammatory, and neuropathic pain.
Materials and Methods
Yeast two-hybrid interactor screening
To perform a cGKI interaction screen, a cDNA library of rat spinal cord was constructed using the SuperScript Choice System (Invitrogen, Karlsruhe, Germany). The cDNA library (1 × 106 independent clones) was cloned into the yeast expression plasmid pJG4–5 containing the “acid loop”-DNA activation domain (Ausubel, 2003a,b), yielding pJG4–5/cDNA. cGKIα and cGKIβ cDNAs were used as baits and were inserted into the expression plasmid pEG202 (Ausubel, 2003a,b) in-frame with the DNA-binding domain of LexA, yielding pEG202-LexA/cGKIα or pEG202-LexA/cGKIβ. The reporter plasmid pSH18–34 (Ausubel, 2003a,b), the bait pEG202-LexA/cGKI, and the prey pJG4–5/cDNA were cotransformed in the yeast strain EGY48 using the lithium acetate method (Gietz et al., 1995). Interacting clones were identified by their blue color on 5-bromo-4-chloro-3-indolyl-β-d-galactoside-containing agar plates and their ability to grow on agar plates lacking leucine, tryptophan, histidine, and uracil. The cDNA of positive yeast clones was amplified with PCR and identified by sequence analysis. In a rescreen, the cDNA of positive clones was cotransformed into yeast with bait, without bait, or with an unrelated bait.
CRP2−/− mice
Targeted deletion of CRP2 in mice was performed using the common Cre/loxP system. Briefly, the targeting vector was constructed such that the exons 2–7 were flanked by a single loxP site and a floxed neo/tk cassette. This strategy allows an excision of the neo/tk cassette and the exons 2–7 yielding L1/+ clones. Correctly targeted L1/+ clones were injected into C57BL/6 blastocysts, and the resulting chimeras were mated with 129/Sv mice. Heterozygous offspring were intercrossed to produce wild-type (WT) and CRP2−/− mice on an inbred 129/Sv background. Mice were bred and maintained at the animal facility of the Institute of Pharmacy, Department Pharmacology and Toxicology, University of Tübingen (Tübingen, Germany). All experiments were approved by the local Ethics Committee for Animal Research.
Immunohistochemistry
Brain tissue.
Immunohistochemistry was performed on 10 μm cryosections from WT and CRP2−/− brains perfusion fixed with 2% paraformaldehyde. After preincubation with 10% normal donkey serum [in 1% BSA, 0.5% Triton X-100, and 0.05 m Tris-buffered saline (TBS)] and rinsing with TBS, serial slices were incubated with rabbit anti-CRP2 (Huber et al., 2000) (1:1000 in 1% BSA, 0.5% Triton X-100, 0.05 m TBS) overnight and tagged with Alexa 555-conjugated donkey anti-rabbit IgG (1:1000 in 1% BSA, 0.5% Triton X-100, 0.05 m TBS) after rinsing with TBS. CRP2 immunofluorescence was analyzed using a Bio-Rad (Hercules, CA) MRC1000 confocal laser scanning microscope equipped with a krypton-argon laser and attached to Nikon (Düsseldorf, Germany) Diaphot 300. Specificity of anti-CRP2 was confirmed using slices from CRP2−/− tissues.
Spinal cord and dorsal root ganglia.
Mice were intracardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.1 m PBS, pH 7.4, under deep ketamine/xylazine anesthesia. The lumbar spinal cords were dissected, postfixed in the same fixative for 2.5 h, and cryoprotected in 30% sucrose overnight. Tissues were frozen in tissue-freezing medium on dry ice and cryostat sectioned at a thickness of 8–16 μm. Sections were permeabilized for 5 min in PBST (0.1% Triton X-100 in PBS), blocked for 1 h in blocking buffer (3% bovine serum albumin in PBST) containing 10% normal goat serum, and incubated overnight at 4°C with rabbit anti-CRP2 [1:500 (Huber et al., 2000)], rabbit anti-cGKI [1:100; recognizes the α and β isoform of cGKI (Pfeifer et al., 1998)], mouse anti-calcitonin gene-related peptide (CGRP; 1:500; Sigma-Aldrich, Munich, Germany), mouse anti-nuclear neuronal protein (NeuN; 1:1000; Millipore, Billerica, MA), mouse anti-glial fibrillary acidic protein (GFAP; 1:1000; Millipore), rat anti-mouse CD11b (1:200; Serotec, Düsseldorf, Germany) dissolved in blocking buffer, or with fluorescein isothiocyanate (FITC)-conjugated Griffonia simplicifolia isolectin B4 (IB4; 10 μg/ml; Sigma-Aldrich) dissolved in PBST. In double-labeling experiments, primary antibodies were consecutively incubated overnight. After washing in PBS, sections were incubated for 2 h at room temperature with species specific secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen) or Cy3 (Sigma-Aldrich). After immunostaining, slides were immersed for 5 min in 0.06% Sudan black B (in 70% ethanol) to reduce lipofuscin-like autofluorescence (Schnell et al., 1999; Schmidtko et al., 2005), rinsed in PBS, and coverslipped in Fluoromount G (Southern Biotech, Birmingham, AL). Images were obtained using an Eclipse E600 microscope (Nikon) equipped with a Kappa DX 20 H camera and Kappa ImageBase software (Kappa, Gleichen, Germany). Adjustment of brightness and contrast and preparation of figures was performed with Adobe (San Jose, CA) Photoshop CS software. Controls were performed by omitting the first and/or the second primary antibodies and by incubating spinal cords of CRP2−/− mice.
Multiepitope ligand cartography
Multiepitope ligand cartography (MELC) is an automated imaging technology using FITC-labeled antibodies. Each labeled antibody is applied sequentially on the same sample followed by an imaging step before bleaching the fluorescence signal, which allows immunohistochemical staining of one sample with multiple antibodies, independent from the antibody host (Schubert et al., 2006). The lumbar spinal cords of three wild-type mice were removed, and transverse slices were sectioned (12 μm in thickness) in a cryostat, fixed in acetone at −20°C for 10 min, and stored at −80°C. Before use in the MELC system (MelTec, Magdeburg, Germany) sections were rehydrated in PBS at 20°C, incubated with normal goat serum for 30 min, and washed again in PBS. Slides were placed on the stage of an inverted wide-field fluorescence microscope equipped with fluorescence filters for FITC. Labeled rabbit antibodies [against CRP2 (Huber et al., 2000), cGKI (Pfeifer et al., 1998), or syntaxin 1A (Synaptic Systems, Göttingen, Germany)], propidium iodide, and wash solutions were added and removed robotically. After incubation for 15 min, phase-contrast and fluorescence images of the superficial dorsal horn of the spinal cord were acquired. Sections were then washed and bleached at the excitation wavelength. After acquisition of postbleaching images, the next cycle was started. Different-colored images for each antibody were processed and superimposed using the software of the MELC system.
Combined in situ hybridization and immunohistochemistry
A cDNA was reverse transcribed from mouse spinal cord with random primers. The primers 5′-cctccaagtgtcccaagtgt-3′ and 5′-aggctgaactgtgccttcc-3′ were used to synthesize a fragment corresponding to nucleotides 61–680 of CRP2 mRNA (accession number D17512) by PCR (Okano et al., 1993). Agarose gel electrophoresis presented a single band with the expected size. The fragment was cloned into the pCR-TOPO plasmid vector (Invitrogen) and amplified using Escherichia coli DH5α. Isolated plasmid was linearized with NotI and PmeI, and digoxigenin-labeled sense and antisense probes were transcribed using T3 or T7 RNA polymerases, respectively (Roche Diagnostics, Mannheim, Germany).
In situ hybridization was performed before immunohistochemistry.
DRGs (L3–L5) were dissected and immediately frozen in tissue-freezing medium on dry ice. Cryostat sections were cut at a thickness of 16 μm. Sections were fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, for 10 min, rinsed in PBS, acetylated with acetic anhydride in 0.1 m triethanolamine for 10 min, prehybridized in hybridization buffer (50% formamide, 5× SSC, 5× Denhardt's solution, 500 μg/ml herring sperm DNA, 250 μg/ml yeast tRNA) at 70°C for 1.5 h, and incubated with CRP2 sense or antisense probes in hybridization buffer (100 ng/ml) at 70°C overnight. After hybridization, the slides were washed in 0.2× SSC and TBS (0.1 m Tris-HCl, 0.15 m NaCl, pH 7.5) and incubated for 1 h in blocking buffer (0.12 m maleic acid, 0.15 m NaCl, pH 7.5; 1% blocking reagent; Roche Diagnostics), followed by incubation of alkaline phosphatase-conjugated anti-digoxigenin antibody (1:1000; Roche Diagnostics) in blocking buffer at 4°C overnight. Sections were then washed in TBS and equilibrated in alkaline buffer (0.1 m Tris-HCl, 0.1 m NaCl, 0.05 m MgCl2, pH 9.5, 2 mm levamisole). A colorimetric reaction using BM Purple AP substrate (Roche Diagnostics) was performed at 37°C for 6–10 h. After rinsing in PBS, sections were incubated with mouse anti-neurofilament 200 (NF200, clone N52; 1:1000; Sigma-Aldrich), mouse anti-peripherin (1:1000; Millipore), or rabbit anti-cGKI [1:50 (Pfeifer et al., 1998)] dissolved in PBST at 4°C overnight. Finally, sections were rinsed in PBS, incubated for 2 h at room temperature with Cy3-conjugated secondary antibodies dissolved in PBST, rinsed again in PBS, and coverslipped in Fluoromount G.
Images were obtained using an Eclipse E600 microscope equipped with a Kappa DX 20 H camera and Kappa ImageBase software. BM Purple-stained CRP2 mRNA was visualized using bright-field illumination. In combined preparations with fluorescent stains to identify cell populations, images were taken separately by switching between bright-field illumination and fluorescence filter blocks. Using Adobe Photoshop CS software, bright-field illumination images were converted to grayscale (command: image > mode > grayscale) and inverted (command: image > adjust > invert) before being overlaid with images of fluorescent stains. Double labeling was quantified in three nonadjacent sections from L3–L5 DRGs from five wild-type mice. At least 300 neurons were counted for each staining set per animal. The intensity of hybridization signals using antisense probe on DRGs from CRP2−/− mice served as background staining to determine CRP2 mRNA-positive neurons. All CRP2-positive profiles with a clear nucleus were analyzed from the images, and the percentage of colocalization was calculated.
Western blot
Tissue samples were homogenized in buffer containing 10 mm Tris/HCl, pH 7.4, 20 mm 3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate, 0.5 mm EDTA, 1 mm DTT, 0.5 mm PMSF, and 1 mm Pefabloc SC (Alexis, Grünberg, Germany). Extracted proteins (30 μg per lane) were separated by SDS-PAGE and transferred onto nitrocellulose membranes by electroblotting. After blocking of nonspecific binding sites with blocking buffer (PBS with 0.2% Tween and 5% low-fat milk), membranes were incubated overnight at 4°C with anti-CRP2 [1:1000 (Huber et al., 2000)] diluted in blocking buffer. Anti-extracellular signal-regulated kinase 2 (ERK-2; 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA) was incubated for 1 h and used as loading control. After incubation with the secondary antibody conjugated with Alexa Fluor 680 or 800 (1:10,000; Invitrogen) for 2 h, blots were visualized on the Odyssey Infrared Imaging System (LI-COR Biosciences, Bad Homburg, Germany). The band densities were quantified by densitometry using the Odyssey software. All CRP2 levels were normalized relative to the level of ERK-2.
In vivo phosphorylation of CRP2
Fourteen left and right DRGs (Th5-L5) from six WT and four CRP2−/− mice were removed. Left and right DRGs from each mouse were separately pooled and incubated for 30 min at 37°C with B27-supplemented Neurobasal medium (Invitrogen) containing 0.1% collagenase (type CLS IV; Biochrom KG, Berlin, Germany). After washing, DRGs were labeled with [33P]-H3PO4 (200 μCi/ml) in B27/Neurobasal medium for 4 h at 37°C. At the end of the labeling period, the phosphatase inhibitor okadaic acid (100 nm) was added, and the left DRGs were treated for 15 min with 100 μm 8-pCPT-cGMP (Biolog, Bremen, Germany), whereas the right DRGs were run as a control. The DRGs were then homogenized in 200 μl of lysis buffer (50 mm Tris, 0.5% SDS, 12.5 mm Na2HPO4, pH 8.0, 100 nm okadaic acid), heated for 5 min at 95°C, and incubated with 150 U of DNase I for 15 min at 25°C. After fourfold dilution with RIPA correction buffer (12.5 mm Na2HPO4, 1.25% Nonidet P-40, 1.25% SDS, 2 mm EDTA, 50 mm NaF, pH 7.2, 1 μg/ml aprotinin, 2 mm benzamidine, 100 μm PMSF, 100 nm okadaic acid), samples were centrifuged at 25,000 × g for 1 h at 4°C. The lysates were immunoprecipitated with CRP2 antibody and protein A-Sepharose beads (Sigma-Aldrich) for 2 h at 4°C. After washing, the immunoprecipitates were eluted with Laemmli buffer, subjected to SDS/PAGE, and blotted onto nitrocellulose membranes. Phosphorylated CRP2 was detected by phosphoimaging, and the immunoprecipitated amount of CRP2 was analyzed as described above.
Behavioral testing
Littermate wild-type and CRP2−/− mice were used in all behavioral tests. Animals were habituated to the experimental room and were investigated by observers blinded for the genotype and treatment of the animals.
Rotarod test.
Motor coordination was assessed with a rotarod treadmill for mice (Ugo Basile, Comerio, Italy) at a constant rotating speed of 32 rpm. All mice had five training sessions before the day of the experiment. The fall-off latency was averaged from five tests. The cutoff time was 120 s.
Hot plate test.
Mice were placed into a Plexiglas cylinder (diameter, 20 cm; height, 18 cm) on a metal surface maintained at 52.0°C (hot plate; Ugo Basile). The time between placement and shaking or licking of the hindpaws was recorded. A 45 s cutoff was used to prevent tissue damage, but it was not reached by any animal. Only one test per animal was performed, because repeated measures cause profound latency changes (Mogil et al., 1999).
Formalin test.
Fifteen microliters of a 5% formaldehyde solution (formalin) were injected subcutaneously into the dorsal surface of one hindpaw. The time spent licking the formalin-injected paw was recorded in 5 min intervals up to 120 min after formalin injection.
Mechanical hyperalgesia.
The mechanical sensitivity of the plantar side of a hindpaw was assessed with an automated testing device (dynamic plantar aesthesiometer; Ugo Basile). A steel rod (2 mm diameter) was pushed against the paw with ascending force (0–5 g over a 10 s period, time resolution, 0.1 s) until a strong and immediate withdrawal occurred. The paw withdrawal latency was taken to be the mean of three consecutive trials with at least 10 s in between. After baseline measurements, 15 μl of a zymosan A suspension (5 mg/ml in 0.1 m PBS, pH 7.4; Sigma-Aldrich) or 20 μl of complete Freund's adjuvant (CFA; containing 1 mg/ml heat-killed Mycobacterium tuberculosis in paraffin oil 85% and mannide monooleate 15%; Sigma-Aldrich) was injected into the plantar subcutaneous space of a hindpaw, and withdrawal latencies were determined at 1, 2, 3, 4, 5, 6, and 8 h after zymosan injection or at 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20 d after CFA injection.
Neuropathic pain.
The “spared nerve injury” (SNI) model was used to investigate neuropathic pain. Mice were anesthetized with isoflurane, and the tibial and common peroneal branches of the sciatic nerve were ligated and sectioned distally, whereas the sural nerve was left intact (Decosterd and Woolf, 2000). Mechanical allodynia was determined with a dynamic plantar aesthesiometer as described above. Cold allodynia was measured by counting the number of flinches of the SNI-operated hindpaw on a 5°C cold plate (AHP-1200CPHC; Teca, Chicago, IL) during an observation period of 90 s. Measurements were conducted at 2 and 1 d before SNI surgery (baseline) and at 7, 14, 21, and 28 d after SNI surgery.
Intrathecal administration of cGMP analogs.
For intrathecal delivery of drugs, a spinal catheter was constructed by inserting a slim polytetrafluoroethylene Sub-Lite Wall Tubing catheter [outer diameter (OD), 0.15 mm; inner diameter (ID), 0.05 mm; length, 2 cm; SUBL-60; Braintree Scientific, Braintree, MA] for 1 cm into a wider polyethylene tube (OD, 0.61 mm; ID, 0.28 mm; length, 9 cm; neoLab, Heidelberg, Germany) and fixation of both tubes with cyanacrylate glue (Stabiloplast; Renfert, Hilzingen, Germany). Saline was injected through the catheter to check for leakage and occlusion. Mice were anesthetized with a mix of isoflurane and carbogen. A 2 cm longitudinal skin incision was made above the pelvic girdle and the muscles were bluntly dissected from the vertebrae to expose the L4 and L5 spinous processes. The L5 spinous process and the intervertebral ligament were cut, and a small hole was made in the dura with a 27 gauge needle. Then the slim part of the catheter (length 1 cm) was inserted, so that the tip reached vertebral level L1, which approximates L4–L5 in the mouse lumbosacral spinal cord. The sign of a sudden tail or hind limb movement verified the right localization of the catheter. Using cyanacrylate glue, the hole on the dura was covered and the catheter was fixed on the bone. After muscle suture, the catheter was tunneled under the skin through a trocar and pulled out from the dorsal neck area. Finally, the skin incision was sutured, and the end of the catheter was sealed by melting. After surgery, mice were caged individually for 3 d to recover. Only mice without obvious neurological deficits were used for behavioral experiments.
The mechanical sensitivity of the plantar side of a hindpaw was assessed with a dynamic plantar aesthesiometer as described above. After baseline measurements, the cGMP analogs 8-Br-cGMP or 8-pCPT-cGMP (Biolog), dissolved in artificial CSF (ACSF; 141.7 mm Na+, 2.6 mm K+, 0.9 mm Mg2+, 1.3 mm Ca2+, 122.7 mm Cl−, 21.0 mm HCO3−, 2.5 mm HPO42−, 3.5 mm dextrose, bubbled with 5% CO2 in 95% O2 to adjust pH to 7.2), were intrathecally injected in a volume of 2.5 μl through the catheter followed by 2.5 μl of ACSF. Based on previous results in mice (Tegeder et al., 2004) and rats (Tegeder et al., 2002), 250 and 25 nmol of 8-Br-cGMP were considered as high and low dose, respectively. The high (25 nmol) and low (1.25 nmol) dose of 8-pCPT-cGMP, which has a higher membrane permeability and phosphodiesterase stability than 8-Br-cGMP (Butt et al., 1992; Schwede et al., 2000) was determined in preliminary experiments. After drug injection, the paw withdrawal latencies were determined for up to 120 min in intervals of 10 (0–60 min) and 20 min (>60 min). Then the mice were killed, and the catheter position was verified by injection of 5 μl of methylene blue. Only animals in which the catheter tip was located between the vertebral segments Th13-L1 and methylene blue marked the lumbar spinal cord were included.
Statistics
Statistical evaluation was done with SPSS 12.0.1 for Windows (SPSS, Chicago, IL). The Kolmogorov–Smirnov test was used to assess normal distribution of data within groups. Normally distributed data of two groups were analyzed with Student's t test and are presented as the mean ± SEM. For the formalin test, the licking times of phases 1, 2, and 3 (1–10, 11–60, and 61–120 min, respectively) were analyzed. Areas under the curve (AUCs) were calculated using the linear trapezoidal role. Rotarod fall-off latencies were analyzed with Mann–Whitney U test and are expressed as median and interquartile range. For all tests, a probability value p < 0.05 was considered statistically significant.
Results
Identification of CRP2 as a cGKI interactor in the spinal cord
To identify cGKI-interacting proteins, we screened a spinal cord cDNA library by yeast two-hybrid analysis using cGKIα and cGKIβ as a bait and found predominantly clones encoding sequences from CRP2 (supplemental Table 1, available at www.jneurosci.org as supplemental material). CRP2, originally identified by Okano et al. (1993), contains two LIM domains, shares 30–45% homology with the other known members of the vertebrate CRP family, CRP1 (Liebhaber et al., 1990), CRP2/SmLIM [in some studies also referred to as CRP2 (Weiskirchen et al., 1995)], and CRP3/MLP (Arber et al., 1994), and was suggested previously as a novel target of cGKI (Huber et al., 2000). However, up to now the function of CRP2 in the spinal cord has not been resolved.
CRP2 expression in the spinal cord and in dorsal root ganglia
CRP2 protein expression in the spinal cord
Immunohistochemical studies of the mouse spinal cord revealed intense CRP2-like immunoreactivity within superficial laminas of the dorsal horn (Fig. 1A) and weak immunoreactivity in blood vessels. No CRP2-like immunoreactivity was observed in neurons or nuclei of supraspinal brain regions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Specificity of anti-CRP2 (Huber et al., 2000) was confirmed by immunohistochemistry in the spinal cord of CRP2−/− mice (Fig. 1B) and by Western blot analyses detecting a single band at the estimated molecular weight of 23 kDa in spinal cord protein extracts of wild-type (WT) mice, but not in extracts derived from CRP2−/− mice (Fig. 1C).
Figure 1.

Expression of CRP2 in the spinal cord. A, B, Immunohistochemical analysis of CRP2 in lumbar spinal cord in wild-type mice (A) and in CRP2−/− mice (B). Scale bar, 250 μm. C, Western blot of CRP2 (23 kDa) with spinal cord homogenates of wild-type mice (left) and CRP2−/− mice (right). ERK-2 (42 kDa) was used as loading control.
To precisely characterize the distribution of CRP2 in the dorsal horn of the spinal cord, double-labeling immunohistochemistry experiments with established neuronal and glial markers were performed. CRP2 colocalized with CGRP-positive (Fig. 2 A–D) and isolectin B4 (IB4)-binding (Fig. 2 E–H) terminals of nociceptive primary afferents in laminas I and II. There was no apparent overlap between CRP2 and markers of neuronal nuclei (NeuN) (Fig. 2 I–L), astrocytes (GFAP) (Fig. 2 M–P), or microglia (CD11b) (data not shown).
Figure 2.

Colocalization of CRP2 with markers in the spinal cord. A, E, I, M, CRP2 immunoreactivity. B, F, J, N, Immunoreactivity for CGRP, IB4, NeuN, and GFAP. C, G, K, O, Merged images with colocalization being indicated in yellow. D, H, L, P, Merged images at a higher magnification. Scale bars: A, 50 μm; D, 20 μm.
Colocalization of CRP2 with cGKI was assessed by separate immunohistochemical analyses on thin adjacent sections. CRP2 and cGKI showed a similar staining pattern in the superficial dorsal horn (Fig. 3A,B). The colocalization was more precisely investigated with MELC (see Materials and Methods) using FITC-labeled primary antibodies. As shown in Figure 3C–F, MELC analyses revealed that CRP2 and cGKI are colocalized within the superficial dorsal horn. CRP2- and cGKI-immunopositive areas also costained for syntaxin 1A, a neuronal protein concentrated on presynaptic plasma membranes (Bennett et al., 1992). These findings implicate that CRP2 and cGKI are localized in terminals of primary afferent nerve fibers, which terminate in the superficial dorsal horn of the spinal cord.
Figure 3.
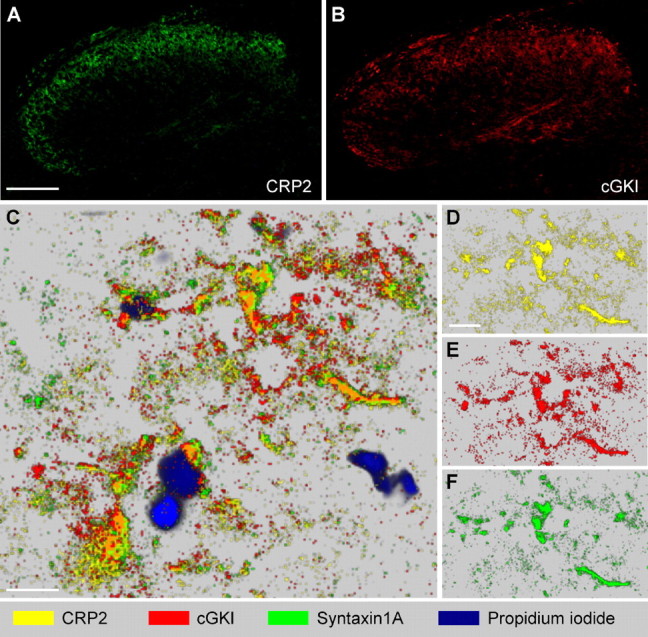
CRP2 and cGKI colocalization in the spinal cord. A, B, CRP2 and cGKI immunoreactivity in adjacent slices reveals a similar staining pattern of both proteins in the dorsal horn. C–F, Protein mapping by MELC in lamina II of the dorsal horn demonstrates colocalization of CRP2 (yellow) with cGKI (red) and syntaxin 1A (green). Extensive colocalization of CRP2 with cGKI appears orange. Propidium iodide (blue) was used to label DNA. Pictures were taken separately from the same slice using sequential rounds of fluorescent detection by the MELC robotic system and mapped using the software of the MELC system. C, Overview. D–F, Partition with single MELC maps of CRP2, cGKI, and syntaxin 1A. Scale bars: A (for A, B), 100 μm; C, D (for D–F), 10 μm. Maximal resolution in C–F was 0.4 μm (pixel size).
CRP2 mRNA expression in DRGs
In contrast to the spinal cord tissue, anti-CRP2 revealed a strong additional band of ∼50 kDa in Western blot analysis of DRG extracts of both wild-type and CRP2−/− mice and no specific CRP2 signal in immunohistochemical analyses of DRG cell bodies (data not shown). We therefore investigated the distribution of CRP2 mRNA in DRGs by in situ hybridization. The CRP2 antisense probe produced intense staining of lumbar DRG cell bodies in WT, but not in CRP2−/− mice (Fig. 4 A–C). Specific CRP2 labeling was observed in 37.5 ± 2.3% of total neuronal profiles. To estimate the degree of CRP2 colocalization with markers of DRG neuron subpopulations and with cGKI, we performed quantitative in situ hybridization of CRP2 mRNA combined with immunohistochemistry (Table 1). The majority (82.9%) of CRP2 mRNA-positive neurons were positive for peripherin (Fig. 4 D–F), which labels the small-diameter, unmyelinated C-fiber DRG cell population (Wallace et al., 2003). Only a small portion (7.8%) of CRP2 mRNA-positive neurons expressed NF200 (clone N52), a marker of large-diameter, myelinated DRG neurons (Fig. 4 G–I). Seventy-two percent of CRP2-positive neurons expressed cGKI, and 48% of cGKI-positive neurons expressed CRP2 (Fig. 4 J–L).
Figure 4.
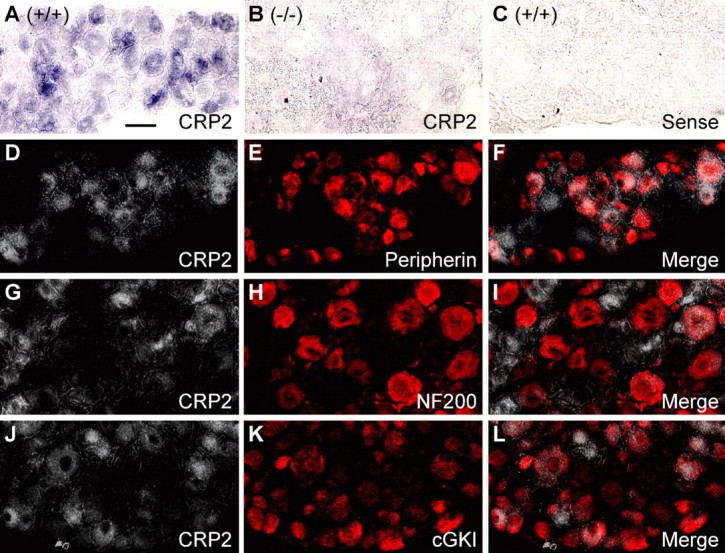
Expression of CRP2 in dorsal root ganglion cells. A–C, In situ hybridization for CRP2 mRNA (purple) in lumbar DRGs of wild-type mice (A) reveals CRP2 expression in a subset of DRG neurons. Specificity of the CRP2 mRNA antisense probe was confirmed by incubating DRGs of CRP2−/− mice with the antisense probe (B) and DRGs of wild-type mice with the respective sense probe (C). Pictures are presented as bright-field photomicrographs. D–L, Double labeling by a combined method of in situ hybridization for CRP2 mRNA and immunohistochemistry for peripherin (D–F), NF200 (G–I), or cGKI (J–L). CRP2 mRNA staining (D, G, J) was recorded as bright-field photomicrographs, converted to grayscale and inverted using Photoshop software. Thus, white indicates the presence of CRP2 mRNA. Immunoreactivity of peripherin (E), NF200 (H), and cGKI (K) was visualized by Cy3 fluorescence and appears in red. Overlays (F, I, L) demonstrate a large number of cells colabeling for CRP2 and peripherin (F) or cGKI (L) and a small number of cells colabeling for CRP2 and NF200 (I). Scale bar, 25 μm.
Table 1.
Percentage of colocalization of CRP2 mRNA and mouse lumbar DRG proteins
| Marker | CRP2 mRNA-positive neurons expressing peripherin, NF200, and cGKI, respectively | Peripherin-, NF200-, and cGKI-positive neurons, respectively, expressing CRP2 mRNA |
|---|---|---|
| Peripherin | 82.9 ± 1.9% | 36.3 ± 3.1% |
| NF200 | 7.8 ± 0.9% | 5.6 ± 1.2% |
| cGKI | 72.0 ± 4.2% | 47.5 ± 3.3% |
cGMP-dependent in vivo phosphorylation of CRP2
To evaluate whether CRP2 is a target of cGKI, we labeled DRGs of wild-type and CRP2−/− mice with [33P]-H3PO4 and stimulated with the cGKI-activating cGMP analog 8-pCPT-cGMP. Immunoprecipitation with anti-CRP2 demonstrated that CRP2 is phosphorylated after activation of cGKI in dorsal root ganglia (Fig. 5).
Figure 5.

In vivo phosphorylation of CRP2 by cGKI in dorsal root ganglia. DRGs of wild-type and CRP2−/− mice were dissected, labeled in vivo with [33P]-H3PO4, incubated with the cGKI activator 8-pCPT-cGMP (100 μm), and immunoprecipitated with the CRP2 antibody. The immunoprecipitates were then subjected to SDS/PAGE and blotted onto nitrocellulose membranes. Top, Phosphoimage analysis detects [33P]-CRP2 in DRGs of wild-type mice after stimulation with 8-pCPT-cGMP (left), but neither in unstimulated DRGs of wild-type mice (middle) nor in stimulated DRGs of CRP2−/− mice (right). Bottom, Western blot analysis with the CRP2 antibody shows a similar amount of immunoprecipitated CRP2 (23 kDa) in DRGs of wild-type mice and no immunoprecipitated CRP2 in DRGs of CRP2−/− mice. The protein band at ∼25 kDa originates from the CRP2 antibody. Representative results of four independent experiments are shown.
CRP2 expression after noxious stimulation
Western blot analyses revealed significantly increased expression of CRP2 protein in the spinal cord and in ipsilateral L4/L5 DRGs after noxious stimulation of a hindpaw by zymosan injection (Fig. 6). The increase was 3.7-fold (24 h) and 4.5-fold (48 h) in the spinal cord, as well as 1.6-fold (24 h) and 2.3-fold (48 h) in the DRGs (p < 0.05 and n = 3 each). Immunohistochemistry revealed that upregulation of CRP2 protein expression in the spinal cord occurred only in CGRP-IR-positive and IB4-binding areas in laminas I and II, but not in other regions (data not shown).
Figure 6.

Increased CRP2 protein expression after noxious stimulation. Western blot of CRP2 (23 kDa) in the mouse lumbar spinal cord and in DRGs after zymosan injection into a hindpaw. Tissue was excised at the indicated time points after zymosan injection. ERK-2 (42 kDa) was used as loading control. Representative results from three animals per group are shown.
Acute and persistent inflammatory pain in CRP2−/− mice
To assess the role of CRP2 in nociceptive processing in vivo, we generated CRP2−/− mice using the Cre/lox system (as described in Materials and Methods) and characterized their nociceptive behavior. Homozygous CRP2−/− mice are viable, fertile, appear normal, and do not show reduced life expectancy. Defects of guidance and connectivity of sensory axons, which have been detected in cGKI−/− mice (Schmidt et al., 2002), are not present in CRP2−/− mice (Schaffer, 2006). Accordingly, no abnormalities in the size and morphology of DRGs and spinal cord were observed in CRP2−/− mice when compared with WT mice. Terminals of nociceptive and thermoreceptive primary afferents in the superficial dorsal horn appear to be normally distributed in CRP2−/− mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Furthermore, CRP2−/− mice had no impairments in the rotarod test, which evaluates motor coordination and balance (Crawley, 2003). The median fall-off latencies were 60.8 s in CRP2−/− mice (interquartile range, 23.9–98.5 s) and 67.0 s in WT mice (interquartile range, 9.8–87.4 s; p = 0.61; n = 8 per group).
We investigated the behavioral responses to noxious stimuli in models of acute and persistent inflammatory pain. In the hot-plate test, no differences in latency times were found between both groups (WT, 33.0 ± 2.4 s; CRP2−/−, 30.3 ± 3.7 s; p = 0.58; n = 8 per group), indicating that the immediate response to noxious thermal stimuli is intact in CRP2−/− mice. To analyze how CRP2 deficiency affects the rapid sensitization in pain pathways, we performed the formalin test (Hunskaar et al., 1985; Tjolsen et al., 1992). Injection of 5% formalin into a hindpaw resulted in a phasic nociceptive response consisting of licking the injected paw. In the first phase (1–10 min), which results from direct activation of primary afferent nociceptors, the licking behavior of CRP2−/− and WT mice was similar (Fig. 7A,B) (p = 0.732). However, the second phase of paw licking that involves a period of sensitization lasted up to 60 min after formalin injection and was significantly increased compared with WT mice (Fig. 7A,B) (p = 0.026). We continued to observe the mice until 120 min after injection, because it has been reported that rodents continue to exhibit behavioral responses after the second phase of the formalin test (Kim et al., 1999; Wei et al., 2001; Porro et al., 2003; Huang et al., 2006). In the third phase (61–120 min), paw licking was observed in both groups. However, CRP2−/− mice again spent significantly more time licking the formalin-injected paw than WT mice (Fig. 7A,B) (p = 0.036).
Figure 7.

Inflammatory pain in CRP2−/− mice. A, B, Formalin test. A, Time course of licking behavior of the formalin-injected hindpaw over the 120 min observation period. B, Sum of paw-licking time in phase 1 (0–10 min), phase 2 (11–60 min), and phase 3 (61–120 min). C, D, Mechanical hyperalgesia after injection of zymosan (C) or CFA (D) into a hindpaw. Time courses of paw withdrawal latency time after mechanical stimulation over the 8 h (zymosan) or 20 d (CFA) observation period are shown. All data are presented as mean ± SEM. *p < 0.05, comparing CRP2−/− and CRP2+/+ mice; n = 7 or 8 for each group.
We then tested the extent of mechanical hyperalgesia evoked by injection of zymosan or CFA into a hindpaw (Meller and Gebhart, 1997; Ferreira et al., 2001). Under baseline conditions, CRP2−/− mice and WT littermates showed nearly identical paw withdrawal latencies after automated mechanical stimulation, indicating a normal basic transmission of mechanical stimuli in CRP2−/− mice (Fig. 7C,D). After zymosan injection into a hindpaw, both genotypes developed mechanical hyperalgesia during the 8 h observation period; however, hyperalgesia was increased in CRP2−/− mice compared with WT mice (Fig. 7C). Statistical analysis of the area under the “latency time” versus “time after zymosan injection” (0–8 h) curve revealed significant differences between CRP2−/− and WT mice (WT, 48.3 ± 4.2; CRP2−/−, 31.6 ± 5.2; p = 0.023). Injection of CFA into a hindpaw produced a pronounced mechanical hyperalgesia, which lasted for several days (Fig. 7D). Similarly to the zymosan model, hyperalgesia induced by CFA was significantly increased in CRP2−/− mice, as assessed by analysis of the area under the “latency time” versus “time after CFA injection” (0–20 d) curve (WT, 118.3 ± 3.2; CRP2−/−, 97.3 ± 5.0; p = 0.003). Together, the data suggest an inhibitory role of CRP2 in nociceptive processing during inflammatory pain.
Neuropathic pain in CRP2−/− mice
The contribution of CRP2 to neuropathic pain was tested using the SNI model (Decosterd and Woolf, 2000). At 7 d after injury to the tibial and common peroneal branches of the sciatic nerve, both CRP2−/− and wild-type mice demonstrated mechanical allodynia in the affected hindpaw (Fig. 8A). The allodynia persisted for the rest of the experiment, which lasted 4 weeks in total. Similarly, both CRP2−/− and WT mice showed increased cold pain sensitivity on a 5°C plate at 7–28 d after SNI surgery (Fig. 8B). A tendency toward exaggerated SNI-induced mechanical allodynia and cold pain was observed in CRP2−/− mice; however, statistical analyses of the area under the “latency time” or “number of flinches” versus “time after SNI” (0–28 d) curve revealed no significant differences between CRP2−/− and WT mice (mechanical allodynia, WT, 130.9 ± 5.5; CRP2−/−, 117.7 ± 5.0; p = 0.099; cold pain, WT, 247.6 ± 39.7; CRP2−/−, 339.9 ± 32.4; p = 0.093). Thus, CRP2 seems not to be critically involved in the development of neuropathic pain after peripheral traumatic axonal injury.
Figure 8.

Pain behavior of CRP2−/− mice in the SNI model of neuropathic pain. A, Mechanical allodynia. Time course of paw withdrawal latency time after mechanical stimulation. B, Cold pain. Number of flinches of the nerve-injured hindpaw during an observation period of 90 s after placement on a 5°C cold plate. All data are presented as mean ± SEM; n = 8 for each group. No significant differences were detected between CRP2−/− and CRP2+/+ mice.
Responses of CRP2−/− mice to cGMP analogs
To assess the impact of CRP2 for cGKI signaling in nociceptive processing, we determined the nociceptive behavior of CRP2−/− and WT mice after administration of cGMP analogs onto the lumbar spinal cord. Previous studies revealed that intrathecal administration of the cGMP analog 8-Br-cGMP elicits a dose-dependent dual effect on the nociceptive behavior in mice and rats; i.e., high doses of 8-Br-cGMP cause cGKI-dependent hyperalgesia, whereas low doses lead to hypoalgesia via a cGKI-independent mechanism (Tegeder et al., 2002, 2004). In the present study, a high dose of 8-Br-cGMP (250 nmol, i.t.) evoked mechanical allodynia in WT, but not in CRP2−/− mice (Fig. 9A). Similar results were obtained after administration of a high dose (25 nmol, i.t.) of the stable and phosphodiesterase-resistant cGMP analog 8-pCPT-cGMP (Fig. 9B). Statistical analysis of the area under the “latency time” versus “time after injection” curve (AUCLTvT) revealed significant differences between both genotypes (8-Br-cGMP, p = 0.003; 8-pCPT-cGMP, p = 0.016). In contrast, low doses of 8-Br-cGMP (25 nmol, i.t.) or 8-pCPT-cGMP (1.25 nmol, i.t.) caused antinociception in both genotypes to a similar extent (Fig. 9C,D). Determination of AUCLTvT showed no statistical differences in the antinociceptive behavior in both groups (8-Br-cGMP, p = 0.639; 8-pCPT-cGMP, p = 0.867). These findings indicate that CRP2 essentially contributes to the pronociceptive effects after cGKI activation, whereas the antinociceptive, cGKI-independent effects of cGMP analogs do not depend on CRP2.
Figure 9.

Mechanical allodynia and antinociception in CRP2−/− mice induced by cGMP analogs. 8-Br-cGMP or 8-pCPT-cGMP was administered onto the lumbar spinal cord via an intrathecal catheter. Thereafter, the withdrawal latency time of a hindpaw after mechanical stimulation by an automated device was measured at the indicated time points. Data were normalized to baseline latency times and are presented as mean percentage ± SEM; n = 6 per compound and genotype. A, “High-dose” 8-Br-cGMP (250 nmol). B, “High-dose” 8-pCPT-cGMP (25 nmol). C, “Low-dose” 8-Br-cGMP (25 nmol). D, “Low-dose” 8-pCPT-cGMP (1.25 nmol).
Discussion
In this study we identified CRP2 as a novel effector of the cGMP/cGKI signaling pathway in spinal nociceptive processing. We found CRP2 to be expressed in small-diameter dorsal root ganglion neurons and their axon terminals in the superficial dorsal horn of the spinal cord, i.e., in areas associated with nociceptive pathways. Analyzing the nociceptive behavior of CRP2-deficient mice points to its contribution in an inhibitory manner to the generation of inflammatory pain, but it does not essentially contribute to acute or neuropathic pain.
Our immunohistochemical experiments revealed the presence of CRP2 protein in the superficial dorsal horn of the spinal cord and a high degree of colocalization with CGRP-IR-positive and IB4-binding fibers. The neuropeptide CGRP and the lectin Griffonia simplicifolia IB4 define two largely nonoverlapping populations of small-diameter, nonmyelinated DRG neurons, whose central axons terminate in laminas I and outer II (CGRP) or inner II (IB4) of the spinal cord (Silverman and Kruger, 1990; Alvarez and Fyffe, 2000). However, CRP2-IR in the spinal cord did not coexist with the neuronal nuclei marker NeuN, nor did it appear to be expressed in astrocytes or microglia, as evidenced by the lack of colocalization with GFAP or CD11b, respectively. We therefore suggest that CRP2 is mainly synthesized by small-diameter DRG neurons. Consistent with this hypothesis, CRP2 mRNA was mainly found in DRG neurons expressing peripherin, which labels small-diameter DRG neurons, but only in relatively few N52-positive large-diameter DRG neurons that innervate low-threshold mechanoreceptors (Goldstein et al., 1991; Averill et al., 1995; Michael et al., 1997). Thus, the high degree of colocalization of CRP2 with CGRP and IB4 in the spinal cord and with peripherin in DRG cells provides strong evidence that CRP2 is highly expressed in primary afferent neurons of the “nociceptive system” that project to laminas I and II of the spinal cord. The fact that noxious stimulation of a hindpaw led to an increased expression of CRP2 in protein extracts from DRGs and spinal cord further indicates an involvement of CRP2 in nociceptive processing.
The study also demonstrates a colocalization of CRP2 and cGKI in the superficial dorsal horn and in DRGs. The fact that the cGKIα isoform is predominantly expressed in these regions, as demonstrated in earlier studies (Qian et al., 1996), and that we found six independent clones containing CRP2 cDNA sequences, which interact with cGKIα in the yeast two-hybrid screen, strongly points to a colocalization of cGKIα and CRP2. In particular, intense cGKI staining was found in small- and medium-diameter DRG neurons and in axonal processes in laminas I and II of the spinal cord (Qian et al., 1996; Sung et al., 2006). However, cGKI-IR was also detected in large-diameter DRG cell bodies, indicating that cGKI, in addition to its well characterized role in nociception, might also be involved in non-nociceptive functions of primary afferent neurons. This could be a reason for our finding that 72% of CRP2-positive DRGs express cGKI, but only 48% of cGKI-positive DRGs express CRP2. Nevertheless, our study revealed an in vivo phosphorylation of CRP2 by cGKI in DRGs. Moreover, our experiments with intrathecally injected cGMP analogs provided evidence that CRP2 is required for the pronociceptive role of cGKI within the spinal cord, because the pronociceptive “high” doses of cGMP analogs increased the nociceptive behavior in wild-type mice, but neither in cGKI−/− (Tegeder et al., 2004) nor in CRP2−/− mice. These data imply that CRP2 might represent an important downstream target of cGKI in spinal nociceptive processing. The finding that the cGMP analogs at “low” doses evoked antinociception in CRP2−/− mice that was indistinguishable from wild-type mice, a phenomenon also observed in cGKI−/− mice (Tegeder et al., 2004), supports the idea that cGMP, dependent on its concentration, exerts a dual effect on nociceptive processing: high concentrations of cGMP in the spinal cord obviously activate the cGKI/CRP2 pathway to produce hyperalgesia, whereas low concentrations have antinociceptive effects that are independent of cGKI and CRP2 (Tegeder et al., 2002, 2004).
Acute nociceptive responses were unaffected in CRP2−/− mice as observed in the hot plate test, after mechanical stimulation of a naive hindpaw, and in the immediate response to formalin injection (phase 1), similar to the findings in cGKI−/− mice (Tegeder et al., 2004). Moreover, the neuropathic pain behavior was not significantly increased in CRP2−/− mice, suggesting that CRP2 does not essentially contribute to the development of hypersensitivity evoked by axonal injury, pointing to a specific role of CRP2 for inflammatory pain. This might reflect the more complex adaptive and maladaptive changes after nerve injury compared with peripheral inflammation, thus mitigating the impact of CRP2 on the behavioral outcome. cGKI−/− mice have not yet been tested in models of neuropathic pain, so it remains to be evaluated whether cGKI also does not affect neuropathic pain or whether cGKI uses targets different from CRP2 after nerve injury.
In contrast to acute and neuropathic pain, CRP2−/− mice demonstrated an increased nociceptive behavior in several inflammatory pain models, including the mechanical hyperalgesia after intraplantar injection of zymosan or CFA and the second phase of the formalin test. Interestingly, phase 3 responses to formalin (i.e., 61–120 min after injection) were also increased in CRP2−/− mice. The vast majority of studies using the formalin test mainly recorded up to 1 h after formalin injection, and therefore the phase 3 has been largely overlooked (Kim et al., 1999). Recent studies suggested that changes of activity pattern in forebrain networks regulate the emerging and subsiding of central sensitization-induced pain behavior in the second phase, and that the attenuated phase 3 responses (compared with phase 2) might reflect an increased activity of inhibitory neurons in the forebrain during ongoing noxious stimulation of primary afferent fibers (Porro et al., 2003; Huang et al., 2006). Because CRP2 is not expressed in supraspinal areas but only in DRGs and the spinal cord, our data indicate that spinal mechanisms also contribute to the inhibition of the nociceptive behavior during phase 3. Further work will be required to resolve the details of this phase.
The increased inflammatory pain behavior in CRP2−/− mice, however, is a surprising result, given that genetic deletion or inhibition of the upstream effector, cGKI, leads to a decreased nociceptive behavior in models of inflammatory pain (Tao et al., 2000; Schmidtko et al., 2003; Tegeder et al., 2004). There are several hypotheses to explain this phenomenon. First, the lack of CRP2 might modulate the expression of other “pain-relevant” genes. We found no alterations in cGKI protein in DRGs and in the spinal cord of CRP2−/− mice as revealed by Western blot analyses (data not shown). However, we cannot exclude that the expression of other genes might be altered in the mutants. Another explanation of the increased nociceptive behavior in CRP2−/− mice might be an inhibitory effect of CRP2 on pronociceptive pathways under resting conditions, and the cessation of this inhibitory effect during the processing of central sensitization that includes phosphorylation of CRP2 by cGKI. This hypothesis, although speculative, would explain (1) that the lack of CRP2 leads to an increased nociceptive behavior after central sensitization, and (2) that deficiency or inhibition of cGKI results in decreased nociceptive behavior, because CRP2 is not phosphorylated by cGKI. The identification of other proteins that interact with CRP2 in the spinal cord and in DRGs is required to elucidate the downstream signaling mechanism by which CRP2 exerts this inhibitory effect in inflammatory hyperalgesia.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (DFG-SFB 553/C6). We thank Clement Kabagema for excellent technical assistance and MelTec (Magdeburg, Germany) for help with the MELC technology.
References
- Alvarez FJ, Fyffe RE. Nociceptors for the 21st century. Curr Rev Pain. 2000;4:451–458. doi: 10.1007/s11916-000-0069-4. [DOI] [PubMed] [Google Scholar]
- Arber S, Halder G, Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79:221–231. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- Ausubel FM. Yeast. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current protocols in molecular biology. New York: Wiley; 2003a. pp. 13.1.1–13.13.7. [Google Scholar]
- Ausubel FM. Analysis of protein interactions. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current protocols in molecular biology. New York: Wiley; 2003b. pp. 20.1.1–20.1.33. [Google Scholar]
- Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci. 1995;7:1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- Butt E, Nolte C, Schulz S, Beltman J, Beavo JA, Jastorff B, Walter U. Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochem Pharmacol. 1992;43:2591–2600. doi: 10.1016/0006-2952(92)90148-c. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of rodents. Comp Med. 2003;53:140–146. [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Campos MM, Pesquero JB, Araujo RC, Bader M, Calixto JB. Evidence for the participation of kinins in Freund's adjuvant-induced inflammatory and nociceptive responses in kinin B1 and B2 receptor knockout mice. Neuropharmacology. 2001;41:1006–1012. doi: 10.1016/s0028-3908(01)00142-3. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- Goldstein ME, House SB, Gainer H. NF-L and peripherin immunoreactivities define distinct classes of rat sensory ganglion cells. J Neurosci Res. 1991;30:92–104. doi: 10.1002/jnr.490300111. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Huang J, Chang JY, Woodward DJ, Baccala LA, Han JS, Wang JY, Luo F. Dynamic neuronal responses in cortical and thalamic areas during different phases of formalin test in rats. Exp Neurol. 2006;200:124–134. doi: 10.1016/j.expneurol.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Huber A, Neuhuber WL, Klugbauer N, Ruth P, Allescher HD. Cysteine-rich protein 2, a novel substrate for cGMP kinase I in enteric neurons and intestinal smooth muscle. J Biol Chem. 2000;275:5504–5511. doi: 10.1074/jbc.275.8.5504. [DOI] [PubMed] [Google Scholar]
- Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Calejesan AA, Li P, Wei F, Zhuo M. Sex differences in late behavioral response to subcutaneous formalin injection in mice. Brain Res. 1999;829:185–189. doi: 10.1016/s0006-8993(99)01353-0. [DOI] [PubMed] [Google Scholar]
- Liebhaber SA, Emery JG, Urbanek M, Wang XK, Cooke NE. Characterization of a human cDNA encoding a widely expressed and highly conserved cysteine-rich protein with an unusual zinc-finger motif. Nucleic Acids Res. 1990;18:3871–3879. doi: 10.1093/nar/18.13.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Peng YB, Wu J, Willis WD. Involvement of cGMP in nociceptive processing by and sensitization of spinothalamic neurons in primates. J Neurosci. 1997;17:3293–3302. doi: 10.1523/JNEUROSCI.17-09-03293.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain. 1993;52:127–136. doi: 10.1016/0304-3959(93)90124-8. [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/s1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80:67–82. doi: 10.1016/s0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- Okano I, Yamamoto T, Kaji A, Kimura T, Mizuno K, Nakamura T. Cloning of CRP2, a novel member of the cysteine-rich protein family with two repeats of an unusual LIM/double zinc-finger motif. FEBS Lett. 1993;333:51–55. doi: 10.1016/0014-5793(93)80373-3. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fassler R, Hofmann F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porro CA, Cavazzuti M, Lui F, Giuliani D, Pellegrini M, Baraldi P. Independent time courses of supraspinal nociceptive activity and spinally mediated behavior during tonic pain. Pain. 2003;104:291–301. doi: 10.1016/s0304-3959(03)00015-0. [DOI] [PubMed] [Google Scholar]
- Qian Y, Chao DS, Santillano DR, Cornwell TL, Nairn AC, Greengard P, Lincoln TM, Bredt DS. cGMP-dependent protein kinase in dorsal root ganglion: relationship with nitric oxide synthase and nociceptive neurons. J Neurosci. 1996;16:3130–3138. doi: 10.1523/JNEUROSCI.16-10-03130.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter M, Strijbos PJ, Neale S, Duffy C, Follenfant RL, Garthwaite J. The nitric oxide-cyclic GMP pathway is required for nociceptive signalling at specific loci within the somatosensory pathway. Neuroscience. 1996;73:649–655. doi: 10.1016/0306-4522(96)00060-7. [DOI] [PubMed] [Google Scholar]
- Schaffer S. Berlin: Freie Universität Berlin; 2006. Investigations about the function of the cGMP-dependent kinase I alpha during the development of the nervous system (in German) [Google Scholar]
- Schmidt H, Werner M, Heppenstall PA, Henning M, More MI, Kuhbandner S, Lewin GR, Hofmann F, Feil R, Rathjen FG. cGMP-mediated signaling via cGKIalpha is required for the guidance and connectivity of sensory axons. J Cell Biol. 2002;159:489–498. doi: 10.1083/jcb.200207058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Ruth P, Geisslinger G, Tegeder I. Inhibition of cyclic guanosine 5′-monophosphate-dependent protein kinase I (PKG-I) in lumbar spinal cord reduces formalin-induced hyperalgesia and PKG upregulation. Nitric Oxide. 2003;8:89–94. doi: 10.1016/s1089-8603(02)00165-9. [DOI] [PubMed] [Google Scholar]
- Schmidtko A, Del Turco D, Coste O, Ehnert C, Niederberger E, Ruth P, Deller T, Geisslinger G, Tegeder I. Essential role of the synaptic vesicle protein synapsin II in formalin-induced hyperalgesia and glutamate release in the spinal cord. Pain. 2005;115:171–181. doi: 10.1016/j.pain.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Bockelmann R, Malykh Y, Gollnick H, Friedenberger M, Bode M, Dress AW. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- Silverman JD, Kruger L. Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. J Neurocytol. 1990;19:789–801. doi: 10.1007/BF01188046. [DOI] [PubMed] [Google Scholar]
- Song XJ, Wang ZB, Gan Q, Walters ET. cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J Neurophysiol. 2006;95:479–492. doi: 10.1152/jn.00503.2005. [DOI] [PubMed] [Google Scholar]
- Sung YJ, Chiu DT, Ambron RT. Activation and retrograde transport of protein kinase G in rat nociceptive neurons after nerve injury and inflammation. Neuroscience. 2006;141:697–709. doi: 10.1016/j.neuroscience.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Tao YX, Johns RA. Activation of cGMP-dependent protein kinase Ialpha is required for N-methyl-d-aspartate- or nitric oxide-produced spinal thermal hyperalgesia. Eur J Pharmacol. 2000;392:141–145. doi: 10.1016/s0014-2999(00)00129-1. [DOI] [PubMed] [Google Scholar]
- Tao YX, Johns RA. Activation and up-regulation of spinal cord nitric oxide receptor, soluble guanylate cyclase, after formalin injection into the rat hind paw. Neuroscience. 2002;112:439–446. doi: 10.1016/s0306-4522(02)00075-1. [DOI] [PubMed] [Google Scholar]
- Tao YX, Hassan A, Haddad E, Johns RA. Expression and action of cyclic GMP-dependent protein kinase Ialpha in inflammatory hyperalgesia in rat spinal cord. Neuroscience. 2000;95:525–533. doi: 10.1016/s0306-4522(99)00438-8. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Schmidtko A, Niederberger E, Ruth P, Geisslinger G. Dual effects of spinally delivered 8-bromo-cyclic guanosine mono-phosphate (8-bromo-cGMP) in formalin-induced nociception in rats. Neurosci Lett. 2002;332:146–150. doi: 10.1016/s0304-3940(02)00938-2. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Del Turco D, Schmidtko A, Sausbier M, Feil R, Hofmann F, Deller T, Ruth P, Geisslinger G. Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc Natl Acad Sci USA. 2004;101:3253–3257. doi: 10.1073/pnas.0304076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Wallace VC, Cottrell DF, Brophy PJ, Fleetwood-Walker SM. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci. 2003;23:3221–3233. doi: 10.1523/JNEUROSCI.23-08-03221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M. Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci. 2001;4:164–169. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, Pino JD, Macalma T, Bister K, Beckerle MC. The cysteine-rich protein family of highly related LIM domain proteins. J Biol Chem. 1995;270:28946–28954. doi: 10.1074/jbc.270.48.28946. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]