

Abstract
The regulation of growth cone actin dynamics is a critical aspect of axonal growth control. Among the proteins that are directly involved in the regulation of actin dynamics, actin nucleation factors play a pivotal role by promoting the formation of novel actin filaments. However, the essential nucleation factors in developing neurons have so far not been clearly identified. Here, we show expression data, and use true loss-of-function analysis and targeted expression of activated constructs to demonstrate that the Drosophila formin DAAM plays a critical role in axonal morphogenesis. In agreement with this finding, we show that dDAAM is required for filopodia formation at axonal growth cones. Our genetic interaction, immunoprecipitation and protein localization studies argue that dDAAM acts in concert with Rac GTPases, Profilin and Enabled during axonal growth regulation. We also show that mouse Daam1 rescues the CNS defects observed in dDAAM mutant flies to a high degree, and vice versa, that Drosophila DAAM induces the formation of neurite-like protrusions when expressed in mouse P19 cells, strongly suggesting that the function of DAAM in developing neurons has been conserved during evolution.
Keywords: axon growth, Formin, dDAAM, filopodia formation, Ena, Profilin, Rac
Introduction
In the developing nervous system, axons are guided to their targets by highly motile growth cones at their distal tips. Directed growth cone motility in response to extracellular cues is produced by the coordinated regulation of peripheral F-actin and central microtubule networks (Dent and Gertler, 2003). The peripheral F-actin is organized into long bundled actin filaments underlying the finger-like filopodia and diffuse networks of shorter actin filaments contained in the veil-like lamellipodia (Luo, 2002; Dent and Gertler, 2003). Previous work identified many immediate regulators of F-actin dynamics in growth cones (Pak et al., 2008), and for some of these, it has been demonstrated that they act downstream of signaling pathways involved in axonal growth regulation (Dickson, 2002; Huber et al., 2003).
Key regulators of actin dynamics are the so called nucleation factors, such as the Arp2/3 complex and formins, which use different mechanisms to seed new actin filaments (Wallar and Alberts, 2003; Quinlan et al., 2005). These types of actin assembly factors are well defined in migrating cells, however, in growth cones the essential nucleators have not been unambiguously identified yet. Although it has been shown that the Arp2/3 complex plays a certain role in some models of neuronal growth (Zallen et al., 2002; Mongiu et al., 2007; Korobova and Svitkina, 2008), not all neurons seem to depend on this form of nucleation (Ng and Luo, 2004; Strasser et al., 2004). Formins would represent an alternative or complement to Arp2/3 activity in the developing nervous system, yet the existing data about formins are rather controversial. Notably, mDia1 has been linked to axonal elongation in cultured cerebellar granule cells (Arakawa et al., 2003), but subsequent work demonstrated that the mDia1 knock-out mice are fully viable and do not exhibit any apparent CNS defects (Eisenmann et al., 2007; Peng et al., 2007; Sakata et al., 2007). Another formin, mDia2 has been shown to rescue filopodia formation defects in cultured Ena/VASP-deficient cortical neurons (Dent et al., 2007), but it does not appear to be expressed in the embryonic mouse cortex at the time of neuritogenesis (Dent et al., 2007). Thus, these data do not support the view that mDia1 or mDia2 are essential regulators of neural development in vivo. Contrasting to this, formin proteins of the DAAM subfamily (disheveled associated activator of morphogenesis) appeared to be promising candidates in this context, since they were reported to be abundantly expressed in the developing nervous system of various species (mouse, chicken, Xenopus) (Kida et al., 2004; Nakaya et al., 2004). However, before our work, compelling evidence for their involvement in neuronal growth was missing.
Here, we used the combination of genetic and cell biological tools to demonstrate that Drosophila DAAM plays a pivotal role in the regulation of axonal growth. We show that dDAAM cooperates with Rac GTPases, profilin and Ena to promote filopodia formation at axonal growth cones. Significantly, we present data obtained form cross-species assays that strongly suggest that the function of DAAM in developing neurons has been conserved during evolution.
Materials and Methods
Fly stocks and genetics.
The dDAAM excision alleles, the UAS-FL-DAAM and UAS-C-DAAM transgenic flies were created as described by Matusek et al. (2006). For the aims of the dominant interaction studies, we first created the following double mutant stocks: (1) dDAAMEx68/FM7c,ftz-lacZ; RhoA72F/CyO,ftz-lacZ; (2) dDAAMEx68/FM7c,ftz-lacZ; chic221/CyO,ftz-lacZ; (3) dDAAMEx68/FM7c,ftz-lacZ; enaGC1/TM3,ftz-lacZ; (4) dDAAMEx68/FM7c,ftz-lacZ; ena02029/TM3,ftz-lacZ; (5) dDAAMEx68/FM7c,ftz-lacZ; ena23/TM3,ftz-lacZ; (6) dDAAMEx68/FM7c,ftz-lacZ; Rac1J11,Rac2Δ/TM3,ftz-lacZ; (7) dDAAMEx68/FM7c,ftz-lacZ; Rac1J10,Rac2Δ, MtlΔ/TM3,ftz-lacZ. Subsequently, to generate dDAAMEx68/YDp(1;Y)Sz280; mutantX/+ males, we crossed the females of the double mutant stocks to dDAAMEx68/YDp(1;Y)Sz280 males. To carry out the dominant genetic interaction studies, we used the following strategy: dDAAMEx68/YDp(1;Y)Sz280; mutantX/+ males were either crossed to dDAAMEx68/+ or mutantX/+ mothers. The progeny of these crosses were compared with that of a dDAAMEx68/+ to dDAAMEx68/YDp(1;Y)Sz280 or a mutantX/+ female to a mutantX/+ male cross, respectively. The ratio of axonal morphogenesis defects was <0.5% when dDAAMEx68/+ or mutantX/+ mothers were crossed to wild type males. The following mutant alleles were tested: chic221; RhoA72F; ena23; ena02029; enaGC1; Rac1J11,Rac2Δ; Rac1J10,Rac2Δ,MtlΔ. For the epistasis analysis elav-Gal4(C155); mutantX/Cy,ftz-lacZ or elav-Gal4(C155); mutantY/TM3,ftz-lacZ females were crossed to UAS-C-DAAM/TM3,ftz-lacZ males. We tested the same set of mutant alleles as for the dominant interaction studies.
In addition, we used the following fly stocks: w1118; Ore-R; dDAAMEx1; dDAAMEx68/FM7c,ftz-lacZ; dDAAMEx68/YDp(1;Y)Sz280/C(1)DX; dDAAMEx68<arm-lacZ/YDp(1;Y)Sz280/C(1)DX; UAS-DADm-DAAM::EGFP; Act-Gal4<UAS-FL-DAAM/CyO,ftz-lacZ; elav-Gal4<UAS-FL-DAAM/CyO,ftz-lacZ; elav-Gal4<UAS-EGFP::mDaam1/CyO,ftz-lacZ;UAS-diaCA and Sca-Gal4.
Molecular biology and immunohistochemistry.
DNA constructs for transgenic flies and transfection experiments were created by standard cloning techniques. Whole-mount embryo RNA in situ hybridization was performed as described by Matusek et al. (2006).
RNA isolation for quantitative reverse transcription (RT)-PCR was performed by NucleoSpin RNA II RNA isolation kit (Macherey-Nagel) according to the manufacturer's instruction. RT reactions were performed in a final volume of 20 μl with 2 μg total RNA using RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas).
Embryonic immunostainings were done as described by Matusek et al. (2006). Drosophila S2 cells and mouse P19 cells were fixed in 4% formaldehyde in PBS for 10 min and permeabilized in PBS + 0.1% Triton X-100 for 3 min before staining. Primary antibodies were applied for 1 h RT, and after 3 × 5 min washing in PBS, cells were incubated with secondary antibodies for another 1 h.
For immunostainings, we used the following primary antibodies: Rb-anti-DAAM 1:3000 (Matusek et al., 2006), M-anti-β-gal: 1:1000 (Promega), M-anti-FasII: 1:50 (DSHB: 1D4), M-anti-BP102: 1:5 (DSHB), Rb-anti-GFP: 1:1000 (Invitrogen), Rb-anti-β-tubulinIII: 1:1000 (Sigma), M-anti-β-tubulinIII: 1:1000 (Millipore), M-anti-β-tubulin (E7): 1:10 (DSHB), M-anti-MC-480: 1:50 (DSHB), M-anti-NF-M: 1:50 (DSHB), M-anti-Engrailed: 1:10 (DSHB: 4D9), Rb-anti-Eve 1:1000 (a gift from M. Frasch, Mount Sinai School of Medicine, New York, NY), M-anti-Ena: 1:500 (DSHB), M-anti-chic: 1:10 (DSHB). For secondary antibodies we used the appropriate Alexa-488, Alexa-546 and Alexa-633 (Invitrogen), actin was stained with Rhodamine-Phalloidin: 1:100 (Invitrogen). For horseradish peroxidase (HRP) staining, we used the Vectastain ABC kit.
Nomarski images were collected on Zeiss Axioskop MOT2 with Axiocam HR. Confocal images were collected with Olympus FV1000 LSM microscope, images were edited with Adobe Photoshop 7.0CE and Olympus FW10-ASW version1.7a.
Tissue culture and embryonic nerve cord culture.
Drosophila S2 cells were transfected with the Effectene transfection kit (Qiagen), and incubated in Drosophila Schneider's medium (Lonza) for 24 h before fixation. P19 cells were transfected with Superfect transfection reagent kit (Qiagen). Stable transfectants were selected by the introduction of 400 μg/ml G418 (Invitrogen). Neuronal differentiation was induced with 10−7 m retinoic acid (RA) as described by Rasko et al. (1993).
Embryonic nerve cord cultures were treated as described by Wills et al. (1999a) and Wang et al. (1998). Nerve cords were fixed 9 h after dissection in 4% formaldehyde in PBS and stained in a similar way as whole mount embryos.
Primary cell cultures are described in supplemental Materials, available at www.jneurosci.org.
Results
dDAAM localizes to neurites and growth cones of developing neurons
While studying the single Drosophila DAAM ortholog (Matusek et al., 2006), we revealed that dDAAM is transcribed pan-neurally from stage 11 of embryogenesis (Fig. 1A). This observation has been confirmed by immunostaining experiments demonstrating that dDAAM is highly enriched in neurites, where it shows a strong colocalization with actin (Fig. 1B–D). The protein can be first detected from stage 12 of embryogenesis onwards indicating that dDAAM is already expressed when neuritogenesis begins in the CNS (data not shown). To extend our studies to the subcellular level, we used cultured primary neurons obtained from stage 11 embryos (see Materials and Methods). We found that the dDAAM protein is strongly expressed in these neurons (Fig. 1E–G). It colocalizes with F-actin rich structures along the neurite and, in particular, at the growth cone where it can be observed as bright dots along filopodia (Fig. 1E–G). Remarkably, the protein often accumulated in the filopodial tips (Fig. 1E–G), i.e., the places where the fast growing barbed ends of the actin filaments face the cell membrane. Also growth cones of regenerating neurites, that appear similar in morphology to those described in isolated neurons (Wang et al., 1998), displayed a similar kind of dDAAM protein localization (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Thus, dDAAM is clearly present in the nervous system in subcellular positions that would be expected of an actin assembly factor essential for neurite growth.
Figure 1.

dDAAM expression in the embryonic CNS. A, RNA in situ hybridization of dDAAM reveals a strong expression in the developing embryonic CNS from stage11 onwards. B–D, The anti-dDAAM staining (in red) shows a ladder-like pattern in a wild-type ventral nerve cord, corresponding to the major axonal tracts, where the protein displays a strong colocalization with that of actin (green). E–G, Wild-type cultured embryonic neurons. dDAAM (in red) is enriched along the axonal shaft, and also in the growth cone filopodia (visualized with actin staining in green), often at their tips (arrows). The central growth cone region is labeled with β-tubulin (blue). H, The dDAAM cDNA encodes a protein that contains several conserved homology domains including GBD, DID, dimerization domain (DD), coiled-coil region (CC), formin homology domain 1 (FH1), FH2, and DAD. The position of the dDAAMEx1 and dDAAMEx68 alleles, and the structure of the C-DAAM and DADm-DAAM isoforms, are indicated below the cDNA. Scale bars, 50 μm.
dDAAM regulates neuronal growth and filopodia formation
To investigate dDAAM function in the embryonic CNS, we first analyzed the homozygous null mutant dDAAMEx68 allele (Fig. 1H) by using the neuropile specific BP102 and anti-FasII markers (Fig. 2). The dDAAMEx68 homozygous mutant embryos displayed subtle defects in the neuropile, such as thinning of or gaps in the connectives (in 1.5% of the embryos) (Fig. 2B), or partial lack of the lateral most FasII positive tract (Fig. 2E). Because the maternal dDAAM product is present in the embryos (Matusek et al., 2006), next we generated dDAAM maternal and zygotic null mutant embryos. However, these embryos failed to cellularize properly and could not be used for analyses. Therefore, to reduce the maternal component, we used females homozygous for the viable hypomorphic allele dDAAMEx1 (Fig. 1H), and crossed them to dDAAMEx68/YDp(1;Y)Sz280 males to generate dDAAMEx1/dDAAMEx68 embryos (subsequently referred as dDAAMmat/zyg embryos) in which maternal and zygotic dDAAM functions are both impaired. The dDAAMmat/zyg mutant embryos show enhanced CNS phenotypes, but not the progeny of a reciprocal cross (maintaining maternal component). Thirty-four percent of the dDAAMmat/zyg embryos exhibited severe morphogenetic aberrations, partly with completely disorganized nerve cord. Another 35% displayed frequent breaks in connectives and commissures, misrouted axons, or failures in commissure separation (Fig. 2F–H), and, in extreme cases, an almost complete lack of axon bundles (Fig. 2H). Tests with neuron-specific markers, such as anti-Engrailed (Patel et al., 1989b) and anti-Even-skipped (Patel et al., 1989a), did not reveal any obvious alterations in neuron numbers in dDAAMmat/zyg mutant CNSs (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), suggesting that the CNS phenotypes are caused by defects in neurite growth rather than aberrant lineage formation. Additionally, to exclude the possibility that the CNS defects observed in dDAAMmat/zyg mutant embryos are due to degeneration, we examined stage 12 and 13 embryos. In such embryos axonal development has just commenced, therefore potential axonal growth defects would be unlikely to be caused by degeneration. As shown in supplemental Figure S3, available at www.jneurosci.org as supplemental material, axonal growth defects already occur at these early stages, demonstrating that dDAAM has a specific role in axonal development. The CNS defects revealed in dDAAMmat/zyg mutant embryos were clearly rescued when expressing the UAS-FL-DAAM rescue construct (Matusek et al., 2006) with the pan-neuronal elav-Gal4 (100% rescue) or the ubiquitous Act-Gal4 (95.8% rescue) (Fig. 2I) drivers, demonstrating that the observed phenotypes are specific to the loss of dDAAM function.
Figure 2.

Axonal morphogenesis defects in dDAAM mutant embryos. A, In a wild-type embryonic ventral nerve cord, BP102 stains the complete neuropile which forms a ladder-like structure composed of the longitudinal connectives and two commissures per segment, whereas anti-FasII stains a characteristic subset of the longitudinal fascicles in the connectives (C, D). dDAAMEx68 zygotic null mutant embryos display mild CNS morphology defects including thinning of the connectives (arrows in B) and gaps in the lateral most FasII positive tracts (arrowheads in E). F–H, The three classes of CNS morphology defects exhibited in dDAAMmat/zyg maternal and zygotic mutant embryos are shown on these panels. Class I, weak; class II, moderate; class III, strong phenotypic category. I, Quantification of the penetrance of the CNS phenotypes in different dDAAM loss-of-function allelic combinations and the rescue of dDAAMmat/zyg with UAS-FLDAAM and UAS-EGFP::mDaam1. n = the number of CNSs examined. Scale bars, 50 μm.
To address the subcellular function of dDAAM, we generated primary neuronal cultures from dDAAMmat/zyg mutant embryos. While these neurons were able to develop axons of similar length as their wild-type counterparts, filopodia of mutant neurons were reduced in number by 62% and in length by 26% (both changes are highly significant) (Fig. 3A,B,P). As a second approach to deplete the maternal dDAAM pool, we examined neurons from dDAAMEx68 homozygous null mutant embryos that were incubated for 3 d at 26°C before plating. The severity of the filopodia phenotypes in these neurons was identical to that of dDAAMmat/zyg mutant neurons (Fig. 3P), suggesting that both strategies reduce dDAAM levels below functional thresholds. Thus, these loss-of-function experiments show that dDAAM regulates filopodium formation in growth cones, which is likely to be the subcellular cause for the axonal growth phenotypes observed in vivo.
Figure 3.
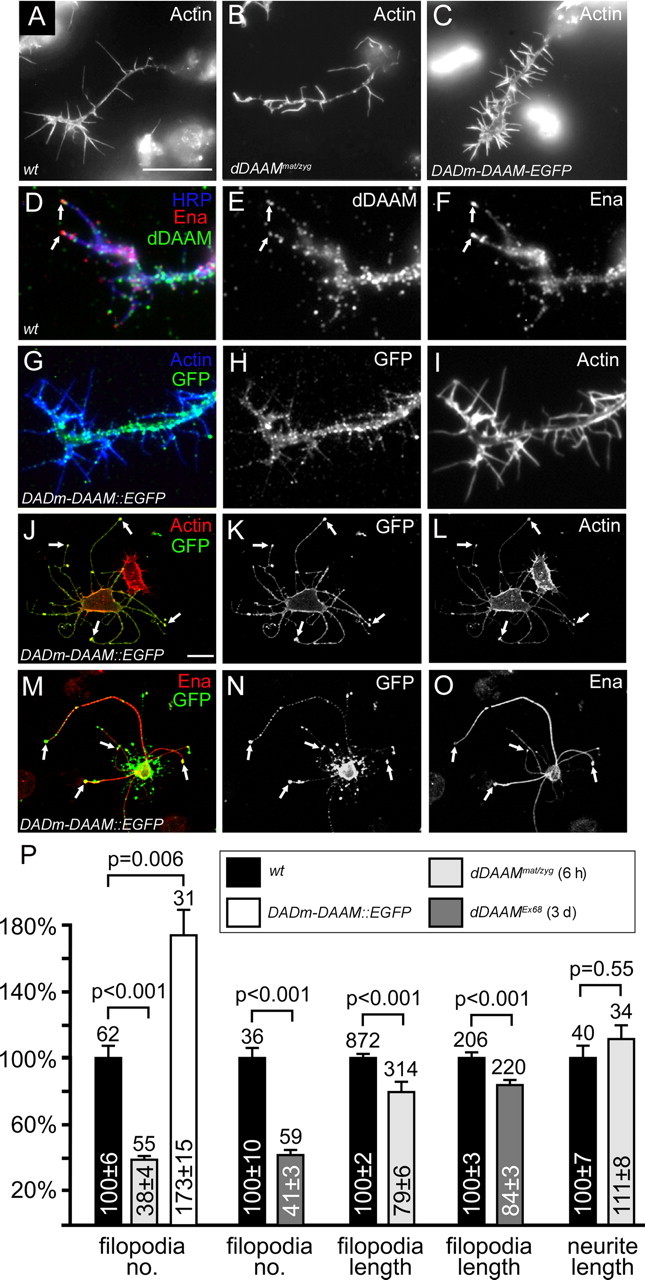
dDAAM is required for filopodia formation in cultured embryonic neurons. A–I, Primary neuronal cultures obtained from wild-type, dDAAMmat/zyg or Sca-Gal4; UAS-DADm-Daam::EGFP embryos, visualized with actin (white in A–C, I; blue in G), anti-Ena (red in D, white in F), anti-Daam (green in D, white in E), anti-HRP (blue in D) or anti-GFP (green in G, white in H). Compared with wild-type (A), dDAAMmat/zyg mutant neurons (B) display fewer number of and shorter filopodia, whereas their number is increased in Sca-Gal4; UAS-DADm-Daam::EGFP neurons (C). D–F, Endogenous dDAAM localizes along the axon shaft, but also along filopodia, often at their tips (arrows), where it tends to colocalize with Ena. G–I, DADm-Daam::EGFP displays a similar localization pattern as the wild-type protein. J–O, When DADm-Daam::EGFP (green in J and M, white in K and N) is expressed in S2 cells, it induces the formation of long axon-like protrusions. The protein is enriched in the protrusions, particularly, in their tip complex (arrows in J and K), where it is often colocalized with Ena (arrows in M–O; red in M, white in O). P, Statistical evaluation of the axon and filopodia phenotypes exhibited in wild-type, Sca-Gal4;UAS-DADm-DAAM, dDAAMmat/zyg and dDAAMEx68 cultured neurons. Bars represent mean values with respective SEs in percent, normalized to wild-type controls; numbers above bars represent sample numbers; p values on top were calculated via Mann–Whitney Rank Sum Tests. The color code of the genotypes is indicated in the boxed legend. All cultures were grown for 6 h after plating, only dDAAMEx68 neurons were dispersed after 3 d in preculture and grown for another 6 h to deplete maternal component. Scale bars, 5 μm.
Activated dDAAM disrupts the fasciculation pattern in the CNS and increases the number of neuronal protrusions
Formins are multidomain proteins defined by the highly conserved formin homology domains, FH1 and FH2 (Fig. 1H). Additionally, formins contain several other conserved domains that are thought to be involved in the regulation of the actin nucleation activity provided by their FH2 domain. Notably, it has been shown that the Dia, FRL and DAAM subfamily members are regulated by auto-inhibition through interaction between the diaphanous inhibitory domain (DID) and diaphanous auto-regulatory domain (DAD) (Fig. 1H), that can be relieved upon Rho-GTP binding to the GTPase-binding domain (GBD) (Wallar and Alberts, 2003). In agreement with this, we have previously found that C-DAAM (Fig. 1H), a truncated dDAAM isoform lacking the N-terminal regulatory domains, behaves as a constitutively active protein when expressed in the tracheal system (Matusek et al., 2006). Here, we report that elav-Gal4 driven C-DAAM expression in the embryonic CNS results in severe fasciculation defects and embryonic lethality. The majority of embryos (78%) display a more prominent neuropile than observed in the wild type situation (Fig. 4A–F), especially commissures and nerve roots appear thicker (Fig. 4B,F). These phenotypic traits are opposite to the ones observed upon loss of dDAAM function described above. Additionally, many axons are misrouted, and either cross the midline or exit the bundles into lateral directions (Fig. 4D). To address the specificity of this phenotype, we tested whether a constitutively activated form of dia (diaCA), the Drosophila formin most closely related to dDAAM, would cause similar effects. When expressing DiaCA pan-neuronally with elav-Gal4, normal CNS fasciculation patterns are clearly disrupted, and thinning of commissures and breaks in connectives can be observed (Fig. 4G). These are the opposite effects to C-DAAM suggesting that only activated dDAAM, but not its most similar paralogue dia, seems to possess the ability to enhance axonal growth.
Figure 4.

Activated dDAAM impairs axon growth and guidance. Compared with a wild-type dissected nerve cord (A, E), C-DAAM overexpression in the CNS impairs axonal morphogenesis. Nerve cords from an elav-Gal4; UAS-C-DAAM/+ embryo (B, F) display more compact axonal tracts than wild-type (compare A and B, and E and F), thicker commissural tracts (arrows in B and F) and increased number of axons exiting the CNS (arrowheads in B and F). Visualization of the FasII positive axons in wild-type (C) and in elav-Gal4; UAS-C-DAAM/+ (D) embryos revealed that activated dDAAM impairs the organization of these longitudinal tracts, the occurrence of tract fusions, midline crossing and increased number of axons exiting the CNS into lateral direction, are evident (D). G, The CNS specific overexpression of diaCA mainly induces breaks in the longitudinal and commissural axon bundles (arrows). Scale bars, (A, B and G) 50 μm; (E, F) 25 μm.
To collect additional evidences that activated dDAAM has the potential to increase neurite number, we examined embryonic nerve cord cultures expressing C-DAAM or DADm-DAAM (an isoform that lacks the DAD domain and would also be expected to behave as an activated form) (Fig. 1H). During these experiments, nerve cords were dissected out from embryos at stage 13 and cultured on poly-l-lysine coated coverslips. After 9 h, wild-type nerve cords grown under these conditions display a moderately dense network of axons (Fig. 5A). In contrast, ventral nerve cords expressing activated dDAAM exhibit a much denser neuritic meshwork and grow more extended axons than their wild type counterparts (Fig. 5B,C). Consistent with the effect on neuronal protrusions, DADm-DAAM::EGFP is enriched in the growth cones of these regenerating neurons and displays a strong accumulation in the filopodial tip complexes (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 5.

Activated dDAAM promotes neurite outgrowth in nerve cord cultures. A, Regenerating motor axons (labeled with anti-FasII) are visible in a moderate density from a wild-type nerve cord grown on a poly-l-lysine coated glass sheet. B, C, Axon growth from elav-Gal4; UAS-C-DAAM/+ or elav-Gal4; UAS-DADm-DAAM::EGFP/+ embryos leads to the formation of increased number of axons with increased length, which is particularly evident in case of elav-Gal4; UAS-DADm-DAAM::EGFP. Scale bars, 25 μm.
To test the effect of dDAAM activation at the subcellular level, we expressed DADm-DAAM::EGFP in cultured primary neurons or S2 cells. In growth cones of cultured primary neurons, the activated protein exhibited a very similar localization pattern as the wild type protein (Fig. 3G–I), and induced a 35% increase in axonal filopodia number (Fig. 3C,P). In Drosophila S2 cells, the activated protein induced characteristic cell shape changes in ∼73% of the transfected cells including of the formation of long neurite-like protrusions stiffened by actin and microtubules (Fig. 3J–L; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The activated DAAM protein is enriched at the cell cortex in these cells and also in the long protrusions including their distal tips (Fig. 3J–L). Thus, the gain-of-function studies demonstrated that activated dDAAM is sufficient to enhance neuronal growth. The formation of axon-like protrusions in the non-neuronal S2 cells suggests that dDAAM might be an important determinant of neuronal morphology.
dDAAM interacts with Rac GTPases, chic and ena
Work in other systems has revealed a number of factors cooperating with or regulating formins during morphogenetic processes. Interestingly, some of these factors have also been implicated in neuronal growth regulation. Therefore, we tested a set of candidate genes to investigate the regulatory context in which dDAAM operates within the CNS. To this end, candidate genes were analyzed in genetic interaction assays, axon growth defects were quantified by counting the number of breakages in the major axonal tracts after BP102 staining. The activity of the Dia, FRL and DAAM subfamily formins are thought to be regulated by Rho GTPase binding (Higgs, 2005). Our previous work suggested that RhoA is an upstream regulator of dDAAM in the developing tracheal cells (Matusek et al., 2006). However, in the context of neuronal growth, we found that RhoA is only a weak enhancer of the zygotic dDAAMEx68 null mutant phenotype (Fig. 6A), suggesting that dDAAM is either not regulated by RhoA, or RhoA is not the only activator of dDAAM in the CNS. Potential candidates would be the three Drosophila Rac GTPases, Rac1, Rac2 and Mtl, all of which were shown to be essential for axon growth with largely overlapping functions (Hakeda-Suzuki et al., 2002; Ng et al., 2002). The reduction of Rac function clearly enhanced the dDAAMEx68 mutant phenotype, the severity of which correlated with the number of Rac gene copies removed (Fig. 6A). Vice versa, the removal of one dDAAM copy strongly enhanced the axonal growth defects exhibited by Rac mutant embryos (Fig. 6B). To investigate the regulatory relationship between dDAAM and the Rac GTPases further, we examined if the elav-Gal4/UAS-C-DAAM gain-of-function phenotype (i.e., the appearance of thicker commissures and nerve roots) was sensitive to the gene dose of the Rac genes. This epistasis analysis indicated that the Rac mutations did not modify the effect of C-DAAM overexpression (Fig. 6C), consistent with the idea that the Rac GTPases act upstream of dDAAM in the embryonic CNS.
Figure 6.

Genetic and biochemical interaction studies with dDAAM. A, Quantification of the axonal growth defects (breakages in the neuropile) in embryos that are homozygous for dDAAMEx68 in the presence of one mutant copy of RhoA, Rac (Rac1, Rac2, Mtl), chic or ena. The number breakages was counted in every nerve cord, the severity of the phenotype was classified as weak, moderate or strong. B, Quantification of the axonal growth defects (breakages in the neuropile) in embryos that are homozygous for the mutations shown in A or homozygous for the same mutations in a dDAAMEx68/+ genetic background. C, Quantification of the CNS defects (the appearance of thicker commissures and nerve roots) exhibited in elav-Gal4; UAS-C-DAAM/+ embryos in the presence of one mutant copy of the genes shown in A. A–C, n indicates the number of CNS examined. D, Immunoprecipitation (IP) of lysates from S2 cells transfected with DADm-DAAM::EGFP using anti-chic and probed with anti-dDAAM (left lanes). IP with anti-ena, probed with anti-dDAAM (right lanes) and anti-chic (middle lanes). Chic coimmunoprecipitates DADm-DAAM::EGFP, whereas Ena coimmunoprecipitates DADm-DAAM::EGFP and Chic.
In addition to the Rho family GTPases, the actin monomer binding protein profilin, the Src kinases and some members of the Ena/VASP protein family have also been shown to bind formin molecules (Wallar and Alberts, 2003; Schirenbeck et al., 2006). Of these, Drosophila profilin (encoded by chickadee, chic) and Ena, have been shown to be essential for axon growth and guidance in vivo (Wills et al., 1999a,b; Ng and Luo, 2004). We found that both chic and ena show a strong dominant genetic interaction with dDAAMEx68 (Fig. 6A). Because the elav-Gal4/UAS-C-DAAM gain-of-function phenotype is not modified by chic (Fig. 6C), it seems likely that profilin concentration is not rate limiting for actin polymerization in this situation. In contrast, the ena loss-of-function alleles act as strong suppressors of the C-DAAM gain-of-function phenotype (Fig. 6C), suggesting that ena acts downstream of dDAAM. In support of the genetic interactions, dDAAM tends to colocalize with Ena along filopodia in primary neurons, including their filopodial tip complexes (Fig. 3D–F). Similarly, Ena and Chic are highly enriched in the cellular protrusions induced by activated dDAAM in S2 cells, where both proteins show a colocalization with activated dDAAM (Fig. 3M–O; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). To verify that these proteins associate in vivo, coimmunoprecipitation experiments were carried out. When Ena or profilin was immunoprecipitated from lysates of S2 cells transfected with DADm-DAAM::EGFP, some of this activated dDAAM, but not the full-length (presumably autoinhibited) endogenous protein, was found to be associated with Ena and profilin (Fig. 6D). Additionally, anti-Ena clearly pulled down profilin (Fig. 6D), in agreement with their previously demonstrated interaction in vitro (Ahern-Djamali et al., 1999). Thus, taken it together with our genetic interaction and localization data, these results indicate that dDAAM operates in close association with Ena and profilin in vivo.
Evolutionary conservation
Recent works investigated the embryonic expression pattern of the mouse, chick and Xenopus DAAM orthologs and came to the conclusion that the major expression domain of the vertebrate DAAM family members is the embryonic CNS (Kida et al., 2004; Nakaya et al., 2004). These observations pointed to the possibility that the dDAAM CNS function revealed during our work represents an evolutionarily conserved feature of this formin subfamily. To test this hypothesis, we performed a series of cross-species experiments using expression of dDAAM and murine Daam1 (mDaam1) in a mouse cell line and Drosophila embryos.
P19 is a pluripotent cell line that clearly has the potential to differentiate toward the neuronal fate, for instance, upon RA treatment, cells differentiate into a population mainly consisting of neurons and glial cells (McBurney et al., 1982). When P19 cells were stably transfected with a full-length dDAAM expressing construct, 21% of the cells exhibited cell shape changes, including the formation of neurite-like protrusions (Fig. 7A–G). Moreover, the cell shape changes induced by dDAAM were accompanied by a significant increase in the expression level of NF-M and β-tubulinIII (Fig. 7H), two cytoskeletal proteins that are known to display an increased expression level if cells are exposed to RA (Paterno et al., 1997; Fanarraga et al., 1999). Consistent with these results, RA treatment of dDAAM expressing cells induced an even higher NF-M expression level than RA induction in normal P19 cells (Fig. 7H), whereas β-tubulinIII exhibited a similar expression level as control RA treated cells (Fig. 7H). Interestingly, we also noticed that the expression level of endogenous mDaam1 increases by sixfold upon RA induction (Fig. 7H), suggesting that mDaam1 may play a role during this type of neuronal differentiation.
Figure 7.

dDAAM expression in P19 cells, and mDaam1 expression in the Drosophila embryonic CNS. A–C, Undifferentiated P19 cells express the SSEA-1 (green in A) marker but not NF-M (green in B) or β-tubIII (green in C). D–G, Most cells expressing Drosophila FL-DAAM lose the expression of SSEA-1, while a subset of them expresses NF-M (green in E and G) and/or β-tubIII (red in F and G). Note that some of the NF-M and/or β-tubIII positive cells extend long, axon-like protrusions. Nuclei in A–D are labeled with DAPI (blue). H, Quantification of NF-M, β-tubIII and mDaam1 relative mRNA levels in control P19 cells, P19 cells treated with 10−7 M RA, cells expressing Drosophila FL-DAAM, cells expressing Drosophila FL-DAAM treated with RA, and cells transfected with an EGFP expressing vector. I–K, The expression pattern of a UAS-EGFP::mDaam1 (green in J and K) transgene is shown in the embryonic CNS, note the strong colocalization with that of BP102 (red in I and K) along the major axonal tracts. Scale bar, 50 μm.
In a second set of experiments, we expressed EGFP-tagged mDaam1 in the Drosophila embryonic CNS. Similarly to the endogenous Drosophila DAAM protein, mDaam1 is highly enriched in the embryonic neurites (Fig. 7I–K), and the expression of EGFP::mDaam1 in dDAAMmat/zyg mutant embryos produced a rescue of the axon defects in 70% of the progeny (Fig. 2I). Together, these experiments revealed that dDAAM and mDaam1 share evolutionary conserved features evident at the level of subcellular localization as well as in their ability to functionally replace each other.
Discussion
dDAAM is an essential regulator of actin assembly during axonal growth
Proper axonal navigation requires actin dynamics in the peripheral growth cone region exhibiting actin-rich filopodia and lamellipodia. While it has been well established that filopodia contain bundled actin filaments, whereas the lamellipodial veils are comprised of a meshwork of actin filaments (Svitkina and Borisy, 1999; Strasser et al., 2004), the origin of these different filaments was unclear. Although several actin assembly factors have been described to date, their role during axonal growth remained a controversial issue. For example, the Arp2/3 complex has been implicated in axonal morphogenesis in several neuronal cell types (Zallen et al., 2002; Mongiu et al., 2007; Korobova and Svitkina, 2008), but in other neurons it had little if any effect on growth cone morphology or filopodia formation (Ng and Luo, 2004; Strasser et al., 2004). Two members of the formin family, mDia1 and mDia2, have also been implicated in neuronal growth regulation (Arakawa et al., 2003; Dent et al., 2007); however, the phenotype of the mDia1 knock-out mice (Eisenmann et al., 2007; Peng et al., 2007; Sakata et al., 2007) and the fact that mDia2 is not expressed in the mouse embryonic cortical neurons at the time of neuritogenesis (Dent et al., 2007), strongly argue against a role during in vivo neurite formation. Thus, it has remained largely unresolved which assembly factors are the key players at neuronal growth cones. Here, we used a number of different model systems to provide compelling evidence that formin proteins of the DAAM subfamily play a crucial role during axonal growth regulation. We report that, unlike the other Drosophila formins, dDAAM is strongly expressed in the developing nervous system. The protein localizes in dots throughout the F-actin rich growth cones, partially at the tip of filopodia. The loss of dDAAM function in embryonic neurons results in reduced neurite densities and abnormal axonal pathfinding. These phenotypes are consistent with aberrant actin dynamics and, indeed, F-actin dependent filopodia are shorter and reduced in number in dDAAM mutant primary neurons. The constitutively activated versions of dDAAM promote filopodia and neurite formation, i.e., cause the opposite phenotypes to the loss-of-function analyses in all assays we used. Because the FH2 domain of dDAAM is a potent actin nucleation factor in vitro (M. Nyitrai and J. Mihály, unpublished results), these observations together, strongly suggest that dDAAM plays a major role in the regulation of actin assembly during axonal growth.
Axonal growth regulation appears to be an evolutionary highly conserved process regulated by a very similar set of molecules in each species investigated. Consistent with this, here we have shown that dDAAM promotes the formation of neurite-like protrusions when expressed in mouse P19 cells, whereas murine Daam1 can functionally replace dDAAM in Drosophila. Significantly, Daam has been demonstrated to be a highly abundant formin in the developing nervous system of various vertebrate species (Kida et al., 2004; Nakaya et al., 2004). Although mutant analysis has not been reported for the vertebrate Daam orthologs, these data suggest that the regulation of actin assembly during axon growth is very likely to signify an evolutionary conserved DAAM function.
Rac not Rho seems to regulate dDAAM in the developing nervous system
The severe neuronal phenotypes demonstrate that dDAAM is an important molecular effector of axon growth that is likely to receive regulatory input from a large number of differentiation, growth and guidance cues. While the identification of the signals and the signaling pathways impinging on dDAAM activity awaits future investigations, our genetic interaction studies provide support for Rac but not Rho GTPases being the key regulators of dDAAM activity in the embryonic CNS. This idea is in good agreement with data demonstrating that Rac GTPases regulate axon growth in Drosophila (Hakeda-Suzuki et al., 2002; Ng et al., 2002) and in other organisms (Govek et al., 2005), and that also other members of the DAAM subfamily have been shown to bind different members of the Rho GTPase family (Aspenstrom et al., 2006; Liu et al., 2008).
Work in Drosophila MB neurons suggested that Rho GTPases regulate axon growth through several signaling pathways (Ng and Luo, 2004). Surprisingly, however, these studies led to the suggestion that Rac GTPases may promote axon growth not through the actin polymerization pathway, but through the regulation of microtubule dynamics and/or vesicle trafficking, that seems to contradict our finding implying a critical role for Rac/dDAAM mediated actin assembly in axon growth. The most likely explanation for this apparent contradiction is that Rac acts in more than one axon growth promoting pathway, in parallel to the LIM kinase dependent growth inhibiting pathway. Such a model is much easier to reconcile with several lines of evidences implicating Rac GTPases in actin cytoskeleton regulation (Govek et al., 2005), and we propose that the link to dDAAM established here is likely to represent one of the molecular mechanisms underlying Rac activity. We note that this hypothesis by no means exclude the possibility that Rac GTPases in growth cones are involved in the regulation of processes other than actin dynamics.
dDAAM function in growing neurons is linked to Ena and profilin
Our genetic interaction studies identified chic and ena as strong dominant enhancers of the dDAAM induced CNS defects, and likewise, we have shown that dDAAM is a dominant enhancer of the weak axonal morphogenesis defects exhibited in homozygous chic and ena mutant embryos (Fig. 6B). Additionally, we found that the activated dDAAM protein forms a complex with Ena and profilin. These results are in good agreement with reports for other formins which have been shown to bind profilin via FH1 domains (Wallar and Alberts, 2003) and Ena/VASP family members via FH2 domains (Schirenbeck et al., 2006). Thus, our data suggest that dDAAM works together with Ena and profilin to regulate axon growth, potentially involving their physical interaction.
What is the subcellular function of this protein complex during axon growth? One aspect clearly shared by profilin, dDAAM and Ena, is that all of them regulate filopodia formation in growth cones. The dDAAM data are presented here, severe reduction of filopodia in the absence of Ena/VASP function has been shown for a number of species, including Drosophila (Ng and Luo, 2004; Dent et al., 2007), while profilin is required for filopodia formation in cultured primary neurons (C. G. Pimentel, N. Sánchez-Soriano, and A. Prokop, unpublished results), and the same seems to be true in developing MB neurons (Ng and Luo, 2004). Additionally, the Ena, profilin and dDAAM proteins all have the ability to enrich at the filopodial tips, suggesting that these proteins are important to the formation of functional tip complexes. The Ena/VASP proteins are known to protect actin filament barbed ends from capping proteins (Bear et al., 2002), and also reported to bundle actin filaments (Bachmann et al., 1999; Schirenbeck et al., 2006), whereas formins act as processive cappers and promote filament elongation (Wallar and Alberts, 2003; Higgs, 2005). Based on these biochemical properties and our epistasis analysis, we propose that profilin promotes filopodia formation by providing actin monomers for a dDAAM induced actin filament assembly coupled to the bundling activity of Ena. Although it is possible that the profilin/dDAAM/Ena module promotes filopodia formation through a more complex molecular mechanism (i.e., feedback regulatory processes and link to microtubule dynamics, are anticipated), we note that this model is entirely consistent with the notion that a similar, formin/VASP dependent mechanism, regulates filopodia formation in Dictyostelium (Schirenbeck et al., 2006).
In the absence of dDAAM, filopodia are reduced both in number and length, indicating that dDAAM is required for filament elongation. Formins are, however, capable of de novo actin nucleation as well, therefore, it is an important question whether dDAAM nucleates novel filaments during filopodia initiation or the main function is to promote elongation. Because the number of filopodia is severely reduced in dDAAM mutant primary neurons, these data would be consistent with a predominant role in nucleation. Interestingly, recent work revealed that another nucleation factor, the Arp2/3 complex, is required for filopodia formation in several neuronal cell types (Korobova and Svitkina, 2008), but it has also been found that some filopodial actin filaments display free “pointed” ends, and thus, might be nucleated by formins. Together, with respect to the currently discussed mechanisms of filopodia formation, these data are most consistent with models in which the Arp2/3 and formin dependent filaments are both required for filopodia formation. On the contrary, it seems unlikely that either the Arp2/3 dependent “convergent elongation model” or the formin based “de novo filament elongation model” (Mattila and Lappalainen, 2008), represents the major mechanism of filopodia formation in neurons. However, whether both types of nucleation activities are required for axon growth, or the nucleation activity of Arp2/3 is coupled to the elongation activity of dDAAM, awaits future investigations.
Footnotes
This work was supported by Wellcome Trust Grant 077748/Z/05/Z (N.S.S, A.P.), and a European Molecular Biology Organization/Howard Hughes Medical Institute Scientist grant (J.M.). We thank B. Dickson, P. Rorth, J. Zallen, T. Yamaguchi, G. Ádám, M. Frasch, the Developmental Studies Hybridoma Bank, and the Bloomington and Szeged Stock Centers for fly stocks and reagents. We are grateful to H. Gyurkovics and P. Vilmos for critical reading and helpful comments on this manuscript. We are particularly grateful to Éva Kurucz for advice on the co-IP experiments, and Anna Rehák, Edina Ördög, Anikó Berente, Margit Pál, Lehőcz Istvánné, and Mária Dudás for technical assistance.
References
- Ahern-Djamali SM, Bachmann C, Hua P, Reddy SK, Kastenmeier AS, Walter U, Hoffmann FM. Identification of profilin and src homology 3 domains as binding partners for Drosophila enabled. Proc Natl Acad Sci U S A. 1999;96:4977–4982. doi: 10.1073/pnas.96.9.4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa Y, Bito H, Furuyashiki T, Tsuji T, Takemoto-Kimura S, Kimura K, Nozaki K, Hashimoto N, Narumiya S. Control of axon elongation via an SDF-1alpha/Rho/mDia pathway in cultured cerebellar granule neurons. J Cell Biol. 2003;161:381–391. doi: 10.1083/jcb.200210149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspenström P, Richnau N, Johansson AS. The diaphanous-related formin DAAM1 collaborates with the Rho GTPases RhoA and Cdc42, CIP4 and Src in regulating cell morphogenesis and actin dynamics. Exp Cell Res. 2006;312:2180–2194. doi: 10.1016/j.yexcr.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Bachmann C, Fischer L, Walter U, Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J Biol Chem. 1999;274:23549–23557. doi: 10.1074/jbc.274.33.23549. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, Alberts AS, Mori S, Gertler FB. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- Eisenmann KM, West RA, Hildebrand D, Kitchen SM, Peng J, Sigler R, Zhang J, Siminovitch KA, Alberts AS. T cell responses in mammalian diaphanous-related formin mDia1 knock-out mice. J Biol Chem. 2007;282:25152–25158. doi: 10.1074/jbc.M703243200. [DOI] [PubMed] [Google Scholar]
- Fanarraga ML, Avila J, Zabala JC. Expression of unphosphorylated class III beta-tubulin isotype in neuroepithelial cells demonstrates neuroblast commitment and differentiation. Eur J Neurosci. 1999;11:517–527. [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;416:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Kida Y, Shiraishi T, Ogura T. Identification of chick and mouse Daam1 and Daam2 genes and their expression patterns in the central nervous system. Brain Res Dev Brain Res. 2004;153:143–150. doi: 10.1016/j.devbrainres.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell. 2008;19:1561–1574. doi: 10.1091/mbc.E07-09-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, Habas R. Mechanism of activation of the Formin protein Daam1. Proc Natl Acad Sci U S A. 2008;105:210–215. doi: 10.1073/pnas.0707277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- Matusek T, Djiane A, Jankovics F, Brunner D, Mlodzik M, Mihály J. The Drosophila formin DAAM regulates the tracheal cuticle pattern through organizing the actin cytoskeleton. Development. 2006;133:957–966. doi: 10.1242/dev.02266. [DOI] [PubMed] [Google Scholar]
- McBurney MW, Jones-Villeneuve EM, Edwards MK, Anderson PJ. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature. 1982;299:165–167. doi: 10.1038/299165a0. [DOI] [PubMed] [Google Scholar]
- Mongiu AK, Weitzke EL, Chaga OY, Borisy GG. Kinetic-structural analysis of neuronal growth cone veil motility. J Cell Sci. 2007;120:1113–1125. doi: 10.1242/jcs.03384. [DOI] [PubMed] [Google Scholar]
- Nakaya MA, Habas R, Biris K, Dunty WC, Jr, Kato Y, He X, Yamaguchi TP. Identification and comparative expression analyses of Daam genes in mouse and Xenopus. Gene Expr Patterns. 2004;5:97–105. doi: 10.1016/j.modgep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Ng J, Luo L. Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron. 2004;44:779–793. doi: 10.1016/j.neuron.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Ng J, Nardine T, Harms M, Tzu J, Goldstein A, Sun Y, Dietzl G, Dickson BJ, Luo L. Rac GTPases control axon growth, guidance and branching. Nature. 2002;416:442–447. doi: 10.1038/416442a. [DOI] [PubMed] [Google Scholar]
- Pak CW, Flynn KC, Bamburg JR. Actin-binding proteins take the reins in growth cones. Nat Rev Neurosci. 2008;9:136–147. doi: 10.1038/nrn2236. [DOI] [PubMed] [Google Scholar]
- Patel NH, Schafer B, Goodman CS, Holmgren R. The role of segment polarity genes during Drosophila neurogenesis. Genes Dev. 1989a;3:890–904. doi: 10.1101/gad.3.6.890. [DOI] [PubMed] [Google Scholar]
- Patel NH, Martin-Blanco E, Coleman KG, Poole SJ, Ellis MC, Kornberg TB, Goodman CS. Expression of engrailed proteins in arthropods, annelids, and chordates. Cell. 1989b;58:955–968. doi: 10.1016/0092-8674(89)90947-1. [DOI] [PubMed] [Google Scholar]
- Paterno GD, Gillespie LL, Julien JP, Skup D. Regulation of neurofilament L, M and H gene expression during retinoic acid-induced neural differentiation of P19 embryonal carcinoma cells. Brain Res Mol Brain Res. 1997;49:247–254. doi: 10.1016/s0169-328x(97)00157-5. [DOI] [PubMed] [Google Scholar]
- Peng J, Kitchen SM, West RA, Sigler R, Eisenmann KM, Alberts AS. Myeloproliferative defects following targeting of the Drf1 gene encoding the mammalian diaphanous related formin mDia1. Cancer Res. 2007;67:7565–7571. doi: 10.1158/0008-5472.CAN-07-1467. [DOI] [PubMed] [Google Scholar]
- Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Drosophila Spire is an actin nucleation factor. Nature. 2005;433:382–388. doi: 10.1038/nature03241. [DOI] [PubMed] [Google Scholar]
- Rasko I, Georgieva M, Farkas G, Santha M, Coates J, Burg K, Mitchell DL, Johnson RT. New patterns of bulk DNA repair in ultraviolet irradiated mouse embryo carcinoma cells following differentiation. Somat Cell Mol Genet. 1993;19:245–255. doi: 10.1007/BF01233072. [DOI] [PubMed] [Google Scholar]
- Sakata D, Taniguchi H, Yasuda S, Adachi-Morishima A, Hamazaki Y, Nakayama R, Miki T, Minato N, Narumiya S. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J Exp Med. 2007;204:2031–2038. doi: 10.1084/jem.20062647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirenbeck A, Arasada R, Bretschneider T, Stradal TE, Schleicher M, Faix J. The bundling activity of vasodilator-stimulated phosphoprotein is required for filopodium formation. Proc Natl Acad Sci U S A. 2006;103:7694–7699. doi: 10.1073/pnas.0511243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallar BJ, Alberts AS. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 2003;13:435–446. doi: 10.1016/s0962-8924(03)00153-3. [DOI] [PubMed] [Google Scholar]
- Wang YE, Chandler R, Lau P, Bieber AJ. Drosophila nerve cord culture: a tool for studying neural development. J Neurosci Methods. 1998;85:21–26. doi: 10.1016/s0165-0270(98)00107-1. [DOI] [PubMed] [Google Scholar]
- Wills Z, Marr L, Zinn K, Goodman CS, Van Vactor D. Profilin and the Abl tyrosine kinase are required for motor axon outgrowth in the Drosophila embryo. Neuron. 1999a;22:291–299. doi: 10.1016/s0896-6273(00)81090-9. [DOI] [PubMed] [Google Scholar]
- Wills Z, Bateman J, Korey CA, Comer A, Van Vactor D. The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron. 1999b;22:301–312. doi: 10.1016/s0896-6273(00)81091-0. [DOI] [PubMed] [Google Scholar]
- Zallen JA, Cohen Y, Hudson AM, Cooley L, Wieschaus E, Schejter ED. SCAR is a primary regulator of Arp2/3-dependent morphological events in Drosophila. J Cell Biol. 2002;156:689–701. doi: 10.1083/jcb.200109057. [DOI] [PMC free article] [PubMed] [Google Scholar]