

Abstract
Localized increases in synaptic strength constitute a synaptic basis for learning and memory in the CNS and may also contribute to the maintenance of neuropathic pain after spinal cord injury (SCI) through the de novo formation or elaboration of postsynaptic dendritic structures. To determine whether SCI-induced dendritic spine remodeling contributes to neuronal hyperexcitability and neuropathic pain, we analyzed spine morphometry, localization, and functional influence in dorsal horn (DH) neurons in adult rats 1 month after sham surgery, contusion SCI, and SCI treated with a selective inhibitor of Rac1 activation, NSC23766. After SCI, DH neurons located in lamina IV–V exhibited increased spine density, redistributed spines, and mature spines compared with control neurons, which was associated with enhancement of EPSCs in computer simulations and hyperexcitable responsiveness to innocuous and noxious peripheral stimuli in unit recordings in vivo. SCI animals also exhibited symptoms of tactile allodynia and thermal hyperalgesia. Inhibition of the small GTP-binding protein Rac1 ameliorated post-SCI changes in spine morphology, attenuated injury-induced hyperexcitability of wide-dynamic range neurons, and progressively increased pain thresholds over a 3 d period. This suggests that Rac1 is an important intracellular signaling molecule involved in a spinal dendritic spine pathology associated with chronic neuropathic pain after SCI. Our report provides robust evidence for a novel conceptual bridge between learning and memory on the one hand, and neuropathic pain on the other.
Keywords: spinal cord injury, Rac1, dendritic spine, neuropathic pain, dorsal horn, pain, synaptic plasticity
Introduction
Spinal cord injury (SCI) adversely modifies the way neurons receive, process, and relay sensory information, and, by producing a long-lasting potentiation of nociceptive signals in the CNS (i.e., central sensitization) (Hains et al., 2003; Ji et al., 2003; Waxman and Hains, 2006), can result in the development of neuropathic pain (Finnerup et al., 2001; Siddall et al., 2003). Current therapies have limited effectiveness in alleviating SCI-induced neuropathic pain.
Neuropathic pain shares mechanistic similarities with learning and memory (Ji et al., 2003; Sandkühler, 2007). Activity-dependent synaptic plasticity underlies how the nervous system rewires connections to learn, store, and modify information (Malenka, 2003). Such dynamic synaptic modifications also occur in the spinal cord in association with pain processing (Ji et al., 2003). High-frequency tetanic stimulation (Ikeda et al., 2000) and low-frequency barrage from acute inflammation (Ikeda et al., 2006) induce synaptic strengthening between c-fiber afferents and dorsal horn nociceptive neurons (Svendsen et al., 1999). Central or peripheral injury (i.e., SCI or axotomy), alternatively, may also lead to synaptic plasticity in the spinal cord dorsal horn and contribute to pain maintenance (Woolf et al., 1992; Ji et al., 2003; Kim et al., 2006).
A synaptic model similar to that involved in long-term memory storage may explain the persistent nature of neuropathic pain. Late phases of cortical long-term potentiation (LTP) require protein synthesis, cytoskeletal rearrangement, and the de novo formation or elaboration of dendritic spines (Yuste and Bonhoeffer, 2001; Chen et al., 2007), which are major sites for excitatory input. Spine geometry regulates neuronal responsiveness and synaptic input integration, and mature spines associate with increased magnitude and faster latency of evoked postsynaptic potentials, which correlate with enhanced synaptic efficacy after memory formation (Lüscher et al., 2000). The location of spines on the dendrite relative to the soma also has significant functional implications (Rall et al., 1967).
Activity of the small GTP-binding protein Rac1 regulates dendritic spine morphology in hippocampal neurons through actin cytoskeleton reorganization and promotes clustering of glutamate AMPA receptors in spines (Nakayama et al., 2000; Tashiro et al., 2000; Tashiro and Yuste, 2004). Importantly, SCI increases Rac1 mRNA expression, which remains elevated for up to 3 months (Erschbamer et al., 2005), and activates other protein members of the GTPase family involved in SCI pathologies (Dergham et al., 2002; Dubreuil et al., 2003). A role of Rac1, however, in neuropathic pain after SCI has not been established.
Here, we asked whether dendritic spine remodeling contributes to the maintenance of chronic neuropathic pain after SCI. SCI animals demonstrated altered spine morphologies and distribution, which contribute to increased neuronal excitability. Because in vitro and in vivo studies demonstrate that Rac1 can modulate dendritic spine morphology and function (Nakayama et al., 2000; Wiens et al., 2005), we disrupted spine structure with the Rac1-specific inhibitor NSC23766, which blocks guanine exchange factors (GEFs), Trio and Tiam1. Inhibition of the Rac1 signaling cascade ameliorated the development of abnormal spine morphologies, reduced neuronal excitability, and normalized nociceptive thresholds. Together, our results identify a molecular correlate of spinal “pain memory” involving Rac1-regulated dendritic spine remodeling after SCI.
Materials and Methods
Animals.
Experiments were performed in accordance with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Yale University Institutional Animal Use Committee. Adult male Sprague Dawley rats (175–200 g) were used for this study. Animals were housed under a 12 h light/dark cycle in a pathogen-free area with water and food given ad libitum.
Spinal cord injury.
Animals were anesthetized with a mixture of ketamine and xylazine (80/5 mg/kg, i.p.) and subjected to low thoracic (T9) spinal cord contusion injury. Briefly, SCI was produced at spinal segment T9 using the NYU/MASCIS (New York University/Multicenter Animal Spinal Cord Injury Study) impact injury device (Gruner, 1992; Hains and Waxman, 2006). After laminectomy, a 2.0-mm-diameter rod (10 g) was dropped from a 25 mm height onto the exposed spinal cord (n = 32). Intact animals (n = 14) underwent laminectomy only. Postoperative treatments included twice daily subcutaneous injections of 0.9% saline solution for rehydration (2.0 cc) and Baytril (0.3 cc; 22.7 mg/ml) to prevent bladder infection. Bladders were manually expressed twice daily until reflex bladder function returned, usually within 10 d after injury.
Intrathecal catheterization and drug delivery.
After 4 weeks post-SCI, under ketamine/xylazine anesthesia, a sterile 32 gauge intrathecal catheter (ReCathCo) was inserted through a slit in the atlanto-occipital membrane, between the base of the skull and spinal vertebra C1, threaded caudally to the lumbar enlargement and secured by suturing. The rostral opening was heat sealed to prevent CSF leakage. Tip location was confirmed after the animals were killed. Under brief halothane sedation (<2 min; 3% by chamber), either control vehicle (veh) (0.9% saline; n = 16) or NSC23766 (1 mg/ml; n = 16), a Rac1 GTPase-specific inhibitor (Gao et al., 2004), were infused (5 μl volume; twice daily; 3 d), followed by sterile saline flush (20 μl).
Histology.
Intact (n = 6), SCI plus veh (n = 8), and SCI plus NSC23766 (n = 8) rats were killed 31 d after SCI for Golgi-cox staining using a FD Rapid GolgiStain kit (FD Neurotechnologies). Briefly, fresh unfixed spinal cord tissue from the lumbar enlargement (L3–L6) was rapidly removed, washed in distilled water, and immersed in the impregnation solutions. After a series of steps described by the manufacturer's instructions, 200-μm-thick tissue sections were cut on a vibratome and mounted on gelatinized glass slides. Sections were stained, rinsed in distilled water, dehydrated, cleared, and coverslipped.
Behavioral assays.
Animals were tested for tactile allodynia and thermal hyperalgesia daily from days 29–31 after SCI or “intact” surgery (intact, n = 6; SCI plus veh, n = 6; SCI plus NSC23766, n = 6). A blinded observer performed all tests. Baseline thresholds were established before surgery, and then performed after SCI and injections. Mechanical thresholds were measured by hindpaw withdrawal to von Frey filaments (Stoelting) (0.4–26 g) applied to the midplantar region of the hindpaws and a modified “up–down” method to accurately determine nociceptive thresholds (Chaplan et al., 1994). Thermal sensory thresholds were measured by latency of paw withdrawal in response to a calibrated radiant heat source (4.7 amps) of the plantar surface of the paw (Dirig et al., 1997). Another subset of animals were tested for their sensory-motor ability 31 d after SCI (intact, n = 5; SCI plus veh, n = 5; SCI plus NSC23766, n = 5) using a motorized rotor rod system and Rotamex software package, version 1.32A (Columbus Instruments). The rod (diameter, 7.0 cm) was accelerated at 0.1 rpm/1 s. A total of seven trials were performed with the first two trials considered “training.” The remaining five trials were used for analysis and the revolutions per minute of the rod when the animal fell was compared between groups.
Electrophysiological recordings.
Thirty-one days after SCI or intact surgery, animals (n = 4/group) were anesthetized with sodium pentobarbital (40 mg/kg, i.p.). Extracellular single-unit recordings and identification of wide-dynamic range (WDR) neurons were described by Hains et al. (2003). Units were isolated from L3–L5 near the dorsal root entry zone up to a depth of 1000 μm in the dorsal horn. Recordings were made with a low impedance 5 MΩ tungsten-insulated microelectrode (A-M Systems) after a cell was located and its receptive field mapped. Six mechanical stimuli were applied: (1) phasic brush (PB) stimulation of the skin with a cotton applicator, (2) pressure (144 g/mm2) by attaching a large arterial clip with a weak grip to a fold of the skin, (3) pinch (583 g/mm2), (4) 0.38 g von Frey filament, (5) 1.01 g von Frey filament, and (6) 20.8 g von Frey filament.
Dendritic spine/image analysis.
Golgi-stained coronal spinal cord tissue sections taken from the lumbar enlargement were examined under transmitted light microscopy. Five criteria allowed us to sample and analyze whole cells with morphology similar to those observed for WDR neurons identified by Woolf (Woolf and King, 1987): (1) neurons were located within lamina 4 and 5; (2) Golgi-stained neurons must have had dendrites and spines that were completely impregnated, appearing as a continuous length; (3) at least one dendrite extended into an adjacent lamina relative to the origin of the cell body; (4) at least one-half of the primary dendritic branches remained within the thickness of the tissue section, such that their endings were not cut and instead appeared to taper into an ending; and (5) the cell body diameter fell between 20 and 50 μm. As a note, these criteria do not imply the physiological characterization of the neurons we analyzed, and instead control for the morphological diversity of spinal cord dorsal horn neurons. A total of 127 neurons (intact, n = 38; SCI plus veh, n = 49; SCI plus NSC23766, n = 40) was identified in this manner and included in our analysis.
Dendritic spines were identified using criteria described by Kim et al. (2006). Protrusions from the dendrite branch with a clearly recognizable neck were counted as spines (Kim et al., 2006). When protrusions from the dendrite branch did not have an apparent neck structure, the protrusion was only considered a spine if there was a visible indention on either side of junction of the protrusion and the dendritic branch. Dendritic spines were classified into two types: thin and mushroom-shaped spines. Thin spines were considered spines that had head-like enlargements with diameters less than the length of the neck. Mushroom spines were spines whose head diameter was greater than the length of the neck. We defined the spine neck as the structure between the base of the spine, at the juncture to the parent dendritic branch, and the base of the spine head where the appearance of the spine begins to swell distally. The appearance of the spine head varied but in general was observed as bulb-like structure at the distal end of the spine protrusion. We felt that these two classifications were valid for three reasons. First, having only two spine shape categories allowed us to use simple but strict rules in classifying spine shape. Second, the number of spines we included in our study (total spines counted, 50,485) precluded discrimination of the subtle variations in spine shape. Third, there is a large body of literature describing the different physiological characteristics associated with the two morphologies of thin and mushroom-shaped spines (Calabrese et al., 2006).
To analyze and reconstruct the complete appearance of the dorsal horn neurons, we used Neurolucida software (MicroBrightField), and the resulting three-dimensional reconstructions of dorsal horn neurons were analyzed for spine density and spine distribution. Two Neurolucida traces were made for each cell. First, a contour trace of the perimeter of coronal spinal cord section and the dorsal horn containing each identified cell was made. The locations of these cells were then marked on the spinal cord contour maps. Three-dimensional reconstructions of these dorsal horn neurons were created by tracing through the x-, y-, and z-axis. Dendritic spine type (thin spines, blue; mushroom spines, red) and location were marked on these three-dimensional reconstructions.
Spine density was expressed as spine number/micrometer dendritic length. To determine changes in spine distribution relative to the cell body between groups, a modified Sholl's analysis was performed using the Neurolucida software. Seven 50-μm-wide spherical bins were formed around the cell body, spine density was measured within each bin, and data in each bin were averaged within a treatment group and the means were compared against equivalent bins across groups.
To determine changes in spine dimensions, five neurons were arbitrarily chosen from each treatment group, and visible spines were measured for spine length and spine head diameter. The length of a spine was defined as the distance from the tip of the spine to the junction of the spine and the parent dendritic branch. The spine head diameter was defined as the longest line drawn normal to the length of the parent dendritic branch.
Images shown in Figure 2 were captured with a Nikon Eclipse E800 microscope with a HQ Coolsnap camera (Roper Scientific).
Figure 2.

Coronal sections of spinal cord tissue from the lumbar enlargement were Golgi-stained and examined for dendritic spines. A, A representative neuron whose soma (black arrow) is located in lamina 5. B, A magnified view of the neuron identified in A (inset). C, A ∼50 μm dendritic segment from the intact group (see C′, inset). D, After SCI, there is an increase in spine density and the appearance of larger spine head structures. E, NSC23766 reduces the number of spines and decreases the overall dimension of spines after SCI. F, High-power images of two representative thin-shaped spines found on deep lamina neurons in the spinal cord dorsal horn. G, Mushroom spines appeared with swollen head structures with narrow spine necks. C′–E′, Inset from white boxes shows visible thin and mushroom spines distributed along a 10 μm span of the dendrite from each treatment group. Scale bar is the same for C–E and C′–E′. H, All dendritic spines from five neurons randomly chosen from each treatment group were measured for spine length. SCI plus veh treatment increased the length of spines compared with intact animals; spine length was significantly decreased with NSC23766 treatment (*p < 0.001). I, SCI plus veh treatment also increased the spine head diameter of dendritic spines compared with intact animals. NSC23766 treatment significantly decreased the spine head diameter compared with both SCI plus veh treated animals and intact, uninjured animals (*p < 0.001). Scale bars: A, 500 μm; B, 100 μm; C–E, 10 μm; C′–E′, 5 μm; F, G, 1 μm.
Neuron simulation environment.
We used the NEURON simulation environment, a freely available software package (www.neuron.yale.edu/neuron) to model the in silico effects of spine shape, density, and distribution on the transduction of signals onto a postsynaptic neuron. We modeled the presynaptic input onto dendritic spines as an α-function with kinetics similar to excitatory AMPA synapses (τ = 5 ms; gmax = 0.05 μS) (Destexhe et al., 1994) (shown in Fig. 5A). We used a two-part compartment model (spine neck and head), assumed equal spine volume across treatment groups (to examine shape effects only), and included previously described (Segev and Rall, 1988; Rusakov et al., 1996) membrane properties to study spine physiology (see Fig. 5D).
Figure 5.

NEURON simulation demonstrates that SCI-induced dendritic spine remodeling (E) alters the input–output function of a postsynaptic neuron. A, Presynaptic input onto a dendritic spine is modeled using an α-function to simulate the kinetics of an excitatory AMPA synapse (τ max conductance, 5 ms; gmax, 0.05 μS). B, The shape of the spine influences the EPSP recorded at the base of the dendritic spine neck (as shown in D) in intact, SCI plus veh, and SCI plus NSC23766 (as shown in D; H-H channels in gray region head compartment). C, Post-SCI changes in spine density and distribution (Fig. 4) result in a second action potential. NSC23766 decreased spine density and resulted in output similar to output cells from intact animals.
We used experimentally measured spine lengths and spine head diameter values, and varied diameter values for the spine neck and length values for the spine head (to maintain constant spine volume). The spine head was modeled as a sphere (i.e., a cylinder with equal diameter and length) whose diameter was equal to the experimental mean spine head diameter. The spine neck membrane was modeled with passive properties with transmembrane resistivity Rm = 10 kΩ · cm2 and capacity Cm = 1.0 μF/cm2. The spine head had active membrane properties and contained Hodgkin–Huxley (H-H) channels (gNa = 1.2 S/cm2; gK = 0.36 S/cm2; gL = 3 mS/cm2). The cytoplasmic resistivity of the entire spine was Ra = 200 Ω · cm to account for dense “packing” of cellular organelles such as the spine apparatus (Segev and Rall, 1988). The soma (diameter, 30 μm) contained H-H active membrane properties (gNa = 0.12 S/cm2; gK = 0.036 S/cm2; gL = 0.3 mS/cm2). The initial segment (d = 2 μm; l = 50 μm) contained active membranes with twice the sodium conductance as the soma (gNa = 0.24 S/cm2) to account for a greater density of sodium channels. The axon hillock (d = 2.5 μm; l = 50 μm) and dendritic branch were modeled with passive membrane properties (Rm = 10 kΩ · cm2; capacity Cm = 1.0 μF/cm2). The cytoplasmic resistivity of the neuron (except for the spines) was Ra = 100 Ω · cm.
The simplified neuron was also modeled in a compartmental manner (see Fig. 5E), including soma, initial segment, axon hillock, a single dendritic branch, and spines. The dendritic branch was divided into nine compartments, each of the same length and diameter (d = 2 μm; l = 50 μm). The first compartment closest to the soma contained no spines. The dendritic compartment modeled the relative spine density and distribution measured from our Neurolucida reconstructions (see Results) across treatment groups. In the simplified neuronal model (see Fig. 5) for intact neurons, the total number of spines was six spines distributed across a 350 μm length of dendrite. For SCI neurons, nine spines were distributed along the same length of dendrite; the first two dendrite length compartments contained twice the spine density compared with the intact neuron, to correspond with the approximate relative spine distribution obtained from the Neurolucida analysis. For SCI plus NSC23766 treatment, five neurons were distributed accordingly. All spines were modeled similarly, with electrophysiological and morphological properties similar across treatment groups, the latter using intact spine parameters measured in intact rats (see Results). To execute the simulation, all spines were simultaneously stimulated, and the resultant activity was recorded from the soma.
Western blot analyses.
Spinal cord tissue from lumbar level 3–6 were freshly extracted from a subset of animals (n = 10), snap frozen in liquid nitrogen, and homogenized in lysis buffer (125 mm HEPES, pH 7.5, 750 mm NaCl, 5% IGEPAL CA-630, 50 mm MgCl2, 5 mm EDTA, and 10% glycerol). Portions of lysate were taken for protein quantification using a Bio-Rad protein assay kit (Bio-Rad Laboratories). For analyses, lysate (50–100 μg) was subjected to electrophoresis (4–12% polyacrylamide gel), transferred to nitrocellulose membranes (Invitrogen), and incubated in appropriate primary and secondary antibodies. The blots were incubated in 10% dry milk for 1 h followed by overnight incubation at 4°C in primary antibodies. After washing, sections were incubated for 1 h at room temperature in species-appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Dako). Blots were developed using chemiluminescence markers (PerkinElmer Life and Analytical Sciences). The blots were apposed to photographic film and the film was developed (Kodak). The following primary antibodies were used: rabbit anti-phospho Rac1 plus Cdc42 (Abcam; 1:250), mouse anti-Rac1 (Millipore), rabbit anti-cortactin (Santa Cruz Biotechnologies; 1:2000), mouse anti-PSD-95 (Abcam; 1:2000), rabbit anti-synaptophysin (GeneTex), and rabbit anti-β-actin (Abcam; 1:5000). The following secondaries were used: goat anti-mouse HRP (1:10,000) and goat anti-rabbit HRP (1:10,000). For quantification of blot intensities, we used ImageJ software (available free at http://rsb.info.nih.gov/ij/index.html). To normalize for variations between Western blots, we set the control, β-actin staining intensity in each lane to 100% and compared proteins of interest between treatment groups.
Statistical analysis.
All statistical tests were performed at the α-level of significance of 0.05 for all analyses using either parametric and nonparametric tests, including one-way ANOVA pairwise comparison and repeated-measures ANOVA, followed by Bonferonni's post hoc analysis, and Mann–Whitney rank sum test. Data management and statistical analyses were performed using SigmaStat (version 3.0.1a; Jandel Scientific), SPSS, and Microsoft Office Excel (2003), and plotted as mean ± SEM using SigmaPlot, version 8.02a.
Results
Spinal cord injury triggers Rac1 activation in the lumbar spinal cord tissue
Rac1 contributes to regulating dendritic spine morphology and spine density (Tashiro et al., 2000; Tashiro and Yuste, 2004). In addition, neurotrauma, such as with SCI, activates protein members of a small GTPase family, including Rho, Rac1, and cdc42 (Dergham et al., 2002; Dubreuil et al., 2003; Monnier et al., 2003). It is possible that levels of phosphorylated Rac1 after SCI contributes to dendritic spine remodeling in the dorsal horn neurons. To investigate the levels of endogenous phosphorylated Rac1, we prepared fresh lumbar spinal cord tissue for Western blot analysis. Lumbar dorsal horn tissue from animals 28 d after SCI with vehicle or NSC23766 treatment was compared with tissue from uninjured animals. Western blot analyses (Fig. 1A) showed that spinal cord 28 d after contusion injury contained significantly higher levels of activated Rac1 compared with intact cord (51.3 ± 2.0 vs 34.4 ± 3.8%; p = 0.006). Lumbar infusion of NSC23766 resulted in a reduction in the levels of activated Rac1 compared with SCI animals (27.8 ± 3.8 vs 51.3 ± 2.0%; p = 0.018), to levels that were not statistically significant from Rac1 levels (27.8 ± 6.6%) observed in spinal cord tissue from intact animals (p = 0.22). We did not observe any differences in levels of β-actin (Fig. 1A, bottom) or total Rac protein (Fig. 1C,D), excluding the possibility of unequal protein loading between lanes. These results demonstrate that injury induces Rac1 phosphorylation in the spinal cord.
Figure 1.
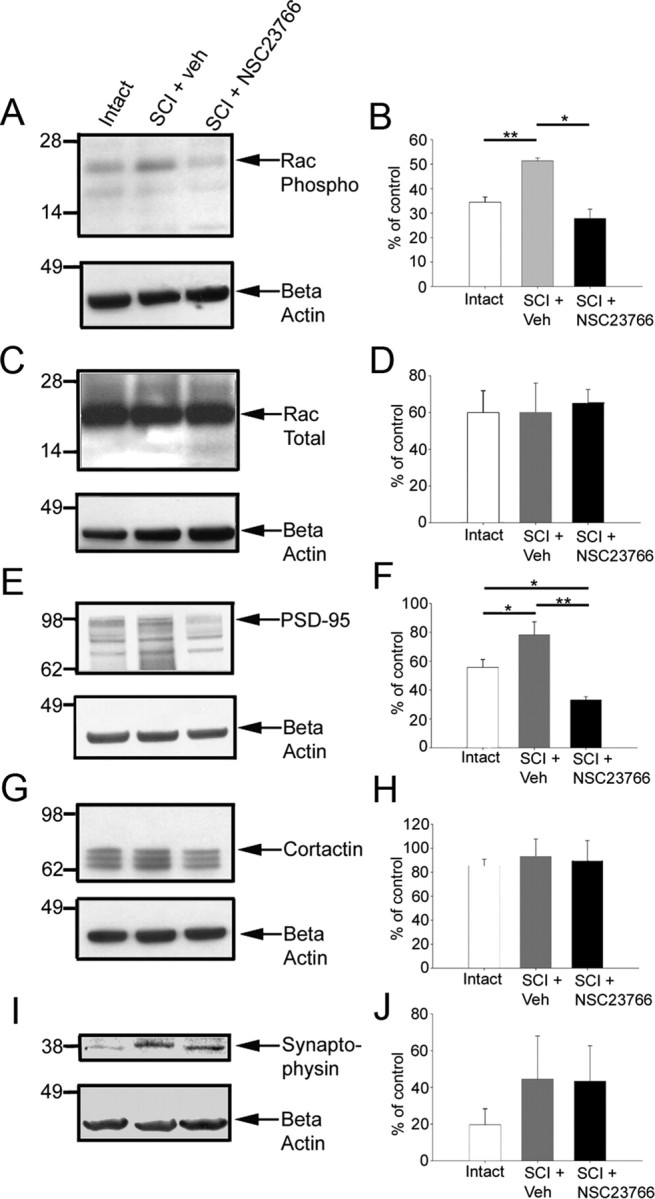
Western blot analysis of synapse-associated proteins in lumbar spinal cord tissue from intact, SCI and control, vehicle- or NSC23766-treated animals. Quantification was performed on three blots for each protein. To normalize for variation between blots, β-actin control protein level was set to 100% in each lane and proteins of interest were compared across treatment groups. A, Phosphorylated Rac1 protein was probed in the three treatment groups with a visible band at ∼21 kDa. B, Significantly increased levels of phosphorylated Rac1 were found after SCI plus veh treatment compared with uninjured controls. Post-SCI levels were reduced with intrathecal NSC23766. C, D, No significant difference in the levels of total Rac (∼21 kDa) was observed between the treatment groups. E, F, PSD-95 band is visible at ∼95 kDa (E), and significantly higher levels were observed in SCI plus veh compared with intact levels (F). NSC23766 treatment also significantly reduced PSD-95 levels below that of SCI plus veh and intact animals. G, A cortactin band was observed at ∼80 kDa along with breakdown products. H, No differences were observed for levels of cortactin between groups. I, J, We also observed a trend, which did not reach statistical significance, toward an increase in the levels of synaptophysin after SCI. Graphs are mean ± SEM. *p < 0.05; **p < 0.01.
SCI leads to more mature synapses
To examine changes in synapse-associated proteins (PSD-95 and cortactin), we performed Western blot analyses on lumbar level spinal cord tissue after SCI (Fig. 1E–J). PSD-95 is a postsynaptic density protein localized to excitatory glutamatergic synapses, which regulates the clustering of AMPA receptors (Chetkovich et al., 2002), and recruits cell adhesion molecules such as neuroligin-1, which are involved in synapse formation (Ichtchenko et al., 1995). Thus, PSD-95 expression is a marker of sites of synapse plasticity. Expression of PSD-95 increased significantly in injured spinal cord tissue compared with uninjured controls (Fig. 1E,F) (79 ± 16.5 vs 55.9 ± 11%; p = 0.039). NSC23766 decreased post-SCI levels of PSD-95 (33.2 ± 4 0.5 vs 79.0 ± 16.5%; p = 0.004). Interestingly, NSC23766 treatment also reduced PSD-95 levels below that of uninjured levels (33.2 ± 4.4 vs 55.0 ± 11.0%; p = 0.023). Cortactin contributes to the stabilization and reorganization of actin filaments (Daly, 2004) and has an essential role in activity-dependent dendritic spine morphogenesis (Hering and Sheng, 2003). Cortactin levels did not significantly change after NSC23766 treatment compared with intact animals (Fig. 1G,H). Synaptophysin is a presynaptic-associated protein associated with synaptic vesicles and is used as a marker for synaptogenesis (Masliah et al., 1991). We observed a trend toward increased spinal cord tissue levels of synaptophysin after SCI, but it did not reach statistical significance (Fig. 1I,J).
Dendritic spines remodel in deep dorsal horn neurons after SCI
To determine whether SCI resulted in changes in dendritic spines, we analyzed Golgi-stained coronal sections of spinal cord at lumbar segments L3–L5 (Fig. 2A,B). WDR neurons were identified on the basis of morphological features (see Materials and Methods) in lamina IV and V (∼400–1200 μm deep) from the dorsal spinal cord surface. The dimensions of cell body, total dendrite length, number of primary dendrites, length of primary dendrites, and the percentage of primary dendrites with secondary branches (Table 1) were not significantly different when compared across treatment groups. Therefore, any observed changes in spine morphologies were not attributable to differences in neuronal sampling. A representative neuron located in lamina V is shown in Figure 2, A and B. We categorized all the observed spines (intact, n = 14,925; SCI plus veh, n = 22,870; SCI plus NSC23766, n = 12,690) into two groups: thin- and mushroom-shaped (see Materials and Methods) (Fig. 2C–G).
Table 1.
Cell body dimensions and dendritic branch morphology comparison
| Dorsal–ventral (μm) | Medial–lateral | Total dendrite length (μm) | No. of primary dendrites | Length of primary dendrite (μm) | Primary dendrites with secondary branches (%) | |
|---|---|---|---|---|---|---|
| Intact | 39.2 ± 8.5 | 24.7 ± 6.8 | 1372 ± 755 | 5.3 ± 2.0 | 50.2 ± 63 | 54.7 ± 28.7 |
| SCI + veh | 38.3 ± 14.2 | 21.5 ± 5.2 | 1419 ± 528 | 4.8 ± 1.2 | 44.7 ± 54.7 | 54.6 ± 26.6 |
| SCI + anti-Rac1 | 38.2 ± 6.5 | 21.7 ± 5.0 | 1550 ± 539 | 4.6 ± 0.9 | 35.6 ± 35 | 62.4 ± 20.1 |
Data are shown as mean ± SD.
To examine the effects of SCI on spine length and spine head diameter, we randomly sampled five neurons from each group and measured spine dimensions from each neuron. Compared with spines in the intact group (n = 1590 spines), spines in SCI plus veh (n = 2280) demonstrated longer lengths (2.17 ± 0.04 vs 2.45 ± 0.03 μm; p < 0.001) (Fig. 2H). Spines in SCI plus NSC23766 (n = 2256) exhibited reduced lengths compared with SCI plus veh (2.15 ± 0.03 vs 2.45 ± 0.03 μm; p < 0.001). There was no significant difference in spine length between intact and SCI plus NSC23766 groups (p = 0.12).
To determine whether SCI influence changes in spine head morphology, we also measured spine head diameter (Fig. 2I). Neurons in the SCI plus veh group exhibited dendritic spines with significantly larger spine head diameters than neurons in the intact group (1.59 ± 0.04 vs 1.89 ± 0.03 μm; p < 0.001). NSC23766 treatment significantly reduced spine head diameter compared with intact (0.85 ± 0.01 vs 1.59 ± 0.04 μm; p < 0.001) and SCI plus veh (0.85 ± 0.01 vs 1.89 ± 0.03 μm; p < 0.001). Together, the results demonstrate that spine length and spine head diameter increase after SCI, but these changes can be reversed with Rac1 inhibition.
Dendritic spine density increases after SCI
To obtain an accurate count of dendritic spines from Golgi-stained spinal cord tissue, we used a Neurolucida reconstruction technique (Fig. 3). The spinal cord and dorsal horn border were traced to indicate the location of the identified neuron (Fig. 3A), and the location of every spine was color coded as either thin (blue dot) or mushroom-shaped (red dot) (Fig. 3B,C) (for more complete three-dimensional rendering of this data, see the supplemental movies, available at www.jneurosci.org as supplemental material). Total spine density was measured and expressed as the number of spines per 10 μm dendritic length. As shown in Figure 4A, the total spine density increased after SCI (3.9 ± 0.14/10 μm dendrite) compared with intact animals (3.13 ± 0.13/10 μm dendrite; p < 0.001). SCI-induced increases in total spine density was reduced with NSC23766 (3.2 ± 0.27/10 μm dendrite; p = 0.027). There was no statistical difference in spine density between intact and SCI plus NSC23766 animals (p = 0.79). We also examined the density of thin (Fig. 4B) and mushroom-shaped spines (Fig. 4C). The density of thin-shaped spines increased after SCI (3.7 ± 0.14/10 μm dendrite) compared with intact (3.0 ± 0.13/10 μm dendrite; p < 0.001). NSC23766 treatment (3.1 ± 0.27/10 μm dendrite) significantly decreased thin-shaped spine density compared with SCI plus veh (p = 0.032). There was no difference in thin-shaped spine density between intact and SCI plus NSC23766 groups. The density of mushroom spines increased approximately twofold after SCI (0.3 ± 0.02/10 μm dendrite) compared with intact (0.18 ± 0.02/10 μm dendrite; p < 0.001) (Fig. 4C). After NSC23766 treatment (0.15 ± 0.02/10 μm dendrite), mushroom spine density decreased significantly (p < 0.001) compared with SCI animals, with a spine density that was not significantly different compared with levels in intact animals (p = 0.33).
Figure 3.
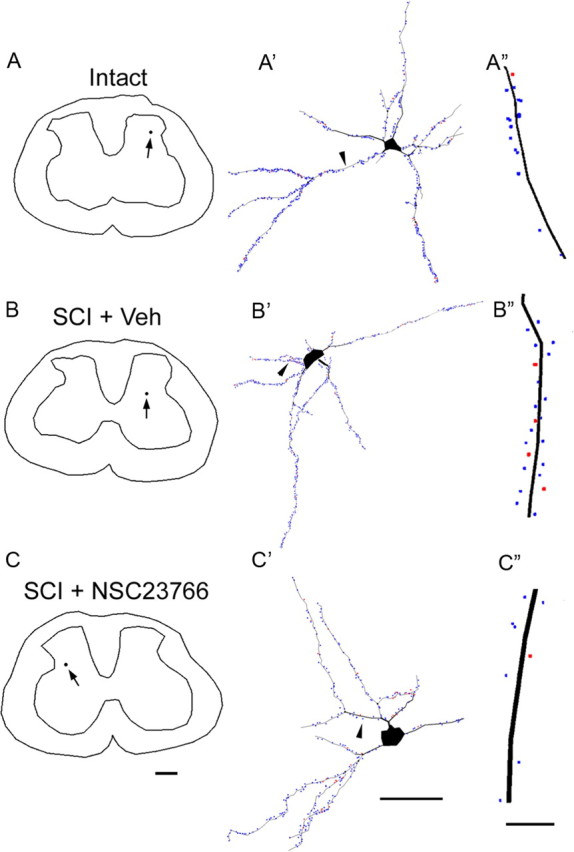
Neurolucida reconstructions reveal the location of identified neurons (A–C), the complete spatial distribution and density of thin- and mushroom-shaped spines. A′, An identified neuron has several primary dendritic branches arising from the soma. A 50 μm length of the primary dendrite from this neuron is shown in A″, which reveals a single mushroom spine (red dot) and multiple thin spines (blue dots) along its length. B′, A neuron from an SCI plus veh treated animal. B″, A sample 50 μm length of the primary dendrite shows a higher density of thin spines and mushroom spines. C′, A neuron reconstructed from an SCI and NSC23766 Rac1 inhibitor-treated animal. C″, A significant decrease in thin and mushroom spine density is observed on the dendrites from NSC23766-treated animals. Dendrites in A″, B″, and C″ are oriented with bottom closest to soma. Scale bars: A–C, 500 μm; A′–C′, 50 μm; A″–C″, 10 μm.
Figure 4.

Analysis of reconstructed neurons reveals differences in spine density (A–C), spine distribution (D–F), and spine shape (G, H) in the three treatment groups. A–C, Total spine density (A), thin-shaped spine density (B), and mushroom-shaped spine density (C) increased after SCI plus veh treatment compared with intact animals. A–C, NSC23766 treatment reduces total spine, thin spines, and mushroom spine density after SCI (*p < 0.05; **p < 0.001). D, The distribution of spines shifts significantly after SCI. Significantly more spines are observed more proximal to the soma after SCI and vehicle treatment compared with intact animal group (*p < 0.001). NSC23766 Rac1 inhibitor treatment significantly reduced the spine density at the 350 μm distance compared with intact animals (**p < 0.001). E, Similarly, thin spines increase in density in regions more proximal to the soma after SCI plus veh treatment compared with intact animals. NSC23766 decreases this density distribution so that it is not significantly different compared with intact animals. F, SCI results in a significant increase in mushroom spines in two region, 50 and 100 μm, distances from the soma compared with intact animals (*p < 0.001). No significant differences were observed between intact and SCI plus NSC23766-treated animals. G, Summary diagram demonstrating the increase in spine density and the proximal shift in spine distribution after SCI. Sample size for neurons analyzed for each spine category in A–F is shown in parentheses in A–C. Graphs are mean ± SEM.
Dendritic spine redistribution after SCI
The location of spines on the dendrite relative to the cell body has functional implications for a neuron (Holthoff et al., 2002; Yuste and Urban, 2004). To determine whether SCI results in changes in spine distribution, we used a modified Sholl's analysis. In SCI plus veh animals, the density of spines significantly shifted toward the cell body compared with the spine density distribution in intact animals. Significantly higher spine density was found in the closest bin, 50 μm from the cell body (4.31 ± 0.18 vs 2.94 ± 0.17/10 μm dendrite; p < 0.001) (Fig. 4D). Interestingly, in SCI animals treated with NSC23766 over 3 d, spine density was significantly reduced below intact levels in the most distal bin (2.12 ± 0.23 vs 2.94 ± 0.17/10 μm dendrite; p < 0.001). The density distribution of thin-shaped spines in animals with SCI plus veh treatment also shifted significantly compared with intact animals (2.88 ± 0.14 vs 4.01 ± 0.18/10 μm dendrite; p < 0.001), with a higher density of thin spines found in the bin located 50 μm from the soma. In addition, there was a significantly higher density of mushroom-shaped spines in the two bins closest to the cell body: 50 μm (0.44 ± 0.05 vs 0.16 ± 0.02/10 μm dendrite; 35% post-SCI increase; p < 0.001) and 100 μm (0.33 ± 0.03 vs 0.19 ± 0.02/10 μm dendrite; 59% post-SCI increase; p < 0.001), indicating a shift toward the cell body in animals with SCI plus veh treatment compared with intact animals. Figure 4G summarizes the morphological results for analysis of spine density and distribution.
Remodeling of dendritic spines contributes to neuronal excitability
To examine the contribution of the dendritic spine morphologies observed after SCI toward neuronal function, we created a simple compartmentalized neuronal model using the NEURON simulation environment. Because most dendritic spines are excitatory glutamatergic synapses (Calabrese et al., 2006), we modeled the presynaptic input with kinetics similar to excitatory AMPA synapses (see Materials and Methods). As shown in Figure 5A, this α-function produces a skewed curve, representing the fast release of neurotransmitter and binding to postsynaptic receptors, and the resultant postsynaptic membrane depolarization. This synaptic potential decays slowly back to resting potential (Destexhe et al., 1994).
To examine the effects of spine shape on signal transduction onto the parent dendritic branch, we used a two-part compartment model: spine neck and head. So as to consider the effects of geometric changes only (Segev and Rall, 1998), spine volume across treatment groups remained constant. Because cylinders with equal length and diameter values represent spherical shapes, computationally, we simulated spine heads as cylindrical shapes with both length and diameter values equal to the experimental mean spine head diameter. The spine neck membrane was modeled with passive properties. The spine head contained active membrane with Hodgkin–Huxley channel kinetics (Fig. 5D, gray region) (Miller et al., 1985; Segev and Rall, 1988; Rusakov et al., 1996). EPSPs were recorded from the base of spine neck in response to a single presynaptic stimulus onto the spine head. In the intact spine, presynaptic stimulation resulted in a sharp upshoot waveform with a peak at −19.1 mV (stim-to-peak latency, 2.6 ms) and a gradual decay back to resting membrane potential. The EPSP recorded from the SCI plus veh spine also exhibited a sharp upshoot, maximal depolarization with a shorter latency (stim-to-peak latency, 2.2 ms), and increased amplitude (peak at −17.8 mV) compared with intact. Stimulation of the SCI plus NSC23766 spine did not result in an EPSP recording containing the sharper waveform, and reached a smaller peak (at −27.4 mV) with the greatest latency (3.5 ms). Repolarization after the EPSP was fastest with the SCI plus veh (τ = 18.3 ms) spine shape and slowest for the SCI plus NSC23766 spine shape (τ = 21.9 ms). Area under the EPSP curve was comparable for intact and SCI plus NSC23766 (776 vs 778 mV · ms). The SCI plus veh spine attenuated presynaptic depolarizing input arriving onto the dendritic branch, with a decreased EPSP area (718 mV · ms).
To examine the cumulative effects of spine density and spine distribution on the output of a postsynaptic neuron, a simplified compartment model was constructed (Fig. 5E). All spines were stimulated simultaneously and the resultant activity was recorded from the soma. In all three neuron models, a single action potential was produced after stimulating the dendritic spines, although with subtle differences in their peak amplitude (measured from resting potential to the peak) and latency: intact, amplitude, 40.2 mV; stim-to-peak latency, 1.7 ms; SCI plus veh, amplitude, 39.4 mV; stim-to-peak latency, 1.4 ms; SCI plus NSC23766, amplitude, 38 mV; stim-to-peak latency, 1.8 ms. In the intact and SCI plus NSC23766 models, a slow rising depolarizing potential appeared after the first action potential but failed to reach threshold at −40 mV. In SCI plus veh, however, increased dendritic spine density in the proximal compartments produced a second somatic spike (amplitude, −11.8 mV; stim-peak latency, 13.8 ms). The second spike in response to activation remained with the removal of the spines in the sixth compartment (data not shown), to match its absence from both intact and SCI plus NSC23766 models, demonstrating that distal spines are not a prerequisite for production of the secondary spike. The interspike interval between the first and second peak was 12.4 ms, which corresponds with a firing frequency of ∼80.6 impulses/s.
Neuronal hyperresponsiveness accompanies dendritic spine remodeling after SCI in vivo
To assess the responsiveness of WDR neurons, units were sampled from dorsal horn lamina IV–V (490–1259 μm) (n = 13/group). Figure 6A–C shows representative peristimulus time histograms (spikes/1 s bin) for WDR neurons in intact, SCI plus veh, and SCI plus NSC23766 animals. Compared with intact animals, SCI plus veh groups exhibited increased evoked unit activity in response to all peripheral stimuli (Fig. 6D): for phasic brush, 7 ± 1.1 versus 50 ± 7.2 impulses/s, p < 0.001; for pressure, 10.6 ± 2.6 versus 39 ± 4.3 impulses/s, p < 0.001; for pinch, 8.75 ± 1.09 versus 48.3 ± 7.26 impulses/s, p < 0.001; for 0.38 g von Frey filament, 6.67 ± 1.47 versus 17.6 ± 2.0 impulses/s, p < 0.001; for 1.01 g von Frey filament, 7.75 ± 1.69 versus 22.8 ± 4.97 impulses/s, p = 0.012; and for 20.8 g von Frey filament, 9.4 ± 1.89 versus 46.8 ± 7.14 impulses/s, p < 0.001. Interestingly, peripherally evoked responses with NSC23766 remained significantly higher compared with intact animals for phasic brush (20.0 ± 2.32 vs 6.92 ± 1.12 impulses/s; p < 0.001), 1.01 g/mm2 (11.92 ± 1.69 vs 7.75 ± 1.69 impulses/s; p = 0.093), and 20.8 g/mm2 von Frey filament (17.0 ± 2.26 vs 9.41 ± 1.89 impulses/s; p = 0.017).
Figure 6.
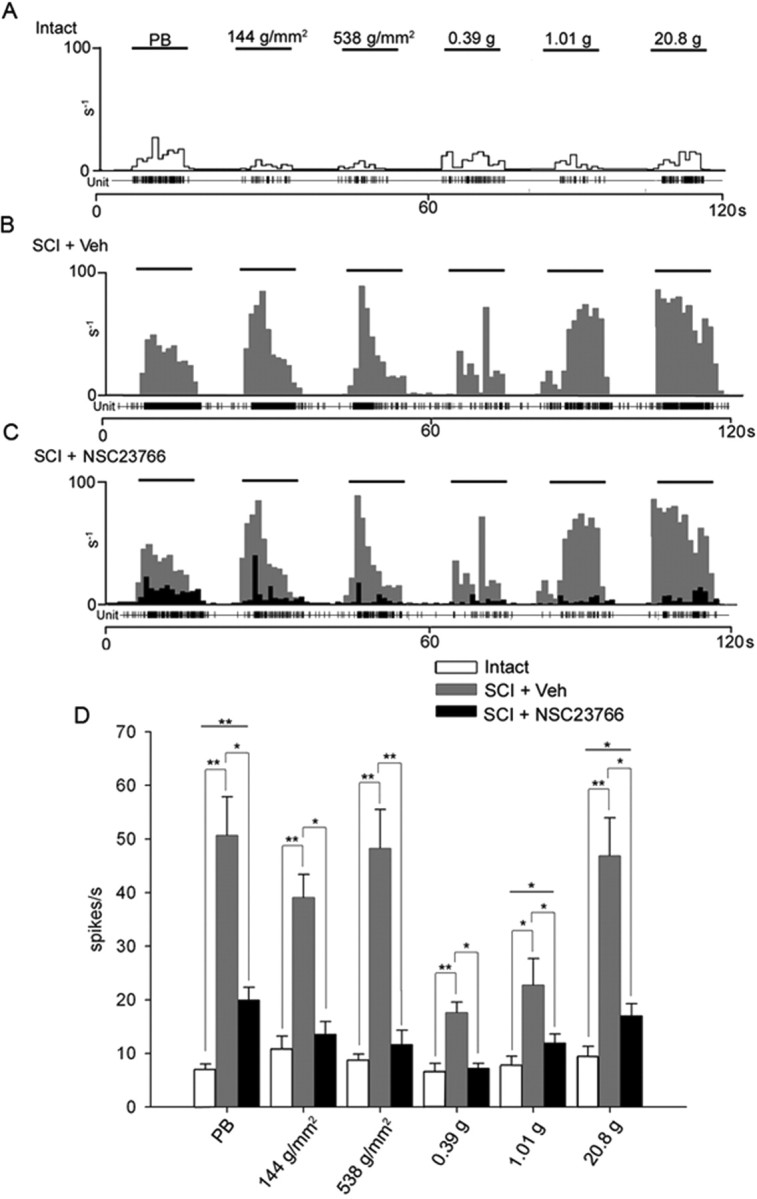
Intrathecal administration of NSC23766 attenuated peripherally evoked activity in WDR units from SCI animals 31 d after injury. A, A representative unit from an intact animal displaying evoked responses to stimuli (PB, 1.44 g/mm2, 538 g/mm2, 0.39 g, 1.01 g, and 20.8 g refer to phasic brush, pressure, pinch, and von Frey filaments of increasing intensities applied for 10 s). B, After SCI, the peristimulus histogram shows increased evoked responses to all peripheral stimuli. Evoked discharged rates were between 45 and 60 impulses/s. Phasic brush, pressure, and pinch resulted in elevated responses. von Frey filament stimulation resulted in a graded increase. C, Peripherally evoked responses of all sampled WDR units decreased significantly after NSC23766 treatment. SCI plus veh WDR unit peristimulus histogram responses (gray) are overlaid with SCI plus NSC23766 peristimulus histogram responses (black). D, SCI significantly (*p < 0.05; **p < 0.001) increased the evoked responses to all peripheral stimuli compared with intact animals. NSC23766 significantly (*p < 0.05; **p < 0.001) reduced SCI-induced increases in all peripherally evoked responses. Evoked responses with NSC23766 remained significantly higher for phasic brush and 1.01 and 20.8 g/mm2 von Frey filament stimulation (*p < 0.05; **p < 0.001) compared with intact. Graphs are mean ± SEM.
Pain-related behavior accompanies SCI-induced dendritic remodeling
After SCI, mechanical paw withdrawal thresholds significantly (p = 0.039) decreased in all groups (group mean, 10.4 ± 1.51 g; n = 10) compared with intact animals (group mean, 21.8 ± 0.33 g; n = 10). SCI plus NSC23766 animals demonstrated progressively increased paw-withdrawal thresholds over the 3 d treatment regimen compared with animals given vehicle (n = 5/group; day 1, 5.96 ± 1.07 g vs 2.6 ± 0.21 g, p = 0.027; day 2, 11.2 ± 1.26 g vs 1.81 ± 0.15 g, p < 0.001; day 3, 14.1 ± 2.19 g vs 2.43 ± 0.19 g, p = 0.003) (Fig. 7A).
Figure 7.

Behavioral assays for pain and locomotor function. Intact animals demonstrated expected levels for tactile allodynia (A), thermal hyperalgesia (B), and displayed competent locomotor function (C). The dashed line represents group mean for intact stimulus thresholds. After SCI, however, stimulus threshold for hindpaw withdrawal decreased (open circles) in response to both tactile and thermal stimulus intensities. Intrathecal NSC23766 administration over 3 d progressively restored tactile and thermal thresholds compared with SCI plus veh (*p < 0.05; **p < 0.01). No significant difference in locomotor competence between SCI plus veh and SCI plus NSC23766 animals (p = 0.266). Compared with intact animals, both performed poorly (**p < 0.001). Graphs are mean ± SEM.
Thermal paw withdrawal thresholds (Fig. 7B) for all groups were significantly lowered (p < 0.001) after SCI compared with intact animals (group mean, 37.3 ± 0.22 vs 43.0 ± 0.15°C). NSC23766 did not change thermal hindpaw withdrawal thresholds on the first day of treatment (37.3 ± 0.20 vs 37.9 ± 0.26°C; p = 0.088), but significantly attenuated thermal hyperalgesia beginning on the second day (41.2 ± 0.27 vs 36.9 ± 0.19°C; p < 0.001) and third day (41.5 ± 0.33 vs 37.1 ± 0.21°C; p < 0.001) of treatment.
To rule out motor compromise as a confounder of withdrawal thresholds, motor coordination and ability were evaluated. Intact animals were able to stay on the rotor rod up to 19.4 ± 8.0 rpm, whereas those animals with SCI plus veh (1.6 ± 0.78 rpm; p < 0.001) or NSC23766 treatment (2.6 ± 0.71 rpm; p < 0.001) demonstrated impaired performance. We found no statistical difference in fall latency among SCI plus veh or NSC23766 (p = 0.266).
Discussion
Our data reveal a molecular substrate for synaptic plasticity that contributes to chronic pain induced by SCI. Here, we show that dendritic spine remodeling (an increase in spine density and redistribution of spine location relative to the cell body, and increases in spine length and head diameter after SCI) occurs after SCI in dorsal horn neurons, and identify Rac1 as critical in maintaining neuropathic pain through its regulation of dendritic spines. Because spines are essential structural elements involved in postsynaptic signal transduction (Yuste and Bonhoeffer, 2004), and spine shape, density, and distribution can directly influence the response of a neuron to excitatory stimuli, we expected that spine remodeling after SCI would be accompanied by adverse physiological and behavioral consequences. Our data confirmed that dorsal horn WDR neurons exhibit hyperexcitability in response to evoked-innocuous and noxious stimulation after SCI, concomitant with increased sensitivity to mechanical stimuli and reduced thermal nociceptive thresholds. Because in vitro and in vivo studies demonstrate that Rac1 modulates dendritic spine morphology and function (Nakayama et al., 2000; Tashiro and Yuste, 2004; Wiens et al., 2005), we disrupted dendritic spine structure with the Rac1-specific inhibitor NSC23766, which blocks guanine exchange factors, Trio and Tiam1. Importantly, NSC23766 is specific for Rac1 and does not interfere with cdc42 or RhoA-GEF or Rac1 binding to its effector PAK1 (Gao et al., 2004). In agreement with other loss-of-function studies (Nakayama et al., 2000; Tashiro et al., 2000; Tashiro and Yuste, 2004; Wiens et al., 2005), we show that NSC23766 Rac1 inhibition decreased spine length, head diameter, and spine density. Except for a single case (Fig. 4D), spine distribution along dendritic processes in the dorsal horn of NSC23766-treated rats after SCI was indistinguishable from the distribution in intact rats. Immunoblot analysis showed that SCI increased the amount of phosphorylated Rac1, which was reduced by the Rac1 inhibitor. Together, these results implicate Rac1 in the regulation of injury-induced dendritic spine remodeling after SCI.
Our findings offer new insight into how the injured nervous system actively maintains pain over long periods of time. In this regard, a synaptic model for long-term memory can explain the maintenance of chronic neuropathic pain. Late-phase cortical LTP involves protein synthesis and cytoskeletal rearrangement that can lead to development of new synaptic connections and the de novo formation or elaboration of dendritic spines (Yuste and Bonhoeffer, 2001; Fukazawa et al., 2003; Carlisle and Kennedy, 2005; Halpain et al., 2005; Calabrese et al., 2006; Bourne and Harris, 2007; Chen et al., 2007). Dendritic spines are microscopic protrusions and composed almost entirely of a filamentous actin cytoskeleton, lacking the intermediate filaments and microtubules found in the parent dendrite. Dendritic spines are major postsynaptic sites for excitatory input and are found primarily on neurons that receive a diverse array of input, such as pyramidal hippocampal neurons and wide-dynamic range neurons in the spinal cord dorsal horn (Calabrese et al., 2006). After a synaptic learning event, dendritic spines remodel; spine density increases, which is an indication that a neuron has entered into a more plastic state, more readily available to form new or stronger synapses. Dendritic spine morphologies also change in correlation with the physiological capabilities of the synapse (Bourne and Harris, 2007). Larger head structures of mushroom-shaped spines permit greater postsynaptic membrane receptor clustering and number (i.e., AMPA receptors) and decrease the resistance load of the dendritic spine head (Wiens et al., 2005). Narrower spine necks act as an electrochemical diffusion barrier and the elaboration of mature spines into mushroom-shaped geometries results in a faster, more efficient postsynaptic membrane depolarization (Rall, 1962). Our simulation data based on observations after SCI demonstrated that an increase in the spine head volume and surface area results in faster and slightly higher amplitude EPSPs compared with spines with a smaller spine head compartment but with equal total spine volume. The increased spine head compartment also resulted in faster repolarization back to resting membrane potential. Our results also show an increase in mushroom spine density on deep dorsal horn neurons, suggesting that a post-SCI shift in the number of mature spines may contribute to the strengthening of synaptic inputs, enhanced transmission fidelity, and potentiation of electrical transduction, which together may lead to a pathological amplification of sensory information after SCI.
Our results indicate that SCI leads to alterations in the synaptic structure underlying input–output functions of neurons involved with nociceptive signaling. Inputs onto dendritic locations more proximal to the soma have a greater nonlinear effect on the somatic EPSP compared with inputs on the distal dendrite (Rall et al., 1967). This is because the spatial origin of inputs significantly affects the temporal dynamics of the synaptic potentials: more distal inputs increase the latency to peak amplitude, decrease maximal amplitude, and lengthen the decay to resting potential. This change in synaptic potential propagation has a strong impact on input summation and coincidence detection (Lev-Tov et al., 1983). Our data show a proximal redistribution of mature spines after SCI, thus increasing the gain of nociceptive information to a greater extent than distal spines and amplifying inputs in a pathological manner. We also observed a decrease in EPSP area and a narrowing of the EPSP curve, which suggests a discretization of excitatory inputs. EPSP discretization reduces spatial interference between adjacent inputs (traveling inputs do not and can allow suprathreshold potentials to propagate more successfully, contributing to excessive firing in hyperexcitable neurons after SCI) (Miller et al., 1985; Segev and Rall, 1988; Matsuzaki, 2007).
We identify Rac1 activation after SCI in both the anatomical and pathophysiological changes that contribute to chronic pain. Constitutively activated Rac1 increases spine turnover rate, increases spine density, and increases spine structure volume (Nakayama et al., 2000; Tashiro and Yuste, 2004; Wiens et al., 2005). In contrast, dominant-negative Rac1 transfection decreased spine density and inhibits spine maturation. Our present results demonstrate that inhibiting Rac1 activity after SCI reduces postsynaptic density protein, PSD-95, a neuronal postsynaptic density-95 (PSD-95)/Discs large (Dlg)/zona occludens-1 (ZO-1) (PDZ) protein associated with AMPA (El-Husseini et al., 2000) and NMDA receptors (Sharma et al., 2006). A PSD-95 protein/PDZ domain interaction with AMPA receptors may be important to the “unsilencing” of silent synapses and sensory synaptic potentiation (Li et al., 1999; Tomita et al., 2001). PSD-95 overexpression in hippocampal neurons also induces the maturation of excitatory synapses, which includes an increased number and size of dendritic spines (El-Husseini et al., 2000). Together, these results show that inhibition of Rac1 activity after SCI can perturb mechanisms of excitatory transmission.
Alterations in sodium channel expression in dorsal horn neurons (Hains et al., 2003), the loss of inhibitory GABAergic inputs (Drew et al., 2004), and injury-induced synaptic plasticity (Ji et al., 2003) contribute to neuronal hyperexcitability associated with post-SCI neuropathic pain. Given our results, we asked whether abnormal spine features after SCI can also contribute to single-unit hyperexcitability. Using a neuronal model based on our anatomical findings, we confirmed the prediction that dendritic spine geometric remodeling can lead to significant changes in the intrinsic computational properties of the postsynaptic neuron after SCI: an increase in synaptic efficacy as a result of subtle geometric changes at individual dendritic spines significantly influences somatic EPSP generation (Rusakov et al., 1996). Within our simulation parameters, SCI-induced spine remodeling contributed to doublet firing with a frequency of 80.6 impulses/s. This interspike interval falls within the range of frequencies we observed in vivo for peripherally evoked responses for WDR neurons after SCI (Hains and Waxman, 2006).
In conclusion, we identify a novel mechanism by which SCI-induced remodeling of dendritic spines powerfully alters the functional properties and excitability of dorsal horn pain-processing neurons. Furthermore, we identify Rac1 as an important intracellular signaling molecule that governs this process. Given the molecular and anatomical similarity between cortical mechanisms of learning and memory, our findings lend additional support to the idea that injury-induced central sensitization is a form of spinal cord pain memory. By targeting this signaling mechanism, it may be possible to more successfully manage chronic pain after SCI.
Footnotes
This work was supported in part by grants from the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs. The Center for Neuroscience and Regeneration Research is a collaboration of the Paralyzed Veterans of America and the United Spinal Association with Yale University. B.C.H. was funded by Pfizer (Scholar's Grant in Pain Medicine) and The Dana Foundation.
References
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (Bethesda) 2006;21:38–47. doi: 10.1152/physiol.00042.2005. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetkovich DM, Chen L, Stocker TJ, Nicoll RA, Bredt DS. Phosphorylation of the postsynaptic density-95 (PSD-95)/discs large/zona occludens-1 binding site of stargazin regulates binding to PSD-95 and synaptic targeting of AMPA receptors. J Neurosci. 2002;22:5791–5796. doi: 10.1523/JNEUROSCI.22-14-05791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly RJ. Cortactin signalling and dynamic actin networks. Biochem J. 2004;382:13–25. doi: 10.1042/BJ20040737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Mainen ZF, Sejnowski TJ. Synthesis of models for excitable membranes, synaptic transmission and neuromodulation using a common kinetic formalism. J Comput Neurosci. 1994;1:195–230. doi: 10.1007/BF00961734. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Drew GM, Siddall PJ, Duggan AW. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain. 2004;109:379–388. doi: 10.1016/j.pain.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Erschbamer MK, Hofstetter CP, Olson L. RhoA, RhoB, RhoC, Rac1, Cdc42, and Tc10 mRNA levels in spinal cord, sensory ganglia, and corticospinal tract neurons and long-lasting specific changes following spinal cord injury. J Comp Neurol. 2005;484:224–233. doi: 10.1002/cne.20471. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Sindrup SH, Bach FW, Jensen TS. Pain and dysesthesia in patients with spinal cord injury: a postal survey. Spinal Cord. 2001;39:256–262. doi: 10.1038/sj.sc.3101161. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–126. doi: 10.1089/neu.1992.9.123. discussion 126–128. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S, Spencer K, Graber S. Dynamics and pathology of dendritic spines. Prog Brain Res. 2005;147:29–37. doi: 10.1016/S0079-6123(04)47003-4. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff K, Tsay D, Yuste R. Calcium dynamics of spines depend on their dendritic location. Neuron. 2002;33:425–437. doi: 10.1016/s0896-6273(02)00576-7. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Asai T, Murase K. Robust changes of afferent-induced excitation in the rat spinal dorsal horn after conditioning high-frequency stimulation. J Neurophysiol. 2000;83:2412–2420. doi: 10.1152/jn.2000.83.4.2412. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol. 2006;198:401–415. doi: 10.1016/j.expneurol.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Lev-Tov A, Miller JP, Burke RE, Rall W. Factors that control amplitude of EPSPs in dendritic neurons. J Neurophysiol. 1983;50:399–412. doi: 10.1152/jn.1983.50.2.399. [DOI] [PubMed] [Google Scholar]
- Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- Masliah E, Fagan AM, Terry RD, DeTeresa R, Mallory M, Gage FH. Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP-43 in the dentate gyrus of the adult rat. Exp Neurol. 1991;113:131–142. doi: 10.1016/0014-4886(91)90169-d. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M. Factors critical for the plasticity of dendritic spines and memory storage. Neurosci Res. 2007;57:1–9. doi: 10.1016/j.neures.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Miller JP, Rall W, Rinzel J. Synaptic amplification by active membrane in dendritic spines. Brain Res. 1985;325:325–330. doi: 10.1016/0006-8993(85)90333-6. [DOI] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci. 2003;22:319–330. doi: 10.1016/s1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W. Theory of physiological properties of dendrites. Ann N Y Acad Sci. 1962;96:1071–1092. doi: 10.1111/j.1749-6632.1962.tb54120.x. [DOI] [PubMed] [Google Scholar]
- Rall W, Burke RE, Smith TG, Nelson PG, Frank K. Dendritic location of synapses and possible mechanisms for the monosynaptic EPSP in motoneurons. J Neurophysiol. 1967;30:1169–1193. doi: 10.1152/jn.1967.30.5.1169. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Stewart MG, Korogod SM. Branching of active dendritic spines as a mechanism for controlling synaptic efficacy. Neuroscience. 1996;75:315–323. doi: 10.1016/0306-4522(96)00253-9. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Understanding LTP in pain pathways. Mol Pain. 2007;3:9. doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev I, Rall W. Computational study of an excitable dendritic spine. J Neurophysiol. 1988;60:499–523. doi: 10.1152/jn.1988.60.2.499. [DOI] [PubMed] [Google Scholar]
- Segev I, Rall W. Excitable dendrites and spines: earlier theoretical insights elucidate recent direct observations. Trends Neurosci. 1998;21:453–460. doi: 10.1016/s0166-2236(98)01327-7. [DOI] [PubMed] [Google Scholar]
- Sharma K, Fong DK, Craig AM. Postsynaptic protein mobility in dendritic spines: long-term regulation by synaptic NMDA receptor activation. Mol Cell Neurosci. 2006;31:702–712. doi: 10.1016/j.mcn.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Svendsen F, Tjølsen A, Gjerstad J, Hole K. Long-term potentiation of single WDR neurons in spinalized rats. Brain Res. 1999;816:487–492. doi: 10.1016/s0006-8993(98)01250-5. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Tomita S, Nicoll RA, Bredt DS. PDZ protein interactions regulating glutamate receptor function and plasticity. J Cell Biol. 2001;153:F19–F24. doi: 10.1083/jcb.153.5.f19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Hains BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci. 2006;29:207–215. doi: 10.1016/j.tins.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Wiens KM, Lin H, Liao D. Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci. 2005;25:10627–10636. doi: 10.1523/JNEUROSCI.1947-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, King AE. Physiology and morphology of multireceptive neurons with C-afferent fiber inputs in the deep dorsal horn of the rat lumbar spinal cord. J Neurophysiol. 1987;58:460–479. doi: 10.1152/jn.1987.58.3.460. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Shortland P, Coggeshall RE. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature. 1992;355:75–78. doi: 10.1038/355075a0. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Yuste R, Urban R. Dendritic spines and linear networks. J Physiol Paris. 2004;98:479–486. doi: 10.1016/j.jphysparis.2005.09.014. [DOI] [PubMed] [Google Scholar]