

Abstract
Reactive oxygen species (ROS) modulate intracellular signaling but are also responsible for neuronal damage in pathological states. Microglia, the resident CNS macrophages, are prominent sources of ROS through expression of the phagocyte oxidase which catalytic subunit Nox2 generates superoxide ion (O2·−). Here we show that microglia also express Nox1 and other components of nonphagocyte NADPH oxidases, including p22phox, NOXO1, NOXA1, and Rac1/2. The subcellular distribution and functions of Nox1 were determined by blocking Nox activity with diphenylene iodonium or apocynin, and by silencing the Nox1 gene in microglia purified from wild-type (WT) or Nox2-KO mice. [Nox1-p22phox] dimers localized in intracellular compartments are recruited to phagosome membranes during microglial phagocytosis of zymosan, and Nox1 produces O2·− in zymosan-loaded phagosomes. In microglia activated with lipopolysaccharide (LPS), Nox1 produces O2·−, which enhances cell expression of inducible nitric oxide synthase and secretion of interleukin-1β. Comparisons of microglia purified from WT, Nox2-KO, or Nox1-KO mice indicate that both Nox1 and Nox2 are required to optimize microglial production of nitric oxide. By injecting LPS in the striatum of WT and Nox1-KO mice, we show that Nox1 also enhances microglial production of cytotoxic nitrite species and promotes loss of presynaptic proteins in striatal neurons. These results demonstrate the functional expression of Nox1 in resident CNS phagocytes, which can promote production of neurotoxic compounds during neuroinflammation. Our study also shows that Nox1- and Nox2-dependent oxidases play distinct roles in microglial activation and that Nox1 is a possible target for the treatment of neuroinflammatory states.
Keywords: microglia, NADPH oxidase, Nox, nitric oxide, interleukin-1, LPS, neuroinflammation
Introduction
Microglia become activated in pathological contexts affecting neural cells. These cells may be neuroprotective through phagocytic elimination of pathogens, dying cells, and neurotoxic compounds, or through production of neurotrophic factors, but they may also promote neurodegeneration by producing proinflammatory or potentially neurotoxic effectors, including interleukin-1β (IL-1β), nitric oxide (NO·), or reactive oxygen species (ROS) (Allan et al., 2005; Block et al., 2007; Hanisch and Kettenmann, 2007). Understanding the mechanisms that control microglial expression of neurotoxic effectors is crucial for the development of new therapeutic approaches to neuroinflammatory or neurodegenerative diseases.
Like other phagocytes, microglia express the phagocyte NADPH oxidase (Sankarapandi et al., 1998; Bianca et al., 1999), an enzyme composed of four regulatory cytosolic components (p47phox, p67phox, p40phox, and rac proteins) and a transmembrane flavocytochrome heterodimer consisting of the p22phox and the gp91phox (Nox2) catalytic subunit. Nox2 generates superoxide ion (O2·−), which ensures the phagocytic neutralization of microorganisms (Nauseef, 2007), but also promotes neural cell death in animal models of neurodegenerative diseases and stroke (Walder et al., 1997; Wu et al., 2003, 2006). O2·− may be neurotoxic by fueling the formation of highly pro-oxidant hydroxyl free radical or peroxynitrite (Halliwell, 2006; Szabó et al., 2007). However, O2·− is also converted to hydrogen peroxide that primarily contributes to intracellular cell signaling processes through redox modulation of ion channels, enzymes, or transcription factors (Dröge, 2002; Veal et al., 2007). Studies using cultured microglia have shown redox modulations of microglial cell proliferation or function, which were attributed to ROS generated by the phagocyte oxidase (Pawate et al., 2004; Mander et al., 2006; Block et al., 2007). However, several homologues of Nox2 encoded by distinct genes (Nox1, Nox3, Nox4, Duox1, and Duox2) have been identified in nonphagocytic rodent and human cells (Bedard and Krause, 2007; Lambeth et al., 2007). Nox1 is predominantly expressed in colon epithelium, but was also detected in other peripheral tissues, including dorsal root ganglia and in vascular cells (Suh et al., 1999; Bedard and Krause, 2007; Ibi et al., 2008). Like Nox2, Nox1 is a transmembrane protein which forms a heterodimer with p22phox and generates O2·− (Ambasta et al., 2004; Kawahara et al., 2005). Nox1 activity requires rac proteins and is optimized in the presence of NOXO1 and NOXA1, two functional homologues of p47phox and p67phox, respectively (Bánfi et al., 2003). Nox1 deletion was reported to increase survival in a mouse model of amyotrophic lateral sclerosis (ALS), but the cellular sources of Nox1 and the mechanisms by which Nox1 may promote neural tissue damages remained undetermined (Marden et al., 2007).
In this study, we first show that microglia express catalytically active Nox1 in addition to the phagocyte oxidase. We then showed that activation of microglia with lipopolysaccharide (LPS), a component of Gram-negative bacteria commonly used to model proinflammatory and neurotoxic activation of microglia (Lehnardt et al., 2003; Qin et al., 2004), and injection of LPS in the brain induce IL-1β and NO· production via Nox1, and favors peroxynitrite-mediated tyrosine nitration as well as loss of synapse proteins.
Materials and Methods
Animals.
Wild-type (WT), CX3CR1-EGFP, Nox1, and gp91phox/Nox2 knock-out (KO) mice (Pollock et al., 1995; Jung et al., 2000; Gavazzi et al., 2006) were on a C57BL/6J background. Nox2-KO mice were obtained from The Jackson Laboratory. WT mice were purchased from Iffa-Credo. Nox1-KO mice (Gavazzi et al., 2006) used in our experiments were derived from at least 10 backcrosses with WT C57BL/6J mice. All procedures were performed in accordance with the guidelines for care and use of experimental animals of the European Economic Community (ref 86/609/CEE) and approved by the ethics committee for animal experimentation in Ile-de-France.
Antibodies.
Rabbit anti-p22phox antiserum (Campion et al., 2007) was a generous gift from Dr. Françoise Morel (GREPI, CNRS, Grenoble, France). Anti-LAMP1 rat monoclonal antibody developed by J. T. August was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa (Department of Biological Sciences, Iowa City, IA). Commercial antibodies used include rat monoclonal anti-EGFP (Nacalai Tesque); anti-F4/80, anti-CD11b (M1/70.1), and anti-CD68 (Serotec); mouse monoclonal anti-β-actin (clone AC15, Sigma); rabbit polyclonal anti-Iba1 (Wako Chemicals); anti-GFAP (DakoCytomation); anti-nitrotyrosine (Ntyr) (Upstate Biotechnology); anti-inducible NO synthase (iNOS) (Transduction Laboratory); and anti-synapsin (Synaptic Systems).
FACS sorting of microglial cells.
Cerebral cortex quickly removed from killed 1-d-old (P1) heterozygous CX3CR1EGFP/+ mice was immersed in 0.1 m ice-cold PBS, pH 7.4, dissected free of meninges, and mechanically dissociated. Viable cells expressing enhanced green fluorescent protein (EGFP) were sorted in a FACSAria analyzer (Becton Dickinson).
Reagents for cell cultures.
Diphenylene iodonium salt (DPI) from Sigma and apocynin from VWR International were dissolved in sterile DMSO at 25 mm and 5 m, respectively. DPI was stored at −20°C. LPS from Salmonella typhimurium (Sigma) was dissolved in sterile PBS at 1 mg/ml and stored at −20°C. Zymosan A (Sigma) was opsonized with fetal calf serum (FCS). All other components of the culture media, including FCS with low endotoxin content (< 0.05 ng/ml), were from Invitrogen. Cells were cultured in plastic dishes (TPP).
Microglial cell cultures.
Cultured microglial cells were derived from the cerebral cortex of 1-d-old C57BL/6J, Nox1-KO, or Nox2-KO mice (P1), according to previously described procedures (Théry et al., 1991). Highly pure (> 99%) microglia were isolated from 2-week-old glial primary cultures grown in DMEM supplemented with 10% FCS. Purified microglia were washed three times in DMEM, seeded in DMEM plus 10% FCS, and cultured for 2 d before any treatment with LPS, FCS-opsonized Zymosan A, DPI, or apocynin.
siRNA and lentiviral shRNA silencing of the Nox1 gene in microglial cells.
BV2 cell clones overexpressing Nox1 were selected by transfection of parental BV2 cells with a pcDNA3.1 plasmid encoding the mouse Nox1 cDNA under the control of the CMV promoter (Bánfi et al., 2003). siRNA oligomers specific for Nox1 (Dharmacon) or luciferase (used as a negative control; Qiagen) were transfected using Jetsi carrier (Qbiogene) and Nox1 gene silencing was assessed by RT-PCR analysis. A silencing siRNA sequence was used to design a short hairpin (sh) RNA specific for Nox1 (shNox1). A control shRNA (shCtrl) was designed from a 3 base-mutated form of the siNox1 sequence. shNox1 and shCtrl sequences were inserted under the control of the human U6 promoter into a pTrip-ΔU3-U6-PGK-EGFP-WPRE vector (Philippe et al., 2006). Lentiviral vectors shNox1 and shCtrl were generated by transfection of HEK293T cells with the vector plasmid (pTrip-shNox1 or pTrip-shCtrl, respectively), the transcomplementation plasmid p8,7 and the pHCMV-G plasmid encoding the vesicular stomatitis virus glycoprotein as described previously (Philippe et al., 2006). Lentiviral vectors shCtrl and shNox1 were applied to freshly purified microglial cells at equivalent numbers of transduction units (TU), as assessed by EGFP labeling.
Detection of superoxide ion.
Cellular production of O2·− was visualized through the O2·−-specific oxidation of hydroethidine (Bindokas et al., 1996). Microglial cells cultured in 8-well Labtek slides were incubated with hydroethidine (10 μm, Molecular Probes) for 45 min, in the presence or the absence of LPS (1 μg/ml) or opsonized zymosan (3 × 106 particles per ml) with or without DPI (1 μm). The cells were washed, fixed with 4% paraformaldehyde (PFA) and counterstained with Hoechst 33342 dye (2 μg/ml in PBS) before labtek slide mounting in Fluoromount G (Southern Biotechnology Associates). Fluorescence images were captured using a Zeiss Axio ImagerZ1 fluorescence microscope (excitation: 530–585 nm/Emission >615 nm) equipped with a 40× NA 0.75 EC Plan-Neo Fluar objective (Zeiss). Fluorescence levels of oxidized hydroethidine were quantified using NIH ImageJ 1.37 software and normalized to the number of Hoechst-stained nuclei (25–40 cells per microscopic fields). Generation of O2·− in microglia engulfing zymosan was also visualized through the O2·−-mediated reduction of nitroblue tetrazolium (NBT). Microglia were incubated with opsonized zymosan for 45 min, then NBT (1.6 mg/ml, Sigma) was added to the medium for the last 15 min, before PFA fixation. Images were captured under differential interference contrast (DIC) or transmitted light using a DMRB microscope (Leica Microsystems) equipped with a 100× NA 1.40 oil-immersion objective.
Microglial production of IL-1β and NO·.
Microglial cells cultured in 96-well plates (5 × 104 cells seeded per well) were treated with or without LPS 100 ng/ml for 16 h and levels of IL-1β in the culture media were determined using commercially available ELISA (R&D Systems). NO· production was assessed by determination of nitrite levels using the colorimetric Griess method.
Assessment of cell survival.
Microglia cultured in 96-well plates were fixed with PFA added directly to the culture medium for 10 min at RT (final concentration 4%) and stained with Hoechst 33342 dye in PBS. Images were captured using an inverted Leica DMRiB fluorescence microscope (Leica Microsystems). The number of morphologically intact cell nuclei was determined by computer-assisted counts using ImageJ software in microscopic fields covering 6% of the well surface.
Immunocytodetection of p22phox, EGFP, and LAMP1.
Cells cultured in 8-well Labtek slide were fixed with 4% PFA for 10 min at RT before sequential incubations (1 h at RT) with rabbit anti-p22phox antiserum diluted 1:300 in PBS, 0.2% BSA, 0.05% saponin, Alexa Fluor 594- or Alexa Fluor 488-conjugated anti-rabbit IgG antibodies (1:1000, Invitrogen), rat monoclonal anti-EGFP antibody (1:1000) or rat monoclonal anti-LAMP1 antibody (1:20), and Alexa Fluor 488- or Alexa Fluor 594-conjugated anti-rat IgG (Invitrogen). Cell nuclei were stained with Hoechst 33342 dye. Slides were mounted in Fluoromount G, and images were acquired at room temperature using an AxioImagerZ1 fluorescence microscope (Zeiss) or using a confocal microscope SP2 AOBS (Leica Microsystems) equipped with a 63× NA 1.40 oil-immersion objective. Controls performed by substituting primary antibodies with rabbit nonimmune serum or unrelated monoclonal antibodies of matched isotypes were negative.
Reverse transcription PCR analyses.
RNA was extracted from pellet of FACS-purified CX3CR1EGFP/+ cells or from cultured microglia using the Absolutely RNA Microprep Kit (Stratagene). RNA from FACS-purified cells was amplified with the Smart mRNA Amplification Kit (Clontech). Reverse transcription using random primers was performed with Superscript II (Invitrogen). Standard PCR amplification was performed using TaqDNA polymerase from Qiagen, oligonucleotides listed in Table 1 and RT2 PCR primer sets purchased from SuperArray Bioscience (proprietary primers; sequence not disclosed) for mouse Nox1 (producing a 90 bp amplicon, RefSeq accession number: NM_172203), NOXO1 (83 bp amplicon, NM_027988), and NOXA1 (103 bp amplicon, NM_172204). PCR conditions were: 2 min at 95°C, amplification by 30–35 cycles of denaturation at 95°C for 45 s, annealing at 53°C to 60°C for 45 s, and extension at 72°C for 45 s, followed by a final elongation step at 72°C for 10 min. PCR products were separated on 1.8% agarose gels and visualized with ethidium bromide.
Table 1.
Primer sequences used for RT-PCR analyses of NADPH oxidases in microglia
| Primer | Sequence (5′ to 3′) | Amplicon (bp) | Accession number |
|---|---|---|---|
| Nox2_for | CGAAGACAACTGGAAGGAA | 956 | NM_007807 |
| Nox2_rev | GCTCCCACTAACATCACCAC | ||
| Nox3_for | TTGTGGCACACTTGTTCAACCTGG | 493 | AY573240 |
| Nox3_rev | TCACACGCATACAAGACCACAGGA | ||
| Nox4_for | ATCACAGAAGGTCCCTAGCA | 615 | NM_015760 |
| Nox4_rev | GGTCCAGAAATCCAAATCCA | ||
| Duox1_for | CACCATTGGGACCCTTTGCTGTTT | 761 | XM_130483 |
| Duox1_rev | AGCCTTTCATGAAGACCACCAGAA | ||
| Duox2_for | AACCACCTATGTGGGCATCATCCT | 491 | XM_619808 |
| Duox2_rev | AGCTGCCATGGATGATGATCAGGA | ||
| Rac1_for | TGTGGTGGTGGGAGACGGAG | 528 | NM_009007.1 |
| Rac1_rev | TTCTCTGTCCCCCTCCTGTC | ||
| Rac2_for | GACAGGAGGGGGACAGAGAA | 254 | NM_009008.3 |
| Rac2_rev | AGCGAGAAGCAGATGAGAAA | ||
| p22phox_for | GCTGCCCTCCACTTCCTGTT | 254 | NM_007806 |
| p22phox_rev | GCTGCCTCCTCTTCACCCTC | ||
| p40phox_for | AGAGCGACTTTGAGCAGCTT | 460 | AB002665 |
| p40phox_rev | TGTGGAGACACACCCTTGAT | ||
| p47phox_for | AGCCTGAGACATACCTGGTG | 446 | AB002663 |
| p47phox_rev | AGACTTCTGCAGATACATGG | ||
| p67phox_for | CAAGGCTACGGTTGTAGCAT | 465 | AB002664 |
| p67phox_rev | ACCTTGAGCATGTAAGGCAT |
Accession numbers of the sequences and size of the corresponding amplicons are indicated. For, Forward; rev, reverse.
Quantitative real-time PCR was performed using RT2 PCR primer sets (SuperArray Bioscience Corporation) for mouse Nox1, NOXO1, NOXA1, IL-1β (accession number: NM_008361), iNOS (NM_010927), and p22phox (NM_007806), and Brilliant QPCR SYBR green Mix in a MX3000P (Stratagene). The samples were amplified by 40 cycles (95°C for 60 s; 60°C for 30 s) and analyzed using the 2−Δ(ΔCt) method. HPRT served as a reference gene using the following primer sequences: forward 5′-CCT AAG ATG AGC GCA AGT TGA A-3′ and reverse 5′-CCA CAG GAC TAG AAC ACC TGC TAA-3′ (accession number: NM_013556.2).
Western blot analyses.
Cultured microglia were lysed in 1% SDS preheated to 95°C. Lysates were further heated to 95°C for 1 min. Protein concentrations were quantified by the bicinchoninic acid assay (Sigma). Total microglial cell extracts were resolved by 7.5% SDS-PAGE, transferred to a nitrocellulose membrane and processed for immunodetection as described previously (S. Qin et al., 2006). Binding of rabbit polyclonal anti-iNOS (diluted 1:1000) and mouse anti-β-actin antibodies (1:4000) were detected by enhanced chemiluminescence (Perbio) using horseradish peroxidase (HRP)-linked anti-mouse IgG or anti-rabbit IgG (GE Healthcare). Western blot bands of iNOS were quantified and normalized against β-actin using ImageJ software.
LPS injection and immunohistofluorescence.
Twenty-one-week-old male mice (25 g weight) obtained from two breeding pairs of Nox1+/− females and Nox1Y/− males were genotyped. Five Nox1Y/+ (WT) and five Nox1Y/− (Nox1-KO) male littermates were used for the experiments. Four micrograms of LPS diluted in 1 μl of PBS were injected unilaterally into the brain of deeply anesthetized mice, using stereotaxic coordinates for the striatum: +0.6 mm anterior–posterior, 1.8 mm lateral, and 3 mm depth relative to the bregma.
Mice were deeply anesthetized and perfused with 4% PFA 4 d after the injections. Brains were postfixed 2 h in the same fixative and processed for immunohistochemistry as described previously (S. Qin et al., 2006). Briefly, coronal sections (16 μm thick) blocked with goat serum were incubated with rabbit polyclonal anti-Iba1 (diluted 1:400), rabbit anti-GFAP antibodies (1:200), or rabbit polyclonal anti-synapsin antibodies (1:400). Bound antibodies were detected with Alexa Fluor 488-conjugated anti-rabbit IgG. For double-immunofluorescence staining of microglia, a combination of rat monoclonal antibodies specific for different macrophage markers was used to optimize visualization of the cells. Goat serum blocked sections were incubated with rabbit polyclonal anti-Ntyr (diluted 1:400), and primary antibodies were revealed with Alexa Fluor 488-conjugated anti-rabbit IgG. Sections were then incubated with a mixture of rat monoclonal anti-F4/80 (1:100), anti-CD11b (1:400), and anti-CD68 (1:400) antibodies before incubation with an Alexa Fluor 594-conjugated goat anti-rat IgG.
Labeled brain sections were mounted in Fluoromount G, and images were captured using a Zeiss AxioImagerZ1 microscope equipped with the apotome system (Zeiss). A 10× NA 0.50 Fluar objective was used to capture images for quantitative analyses. For each animal, the number of immunostained microglial cells and the proportion of microglial cells stained with anti-Ntyr were determined in microscopic fields (9 × 104 μm2 area) acquired in six sections of a cerebrocortical region centered on the lesional tract and extending over 384 μm on the anteroposterior axis.
Synapsin-stained areas were assessed in 6 sections of a striatum region extending over 560 μm in the anteroposterior axis. Square regions (6.25 × 104 μm2 area) in microscopic fields localized 300 μm ventral to the injection site or in the matched contralateral region were used for quantification. Images were acquired with equal exposure time, without saturation. A threshold was assigned for each animal to eliminate background fluorescence before analysis using ImageJ Software. Synapsin stained area in region ipsilateral to the injection site was expressed relative to the matched contralateral region in each animal to correct for any shift in the anteroposterior axis during sampling.
Results
Expression of Nox genes in microglia
To investigate microglial expression of NADPH oxidase-related genes in vivo, we used the CX3CR1-EGFP knock-in mice (Jung et al., 2000), in the CNS of which enhanced green fluorescent protein (EGFP) is selectively expressed by microglia. EGFP-positive microglia were purified by FACS from freshly dissociated forebrain of newborn heterozygotes and the expression of Nox proteins and Nox regulatory subunits was determined by RT-PCR analyses. Both Nox2 and Nox1 transcripts were clearly expressed by microglia from newborn mice, whereas Nox3, Nox4, Duox1, or Duox2 transcripts were not detectable (Fig. 1). In addition to Nox1 and Nox2, FACS-purified microglia expressed the full set of genes required for optimal activity of Nox1, including p22phox, NOXO1, NOXA1, and Rac1/2. We also detected transcripts of the genes encoding cytosolic regulatory units p47phox, p67phox, and p40phox, which associate with Nox2 in the active form of the phagocyte oxidase. To further analyze the expression of Nox genes and determine their function, we used microglia purified from primary cultures, which were prepared from the cerebral cortex of newborn wild-type (WT) or Nox2-deficient mice (Nox2-KO). RT-PCR experiments confirmed that similar to FACS-purified CX3CR1EGFP/+ cells, microglia derived from WT or Nox2-KO mice expressed transcripts for Nox1, NOXO1, NOXA1, and the genes encoding the phagocyte oxidase components. We also confirmed that neither WT nor Nox2-KO microglia expressed Nox3, Nox4, or Duox transcripts (data not shown). Real time-PCR comparisons of transcript levels in WT and Nox2-KO microglia showed that Nox2 gene inactivation had no significant effect on the expression of Nox1, NOXO1, NOXA1, and p22phox transcripts (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus microglia express in vivo and in vitro the genes encoding the components of Nox1- and Nox2-dependent NADPH oxidases.
Figure 1.

Microglia express the genes encoding Nox1- and Nox2-dependent NADPH oxidases. Ethidium bromide-stained agarose gels of reverse transcription-PCR products generated from Nox1, p22phox, NOXO1, NOXA1, Rac1, Rac2, Nox2, p40phox, p47phox, and p67phox mRNA in microglia purified by FACS from CX3CR1EGFP/+ mice or from Nox3, Nox4, Duox1, or Duox2 mRNA in FACS-purified CX3CR1EGFP/+ microglia (M), inner ear from P3 mice (IE), kidney from 16-d-old mice embryos (Kd), or adult mouse thyroid (Th). IE, Kd, and Th were from WT mice and were used as positive controls.
Distribution of Nox-associated p22phox
The association of either Nox1 or Nox2 with p22phox is required for Nox activity and stabilizes the expression of both Nox and p22phox (Parkos et al., 1989; DeLeo et al., 2000; Ambasta et al., 2004; Kawahara et al., 2005). [Nox-p22phox] heterodimers are formed independently of the enzyme activation (Bedard and Krause, 2007) and are therefore expected in unstimulated microglia. To determine the subcellular distribution of functional Nox2 and Nox1 bound to p22phox in microglial cells, we used an antibody specific for p22phox (Campion et al., 2007). In WT microglia, anti-p22phox immunoreactivity was observed at the plasma membrane and on intracellular vesicles, some of which expressed the lysosome marker LAMP1 (Fig. 2A). In Nox2-KO microglia, the plasma membrane was not stained, although p22phox was clearly detected in vesicles, including LAMP1-positive lysosomes (Fig. 2A, white arrowheads), albeit at lower levels than in WT cells. Despite the apparent lack of p22phox immunoreactivity at the plasma membrane, engulfment of opsonized zymosan by Nox2-KO microglia triggered a marked redistribution of p22phox immunoreactivity to the phagosomes (Fig. 2B). The redistribution of p22phox to phagosomes was also obvious in WT microglia engulfing zymosan (data not shown). These results strongly suggested that at least part of microglial Nox2 is localized in the plasma membrane whereas Nox1 is found in intracellular vesicular compartments including lysosomes, and can be recruited to phagosomal membranes.
Figure 2.
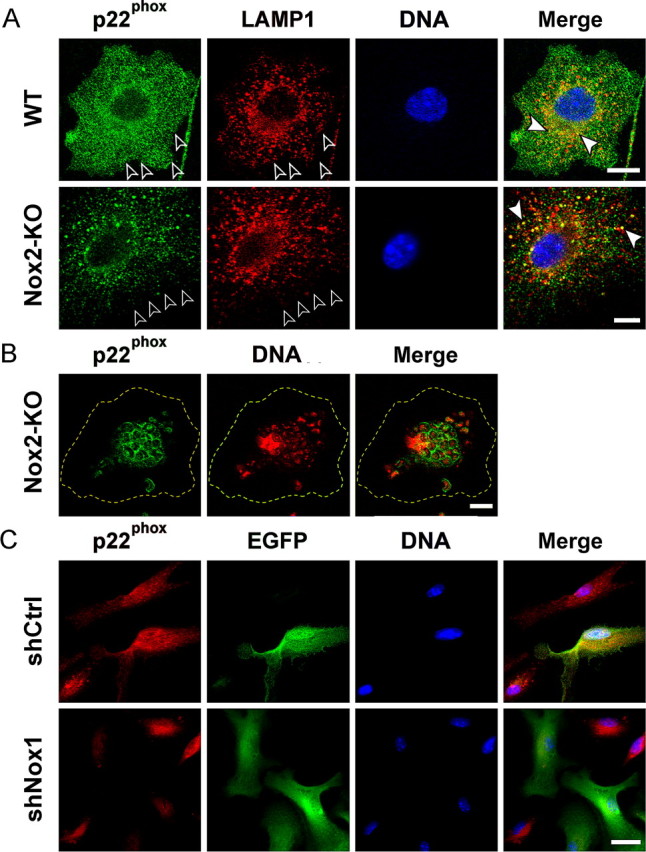
Nox1- and Nox2-dependent localization of p22phox in microglia. A, Codetection of p22phox (anti-p22phox, green), lysosomes (anti-LAMP1, red), and nuclei (Hoechst 33342, blue) in cultured WT or Nox2-KO microglia. Plasma membranes are outlined by open arrowheads. White arrowheads point to lysosomal p22phox in merged views (confocal microscopy). Scale bars, 10 μm. B, Detection of p22phox (green) and nucleic acid (Hoechst staining, blue fluorescence converted to red) in a Nox2-KO murine microglial cell after microglial phagocytosis of yeast particles (zymosan). The plasma membrane is outlined in yellow. Cultured cells were incubated with zymosan for 45 min before fixation and staining. Note the localization of p22phox in phagosome membranes surrounding stained nucleic acid of yeast particles (confocal microscopy). Scale bar, 10 μm. C, Suppression of p22phox expression in Nox2-KO microglial cells transduced with shNox1. Cultures of purified Nox2-KO microglia were transduced with lentiviral shNox1 or shCtrl and cultured for 3 d before fixation and codetection of p22phox (anti-p22phox, red) and EGFP (anti-EGFP, green). Cells transduced with lentiviral shRNA express EGFP. Scale bar, 20 μm.
Antibodies selectively reacting with murine Nox1 are currently unavailable. Therefore, to confirm that p22phox detection in Nox2-KO microglia reflected distribution of [Nox1-p22phox] heterodimers, we investigated how shRNA-based suppression of the Nox1 gene affects the distribution of p22phox. The Nox1 gene was silenced in primary microglia by transduction of the cells with a lentiviral vector encoding a shRNA specific for Nox1 (shNox1) together with an EGFP reporter gene allowing visualization of transduced cells. A lentiviral vector encoding a mutated form of shNox1 (shCtrl) and EGFP was used as a negative control (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Suppression of Nox1 gene expression was shown by the specific reduction in the level of Nox1 transcripts in WT or Nox2-KO microglial cultures transduced with shNox1 (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Lentiviral transduction did not affect microglial cell morphologies examined by phase contrast microscopy (data not shown). Consistent with the detection of p22phox stabilized through association with Nox proteins, staining of [Nox1-p22phox] heterodimers was strongly reduced in shNox1/EGFP-transduced Nox2-KO microglia but not in shCtrl-transduced cells (Fig. 2C). In WT microglia, [Nox2-p22phox] dimers were prominent, preventing clear observation of the loss of p22phox after silencing of the Nox1 gene. It is currently thought that p22phox mRNA is not limiting for p22phox protein expression but that the p22phox monomer is rapidly degraded in the cytosolic proteasome when [Nox-p22phox] heterodimers are not formed (DeLeo et al., 2000; Ambasta et al., 2004; Bedard and Krause, 2007). As expected from the loss of the p22phox resulting from increased degradation of the protein, the levels of p22phox transcripts were not reduced in shNox1-transduced microglia (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
Catalytic activity of Nox1 is stimulated by zymosan and LPS in microglia
Phagocytosis of serum-opsonized zymosan is a potent stimulator of the Nox2 phagocyte oxidase (Sankarapandi et al., 1998). However the redistribution of p22phox to the phagosomal membranes of Nox2-KO microglia suggested that Nox1 is also activated by zymosan phagocytosis and generates O2·− in the phagosome lumen. To detect O2·− generated by phagosomal membranes in Nox2-KO microglia, we used the O2·−-mediated reduction of NBT to insoluble formazan, which labels intracellular sources of O2·− (Serrander et al., 2007). Exposure of engulfing Nox2-KO microglia to NBT triggered formation of a dark precipitate which closely surrounded engulfed zymosan particles (Fig. 3A) and matched with the [Nox1-p22phox] heteromers in phagosomal membranes (Fig. 2B). To show that the phagosomal O2·− was indeed produced by Nox1, engulfing cells were exposed to the flavoprotein inhibitor diphenylene iodonium (DPI), a compound that directly blocks the catalytic activity of Nox proteins (Doussiere et al., 1999). Cell treatment with DPI (1 μm) prevented phagosome staining in Nox2-KO engulfing cells, although it did not block microglial phagocytosis of zymosan (Fig. 3A).
Figure 3.

Detection of phagosomal O2·− in Nox2-KO microglia engulfing zymosan. A, Cultured Nox2-KO microglia were incubated for 30 min with zymosan (Zym) in the absence or the presence of DPI, before addition of NBT to the culture for 15 min. TL, Transmitted light. DIC, Differential interference contrast. White and black arrowheads indicate extracellular and engulfed yeast particles, respectively. The dark blue formazan deposit overlying the phagosomal membrane is not observed in DPI-treated cells. B, Red fluorescence emitted by oxidized hydroethidine (Ox-HE) in a Nox2-KO microglial cell (same cell as in Fig. 2B) after a 45 min incubation with zymosan in the presence of cell-permeant hydroethidine. The DNA of the cell and engulfed zymosan were counterstained with Hoechst 33342 (blue fluorescence converted to green), and the cell nucleus and the plasma membrane are outlined in yellow and white, respectively. Ox-HE fluorescence is colocalized with the DNA of engulfed zymosan (confocal microscopy). Scale bars, 10 μm.
Intracellular production of O2·− in engulfing microglia was quantified at the cellular level by the O2·−-mediated oxidation of hydroethidine (Bindokas et al., 1996), which results in the formation of fluorescent products that bind to DNA. Engulfment of zymosan particles by WT microglia triggered a massive production of O2·− in the phagosome lumen as shown by the strong fluorescence of oxidized hydroethidine in zymosan DNA counterstained with Hoechst dye (Fig. 3B). Forty-five minutes after incorporation of zymosan in the WT microglial cultures, a 15-fold increase in hydroethidine oxidation was observed in cells that had engulfed at least 10 zymosan particles, compared with nonengulfing (control) microglia (Fig. 4A,B). In Nox2-KO cells, there was only a fivefold increase in hydroethidine oxidation. Cell treatment with DPI fully abrogated zymosan-stimulated production of O2·− in both WT and Nox2-KO microglia, indicating that the phagosomal O2·− was indeed produced by Nox1 and Nox2 (Fig. 4A,B).
Figure 4.
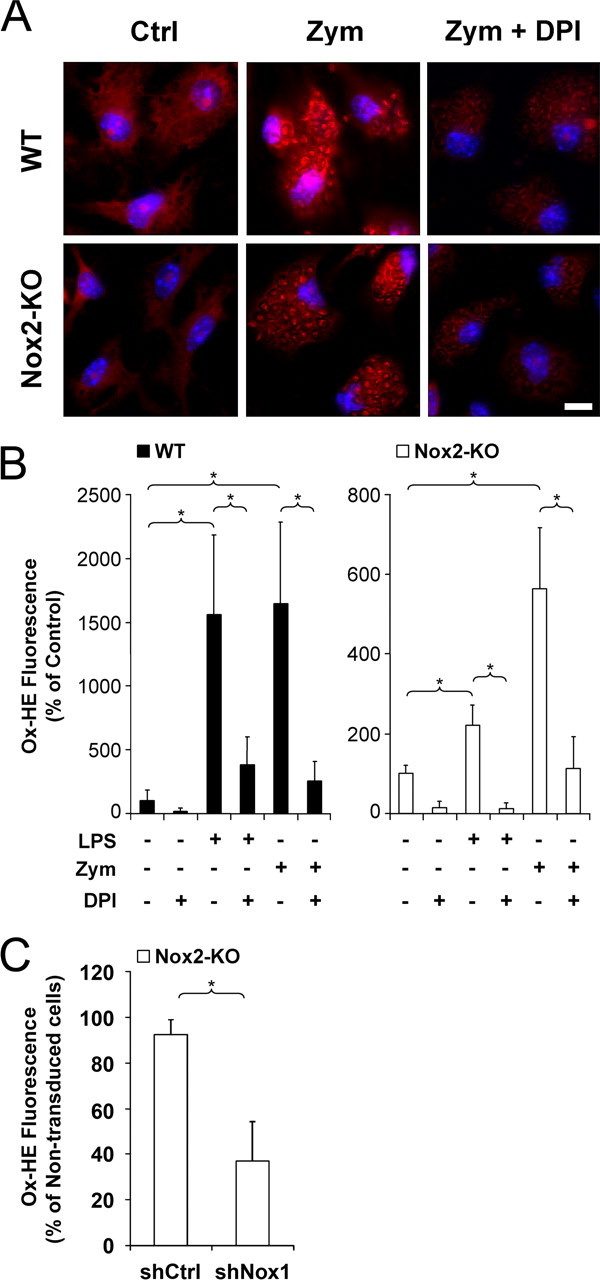
Contributions of Nox1 and Nox2 to intracellular O2·− levels in activated microglia. A, B, Cultured WT and Nox2-KO microglia were incubated for 45 min with cell-permeant hydroethidine in the absence (Ctrl) or the presence of LPS or zymosan and with or without DPI before assessment of hydroethidine oxidation. A, Red fluorescence resulting of hydroethidine oxidation in cells that have ingested zymosan. Strongly enhanced fluorescence is localized in phagosomes containing zymosan in WT or Nox2-KO cells. Fluorescence is strongly reduced when cells are incubated with DPI. Hoechst 33342 dye counterstaining (blue) reveals cell nuclei. Scale bar, 10 μm. B, Levels of oxidized hydroethidine (Ox-HE) were determined by fluorescence intensity normalized to the number of cells and are expressed as the percentage of the mean control value determined in cultures without LPS, zymosan or DPI. In zymosan-treated cultures, Ox-HE levels were determined in cells containing at least 10 zymosan particles. Data are the mean ± SD of two (Nox2-KO cells) or three (WT cells) separate experiments with 5–15 determinations in sister wells per experiment. The asterisk indicates significant differences (*p < 0.01; one-way ANOVA followed by Student-Newman–Keuls multiple-comparisons test). C, Nox2-KO cells were transduced with lentiviral shNox1 or shCtrl before incubation with hydroethidine in the presence of LPS. Levels of oxidized hydroethidine (Ox-HE) were determined in transduced (EGFP-stained) and nontransduced cells and are expressed as the percentage of the mean value of nontransduced cells in each well. A total of 600 cells were assessed in each well. The mean proportion of assessed cells expressing EGFP was 38%. Data are mean ± SD from six determinations in sister wells (*p < 0.001 in Welch's t test).
LPS has been reported to activate NADPH oxidase in different types of phagocytes, including microglia (Qin et al., 2004; Miletic et al., 2007). To investigate the role of Nox1 and Nox2, we assessed O2·− production in LPS-stimulated WT and Nox2-KO microglia. LPS (100 ng/ml) induced a 15-fold increase of hydroethidine oxidation throughout the cytoplasm of WT microglia, similar to levels reached in zymosan-stimulated cells, but only a twofold increase in Nox2-KO microglia (Fig. 4B). This shows that Nox2 plays a prominent role in LPS-triggered microglial generation of O2·−. Consistent with the activation of Nox2, DPI strongly suppressed O2·− production in LPS-stimulated WT microglia. DPI also fully prevented LPS-induction of O2·− in Nox2-KO microglia, indicating that Nox1 is also implicated in LPS-stimulated O2·− production (Fig. 4A,B).
Because Nox2 produced a high level of O2·−, the decrease in Nox1-derived O2·− after silencing of the Nox1 gene in WT microglial is difficult to visualize. However, to further confirm the role of Nox1 in LPS-induced O2·− production, Nox2-KO microglial cultures were transduced with lentiviral vectors shNox1/EGFP or shCtrl/EGFP before LPS stimulation and assessment of hydroethidine oxidation. To allow comparison of EGFP-expressing cells and nontransduced (EGFP-negative) cells in the same culture wells, O2·− production was measured in cultures in which the mean proportions of shNox1- or shCtrl-transduced cells were close to 40%. Hydroethidine oxidation was >60% lower in shNox1-transduced cell than in nontransduced cells, whereas shCtrl-transduction had no significant effect on the level of hydroethidine oxidation (Fig. 4C). LPS treatments and zymosan phagocytosis therefore both activate Nox1 and Nox2 in microglia. However, it cannot be ruled out that the level of Nox1 protein and/or the amount of O2·− produced by Nox1 might differ in WT and in Nox2-KO microglia, even though assessment of Nox1 mRNA levels did not reveal a compensatory upregulation of the Nox1 gene in Nox2-KO microglia (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Nox1 promotes microglial production of IL-1β and NO·
Recent studies have shown that Nox-derived ROS participate in redox modulation of LPS-triggered cell signaling in microglia (Pawate et al., 2004). To determine the respective roles of Nox2 and Nox1, we examined IL-1β and NO· production in WT and Nox2-KO. Microglial cultures were stimulated with LPS at a final concentration of 100 ng/ml that did not alter cell survival over a 16 h period of treatment (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We also verified by RT-PCR that in LPS-treated microglia, Nox1 (WT and Nox2-KO cells) and Nox2 (WT cells) remained expressed, whereas Nox3, Nox4, or Duox transcripts were not induced (data not shown).
LPS strongly stimulated the release of IL-1β by WT and Nox2-KO microglia. The mean levels of IL-1β secretion and their range were very similar in WT and in Nox2-KO microglia treated with LPS, indicating that Nox2 is not necessary for LPS-triggered secretion of IL-1β. (Fig. 5A) (no significant differences in nonparametric Kruskal–Wallis multiple-comparisons test followed by Dunn's test).
Figure 5.

Nox1 promotes microglial production of IL-1β. A, B, ELISA determination of IL-1β levels in the medium of WT or Nox2-KO microglial cultures treated or not with indicated reagents for 16 h. A, Dots represent mean values in eight WT and eight Nox2-KO independent experiments (5–6 determinations in sister wells per experiment). Bold lines indicate the mean value calculated from the means of the independent experiments. B, Apocynin (Apo) was used. For each genotype, data are mean ± SD of six determinations in sister wells from a representative experiment (*p < 0.001, SNK test). C, Real-time PCR determination of IL-1β mRNA levels in cultures treated for 150 min. IL-1β levels are expressed as percentage of the mean value determined in LPS-treated cultures. Data are the mean ± SD of three independent experiments with two determinations in sister wells per experiment (*p < 0.001). D, Microglial cultures transduced with lentiviral shNox1 or shCtrl were treated with LPS for 16 h before ELISA detection of IL-1β. For each genotype, data are mean ± SD of six determinations in sister wells from a representative experiment (*p < 0.001, Student's t test). Mean percentage of transduced microglia exceeds 85% for each genotype (WT or Nox2-KO).
The Nox inhibitor DPI reduced IL-1β secretion by 65% on average in WT and Nox2-KO microglia cultures stimulated with LPS (significant difference between untreated and DPI-treated cultures in each genotype; Dunn's test, p < 0.05). As for untreated cultures, there was no significant difference in IL-1β secretion between WT and Nox2-KO microglia treated with DPI (Fig. 5A). DPI also reduced the levels of IL-1β transcripts in WT and Nox2-KO microglia by >80%, assessed by real-time PCR after 150 min of LPS treatment, when the effect reaches a plateau (Fig. 5C). DPI is a potent blocker of Nox proteins but can also directly inhibit other flavoenzymes (Stuehr et al., 1991; Tew, 1993) that might possibly impact on microglial production of IL-1β. We therefore assessed the effects of apocynin, a compound that inhibits assembly of the phagocyte NADPH oxidase, and has been used to block Nox activities in different cell types, including microglia (Stolk et al., 1994; Mander et al., 2006). Similar to the effect of DPI, apocynin at the optimal concentration of 1 mm reduced IL-1β secretion by >60% in both WT and Nox2-KO microglia (Fig. 5B). Altogether, microglial treatment with NADPH oxidase inhibitors provides evidence that Nox1 activity promotes the LPS-induced IL-1β secretion at the transcriptional level or enhances the stability of IL-1β transcript.
To confirm the role of Nox1 in the induction of IL-1β, WT and Nox2-KO microglial cultures were transduced with the lentiviral vectors shNox1 and shCtrl, then stimulated with LPS for 16 h before assessment of IL-1β. Figure 5D shows the results obtained in cultures in which the mean percentage of transduced microglia was >85% and did not differ significantly between shNox1- and shCtrl-treated cells. The mean IL-1β levels in WT and Nox2-KO microglia was at least 45% lower in shNox1- than in shCtrl-transduced cultures, confirming that Nox1 promotes the production of IL-1β by microglia stimulated with LPS. Noteworthily, the magnitude of DPI-, apocynin-, or shNox1-inhibitory effects were very similar in WT and Nox2-KO microglia (Fig. 5), indicating that the loss of Nox2 does not significantly alter Nox1 regulation of IL-1β.
In contrast to IL-1β, the mean LPS-induced NO· production assessed by nitrite levels was 50% lower in Nox2-KO microglia than in WT cells (Fig. 6A) (Dunn's test, p < 0.05), indicating that Nox2 is implicated. Treatment of microglia with the NO synthase inhibitor N-monomethyl-l-arginine (500 μm) prevented NO· production but did not reduce LPS-triggered IL-1β secretion (data not shown), confirming that Nox2 regulation of NO· (Fig. 6A) can occur without an effect on IL-1β production (Fig. 5A). Apocynin significantly reduced NO· production in LPS-stimulated WT and Nox2-KO microglia (Fig. 6B). Although quite low (20–30% reduction in nitrite levels according to the experiments), this inhibitory effect suggested that not only Nox2 but also Nox1 promoted microglial production of NO·. Because LPS triggers microglial NO· production through induction of the gene encoding inducible NO synthase (iNOS) (Fiebich et al., 1998), a flavoprotein enzyme the activity of which may be directly blocked by DPI (Stuehr et al., 1991), we could not use DPI to determine directly whether Nox1 is also implicated in NO· production. We therefore compared the effect of DPI on iNOS mRNA and iNOS protein levels in LPS stimulated WT and Nox2-KO microglia. DPI strongly reduced iNOS mRNA and protein levels in both WT and Nox2-KO microglia (Fig. 6C,D). Levels of INOS protein after 6 h exposures to LPS were estimated by densitometric analysis of iNOS and β-actin bands in Western blot experiments (Fig. 6). DPI reduced the mean iNOS/β-actin ratio by >80% in both WT and in NOX2-KO microglia (iNOS/β-actin ratio in WT microglia: 7 ± 4% in DPI-treated cells, 100 ± 28% in untreated cells; NOX2-KO microglia: 15 ± 6% in DPI-treated cells, 100 ± 38% in untreated cells; values are expressed relative to the mean ratio in untreated cells set as 100% for each genotype; data are mean ± SD from three independent experiments; differences between DPI-treated and untreated cells are significant (p < 0.05 in Welch's t test). Although flavoenzymes blocked by DPI are not limited to Nox proteins, the microglial responses to DPI and apocynin treatments support the contention that Nox1 promotes iNOS expression and NO· production, at least in Nox2-KO microglia. Furthermore, silencing of Nox1 gene reduced the production of NO· by at least 50% in both WT and Nox2-KO microglial stimulated with LPS (Fig. 6E), showing that Nox1 promotes microglial production of NO· whether or not the cells express functional Nox2. To assess whether the Nox2-promoted NO· production can eventually compensate for a chronic deficiency in Nox1, we compared NO· production in LPS-treated microglia derived from WT or Nox1-KO mice. Despite the variation in absolute nitrite levels in independent experiments, NO· production was consistently lower (23–73%) in Nox1-KO microglia than in WT cells (Table 2). Together with the reduced NO· production in Nox2-KO cells (Fig. 6A), our data indicate that both Nox1 and Nox2 are required to optimize microglial production of NO·.
Figure 6.
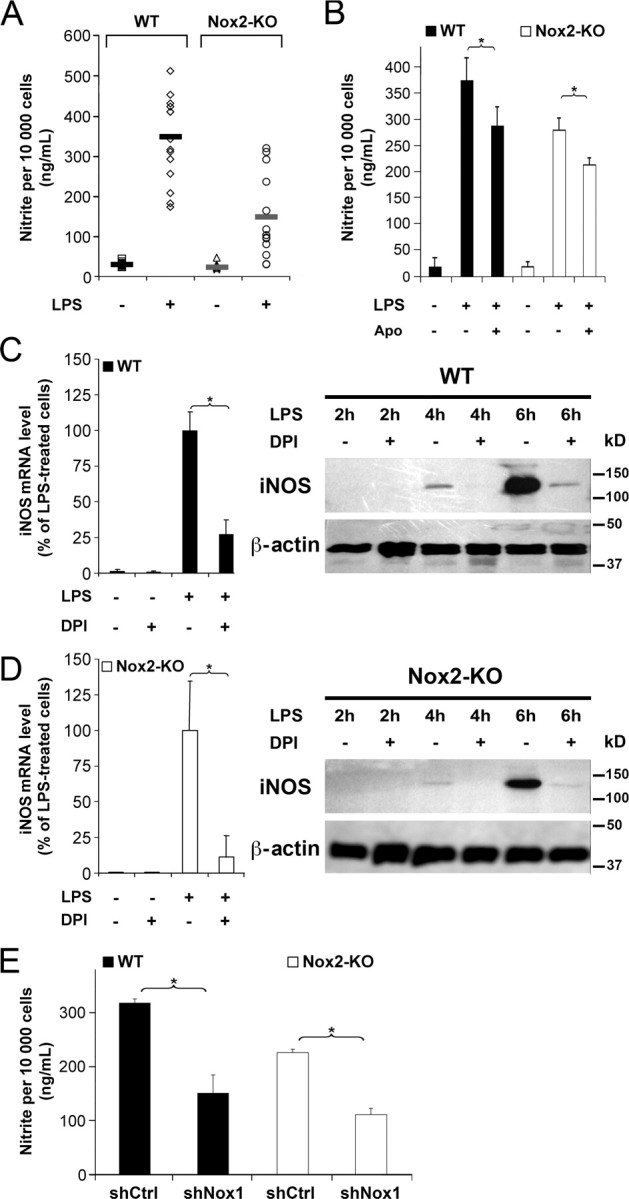
Both Nox1 and Nox2 promote microglial production of NO·. A, B, Cell production of NO· was determined by colorimetric measurement of nitrite levels in the medium of WT or Nox2-KO microglial cultures treated or not with LPS or LPS and apocynin for 16 h. A, Dots represent mean values in 15 WT and 13 Nox2-KO independent experiments (5–6 determinations in sister wells per experiment). Bold lines indicate the mean value calculated from the means of the independent experiments. B, For each genotype, data are mean ± SD of six determinations in sister wells from a representative experiment (*p < 0.01, SNK test). C, D, Real-time PCR determination of iNOS mRNA levels and Western blot detection of INOS protein in WT (C) or Nox2-KO (D) microglial cultures treated or not with LPS and/or DPI. iNOS mRNA levels were assessed in cells exposed 150 min to LPS with or without DPI and are expressed as percentage of the mean value in LPS-treated cultures; data are mean ± SD of three independent experiments with two determinations in sister wells per experiment. Images show Western blot detections of iNOS and β-actin in total protein extracts (7.5 μg/lane) of microglial cultures treated for 2–6 h with LPS in the absence or the presence of DPI. E, Microglial cultures transduced with lentiviral shNox1 or shCtrl were treated with LPS for 16 h before measurement of nitrite levels in the culture medium. For each genotype, nitrite levels are expressed as percentage of the mean value in shCtrl-transduced cultures. For each genotype (WT or Nox2-KO), data are mean ± SD of six determinations in sister wells from a representative experiment (*p < 0.001, Student's t test). Mean percentage of transduced microglia exceeds 85% for each genotype (WT or Nox2-KO).
Table 2.
Nox1-KO microglia stimulated with LPS produce less NO· than WT cells
| Exp number | Nitrite (ng/ml per 10,000 cells) |
Percentage difference | |
|---|---|---|---|
| WT | Nox1-KO | ||
| 1 | 624 ± 46 | 275 ± 32 | −56** |
| 2 | 429 ± 18 | 132 ± 7 | −69** |
| 3 | 409 ± 31 | 110 ± 12 | −73** |
| 4 | 365 ± 37 | 282 ± 19 | −23* |
In each experiment (Exp), both WT and Nox1-KO microglia were treated with LPS for 16 h before assessment of nitrite levels. Data are mean ± SD of six determinations in sister wells. Asterisks indicate significant differences between WT and Nox1-KO cells (*p < 0.01, **p < 0.001, Student's t test).
Nox1 deletion reduces tyrosine nitration and synapsin loss induced by intracerebral injection of LPS
Coactivation of NADPH oxidase and iNOS promote a reaction between O2·− and NO·, which gives rise to highly pro-oxidant peroxynitrite (ONOO−) (Szabó et al., 2007). This compound triggers the formation of nitrotyrosine residues (Ntyr), which provides a tissue marker for reactive nitrogen-related tissue damage (Torreilles et al., 1999).
The preceding experiments show that Nox1 activity generates O2·− and promotes NO· production in purified microglia. To investigate in vivo whether Nox1 promotes the intracerebral formation of deleterious nitrogen species, we injected LPS unilaterally into the striatum of adult WT or Nox1-KO mice from the same litter and we examined microglial activation and Ntyr production by immunohistochemistry in the brains fixed 4 d later. LPS strongly induced a microglial activation shown by the accumulation of amoeboid cells, especially in the vicinity of the needle track in the striatum of WT or Nox1-KO mice. High levels of Ntyr were observed at the level of the microglial reaction, but less in Nox1-KO than in WT mice. The highest levels of Ntyr were found in microglial cell bodies (Fig. 7). Ntyr was not detectable in uninjected brains or in the striatum contralateral to the injection site of WT or Nox1-KO mice. Ntyr and microglial activation were quantified in the cortical area surrounding the needle track, in which the density of activated microglia was lower than in the striatal injection site; thus individualized cells could be counted. The density of microglial cells determined by counts in microscopic fields (see methods) did not differ significantly between WT (101 ± 24 cells per field) and Nox1-KO mice (92 ± 37 cells per field), whereas the proportion of microglial cells stained with anti-Ntyr dropped from 91 ± 9% in WT brain to 47 ± 16% in Nox1-KO mice (data are mean ± SD from 5 animals of each genotype). As in the in vitro experiments, these results indicate that Nox1 promotes the formation of aggressive reactive nitrogen intermediates in activated microglia.
Figure 7.

Nox1 gene deletion reduces the level of tyrosine nitration without affecting recruitment of activated microglia in LPS-injected brain. Brains were fixed 4 d after intrastriatal injection of LPS. Top, Striatal sections in the vicinity of the injection site from WT and Nox1-KO mice double stained with rabbit polyclonal anti-Ntyr (green) and a combination of rat monoclonal anti-F4/80, anti-CD-68, and anti-CD11b antibodies that bind to microglial cells (Microglia, red). Scale bar, 100 μm. Bottom, High-power view (apotome) of a microglial cell body stained with anti-Ntyr. Hoechst staining of the nucleus (blue) is shown in the merge view. Scale bar, 10 μm.
In previous studies, LPS caused neuronal death when injected in the striatum of rats and gerbils but not mice, as indicated by silver or cresyl violet staining (Zito et al., 2001; Choi et al., 2007). In agreement with these observations, we detected very few degenerating cells by TUNEL staining of LPS-injected striatum in WT or Nox1-KO mice (data not shown). However, to further investigate the role of Nox1 in inflammation-triggered tissue damage, we looked for synapse alterations in WT and Nox1-KO mice injected with LPS by immunolabeling of synapsins, a family of presynaptic proteins linking synaptic vesicles to cytoskeleton proteins (Ferreira and Rapoport, 2002). Synapsin immunoreactivity was decreased, compared with the contralateral striatum, in the striatal region in which LPS induced both microglial and astroglial reactions (Fig. 8A,B). Quantification in the striatum 300 μm below the injection site showed a 40% decrease in synapsin immunoreactivity compared with the contralateral striatum (Fig. 8C,D). In striking contrast with the intensity of microglial and astroglial reactions, which reached the level of those observed in WT (Fig. 8A,B), there was no loss of synapsin in the striata of LPS-injected Nox1-KO mice (Fig. 8D).
Figure 8.
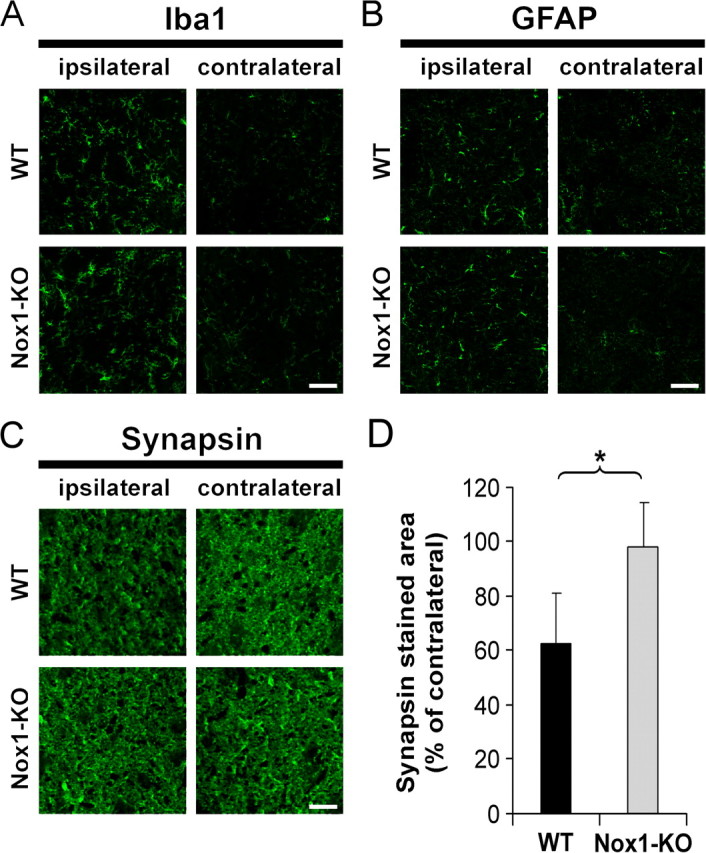
Nox1 deletion prevents loss of synapsin in LPS-injected mouse striatum. A–C, Striatal fields from WT and Nox1-KO mice stained with rabbit polyclonal anti-Iba1 (microglial staining) (A), anti-GFAP (astrocyte staining) (B), or anti-synapsin (C) ipsilateral or contralateral to the lesional tract and localized 300 μm ventral to the injection site. Scale bars, 50 μm. D, Data represent the area of synapsin staining in ipsilateral striata normalized to the contralateral region for each genotype (mean ± SD from 5 animals of each genotype, *p < 0.01, Student's t test).
Discussion
Previous studies have shown that microglia express the full set of specific proteins that compose the phagocyte oxidase (Nox2, p22phox, p47phox, p40phox, p67phox, and Rac) (Bianca et al., 1999) and release O2·− or O2·−-derived hydrogen peroxide when exposed to LPS, opsonized zymosan, or aggregated proteins that accumulate in the brain during neurodegenerative diseases (Sankarapandi et al., 1998; Zhang et al., 2005; B. Qin et al., 2006; Wilkinson et al., 2006). We have shown here, for the first time, that in addition to the phagocyte oxidase, microglia express the genes encoding all the components (Nox1, p22phox, NOXO1, NOXA1, and rac) of a nonphagocyte NADPH oxidase. This finding implicated that characterization of Nox functions in microglia required analyses of single and double Nox deficiencies. In these studies, we used pharmacological inactivation of Nox activity or specific shNox1-mediated gene silencing in combination with Nox2-KO mice to overcome the difficulty of producing double KO animals by cross-breeding given the localization of both the Nox1 and Nox2 genes on chromosome X.
Our experiments have shown that activated Nox1 catalyzes the formation of O2·− in response to engulfment of yeast particles and LPS stimulation. The O2·− generated by Nox1 favors microglial production of IL-1β, NO· and highly deleterious ONOO− triggered by LPS, and causes synaptic damage in a LPS-triggered neuroinflammatory lesion.
The distribution of p22phox in WT and Nox2-KO microglia indicates that [Nox1-p22phox] dimers are restricted to intracellular vesicles, some of which are lysosomes, whereas part of the [Nox2-p22phox] flavocytochromes are located in the plasma membrane. Because O2·− diffuses poorly through phospholipid bilayers, this result suggests that extracellular release of Nox1-derived O2·− is very limited. Accordingly, formation of extracellular O2·− was not detected in LPS-stimulated microglial cultures from Nox2-KO mice (Qin et al., 2004). However, the detection of O2·− and p22phox in microglia that have engulfed yeast particles shows that phagocytosis triggers redistribution of [Nox1-p22phox] dimers to phagosome membranes and that Nox1 significantly contributes to O2·− accumulation in phagosomes. Phagosomal Nox1 most likely originates from the fusions of lysosomes and phagosomes, whereas phagosomal Nox2 might derive at least in part from the plasma membrane incorporated in the primary phagosome.
Phagocyte oxidase is primarily involved in the immune defense against microorganisms ingested by circulating or tissue phagocytes. The O2·− produced in phagolysosomes indirectly activates proteases (Reeves et al., 2002) and gives rise to halide derivatives (Nauseef, 2007), which eventually destroy ingested bacteria or fungi. It is therefore possible that microglial Nox1 contributes to an immune defense against microorganisms that threaten the CNS (Rock et al., 2004). The cytotoxic activity of phagosomal O2·− also participates in normal development, where engulfing microglia cause neuronal apoptosis (Marín-Teva et al., 2004; Wakselman et al., 2008). Our study shows that Nox1, expressed in microglia in the developing brain, might play a key role in this mechanism of neuronal death.
In neuroinflammatory states, activated microglia may be detrimental for neural cells. LPS-activated microglia kill neurons or oligodendrocyte progenitors directly in cell culture via microglial production of IL-1β, NO·, or Nox2-derived ROS (Ma et al., 2002; Li et al., 2005). These same compounds have also been implicated in the neural tissue damage induced by injection of LPS in rodent brain (Iravani et al., 2002; Cai et al., 2003; Arai et al., 2004; Qin et al., 2004). We now show that Nox1 activity markedly promotes microglial production of NO· and IL-1β triggered by LPS. We also provide evidence for the implication of microglial Nox1 in the generation of neurotoxic ONOO− and synapse loss after intracerebral injection of LPS.
Interestingly, Nox1, but not Nox2, was implicated in microglial LPS-stimulated IL-1β production, whereas both oxidases upregulated microglial O2·− and NO· production in response to LPS. Despite this common effect on NO· production, the functions of Nox1 and Nox2 were not redundant, because microglial production of NO· was reduced by inactivating either the Nox2 or Nox1 gene. Furthermore, inhibition of Nox1 expression or activity in Nox2-KO microglia showed that O2·− generated by Nox1 are sufficient to promote iNOS gene expression and NO· production in the absence of Nox2 activity. It was previously shown with Nox inhibitors and ROS scavengers that Nox was implicated in LPS-triggered rat microglial production of NO· (Pawate et al., 2004). We verified that purified rat primary microglia, like mouse cells, express Nox1 and Nox2 (data not shown). Therefore, both of the oxidases probably regulate NO· production in rat microglia. However, a recent study suggests that cultured rat microglia express Nox4 (Harrigan et al., 2008), unlike the mouse microglia investigated here. LPS stimulation of Nox1-expressing Nox2-KO microglia was reduced by apocynin, a compound thought to act by blocking translocation of cytosolic p47phox to [Nox-p22phox] dimers (Stolk et al., 1994). This suggests that in addition to NOXO1/NOXA1-regulation, microglial Nox1 may also be activated through p47phox/p67phox binding, as documented in other cells (Bedard and Krause, 2007).
LPS signals through the toll-like receptor 4 (TLR4), which promotes LPS-triggered microglial neurotoxicity (Lehnardt et al., 2003), although this receptor is not required for LPS activation of microglial Nox2 (Qin et al., 2005). TLR4 activates multiple intracellular signaling pathways leading to transcription of genes, such as iNOS and IL-1β (O'Neill and Bowie, 2007). Nox1-dependent increase of IL-1β and iNOS transcripts might result from the oxidative modulation of a variety of redox-sensitive signaling proteins, such as MAP kinase family members, Tyrosine protein phosphatases or transcription factors such as NFκB or AP1 (Dröge, 2002; Veal et al., 2007). Although both Nox1 and Nox2 were activated by LPS, differential effects of Nox proteins in microglial expression of IL-1β and iNOS genes suggest that microglial Nox1 and Nox2 target different redox-sensitive signaling compounds, possibly because of differences in the subcellular distributions of catalytically active Nox and LPS-signaling components (Hunter, 2000). Consistent with this hypothesis, increasing evidence indicates that intracellular ROS signaling is spatially restricted by the compartmentalization of peroxide-decomposing enzymes such as catalase, glutathione peroxidase, and thioredoxin peroxidase (Veal et al., 2007).
O2·− and NO· combine at diffusion-limited rates to form highly reactive ONOO− that has been implicated in the death of cultured neurons or oligodendrocyte progenitors exposed to LPS-activated microglia (Xie et al., 2002; Li et al., 2005). Tyrosine nitration, a marker of ONOO− activity, has been correlated with tissue damages in human neuropathologies such as multiple sclerosis, ALS, and Parkinson's and Alzheimer's diseases, as well as in animal models of these diseases, including LPS-triggered neuroinflammation (Torreilles et al., 1999; Hill et al., 2004; Tomás-Camardiel et al., 2004). Although Nox2 may contribute to ONOO− formation in part through increased microglial production of NO· (Li et al., 2005; this study), we provide three lines of evidence supporting the hypothesis that microglial Nox1 plays an important role in the generation of neurotoxic ONOO−. (1) Nox1 promotes LPS-triggered expression of iNOS and NO· production in microglia. (2) Nox1 produces O2·−, which may react with NO· formed in microglia. (3) Striatal injection of LPS was followed by a much larger increase in Ntyr formation in WT than in Nox1-KO mice. Consistent with the cell autonomous regulation of microglial iNOS by Nox1, the increase in tyrosine nitration clearly occurred within activated microglia.
Cell death is very limited in LPS-injected mouse striatum. However, the extensive formation of Ntyr in microglia suggests that activated microglia may cause oxidative damage to neighboring neurons through the diffusion of ONOO−, which is known to induce structural and functional alterations of synapse components (Zaidi and Michaelis, 1999). Loss of synaptic proteins associated with microglial activation has been observed in neuroinflammatory states such as multiple sclerosis and Alzheimer's disease (Lue et al., 1996; Vercellino et al., 2007). In Alzheimer's disease, loss of synaptic proteins has been correlated with neurological decline (Terry et al., 1991), whereas both iNOS and IL-1β signaling have been implicated in the loss of synaptic proteins in mouse models of Alzheimer's disease (Craft et al., 2005; Medeiros et al., 2007). Here we show that injection of LPS into the mouse striatum resulted in a marked loss of synapsin. Although the precise mechanism of this synaptic alteration remains to be determined, we have shown that it can be prevented by neutralization of the Nox1 gene. This finding is consistent with the Nox1-enhanced microglial expression of IL-1β and iNOS and establishes that Nox1 may cause synaptic damage in a neuroinflammatory lesion. Recent studies in a mouse model of human ALS in which motoneurons progressively degenerate show that activated microglia accelerate disease progression (Boillée et al., 2006), whereas invalidation of Nox1 retards progression (Marden et al., 2007). We speculate that Nox1 may favor neural cell damage in ALS or other CNS diseases by promoting microglial production of cytotoxic effectors.
In conclusion, this study shows that in addition to the Nox2-dependent phagocyte oxidase, microglia express a Nox1 oxidase that catalyzes intracellular and intraphagosomal production of O2·−. Nox1 and Nox2 differentially promote microglial production of proinflammatory and potentially cytotoxic compounds. Activation of microglial Nox1 may therefore contribute to immune defenses, neuroregressive events during neurogenesis, and the neural tissue damage that occurs in neurodegenerative or inflammatory diseases. These issues deserve further investigation.
Footnotes
This work was supported by Inserm, Université Pierre et Marie Curie, and grants from European Leukodistrophy Association and Fédération pour la Recherche sur le Cerveau. We thank Dr. Françoise Morel for her generous gift of anti-p22phox antibodies, Dr. Chamsy Sarkis for stimulating discussion, and Drs. Merle Ruberg and Séverine Boillée for critical reading of this manuscript.
References
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J Biol Chem. 2004;279:51647–51653. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- Bánfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bianca VD, Dusi S, Bianchini E, Dal Prà I, Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pang Y, Lin S, Rhodes PG. Differential roles of tumor necrosis factor-alpha and interleukin-1 beta in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res. 2003;975:37–47. doi: 10.1016/s0006-8993(03)02545-9. [DOI] [PubMed] [Google Scholar]
- Campion Y, Paclet MH, Jesaitis AJ, Marques B, Grichine A, Berthier S, Lenormand JL, Lardy B, Stasia MJ, Morel F. New insights into the membrane topology of the phagocyte NADPH oxidase: characterization of an anti-gp91-phox conformational monoclonal antibody. Biochimie. 2007;89:1145–1158. doi: 10.1016/j.biochi.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–4968. doi: 10.1523/JNEUROSCI.5417-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi: 10.1186/1742-2094-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem. 2000;275:13986–13993. doi: 10.1074/jbc.275.18.13986. [DOI] [PubMed] [Google Scholar]
- Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–3703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci. 2002;59:589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebich BL, Butcher RD, Gebicke-Haerter PJ. Protein kinase C-mediated regulation of inducible nitric oxide synthase expression in cultured microglial cells. J Neuroimmunol. 1998;92:170–178. doi: 10.1016/s0165-5728(98)00201-x. [DOI] [PubMed] [Google Scholar]
- Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harrigan TJ, Abdullaev IF, Jourd'heuil D, Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem. 2008;106:2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KE, Zollinger LV, Watt HE, Carlson NG, Rose JW. Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: distribution, cellular expression and association with myelin damage. J Neuroimmunol. 2004;151:171–179. doi: 10.1016/j.jneuroim.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe-Nishimura C. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28:9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iravani MM, Kashefi K, Mander P, Rose S, Jenner P. Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience. 2002;110:49–58. doi: 10.1016/s0306-4522(01)00562-0. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–1088. [PubMed] [Google Scholar]
- Ma XC, Gottschall PE, Chen LT, Wiranowska M, Phelps CP. Role and mechanisms of interleukin-1 in the modulation of neurotoxicity. Neuroimmunomodulation. 2002;10:199–207. doi: 10.1159/000068322. [DOI] [PubMed] [Google Scholar]
- Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol. 2006;176:1046–1052. doi: 10.4049/jimmunol.176.2.1046. [DOI] [PubMed] [Google Scholar]
- Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, Engelhardt JF. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest. 2007;117:2913–2919. doi: 10.1172/JCI31265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Prediger RD, Passos GF, Pandolfo P, Duarte FS, Franco JL, Dafre AL, Di Giunta G, Figueiredo CP, Takahashi RN, Campos MM, Calixto JB. Connecting TNF-α signaling pathways to iNOS expression in a mouse model of Alzheimer's disease: relevance for the behavioral and synaptic deficits induced by amyloid β protein. J Neurosci. 2007;27:5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletic AV, Graham DB, Montgrain V, Fujikawa K, Kloeppel T, Brim K, Weaver B, Schreiber R, Xavier R, Swat W. Vav proteins control MyD88-dependent oxidative burst. Blood. 2007;109:3360–3368. doi: 10.1182/blood-2006-07-033662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Parkos CA, Dinauer MC, Jesaitis AJ, Orkin SH, Curnutte JT. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood. 1989;73:1416–1420. [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Philippe S, Sarkis C, Barkats M, Mammeri H, Ladroue C, Petit C, Mallet J, Serguera C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc Natl Acad Sci U S A. 2006;103:17684–17689. doi: 10.1073/pnas.0606197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2006;27:1577–1587. doi: 10.1016/j.neurobiolaging.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, Block ML. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-β peptide 1–40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys. 1998;353:312–321. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, Nathan CF. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991;5:98–103. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tew DG. Inhibition of cytochrome P450 reductase by the diphenyliodonium cation. Kinetic analysis and covalent modifications. Biochemistry. 1993;32:10209–10215. doi: 10.1021/bi00089a042. [DOI] [PubMed] [Google Scholar]
- Théry C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing neurons. Eur J Neurosci. 1991;3:1155–1164. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- Tomás-Camardiel M, Rite I, Herrera AJ, de Pablos RM, Cano J, Machado A, Venero JL. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol Dis. 2004;16:190–201. doi: 10.1016/j.nbd.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Torreilles F, Salman-Tabcheh S, Guérin M, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev. 1999;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, Mutani R, Giordana MT, Cavalla P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol. 2007;66:732–739. doi: 10.1097/nen.0b013e31812571b0. [DOI] [PubMed] [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CY, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281:20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1–42- and lipopolysaccharide-activated microglia. J Neurosci. 2002;22:3484–3492. doi: 10.1523/JNEUROSCI.22-09-03484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi A, Michaelis ML. Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free Radic Biol Med. 1999;27:810–821. doi: 10.1016/s0891-5849(99)00128-8. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- Zito MA, Koennecke LA, McAuliffe MJ, McNally B, van Rooijen N, Heyes MP. Depletion of systemic macrophages by liposome-encapsulated clodronate attenuates striatal macrophage invasion and neurodegeneration following local endotoxin infusion in gerbils. Brain Res. 2001;892:13–26. doi: 10.1016/s0006-8993(00)03135-8. [DOI] [PubMed] [Google Scholar]